

Abstract
DNA damage tolerance (DDT) mechanisms facilitate replication resumption and completion when DNA replication is blocked by bulky DNA lesions. In budding yeast, template switching (TS) via the Rad18/Rad5 pathway is a favored DDT pathway that involves usage of the sister chromatid as a template to bypass DNA lesions in an error‐free recombination‐like process. Here, we establish that the Snf2 family translocase Irc5 is a novel factor that promotes TS and averts single‐stranded DNA persistence during replication. We demonstrate that, during replication stress, Irc5 enables replication progression by assisting enrichment of cohesin complexes, recruited in an Scc2/Scc4‐dependent fashion, near blocked replication forks. This allows efficient formation of sister chromatid junctions that are crucial for error‐free DNA lesion bypass. Our results support the notion of a key role of cohesin in the completion of DNA synthesis under replication stress and reveal that the Rad18/Rad5‐mediated DDT pathway is linked to cohesin enrichment at sites of perturbed replication via the Snf2 family translocase Irc5.
Keywords: cohesin, cohesin loading, DNA damage tolerance, replication stress, template switching
Subject Categories: Cell Cycle; DNA Replication, Repair & Recombination
Introduction
Faithful chromosome duplication requires complete copying of the genetic material once per cell cycle. However, DNA replication can be challenged by many impediments, including limiting levels of nucleotides and proteins required for DNA synthesis, topological constrains, and DNA lesions that arise from both endogenous and exogenous sources. Failure to timely restart replication or usage of inappropriate repair pathways can lead to genomic instability and chromosomal rearrangements, which are hallmarks of cancer and developmental diseases (Branzei & Foiani, 2010; Zeman & Cimprich, 2014; Lee et al, 2016).
Replication fork stalling results in exposure of single‐strand DNA (ssDNA) that activates DNA damage tolerance (DDT) pathways to allow bypass of the blocking lesion and replication restart. This mechanism not only ensures completion of DNA synthesis but also prevents fork collapse as well as DNA double‐strand break (DSB) formation (Sale, 2012; Branzei & Psakhye, 2016). Regions of ssDNA, formed at replication forks or behind the forks due to repriming, are immediately coated by the replication protein A (RPA) complex that recruits the ubiquitin ligase Rad18, which, together with the ubiquitin‐conjugating enzyme Rad6, mediates proliferating cell nuclear antigen (PCNA) monoubiquitylation at Lys164. This enables translesion synthesis (TLS) polymerases to bind to PCNA and replicate across the damaged DNA template but often at the cost of fidelity (Hoege et al, 2002; Stelter & Ulrich, 2003; Bienko et al, 2005; Davies et al, 2008). However, in S phase the DDT pathway of choice seems to be a recombination‐like and error‐free process called template switching (TS) that uses the newly synthesized and undamaged sister strand as a template to bypass the DNA lesion (Zhang & Lawrence, 2005; Branzei et al, 2008; Huang et al, 2013; Giannattasio et al, 2014; Branzei & Szakal, 2016a). Activation of TS requires the ubiquitin ligase Rad5 and the ubiquitin‐conjugating complex Mms2‐Ubc13 that together mediate Lys63‐linked polyubiquitination of monoubiquitinated PCNA at Lys164 (Ulrich & Jentsch, 2000; Hoege et al, 2002). In the presence of polyubiquitinated PCNA, Rad51/Rad52‐dependent recombination‐like structures called sister chromatid junctions (SCJs) are created at the rear of replication forks that are subsequently resolved by the Sgs1/Top3/Rmi1 (STR) complex (Liberi et al, 2005; Branzei et al, 2008). Although homologous recombination (HR) at replication forks is actively prevented by the Srs2 helicase that is recruited by SUMO‐modified PCNA (Papouli et al, 2005; Pfander et al, 2005), in certain cases collapsed or broken forks can be rescued by canonical HR (Carr & Lambert, 2013; Branzei & Szakal, 2016a). However, HR‐mediated fork reactivation may lead to increased mutagenesis and chromosomal rearrangements (Deem et al, 2011; Carr & Lambert, 2013; Lambert & Carr, 2013; Mayle et al, 2015).
Recently, cohesin has emerged as an important factor that promotes the TS pathway (Tittel‐Elmer et al, 2012; Fumasoni et al, 2015; Branzei & Szakal, 2016b). Cohesin is a multiprotein complex that is composed of three essential core components: two structural maintenance of chromosome (SMC) proteins, Smc1 and Smc3, and the non‐SMC protein Scc1 (Marston, 2014). Cohesin forms a ring‐like structure that topologically entraps sister chromatids and facilitates cohesion when additional modifications of cohesin are present (Haering et al, 2008; Gligoris et al, 2014; Murayama & Uhlmann, 2014; Srinivasan et al, 2018). Under normal conditions, cohesin complexes are loaded onto chromatin in late G1/early S phase by the Scc2/Scc4 complex that is proposed to create an entry gate for the DNA (Ciosk et al, 2000; Gruber et al, 2006; Murayama & Uhlmann, 2015). The cohesin complexes are also recruited de novo to DSBs in G2/M, in a manner that depends on the Scc2/Scc4 complex and DNA damage checkpoint proteins, facilitating DNA repair by HR (Ström et al, 2004, 2007; Heidinger‐Pauli et al, 2008). Importantly, cohesin associates transiently with replication sites under normal conditions and accumulates at stalled replication forks promoting replication resumption and completion (Tittel‐Elmer et al, 2012; Frattini et al, 2017).
We have recently shown that the Snf2 family translocase Irc5 contributes to cohesin association with chromatin by facilitating Scc2/Scc4 interaction with both chromatin and cohesin under normal conditions. Interestingly, the irc5‐Δ1 mutant also shows increased sensitivity to the alkylating agent methyl methanesulfonate (MMS) that blocks replication (Litwin et al, 2017). We hypothesized that Irc5 may contribute to cohesin loading at blocked replication forks to facilitate lesion bypass and replication completion. Here, we found that during MMS exposure cells lacking Irc5 accumulate DNA damage and exhibit delay in both S phase progression and DNA replication completion. Genetic analysis suggests that Irc5 is linked to the Rad18‐Rad5‐Sgs1‐dependent error‐free DDT pathway, but acts in parallel with canonical HR and TLS. We tested this directly by showing reduction in the SCJ levels forming in the sgs1Δ mutant when Irc5 is absent. Next, we showed that Irc5 contributes to Scc2/Scc4‐mediated cohesin loading at stalled replication forks, thereby facilitating usage of the newly replicated sister chromatid during recombination‐mediated DNA damage bypass.
Results
Irc5 limits DNA damage during MMS treatment
We have previously shown that the complete deletion of the IRC5 open reading frame results in reduced expression of the essential RSC8 gene located 194 bp downstream from IRC5. As a result of this, the irc5Δ strain displays phenotypes of slow growth and increased sensitivity to DNA‐damaging agents that are not complemented by the wild‐type IRC5. To overcome this problem, we deleted the 3′ end of the IRC5 gene, containing both SNF2_N and Helic C domains, generating the irc5‐Δ1 allele. The irc5‐Δ1 mutant grew normally and exhibited wild‐type levels of RSC8 transcript. Interestingly, irc5‐Δ1 cells showed increased sensitivity to MMS (Litwin et al, 2017), so we decided to investigate which aspect(s) of the replication stress response is defective in this mutant.
As a result of replication fork stalling and bypass of DNA lesions, accumulation of ssDNA regions is observed. It triggers activation of the DNA damage checkpoint pathway leading to hyperphosphorylation of the checkpoint effector kinase Rad53 (Pellicioli et al, 1999). It has been reported that the SWI/SNF chromatin remodeling complex is important for full activation of Rad53 under hydroxyurea (HU)‐induced replication stress (Kapoor et al, 2015). On the other hand, Ino80 and Isw2 chromatin remodeling factors have been shown to limit HU‐ and MMS‐induced checkpoint activation as well as to promote checkpoint deactivation during the recovery period (Au et al, 2011; Lee et al, 2015). To examine the potential involvement of Irc5 in regulation of the DNA damage checkpoint pathway, wild‐type and irc5‐Δ1 cells were synchronized in G1 and released into medium containing 0.03% MMS to monitor the phosphorylation status of Rad53. We found no significant differences in Rad53 activation between the irc5‐Δ1 mutant and wild type suggesting that Irc5 is not required for checkpoint activation in response to MMS‐induced DNA damage (Fig 1A). Next, we investigated whether lack of Irc5 disturbs the checkpoint recovery process. To test this, G1‐synchronized cells were first exposed to 0.03% MMS for 2 h, then extensively washed, and transferred to YPD medium for recovery. In wild‐type cells, the level of Rad53 phosphorylation declined successively starting from the second hour of the recovery period. In contrast, in the irc5‐Δ1 mutant, we observed high levels of Rad53 phosphorylation throughout the experiment (Fig 1B). This observation indicates either perturbations in checkpoint deactivation or generation of higher levels of DNA damage that causes persistence of checkpoint activation.
Figure 1. Irc5 prevents DNA damage accumulation during replication stress.

- MMS‐induced checkpoint activation is not altered in irc5‐Δ1 cells. The level of Rad53 phosphorylation was analyzed in wild‐type (W303‐1a) and irc5‐Δ1 (IL012) cells by Western blot with an anti‐Rad53 antibody.
- Prolonged checkpoint activation in irc5‐Δ1 cells during recovery from MMS treatment. Rad53 phosphorylation status was analyzed in wild‐type (W303‐1a) and irc5‐Δ1 (IL012) cells by Western blot with an anti‐Rad53 antibody. M, MMS.
- Lack of Irc5 results in an elevated number of cells with Rad52‐YFP foci. At indicated time points, samples of wild‐type (W3749‐14C) and irc5‐Δ1 (TB039) cells were collected, washed, and processed for microscopic analysis. Error bars represent mean value ± standard deviations of mean (n = 5). Unpaired t‐test was used to calculate the P‐value. Representative microscopic photographs of cells are shown. Scale bars: 5 μm.
- Disruption of IRC5 leads to accumulation of ssDNA‐containing lesions. ChEC analysis of wild‐type (BYR52MN) and irc5‐Δ1 (TB040) cells cultured in the presence or absence of 0.03% MMS for 2 h (n = 3).
- Quantification of data presented in (D) (n = 3).
- Increased levels of bright Rfa1‐YFP foci in cells lacking Irc5. At indicated time points, culture samples of wild‐type (W3775‐12C) and irc5‐Δ1 (TB041) cells were collected, washed, and processed for microscopic analysis. Error bars represent mean value ± standard deviations of mean (n = 5). Unpaired t‐test was used to calculate the P‐value. Representative microscopic photographs of cells are shown. Scale bars: 5 μm.
In budding yeast, Rad52 is the main mediator protein involved in both HR and DDT (Krogh & Symington, 2004; Zhang & Lawrence, 2005; Vanoli et al, 2010). In response to DSBs and MMS‐induced DNA damage, Rad52‐YFP molecules form distinct nuclear foci, representing DNA repair centers (Lisby et al, 2001, 2003; González‐Prieto et al, 2013). If prolonged Rad53 activation was a result of improper checkpoint regulation but not accumulation of DNA damage, no increase in level of Rad52‐YFP foci in irc5‐Δ1 should be detected. To test this, we synchronized wild‐type and irc5‐Δ1 cells in G1 and transferred them to fresh medium with or without MMS. After 2 h, the cells were washed and released for recovery. The irc5‐Δ1 mutant exhibited higher incidence of Rad52‐YFP foci than the wild type both in the presence of MMS and during the recovery period (Fig 1C). These data suggest that disruption of IRC5 leads to accumulation of DNA damage and an increased requirement for Rad52‐mediated DNA repair.
It has been shown that, during replication stress, HR and DDT mutants (e.g., rad18Δ, rad51Δ, and rad52Δ) accumulate unrepaired ssDNA gaps behind the forks (Hishida et al, 2009; Hashimoto et al, 2010). To determine whether IRC5 disruption also results in accumulation of ssDNA lesions, we performed chromatin endogenous cleavage (ChEC) analysis (Schmid et al, 2004). We used a yeast strain expressing the Rad52 protein fused with the micrococcal nuclease (MN) at its C‐terminus (Rad52‐MN). Recruitment of Rad52‐MN to ssDNA‐containing structures causes Ca2+‐dependent MN activation that results in ssDNA digestion and generation of DSBs that can be monitored by electrophoresis of genomic DNA (González‐Prieto et al, 2013). Wild‐type and irc5‐Δ1 cells were arrested in G1 and transferred to medium with or without 0.03% MMS. Two hours after release, cells were collected and prepared for ChEC analysis. In the absence of Ca2+, only a single high molecular band was detected. In the presence of Ca2+, most of the DNA from untreated cells migrated as a high molecular band with some DNA smear below. Interestingly, compared to wild type, MMS treatment caused stronger DNA digestion in irc5‐Δ1 cells resulting in the high molecular band disappearing and the appearance of low molecular DNA (Fig 1D and E). These data indicate that cells lacking IRC5 accumulate ssDNA‐containing lesions in the presence of MMS.
The ssDNA regions formed during replication are coated by RPA, composed of three Rfa1‐3 subunits, and can be detected using the Rfa1‐YFP fusion protein that forms either multiple faint foci or 1–2 large, bright foci associated with replication and DNA repair, respectively (Lisby et al, 2004; Burgess et al, 2009). To confirm the data obtained with ChEC analysis, we examined Rfa1‐YFP foci during MMS treatment and the recovery period. We found that MMS exposure led to increased incidence of bright Rfa1‐YFP foci in the irc5‐Δ1 mutant compared to the wild type in both conditions (Fig 1F). This suggests that Irc5 prevents accumulation/persistence of MMS‐induced DNA damage, probably in the form of ssDNA gaps.
Irc5 ATPase activity is required for efficient completion of alkylated DNA replication
MMS causes replication fork stalling that leads to slower S phase progression (Tercero & Diffley, 2001). Moreover, proteins involved in DDT, such as Rad5, Rad51, and Rad52, are required for replication progression in the presence of MMS‐induced DNA damage (Vázquez et al, 2008; Alabert et al, 2009; Minca & Kowalski, 2010; Ortiz‐Bazán et al, 2014). To test the involvement of Irc5 in replication progression through DNA containing bulky lesions, we examined DNA synthesis in wild‐type and irc5‐Δ1 cells in the presence of MMS by FACS. No difference in cell cycle progression was observed between wild type and irc5‐Δ1 under normal conditions. In contrast, when cells were treated with MMS, the irc5‐Δ1 mutant progressed more slowly through S phase compared to the wild type (Fig 2A). Pulsed‐field gel electrophoresis (PFGE) allows separation of yeast chromosomes into a characteristic ladder of bands. However, incompletely replicated chromosomes do not enter the gel and remain in the wells (Hennessy et al, 1991). To determine the kinetics of DNA replication completion in the presence of DNA damage, wild‐type and irc5‐Δ1 cells were arrested in G1, released in the presence of 0.03% MMS for 2 h, and then allowed to recover in MMS‐free media. In both strains, exposure to MMS caused retention of chromosomes in wells because of incomplete replication. During the recovery period, however, wild‐type cells quickly resumed replication as indicated by the gradual reentry of chromosomes into the gel starting 30 min after release to fresh medium. Interestingly, chromosomes from the irc5‐Δ1 mutant were retained in the wells longer, suggesting a delay in DNA replication completion (Fig 2B and C). These data were confirmed by FACS analysis showing a slight delay in S phase progression in irc5‐Δ1 cells during recovery from MMS treatment (Fig 2D).
Figure 2. Irc5 is required for S phase progression in the presence of MMS‐induced DNA damage.
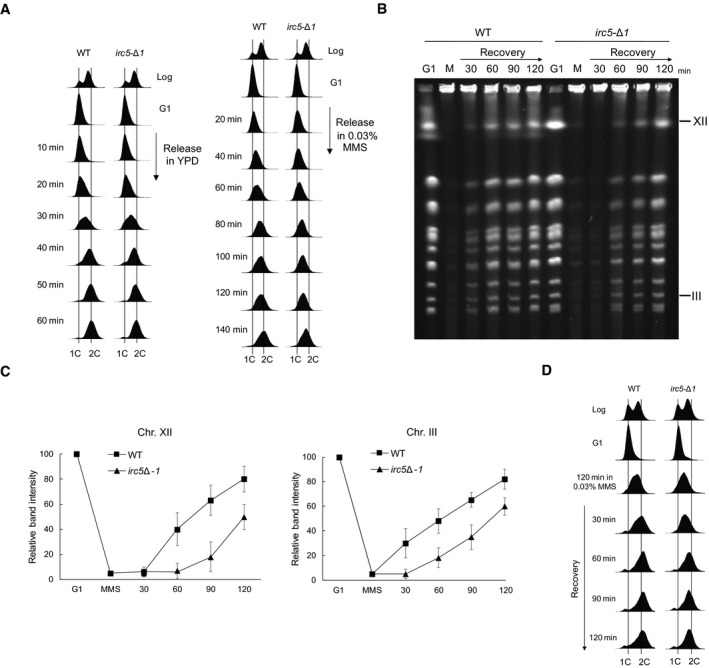
- The irc5‐Δ1 mutant exhibits S phase delay in the presence of MMS. DNA content of wild‐type (W303‐1a) and irc5‐Δ1 (IL012) cells was measured by FACS and plotted as histograms.
- Completion of DNA replication is delayed in the irc5‐Δ1 mutant after MMS treatment. Intact chromosomes were isolated from wild‐type (W303‐1a) and irc5‐Δ1 (IL012) cells and processed for PFGE. M, 0.03% MMS.
- Quantification of data presented in (B). Error bars represent mean value ± standard deviations of mean for the intensity values of three distinct chromosome bands representing chromosome XII or III (n = 3). Values for G1 were set to 100.
- Samples used for PFGE in (B) were processed for FACS analysis.
Source data are available online for this figure.
We have previously shown that irc5 DAEA cells, mutated in a conservative SNF2_N domain, are sensitive to MMS suggesting that ATP‐dependent translocase activity of Irc5 is required for MMS tolerance (Litwin et al, 2017). To evaluate the role of the Irc5 ATPase activity in the replication stress response, we first analyzed the levels of MMS‐induced Rfa1‐YFP and Rad52‐YFP nuclear foci in the irc5 DAEA mutants. Similarly to the irc5‐Δ1 mutant, irc5 DAEA cells exhibited both higher incidence of DNA repair foci during MMS exposure and foci persistence in the course of recovery (Fig 3A and B). Next, we analyzed the replication status of chromosomes in wild‐type and irc5 DAEA cells using PFGE and the conditions described above. The quantifications of chromosome reconstitution and S phase progression during recovery from MMS treatment showed that the irc5 DAEA variant behaved similarly to the irc5‐Δ1 mutant (Fig 3C–E). Taken together, these results suggest that Irc5 contributes, via its ATPase activity, to timely completion of DNA replication in the presence of MMS‐induced damage.
Figure 3. Translocase activity of Irc5 protects against DNA damage accumulation and enables replication completion.
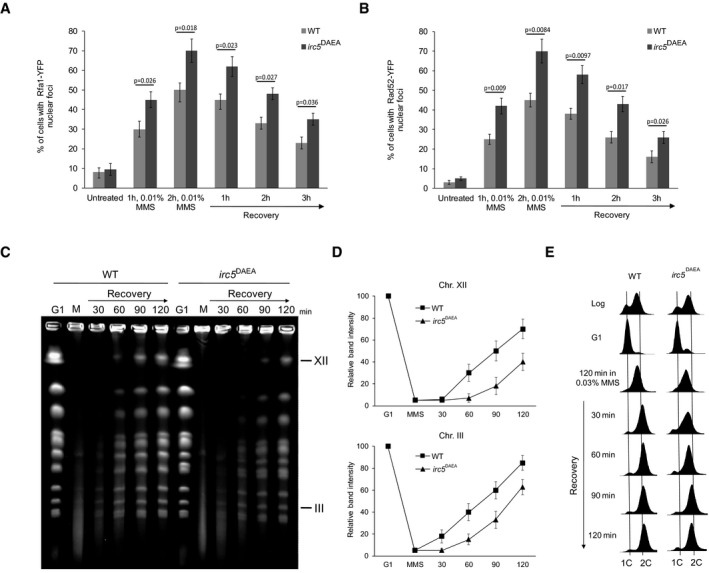
- Lack of Irc5 translocase activity results in elevated number of cells with Rfa1‐YFP foci. At indicated time points, samples of wild‐type (TB042) and irc5 DAEA (TB043) cells were collected, washed, and processed for microscopic analysis. Error bars represent mean value ± standard deviations of mean (n = 5). Unpaired t‐test was used to calculate the P‐value.
- Increased levels of bright Rad52‐YFP foci in cells expressing irc5 DAEA lacking ATPase activity. At indicated time points, samples of wild‐type (TB044) and irc5 DAEA (TB045) cells were collected, washed, and processed for microscopic analysis. Error bars represent mean value ± standard deviations of mean (n = 5). Unpaired t‐test was used to calculate the P‐value.
- Translocase activity of Irc5 is important for completion of DNA replication after MMS treatment. PFGE was performed as in Fig 2B using wild‐type (EMD09) and irc5 DAEA (EMD10) strains.
- Quantification of data presented in (C). Error bars represent mean value ± standard deviations of mean for the intensity values of three distinct chromosome bands representing chromosome XII or III (n = 3). Values for G1 were set to 100.
- Samples used for PFGE in (C) were processed for FACS analysis.
Source data are available online for this figure.
Irc5 promotes sister chromatid junction‐mediated damage bypass
DDT can be genetically divided into two pathways: HR that depends on the RAD51‐RAD52 epistasis group and the RAD6‐RAD18 pathway. The latter is further divided into the TLS branch relying on genes encoding TLS polymerases (i.e., REV3, REV7, REV1, and RAD30) and RAD5‐dependent template switching (TS) that also requires RAD52 and RAD51 (Branzei et al, 2008; Minca & Kowalski, 2010; Vanoli et al, 2010; Branzei & Szakal, 2016a). To determine to which DDT pathway IRC5 may belong to, we performed epistasis analysis between irc5‐Δ1 and gene deletions in HR and DDT pathways. Disruption of IRC5 in the rad51Δ or rad52Δ background conferred additive sensitivity to MMS indicating that Irc5 has independent functions from canonical HR (Fig 4A and Appendix Fig S1A and B). Interestingly, irc5Δ‐1 rad18Δ and irc5Δ‐1 rad5Δ double mutants were no more sensitive to MMS than rad18Δ and rad5Δ single mutants (Fig 4B and Appendix Fig S1C and D). Moreover, lack of IRC5 in rad18Δ did not cause additive accumulation of DNA repair foci (Appendix Fig S2A and B). In contrast, irc5Δ‐1 rev3Δ double mutant was more sensitive to MMS and accumulated more Rfa1‐YFP and Rad52‐YFP foci during MMS treatment than the corresponding single mutants (Fig 4B, Appendix Fig S1E, Appendix Fig S2C and D). These data suggest that IRC5 contributes to the RAD18 and RAD5‐dependent error‐free DDT.
Figure 4. Irc5 contributes to template switching.
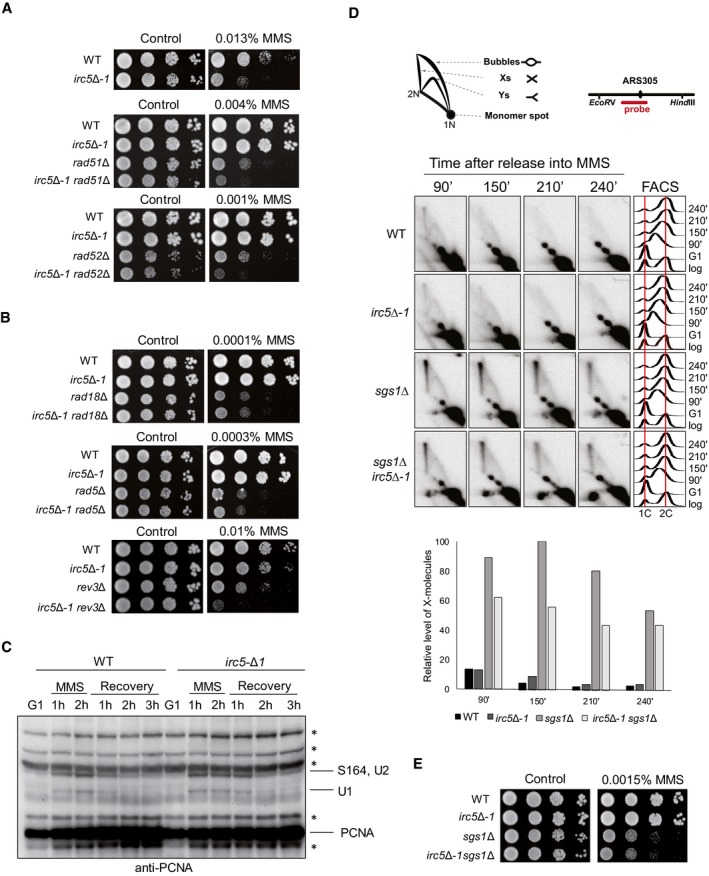
- Genetic interactions between irc5‐Δ1 and HR mutants. Logarithmically growing cultures of wild‐type (W303‐1a), irc5‐Δ1 (IL012), rad51Δ (MC002), irc5‐Δ1 rad51Δ (TB046), rad52Δ (MC006), and irc5‐Δ1 rad52Δ (TB047) strains were 10‐fold serially diluted and plated onto YPD plates with or without MMS.
- Disruption of IRC5 is epistatic to rad18Δ and rad5Δ mutations but not to rev3Δ. Logarithmically growing cultures of wild‐type (W303‐1a), irc5‐Δ1 (IL012), rad18Δ (MC019), irc5‐Δ1 rad18Δ (TB048), rad5Δ (MC018), irc5‐Δ1 rad5Δ (TB049), rev3Δ (MC021), and irc5‐Δ1 rev3Δ (TB050) strains were 10‐fold serially diluted and plated onto YPD plates with or without MMS.
- The effect of irc5Δ‐1 mutation on PCNA posttranslational modifications. PCNA and its modified forms were detected using a polyclonal anti‐PCNA antibody. Asterisks indicate cross‐reacting bands.
- Lack of Irc5 decreases formation of recombination intermediates during TS. Wild‐type (W303‐1a), irc5‐Δ1 (IL012), sgs1Δ (MC012), and irc5‐Δ1 sgs1Δ (TB051) cells were synchronized in G1 with alpha factor and released in media containing 0.03% MMS. The amount of replication intermediates at ARS305 was analyzed by 2D gel. Schematic representation of major 2D gel signals and DNA fragment analyzed is presented. Measurement of DNA content by FACS and X‐molecule quantification is displayed (n = 2).
- irc5‐Δ1 is epistatic to sgs1Δ. Wild‐type (W303‐1a), irc5‐Δ1 (IL012), sgs1Δ (MC012), and irc5‐Δ1 sgs1Δ (TB051) strains were 10‐fold serially diluted and plated onto YPD plates with or without MMS.
TS is activated by polyubiquitination of PCNA mediated by the sequential activity of Rad18 and Rad5 ubiquitin ligases (Hoege et al, 2002). Since two chromatin remodeling complexes, INO80 and RSC, have been shown to regulate DDT by promoting PCNA ubiquitination (Falbo et al, 2009; Niimi et al, 2012), we first addressed whether Irc5 modulates posttranslational modifications of PCNA. G1‐synchronized wild‐type and irc5‐Δ1 cells were released in MMS‐containing media for 2 h and then transferred to fresh medium for recovery. At indicated time points, protein samples were prepared to monitor modified PCNA species by Western blot (Fig 4C). We found that disruption of IRC5 leads to slightly higher and prolonged PCNA ubiquitination levels compared to wild type, probably as a result of increased DNA damage accumulation observed in irc5‐Δ1 (see Fig 1). Thus, in contrast to INO80 and RSC, Irc5 does not promote DDT at the level of PCNA posttranslational modifications.
The final step of TS is the Sgs1‐dependent resolution of recombination intermediates formed between sister chromatids (Liberi et al, 2005; Zhang & Lawrence, 2005; Branzei et al, 2008). In the presence of MMS, cells lacking Sgs1 accumulate X‐shaped intermediates that correspond to SCJs and can be visualized by 2D gel electrophoresis. On the other hand, mutations in genes contributing to TS decrease accumulation of X‐molecules in the sgs1Δ background (Branzei et al, 2008; Vanoli et al, 2010; Karras et al, 2013; Gonzalez‐Huici et al, 2014; Fumasoni et al, 2015). Thus, to test directly the hypothesis about the involvement of Irc5 in TS, we analyzed the accumulation of X‐molecules by monitoring the profile of replication intermediates formed during exposure to MMS at the early replicating origin ARS305 and a late region, ARS301, replicated passively from ARS305 (Fig 4D and Appendix Fig S3). In wild‐type cells, the level of X‐shaped structures was low because of their rapid dissolution mediated by the STR complex. Similar levels of X‐molecules were observed in irc5‐Δ1 cells. As expected, high accumulation of recombination intermediates was detected in sgs1Δ cells. Remarkably, disruption of IRC5 in the sgs1Δ background resulted in decreased levels of X‐molecules (Fig 4D and Appendix Fig S3). Thus, Irc5 contributes to the formation of MMS‐induced SCJs. Consistently, the irc5‐Δ1 sgs1Δ mutant was no more sensitive to MMS than sgs1Δ (Fig 4E and Appendix Fig S1F). Furthermore, disruption of IRC5 in the sgs1Δ background did not result in additive accumulation of Rfa1‐YFP and Rad52‐YFP repair foci (Appendix Fig S2E and F). Taken together, our results reveal that Irc5 is a novel factor facilitating error‐free DDT.
Irc5 is required for cohesin accumulation at stalled replication forks
We have previously reported that Irc5 contributes to cohesin deposition onto chromatin in unstressed conditions (Litwin et al, 2017). Importantly, cells expressing defective cohesin exhibit increased sensitivity to HU and MMS, delayed completion of replication, and accumulation of ssDNA gaps in the presence of MMS as well as decreased levels of X‐molecules in the sgs1Δ background (Tittel‐Elmer et al, 2012; Fumasoni et al, 2015). Moreover, cohesin‐mediated sister chromatid cohesion facilitates recombination‐mediated repair of ssDNA gaps by keeping sister chromatids in close proximity (Fumasoni et al, 2015; Branzei & Szakal, 2016b). This prompted us to investigate the potential role of Irc5 in cohesin loading onto chromatin in response to MMS‐induced replication fork stalling.
To test this, we compared association of PK‐tagged cohesin subunit Scc1 at two early firing origins, ARS305 and ARS607, by chromatin immunoprecipitation (ChIP) in wild‐type and irc5‐Δ1 cells during MMS treatment (Fig 5A and B). To monitor replisome progression under the same conditions, we also performed ChIP of HA‐tagged Pol2, the catalytic subunit of polymerase ε (Fig 5C and D). Fifteen minutes after release of G1‐synchronized cells to MMS, Pol2 started to accumulate at ARS305 and ARS607 in both strains (Fig 5C and D). At the same time point, there was no increase in Scc1 levels in wild‐type or irc5‐Δ1 cells (Fig 5A and B). Thirty minutes after release, Pol2 was highly enriched at ARS305 and ARS607 in both strains (Fig 5C and D). Interestingly, the level of Scc1 in wild‐type cells strongly increased, while in irc5‐Δ1 cells less Scc1 was detected at both early replication origins (Fig 5A and B). Starting from 45‐min time point, Pol2 levels decreased in both strains (Fig 5C and D). In contrast, 45 and 60 min after release, cohesin accumulated even more in wild‐type cells at ARS305 and ARS607 (Fig 5A and B). Similar trends of Scc1 increase at both early replication origins were observed in irc5‐Δ1 cells, but the levels of Scc1 were clearly lower than in the wild‐type (Fig 5A and B). No enrichment of Scc1 or Pol2 was detected at not actively replicated control sites, i.e., 7 kb upstream of ARS305 and the cohesin binding region of the POA1 locus. These results indicate that Irc5 is important for cohesin accumulation at stalled replication forks.
Figure 5. Irc5 promotes cohesin accumulation at stalled replication forks.
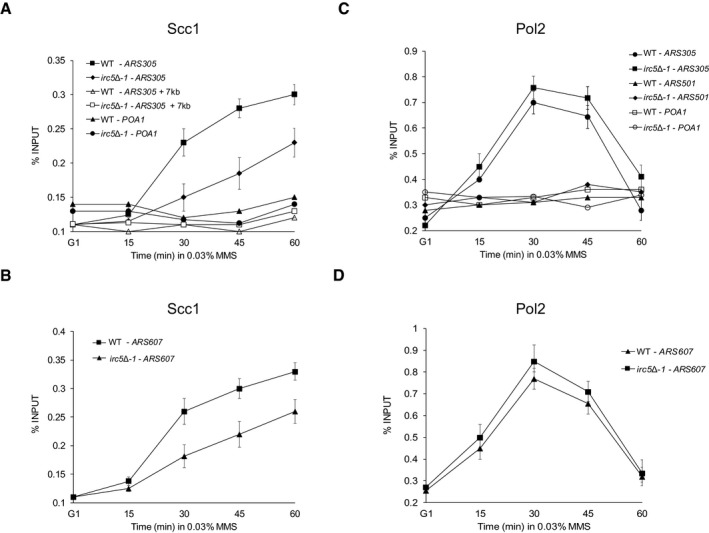
-
A, BChIP–qPCR analysis of Scc1 association with early replication origin ARS305 or ARS607. PK‐tagged Scc1 interaction with chromatin was monitored in wild‐type (JC1513) and irc5Δ‐1 (TB064) cells. Error bars represent mean value ± standard deviations of mean (n = 3). ARS305 + 7 kb locus was used as a control for cohesin‐free chromosomal region. POA1 was used as a control locus for the chromosomal region that is bound by cohesin but is located ˜30,000 bp from the nearest replication origin.
-
C, DChIP–qPCR analysis of Pol2 association with early replication origin ARS305 or ARS607 as well as late replication origin ARS501. HA‐tagged Pol2 interaction with chromatin was monitored in wild‐type (TB065) and irc5Δ‐1 (TB066) cells. Error bars represent mean value ± standard deviations of mean (n = 3).
Cohesin loading complex is required for replication completion in the presence of MMS‐induced DNA damage
Under normal conditions, the cohesin complex is loaded onto chromatin by the Scc2/Scc4 complex in late G1/early S phase (Gruber et al, 2006; Murayama & Uhlmann, 2015). Scc2/Scc4 mediates cohesin association with chromatin also in G2/M phase in response to DSBs (Ström et al, 2004). This prompted us to determine the contribution of Scc2/Scc4 to cohesin loading at early firing origins during MMS exposure. As deletion of SCC2 is lethal, we used the temperature‐sensitive allele scc2‐4 in our assay (Michaelis et al, 1997). Wild‐type and scc2‐4 cells were arrested in G1 at 25°C and then released to media containing MMS at 30°C. Samples were taken after 15, 30, 45, and 60 min to examine Scc1 and Pol2 association with ARS305 and ARS607 by ChIP. As shown before (Fig 5A and B), 30 min after release to MMS, the level of Scc1 in wild‐type cells rapidly increased reaching the maximal level at the 60‐min time point, while in scc2‐4 very little Scc1 was detected at both early replication origins throughout the time course (Fig 6A and B). As in previous experiments (Fig 5C and D), in wild‐type cells Pol2 was highly enriched at both early replication origins 30 min after release to MMS, but declined at 60 min, when the region was presumably replicated (Fig 6C and D). Even more Pol2 was recovered from scc2‐4 cells compared to wild‐type cells, and the levels remained high even at 60 min (Fig 6C and D), indicating that DNA replication is strongly impeded in scc2‐4 cells. These data show that accumulation of cohesin at stalled replication forks requires de novo loading mediated by Scc2/Scc4.
Figure 6. Cohesin loading complex allows replication completion in the presence of MMS‐induced DNA damage by facilitating cohesin association with chromatin at stalled replication forks.

-
A, BChIP–qPCR analysis of Scc1 association with early replication origin ARS305 or ARS607. HA‐tagged Scc1 interaction with chromatin was monitored in wild‐type (Y39) and scc2‐4 (Y4366) cells. Error bars represent mean value ± standard deviations of mean (n = 3).
-
C, DChIP–qPCR analysis of Pol2 association with early replication origin ARS305 or ARS607. HA‐tagged Pol2 interaction with chromatin was monitored in wild‐type (TB067) and scc2‐4 (TB068) cells. Error bars represent mean value ± standard deviations of mean (n = 3).
-
ECohesin loading complex is required for completion of chromosome replication after MMS treatment. Intact chromosomes were isolated from wild‐type (Y39) and scc2‐4 (Y4366) cells cultivated at permissive temperature (25°C) and processed for PFGE. M, MMS.
-
FQuantification of data presented in (E). Error bars represent mean value ± standard deviations of mean for the intensity values of three distinct chromosome bands representing chromosome XII or III (n = 3). Values for G1‐synchronized cells were set to 100.
-
GSensitivity of scc2‐4 to DNA‐damaging agents at permissive temperature (25°C) or semi‐permissive temperature (30°C). Logarithmically growing cultures of wild‐type (Y39) and scc2‐4 (Y4366) strains were 10‐fold serially diluted and plated onto solid YPD containing indicated concentrations of DNA‐damaging agents.
As we observed replisome stalling in the absence of functional Scc2/Scc4 complex (Fig 6C and D), it prompted us to investigate a role of Scc2/Scc4 during replication through alkylated DNA. First, we examined the dynamics of damaged chromosomal DNA replication in scc2‐4 cells by PFGE. Wild‐type and scc2‐4 cells were first synchronized in G1 and then released in medium containing 0.02% MMS at a permissive temperature for 2 h. Next, cells were extensively washed and released into fresh medium at 25°C for recovery. Under these conditions, wild‐type cells rapidly resumed replication while most of the chromosomal DNA from scc2‐4 cells remained in the wells, indicating severe defects in completing replication (Fig 6E and F). These data show that the Scc2/Scc4 complex enables replication through alkylated DNA. Next, we analyzed scc2‐4 sensitivity to DNA‐damaging agents. Under permissive conditions (25°C), scc2‐4 grew as a wild‐type strain on a control plate but already showed weak sensitivity to phleomycin (PM), camptothecin (CPT), UV, HU, and MMS (Fig 6G). In semi‐permissive conditions (30°C), scc2‐4 showed a modest growth defect on control plates, but exhibited severe sensitivity to all DNA‐damaging agents tested (Fig 6G). These data suggest that proper loading and the appropriate amount of cohesin on chromatin are crucial for cell viability in the presence of a wide range of genotoxins.
Irc5 and cohesin function in a common pathway promoting template switch
We have previously shown that under normal conditions Irc5 supports Scc2/Scc4 association with chromatin and the Scc2/Scc4 interaction with Scc1. We have also revealed that viability of cells bearing scc2‐4 allele grown at semi‐permissive temperature relies on Irc5 presence (Litwin et al, 2017). Here, we showed that both Irc5 and cohesin loading complex promote cohesin accumulation at stalled replication forks (Figs 5A and B, and 6A and B). This prompted us to investigate genetic interactions between IRC5 and SCC2 during replication stress. Disruption of IRC5 in the scc2‐4 background conferred additive sensitivity to MMS, CPT, and PM compared to the scc2‐4 single mutant (Fig 7A and Appendix Fig S4A). We also assessed levels of Rfa1‐YFP and Rad52‐YFP foci in scc2‐4 and irc5‐Δ1 scc2‐4 mutants. We found that the irc5‐Δ1 scc2‐4 mutant exhibited similar levels of DNA repair foci compared to single mutants (Fig 7B and C). These data suggest that increased sensitivity of the irc5‐Δ1 scc2‐4 mutant to MMS is not a result of additive accumulation of DNA damage. However, this also indicates that Irc5 and Scc2/Scc4 exhibit both overlapping and independent functions in response to MMS treatment.
Figure 7. Irc5 and cohesin work in a common pathway during MMS‐induced replication stress.
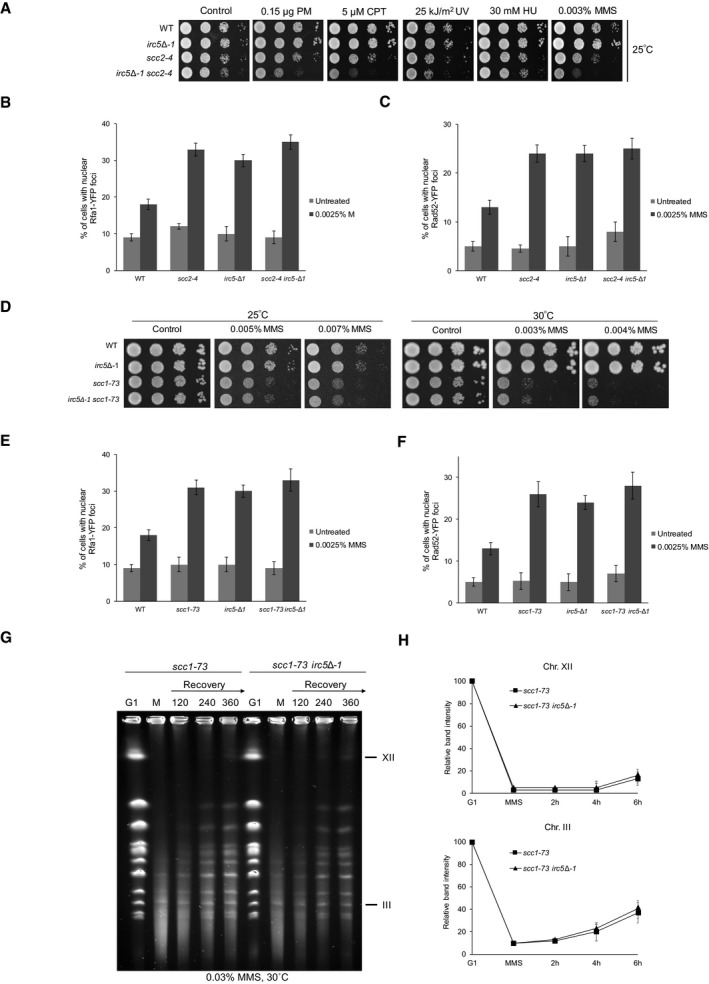
-
ASensitivity of scc2‐4 and scc2‐4 irc5‐Δ1 mutants to DNA‐damaging agents at permissive temperature 25°C. Logarithmically growing cultures of wild‐type (W303‐1a), irc5‐Δ1 (IL012), scc2‐4 (TB069), and irc5‐Δ1 scc2‐4 (TB070) strains were 10‐fold serially diluted and plated onto solid YPD containing indicated concentrations of DNA‐damaging agents.
-
B, CLevels of DNA repair foci in scc2‐4 and scc2‐4 irc5‐Δ1 mutants. Logarithmically growing cultures of scc2‐4 (TB071 or TB073) and scc2‐4 irc5‐Δ1 (TB072 or TB074) were treated with MMS for 1 h at 25°C. Next, cells were washed and processed for microscopic analysis. Error bars represent mean value ± standard deviations of mean (n = 3).
-
DSensitivity of scc1‐73 and scc1‐73 irc5‐Δ1 mutants to DNA‐damaging agents at permissive temperature 25°C or semi‐permissive temperature 30°C. Logarithmically growing cultures of wild‐type (W303‐1a), irc5‐Δ1 (IL012), scc1‐73 (JC1339), and irc5‐Δ1 scc1‐73 (TB075) strains were 10‐fold serially diluted and plated onto solid YPD containing indicated concentrations of DNA‐damaging agents.
-
E, FLevels of Rfa1‐YFP and Rad52‐YFP foci in scc1‐73 and irc5‐Δ1 scc1‐73 mutants. Logarithmically growing cultures of scc1‐73 (TB076 or TB078) and irc5‐Δ1 scc1‐73 (TB077 or TB079) were treated with MMS for 1 h at 25°C. Next, cells were washed and processed for microscopic analysis. Error bars represent mean value ± standard deviations of mean (n = 3).
-
GReplication completion defect of scc1‐73 is not exacerbated by IRC5 disruption. Intact chromosomes were isolated from scc1‐73 (JC1339) and irc5‐Δ1 scc1‐73 cells (TB075), cultured at 25°C, and processed for PFGE. M, 0.03% MMS.
-
HQuantification of data presented in (G). Error bars represent mean value ± standard deviations of mean for the intensity values of two distinct chromosome bands representing chromosome XII or III (n = 2). Values for G1 were set to 100.
Next, we addressed genetic interactions between IRC5 and SCC1, encoding the subunit of the cohesin complex. Because deletion of any of the core cohesin subunits is lethal, we used a temperature‐sensitive allele of SCC1, scc1‐73 (Michaelis et al, 1997; Haering et al, 2004). The irc5‐Δ1 scc1‐73 double mutant was viable at 30°C and no more sensitive to MMS than the scc1‐73 single mutant (Fig 7D and Appendix Fig S4B). Moreover, disruption of IRC5 had no effect on levels of Rfa1‐YFP and Rad52‐YFP foci in scc1‐73 (Fig 7E and F). Finally, we compared replication kinetics of alkylated DNA in scc1‐73 and irc5‐Δ1 scc1‐73 cells by PFGE and found that both mutants showed a similar delay in replication completion (Fig 7G and H). Taken together, these data suggest that Irc5 works in a common pathway with the cohesin loader complex and cohesin during MMS‐induced replication stress.
Having established that Irc5 is involved in the TS pathway of DDT by promoting cohesin accumulation near stalled replication forks, we decided to perform epistasis analysis between cohesin (scc2‐4, scc1‐73) and DDT (rad18Δ and rev3Δ) mutants. Both scc2‐4 rad18Δ and scc1‐73 rad18Δ double mutants were slightly more sensitive to MMS compared to rad18Δ (Fig 8A and B). In contrast, deletion of REV3 in scc2‐4 or scc1‐73 mutant strains resulted in a large decrease in cell survival after MMS treatment compared to single mutants (Fig 8C and D). Next, we examined levels of Rfa1‐YFP and Rad52‐YFP foci in scc2‐4 rad18Δ and scc1‐73 rad18Δ. Mutation of SCC1 or SCC2 in the rad18Δ background did not increase levels of Rfa1‐YFP and Rad52‐YFP foci when compared to single mutants (Fig 8E–H). In agreement with the MMS sensitivity assay, deletion of REV3 caused additive accumulation of DNA repair foci in both scc2‐4 and scc1‐73 backgrounds (Fig 8E–H). These genetic data are in line with the notion that loading of cohesin near stalled replication facilitates the error‐free pathway of DDT. However, we cannot exclude the possibility that the cohesin pathway may have functions independent of Rad18‐mediated TS that are important for cell survival during MMS treatment.
Figure 8. Genetic interactions between DDT and cohesin mutants.
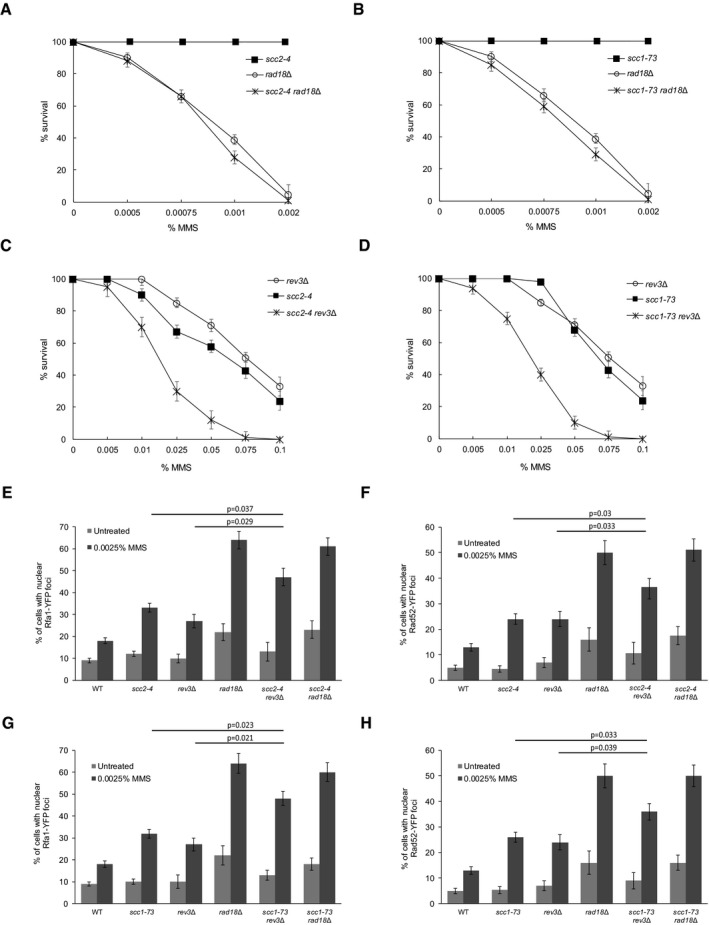
-
A–DSensitivity to acute MMS treatment. Log‐phase cultures of rad18Δ (TB058), rev3Δ (TB060), scc2‐4 (TB073), scc1‐73 (TB078), scc2‐4 rad18Δ (TB082), scc2‐4 rev3Δ (TB083), scc1‐73 rad18Δ (TB086), and scc1‐73 rev3 (TB087) were treated with indicated concentrations of MMS for 1 h. Next, cells were washed and plated on solid YPD. Error bars represent mean value ± standard deviations of mean (n = 3).
-
E–HLevels of MMS‐induced Rfa1‐YFP and Rad52‐YFP foci. Logarithmically growing cultures of rad18Δ (TB052 or TB058), rev3Δ (TB054 or TB060), scc2‐4 (TB071 or TB073), scc1‐73 (TB076 or TB078), scc2‐4 rad18Δ (TB080 or TB082), scc2‐4 rev3Δ (TB081 or TB083), scc1‐73 rad18Δ (TB084 or TB086), and scc1‐73 rev3 (TB085 or TB087) were treated with MMS for 1 h at 25°C. Next, cells were washed and processed for microscopic analysis. Error bars represent mean value ± standard deviations of mean (n = 3).
Discussion
We have previously shown that cells lacking the Snf2 family DNA translocase Irc5 exhibit increased sensitivity to MMS (Litwin et al, 2017). This prompted us to investigate the potential role of Irc5 in coping with replication stress. Here, we provided evidence that, in response to MMS‐induced replication fork stalling, Irc5 promotes DDT by TS. First, cells lacking Irc5 show accumulation of replication‐associated DNA damage and delayed replication of alkylated DNA (Figs 1 and 2). This suggests that Irc5 is required for bypass of DNA lesions and filling of ssDNA gaps. Second, our genetic data clearly indicate that Irc5 plays a role in the Rad18‐Rad5‐dependent TS pathway but not in canonical HR or TLS (Fig 4A and B and Appendix Fig S1). Moreover, simultaneous deletion of RAD18 and IRC5 did not lead to an increase in Rfa1‐YFP and Rad52‐YFP foci levels as would be expected if Irc5 and Rad18 work in two independent pathways for repair of MMS‐induced DNA damage (Appendix Fig S2A and B). Third, lack of Irc5 in the sgs1Δ background resulted in decreased levels of recombination intermediates demonstrating that Irc5 supports formation/stability of SCJs that are the essence of TS (Fig 4D and Appendix Fig S3).
The next question was why disruption of IRC5 leads to reduced levels of SCJs. Here, we showed that, in contrast to RSC or INO80 chromatin remodelers (Falbo et al, 2009; Niimi et al, 2012), Irc5 is not required for the induction of Rad18‐dependent ubiquitination of PCNA that promotes DDT (Fig 4C). However, our previous work revealed that Irc5 facilitates cohesin association with chromatin under normal conditions. Disruption of IRC5 resulted in decreased levels of chromatin‐bound cohesin at centromeres, chromosome arms, and rDNA suggesting that Irc5 contributes to cohesin loading onto chromatin throughout the genome (Litwin et al, 2017). Moreover, it has been recently shown that cohesin complexes accumulate near stalled replication forks and support formation of SCJs in the context of DDT (Tittel‐Elmer et al, 2012; Fumasoni et al, 2015; Frattini et al, 2017). Taking these results into account, we decided to determine whether Irc5 facilitates DDT through cohesin loading during replication stress.
First, we investigated whether Irc5 promotes cohesin association with chromatin at sites of replication arrest. We found that in cells lacking Irc5 cohesin complexes accumulate more slowly and to a lesser extent at stalled replication forks, demonstrating the role of Irc5 in cohesin recruitment to chromatin during replication stress (Fig 5A and B). We posit that the reduced levels of X‐shaped molecules observed in irc5‐Δ1 sgs1Δ cells result from decreased accumulation of cohesin complexes in the rear of blocked replication forks, hindering the use of the undamaged sister chromatid as a template during recombination‐mediated lesion bypass. Indeed, mutations in error‐free DDT and cohesin result in similar phenotypes during MMS exposure, such as decreased survival, S phase progression delay, defects in replication completion, accumulation of ssDNA gaps, and strong reduction or lack of SCJ formation (Chang et al, 2002; Branzei et al, 2008; Hashimoto et al, 2010; Minca & Kowalski, 2010; Tittel‐Elmer et al, 2012; González‐Prieto et al, 2013; Ortiz‐Bazán et al, 2014; Fumasoni et al, 2015). Consistent with the cohesin loading function of Irc5 in DDT, irc5‐Δ1 exhibits similar although milder defects suggesting supportive roles of Irc5 in cohesin‐mediated DDT (Figs 1, 2 and 4).
Cohesin loading is the first stage of cohesin‐mediated processes that spatially and temporally regulate cohesin distribution on chromatin (Litwin & Wysocki, 2018). Cohesin loading requires the Scc2/Scc4 complex which is crucial for cohesin ring opening and sister chromatids entrapment inside the ring (Gruber et al, 2006; Murayama & Uhlmann, 2015). The role of Scc2/Scc4 in cohesin deposition near stalled replication forks has not been investigated in detail yet. Here, we showed that inactivation of the cohesin loading complex strongly decreased cohesin accumulation at stalled replication forks resulting in DNA replication defects and hypersensitivity to DNA‐damaging agents (Fig 6). This confirms that Scc2/Scc4 activity is crucial for cohesin association with chromatin at sites of replication arrest. Previous reports showed that lack of functional cohesin results in decreased survival, defects in replication completion, accumulation of ssDNA gaps, and strong reduction or lack of SCJ formation (Tittel‐Elmer et al, 2012; Fumasoni et al, 2015; Frattini et al, 2017). These results suggest that the cohesin loader and cohesin function in a common pathway with Rad18‐mediated TS during MMS‐induced replication stress. Indeed, simultaneous disruption of RAD18 and SCC2 or SCC1 did not lead to additive accumulation of DNA damage when compared to rad18Δ. Moreover, scc2‐4 rad18Δ and scc1‐73 rad18Δ mutants were almost as sensitive to MMS as the single rad18Δ mutant (Fig 8). The slight increase in MMS sensitivity observed in double mutants likely reflects existence of some independent functions of Rad18, the cohesin loader and cohesin that are important for MMS tolerance. For example, these proteins have been shown to be involved in global gene expression regulation during DNA damage (Fu et al, 2008; Lin et al, 2011; Bose et al, 2012; Lindgren et al, 2014). Importantly, genetic analysis also showed that deletion of REV3 in the scc2‐4 or scc1‐73 background resulted in additive DNA damage accumulation and increased sensitivity to MMS (Fig 8), also suggesting that the cohesin loader and cohesin work in the error‐free DDT pathway and in parallel to translesion synthesis.
Both irc5‐Δ1 and cohesin mutants, scc2‐4 or scc1‐73, showed similar genetic interactions with DDT mutants placing the Irc5 translocase, cohesin loader, and cohesin in the TS pathway of DDT. Consequently, irc5‐Δ1 should also exhibit epistatic interactions with scc2‐4 or scc1‐73. Indeed, lack of IRC5 in the scc1‐73 background had no effect on the levels of DNA repair foci, chromosome replication kinetics, and cell survival compared to single mutants during MMS treatment (Fig 7D–H and Appendix Fig S4B). Similarly, the irc5‐Δ1 scc2‐4 double mutant showed no additive accumulation of MMS‐induced DNA repair foci (Fig 7B and C). However, it is not entirely clear why the irc5Δ‐1 mutation is additive with scc2‐4 in terms of MMS sensitivity (Fig 7A and Appendix Fig S4A). Mutations in the RSC complex, which has a well‐documented role in cohesin loading (Lopez‐Serra et al, 2014), also result in enhanced sensitivity to MMS in combination with scc2‐4 (our unpublished observations). This suggests that the cohesin loader and the RSC complex have both common and independent functions. We propose that increased sensitivity of irc5Δ‐1 scc2‐4 mutant to MMS may reflect the existence of cohesin‐independent functions of the Scc2/Scc4 complex that are important for survival under genotoxic stress. For instance, Scc2/Scc4 contributes to gene expression regulation both in the presence and absence of DNA damage (Lindgren et al, 2014). Importantly, a growing body of evidence seems to support the concept that Scc2/Scc4 can work separately from cohesin (Lindgren et al, 2014; Zuin et al, 2014; Bot et al, 2017; Rhodes et al, 2017).
Previous studies revealed that under normal conditions the Scc2/Scc4 complex is present at centromeres as well as at numerous loci along chromosome arms including telomeres and tRNA genes promoters (Lengronne et al, 2004; D'Ambrosio et al, 2008; Fernius et al, 2013; Lopez‐Serra et al, 2014). Interestingly, no Scc2/Scc4 accumulation at or in proximity to early replication origins has been reported, although cohesin accumulation at these sites was clearly demonstrated (Tittel‐Elmer et al, 2012; Frattini et al, 2017). We were also unable to confirm the presence of Scc2 at early replication origins (our unpublished observations). It is possible that fast turnover of the cohesin loading complex hinders Scc2 ChIP experiments (Fernius et al, 2013; Rhodes et al, 2017). We could not clearly demonstrate the enrichment of Irc5 at stalled replication forks either (our unpublished observations). Possibly, like the Scc2/Scc4 complex, Irc5 is highly dynamic, making its association with chromatin transient.
How the cohesin loader is recruited to chromatin is still largely unknown. It has been recently shown that the Scc2/Scc4 complex localizes to centromeres through interaction with the kinetochore protein Ctf19 phosphorylated by the Cdc7/Dbf4 kinase (DDK) (Hinshaw et al, 2017). However, mutations in the Ctf19 complex decrease cohesin levels only at pericentromeric regions and do not cause lethality suggesting that another pathway promotes Scc2/Scc4 association with chromatin (Fernius et al, 2013; Hinshaw et al, 2015). The latest research suggests that this pathway may depend on chromatin remodelers. The RSC complex was shown to recruit the cohesin loader to both centromeres and chromosome arms probably by creating nucleosome‐free regions at Scc2/Scc4 binding sites. Consequently, disruption of the ATPase subunit of the RSC complex strongly decreases the levels of chromatin‐bound Scc2 resulting in low levels of cohesin complexes associated with chromatin (Lopez‐Serra et al, 2014). Importantly, we have previously reported that Irc5 enables efficient association of the cohesin loading complex with chromatin, allowing productive interaction between the cohesin loader and cohesin (Litwin et al, 2017). How Irc5 enables Scc2/Scc4 interaction with chromatin is not yet known. We have previously shown that ATPase activity of Irc5 is required for a proper level of cohesin on chromatin and viability of the thermosensitive scc2‐4 mutant. We have also demonstrated that irc5 DAEA cells fail to complement irc5Δ‐1 sensitivity to MMS (Litwin et al, 2017). Here, we show that ATP‐dependent translocase activity of Irc5 is also important for completion of DNA replication under MMS stress (Fig 3). We favor the hypothesis that, similar to other Snf2 family proteins, Irc5 may modulate the chromatin environment allowing optimal Scc2/Scc4 binding to chromatin and efficient cohesin loading/binding. What attracts Irc5 to stalled replication forks and the targets for Irc5 translocase activity are remain to be established.
During each cell cycle, replication forks encounter various impediments that obstruct DNA synthesis. Replication fork arrest causes prolonged exposure of ssDNA that is very susceptible to various chemical mutagenic modifications such as deamination, depurination, depyrimidination, or alkylation (Billen, 1990; Lindahl, 1993; Fu et al, 2012). Moreover, replication fork stalling opens up a risk of replication fork collapse that may lead to generation of the most deleterious DNA double‐strand breaks (Zeman & Cimprich, 2014). DDT enables bypass of DNA obstacles allowing restart and completion of DNA replication and therefore disruption of these pathways leads to increased mutagenesis or chromosomal rearrangements that are present in many cancers (Tateishi et al, 2003; Unk et al, 2006, 2008; Alexandrov et al, 2013; Lambert & Carr, 2013). Interestingly, mutations in the SNF2_N or Helic C domain of human Irc5 homolog LSH/HELLS are found in acute myelogenous and lymphoblastic leukemia cells (Lee et al, 2000). Moreover, mutations in cohesin subunits and cohesin regulatory proteins have been found in several cancers (Losada, 2014). It would be interesting to find out whether HELLS is involved in cohesin and DDT regulation in human cells.
Materials and Methods
Yeast strains and growing conditions
Yeast strains used in this study are described in Appendix Table S1. Yeast cells were cultured at 28°C unless stated otherwise. Gene deletion or tagging was performed using a PCR‐based method (Longtine et al, 1998). Multiple mutants were obtained by crossing of relevant haploids followed by diploid dissection. To integrate IRC5‐3HA or irc5 DAEA ‐3HA into the ura3‐1 locus, pRS306 plasmids containing one of the alleles were linearized and transformed into the irc5Δ‐1 background. The resulting integrants were verified by PCR. To assess the sensitivity of relevant strains to genotoxins, mid‐log cultures were 10‐fold serially diluted and spotted on solid media containing indicated concentrations of DNA‐damaging agents. To determine the survival rate of indicated mutant strains after acute MMS treatment, cells were exposed to various concentrations of MMS for 60 min or left untreated, extensively washed, and diluted before plating on YPD medium.
Cell cycle analysis
To synchronize cells in G1 phase mid‐log, cells were cultured for 2 h in the presence of 5 μM α‐factor. G1 block was confirmed by microscopic observations of unbudded cells showing shmoo projections. To measure DNA content, cell samples were collected and fixed with 70% ethanol. Next, cells were washed with water, digested with 0.25 mg/ml RNase for 2 h at 50°C and with 5 mg/ml pepsin for 1 h at 37°C, and sonicated and stained with 1 μM SYTOX Green (Life Technologies) for 30 min before FACS analysis.
Live cell imaging
To assess levels of Rfa1‐YFP and Rad52‐YFP nuclear foci, live cells were observed with an Axio Imager M1 epifluorescence microscope (Carl Zeiss, Germany) equipped with a 100× immersion oil objective (Plan‐Neofluar 1006/1.30), a GFP filter set and differential interference contrast (DIC). Images were collected using an AxioCam MRc digital color camera and processed with AxioVision 4.5 software. Image acquisition times for Rad52‐YFP and Rfa1‐YFP were 400 and 450 ms, respectively.
Chromatin endogenous cleavage (ChEC)
ChEC analysis was performed as described by González‐Prieto et al (2013). Briefly, 108 cells were arrested with 0.1% sodium azide and then permeabilized with 1% digitonin for 5 min. Next, to induce micrococcal nuclease, cells were incubated with 2 mM CaCl2 at 30°C for 30 min. Equal concentrations of total DNA were loaded and resolved on 0.8% agarose gel and stained with ethidium bromide. The DNA intensity in each line was quantified with the Bio‐Rad ChemiDoc MP System and plotted on the histograms with Image Lab software (Bio‐Rad). Each experiment was repeated three times with similar results.
Pulsed‐field gel electrophoresis (PFGE)
Preparation of agarose‐embedded genomic DNA was performed with the CHEF Genomic DNA Plug Kit (Bio‐Rad). To assure equal amount of DNA in the agarose plugs, at each time point, yeast cultures samples were taken, and the number of cells was determined with a hemacytometer and adjusted to 3 × 107 cells per sample. Cells were then embedded in 50 μl of 0.8% low‐melting agarose and digested with 1 mg/ml lyticase for 3 h at 37°C followed by overnight incubation with 1 mg/ml proteinase K at 50°C. Next, plugs were washed four times with washing buffer (20 mM Tris, pH 8, 50 mM EDTA) and loaded onto gel. Electrophoresis was performed using CHEF‐DR III Pulsed Field Electrophoresis Systems (Bio‐Rad). Chromosome samples were resolved in 1% agarose at 6 V/cm for 22 h with a 1‐ to 2‐min switch time ramp and 120° switch angle at 14°C. Afterward, the electrophoresis gel was stained with ethidium bromide. Signal detection and quantification were performed using a Bio‐Rad ChemiDoc MP System and Image Lab software. For quantification, the chromosome III and XII intensities after MMS treatment and during recovery were correlated with intensities obtained for the same chromosomes in G1.
Western blot analysis
To evaluate the level of Rad53 phosphorylation, at each time point 5 × 107 cells were collected and total proteins were prepared by the NaOH/TCA method. Protein extracts were resolved on 8% SDS–PAGE, blotted onto nitrocellulose membranes, and probed with anti‐Rad53 antibody (Santa Cruz, Sc6749). Anti‐G‐6‐PDH (Sigma‐Aldrich, A9521‐1VL) antibody was used to detect the level of glucose‐6‐phosphate dehydrogenase serving as a loading control. To detect PCNA and its modified forms, proteins were extracted as above and resolved on 12% SDS–PAGE, blotted onto nitrocellulose membranes, and probed with rabbit polyclonal anti‐PCNA antibody (Zhang et al, 2000). Chemiluminescence signal detection was performed using the Bio‐Rad ChemiDoc MP System and Image Lab software.
Analysis of replication intermediates
Purification of DNA intermediates and 2D gel analysis were carried out as previously described (Vanoli et al, 2010). Each experiment shown was performed independently at least twice with qualitatively identical results. Genome preparation and signal quantification methods were previously described in detail (Fumasoni et al, 2015). Briefly, quantification of X‐shaped intermediate signals was performed using the Image Quant software (GE Healthcare) as previously described (Vanoli et al, 2010). For each time point, areas corresponding to the monomer spot (M), the X‐signal, and a region without any replication intermediates as a background reference were selected and the signal intensities (SI) in percentage of each signal were obtained. The values for X and the monomer were then corrected by subtracting from the SI value the background value after the latter was multiplied for the ratio between the dimension of the area for the intermediate of interest and for the background. The relative signal intensity for X was determined by dividing the value for X with the sum of the X and monomer values. The resulting values for X signals were then normalized and converted to percentages by using the highest value number of X for each experiment as 100 and normalizing the other values to it.
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed as described by Litwin et al (2017) with minor modifications. 4 × 108 cells were cross‐linked with 1% formaldehyde for 30 min followed by 5‐min incubation with 150 mM glycine. Cells were washed twice with ice‐cold TBS and resuspended in 500 μl of FA lysis buffer (50 mM HEPES–KOH pH 7.5, 140 mM NaCl, 0.5% Triton X‐100, 0.1% sodium deoxycholate, 1 mM EDTA) containing 1 mM PMSF and protease inhibitors (Sigma‐Aldrich, P8215). Next, cells were lysed with glass beads in a bead beater at 4°C. Cell lysates were transferred to new tubes and sonicated to yield an average DNA size of 500 bp followed by centrifugation. Cleared lysates were transferred to new tubes, and an additional 500 μl of FA lysis buffer was added. For IP reactions, 4 μg of anti‐HA (Roche, 12CA5) or anti‐Pk (Serotec, SV5‐Pk1) antibody was added to 500 μl of chromatin lysates and incubated at 4°C overnight. Next, DNA–protein complexes were captured with Protein G Dynabeads (Invitrogen) and sequentially washed with FA lysis buffer, FA‐500 buffer (50 mM HEPES–KOH pH 7.5, 500 mM NaCl, 1% Triton X‐100, 0.1% sodium deoxycholate, 1 mM EDTA), LiCl wash buffer (10 mM Tris–HCl pH 8.0, 250 mM LiCl, 0.5% NP‐40, 0.5% sodium deoxycholate, 1 mM EDTA), and TE (10 mM Tris–HCl pH 7.5, 1 mM EDTA) on a rotator for 5 min each time. Complexes were then eluted with Elution Buffer (50 mM Tris–HCl pH 7.5, 1% sodium dodecylsulfate, 10 mM EDTA) by incubation at 65°C for 10 min. For input samples, 10 μl of cleared chromatin lysate was diluted in 490 μl of TE buffer and together with IP samples treated with 0.2 mg/ml proteinase K for at least 4 h at 42°C followed by overnight incubation at 65°C. All samples were phenol–chloroform purified and ethanol precipitated in the presence of 20 μg/ml of glycogen. qPCRs were performed using both IP and input samples as templates, a 2xPCR Master Mix SYBR Kit (A&A Biotechnology), and the LightCycler 480 System (Roche Applied Science) in a total volume of 15 μl. Primers used for qPCR are listed in Appendix Table S2. Amplifications conditions were as follows: 1 min at 95°C; 45 cycles of 10 s at 95°C, 10 s at 56°C, and 22 s at 72°C. The percentage (% input) value for each sample was calculated according to the formula: ΔCT [normalized ChIP] = CT [ChIP] − {CT [Input] − log2 (dilution factor)} and Input % = 100/2ΔCT [normalized ChIP]. The % input value represents the enrichment of protein at the specific locus and is normalized to the ACT1 reference gene. All ChIP experiments were performed at least three times. qPCRs were performed two times for each sample.
Author contributions
The experiments were designed by IL, RW, and DB, and executed by IL, TB, EP, and EM‐D. BS performed 2D gel electrophoresis and analysis of replication intermediates. IL, RW, and DB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Source Data for Figure 2C
Source Data for Figure 3D
Acknowledgements
We thank J Cobb, F Prado, R Rothstein, and F Uhlmann for providing strains. This work was supported by the National Science Centre (Poland) (2013/11/D/NZ2/02696) to IL and the Italian Association for Cancer Research (IG 18976) and the European Research Council Consolidator Grant (682190) to DB.
The EMBO Journal (2018) 37: e98732
References
- Alabert C, Bianco JN, Pasero P (2009) Differential regulation of homologous recombination at DNA breaks and replication forks by the Mrc1 branch of the S‐phase checkpoint. EMBO J 28: 1131–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik‐Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen‐Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjord JE, Foekens JA, Greaves M et al (2013) Signatures of mutational processes in human cancer. Nature 500: 415–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au TJ, Rodriguez J, Vincent JA, Tsukiyama T (2011) ATP‐dependent chromatin remodeling factors tune S phase checkpoint activity. Mol Cell Biol 31: 4454–4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienko M, Green CM, Crosetto N, Rudolf F, Zapart G, Coull B, Kannouche P, Wider G, Peter M, Lehmann AR, Hofmann K, Dikic I (2005) Ubiquitin‐binding domains in Y‐family polymerases regulate translesion synthesis. Science 310: 1821–1824 [DOI] [PubMed] [Google Scholar]
- Billen D (1990) Spontaneous DNA damage and its significance for the “negligible dose” controversy in radiation protection. Radiat Res 124: 242–245 [PubMed] [Google Scholar]
- Bose T, Lee KK, Lu S, Xu B, Harris B, Slaughter B, Unruh J, Garrett A, McDowell W, Box A, Li H, Peak A, Ramachandran S, Seidel C, Gerton JL (2012) Cohesin proteins promote ribosomal RNA production and protein translation in yeast and human cells. PLoS Genet 8: e1002749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bot C, Pfeiffer A, Giordano F, Manjeera DE, Dantuma NP, Ström L (2017) Independent mechanisms recruit the cohesin loader protein NIPBL to sites of DNA damage. J Cell Sci 130: 1134–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Vanoli F, Foiani M (2008) SUMOylation regulates Rad18‐mediated template switch. Nature 456: 915–920 [DOI] [PubMed] [Google Scholar]
- Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11: 208–219 [DOI] [PubMed] [Google Scholar]
- Branzei D, Psakhye I (2016) DNA damage tolerance. Curr Opin Cell Biol 40: 137–144 [DOI] [PubMed] [Google Scholar]
- Branzei D, Szakal B (2016a) DNA damage tolerance by recombination: molecular pathways and DNA structures. DNA Repair 44: 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Szakal B (2016b) Priming for tolerance and cohesion at replication forks. Nucleus 7: 8–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess RC, Lisby M, Altmannova V, Krejci L, Sung P, Rothstein R (2009) Localization of recombination proteins and Srs2 reveals anti‐recombinase function in vivo . J Cell Biol 185: 969–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr AM, Lambert S (2013) Replication stress‐induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol 425: 4733–4744 [DOI] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW (2002) A genome‐wide screen for methyl methanesulfonate‐sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci USA 99: 16934–16939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciosk R, Shirayama M, Shevchenko A, Tanaka T, Toth A, Shevchenko A, Nasmyth K (2000) Cohesin's binding to chromosomes depends on a separate complex consisting of Scc2 and Scc4 proteins. Mol Cell 5: 243–254 [DOI] [PubMed] [Google Scholar]
- D'Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, Uhlmann F (2008) Identification of cis‐acting sites for condensin loading onto budding yeast chromosomes. Genes Dev 22: 2215–2227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD (2008) Activation of ubiquitin‐dependent DNA damage bypass is mediated by replication protein a. Mol Cell 14: 625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A (2011) Break‐induced replication is highly inaccurate. PLoS Biol 9: e1000594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falbo KB, Alabert C, Katou Y, Wu S, Han J, Wehr T, Xiao J, He X, Zhang Z, Shi Y, Shirahige K, Pasero P, Shen X (2009) Involvement of a chromatin remodeling complex in damage tolerance during DNA replication. Nat Struct Mol Biol 16: 1167–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernius J, Nerusheva OO, Galander S, Alves Fde L, Rappsilber J, Marston AL (2013) Cohesin‐dependent association of Scc2/4 with the centromere initiates pericentromeric cohesion establishment. Curr Biol 23: 599–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini C, Villa‐Hernández S, Pellicanò G, Jossen R, Katou Y, Shirahige K, Bermejo R (2017) Cohesin ubiquitylation and mobilization facilitate stalled replication fork dynamics. Mol Cell 68: 758–772 [DOI] [PubMed] [Google Scholar]
- Fu Y, Zhu Y, Zhang K, Yeung M, Durocher D, Xiao W (2008) Rad6‐Rad18 mediates a eukaryotic SOS response by ubiquitinating the 9‐1‐1 checkpoint clamp. Cell 133: 601–611 [DOI] [PubMed] [Google Scholar]
- Fu D, Calvo JA, Samson LD (2012) Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer 12: 104–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumasoni M, Zwicky K, Vanoli F, Lopes M, Branzei D (2015) Error‐free DNA damage tolerance and sister chromatid proximity during DNA replication rely on the Polα/Primase/Ctf4 Complex. Mol Cell 57: 812–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannattasio M, Zwicky K, Follonier C, Foiani M, Lopes M, Branzei D (2014) Visualization of recombination‐mediated damage bypass by template switching. Nat Struct Mol Biol 21: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligoris TG, Scheinost JC, Bürmann F, Petela N, Chan KL, Uluocak P, Beckouët F, Gruber S, Nasmyth K, Löwe J (2014) Closing the cohesin ring: structure and function of its Smc3‐kleisin interface. Science 346: 963–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Huici V, Szakal B, Urulangodi M, Psakhye I, Castellucci F, Menolfi D, Rajakumara E, Fumasoni M, Bermejo R, Jentsch S, Branzei D (2014) DNA bending facilitates the error‐free DNA damage tolerance pathway and upholds genome integrity. EMBO J 33: 327–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Prieto R, Muñoz‐Cabello AM, Cabello‐Lobato MJ, Prado F (2013) Rad51 replication fork recruitment is required for DNA damage tolerance. EMBO J 32: 1307–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber S, Arumugam P, Katou Y, Kuglitsch D, Helmhart W, Shirahige K, Nasmyth K (2006) Evidence that loading of cohesin onto chromosomes involves opening of its SMC hinge. Cell 127: 523–537 [DOI] [PubMed] [Google Scholar]
- Haering CH, Schoffnegger D, Nishino T, Helmhart W, Nasmyth K, Löwe J (2004) Structure and stability of cohesin's Smc1‐kleisin interaction. Mol Cell 15: 951–964 [DOI] [PubMed] [Google Scholar]
- Haering CH, Farcas AM, Arumugam P, Metson J, Nasmyth K (2008) The cohesin ring concatenates sister DNA molecules. Nature 454: 297–301 [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidinger‐Pauli JM, Unal E, Guacci V, Koshland D (2008) The kleisin subunit of cohesin dictates damage‐induced cohesion. Mol Cell 31: 47–56 [DOI] [PubMed] [Google Scholar]
- Hennessy KM, Lee A, Chen E, Botstein D (1991) A group of interacting yeast DNA replication genes. Genes Dev 5: 958–969 [DOI] [PubMed] [Google Scholar]
- Hinshaw SM, Makrantoni V, Kerr A, Marston AL, Harrison SC (2015) Structural evidence for Scc4‐dependent localization of cohesin loading. Elife 4: e06057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinshaw SM, Makrantoni V, Harrison SC, Marston AL (2017) The kinetochore receptor for the cohesin loading complex. Cell 171: 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hishida T, Kubota Y, Carr AM, Iwasaki H (2009) RAD6‐RAD18‐RAD5‐pathway‐dependent tolerance to chronic low‐dose ultraviolet light. Nature 457: 612–615 [DOI] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6‐dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419: 135–141 [DOI] [PubMed] [Google Scholar]
- Huang D, Piening BD, Paulovich AG (2013) The preference for error‐free or error‐prone postreplication repair in Saccharomyces cerevisiae exposed to low‐dose methyl methanesulfonate is cell cycle dependent. Mol Cell Biol 33: 1515–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor P, Bao Y, Xiao J, Luo J, Shen J, Persinger J, Peng G, Ranish J, Bartholomew B, Shen X (2015) Regulation of Mec1 kinase activity by the SWI/SNF chromatin remodeling complex. Genes Dev 29: 591–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras GI, Fumasoni M, Sienski G, Vanoli F, Branzei D, Jentsch S (2013) Noncanonical role of the 9‐1‐1 clamp in the error‐free DNA damage tolerance pathway. Mol Cell 49: 536–546 [DOI] [PubMed] [Google Scholar]
- Krogh BO, Symington LS (2004) Recombination proteins in yeast. Annu Rev Genet 38: 233–271 [DOI] [PubMed] [Google Scholar]
- Lambert S, Carr AM (2013) Replication stress and genome rearrangements: lessons from yeast models. Curr Opin Genet Dev 23: 132–139 [DOI] [PubMed] [Google Scholar]
- Lee DW, Zhang K, Ning ZQ, Raabe EH, Tintner S, Wieland R, Wilkins BJ, Kim JM, Blough RI, Arceci RJ (2000) Proliferation‐associated SNF2‐like gene (PASG): a SNF2 family member altered in leukemia. Cancer Res 60: 3612–3622 [PubMed] [Google Scholar]
- Lee L, Rodriguez J, Tsukiyama T (2015) Chromatin remodeling factors Isw2 and Ino80 regulate checkpoint activity and chromatin structure in S phase. Genetics 199: 1077–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Choi YL, Kwon M, Park PJ (2016) Mechanisms and consequences of cancer genome instability: lessons from genome sequencing studies. Annu Rev Pathol 11: 283–312 [DOI] [PubMed] [Google Scholar]
- Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, Uhlmann F (2004) Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430: 573–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta‐Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51‐dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Wang M, Jin H, Yu HG (2011) Cohesin plays a dual role in gene regulation and sister‐chromatid cohesion during meiosis in Saccharomyces cerevisiae . Genetics 187: 1041–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T (1993) Instability and decay of the primary structure of DNA. Nature 362: 709–715 [DOI] [PubMed] [Google Scholar]
- Lindgren E, Hägg S, Giordano F, Björkegren J, Ström L (2014) Inactivation of the budding yeast cohesin loader Scc2 alters gene expression both globally and in response to a single DNA double strand break. Cell Cycle 13: 3645–3658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA 98: 8276–8282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Mortensen UH, Rothstein R (2003) Colocalization of multiple DNA double‐strand breaks at a single Rad52 repair centre. Nat Cell Biol 5: 572–577 [DOI] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R (2004) Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118: 699–713 [DOI] [PubMed] [Google Scholar]
- Litwin I, Bakowski T, Maciaszczyk‐Dziubinska E, Wysocki R (2017) The LSH/HELLS homolog Irc5 contributes to cohesin association with chromatin in yeast. Nucleic Acids Res 45: 6404–6416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin I, Wysocki R (2018) New insights into cohesin loading. Curr Genet 64: 53–61 [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wachn A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lopez‐Serra L, Kelly G, Patel H, Stewart A, Uhlmann F (2014) The Scc2‐Scc4 complex acts in sister chromatid cohesion and transcriptional regulation by maintaining nucleosome‐free regions. Nat Genet 46: 1147–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A (2014) Cohesin in cancer: chromosome segregation and beyond. Nat Rev Cancer 14: 389–393 [DOI] [PubMed] [Google Scholar]
- Marston AL (2014) Chromosome segregation in budding yeast: sister chromatid cohesion and related mechanisms. Genetics 196: 31–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayle R, Campbell IM, Beck CR, Yu Y, Wilson M, Shaw CA, Bjergbaek L, Lupski JR, Ira G (2015) Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349: 742–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelis C, Ciosk R, Nasmyth K (1997) Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell 91: 35–45 [DOI] [PubMed] [Google Scholar]
- Minca EC, Kowalski D (2010) Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol Cell 38: 649–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Uhlmann F (2014) Biochemical reconstitution of topological DNA binding by the cohesin ring. Nature 505: 367–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Uhlmann F (2015) DNA entry into and exit out of the cohesin ring by an interlocking gate mechanism. Cell 163: 1628–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi A, Chambers AL, Downs JA, Lehmann AR (2012) A role for chromatin remodellers in replication of damaged DNA. Nucleic Acids Res 40: 7393–7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz‐Bazán MÁ, Gallo‐Fernández M, Saugar I, Jiménez‐Martín A, Vázquez MV, Tercero JA (2014) Rad5 plays a major role in the cellular response to DNA damage during chromosome replication. Cell Rep 9: 460–468 [DOI] [PubMed] [Google Scholar]
- Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19: 123–133 [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M (1999) Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 18: 6561–6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S (2005) SUMO‐modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436: 428–433 [DOI] [PubMed] [Google Scholar]
- Rhodes J, Mazza D, Nasmyth K, Uphoff S (2017) Scc2/Nipbl hops between chromosomal cohesin rings after loading. Elife 15: e30000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sale JE (2012) Competition, collaboration and coordination–determining how cells bypass DNA damage. J Cell Sci 125: 1633–1643 [DOI] [PubMed] [Google Scholar]
- Schmid M, Durussel T, Laemmli UK (2004) ChIC and ChEC; genomic mapping of chromatin proteins. Mol Cell 16: 147–157 [DOI] [PubMed] [Google Scholar]
- Srinivasan M, Scheinost JC, Petela NJ, Gligoris TG, Wissler M, Ogushi S, Collier JE, Voulgaris M, Kurze A, Chan KL, Hu B, Costanzo V, Nasmyth KA (2018) The cohesin ring uses its hinge to organize DNA using non‐topological as well as topological mechanisms. Cell 173: 1508–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelter P, Ulrich HD (2003) Control of spontaneous and damage‐induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425: 188–191 [DOI] [PubMed] [Google Scholar]
- Ström L, Lindroos HB, Shirahige K, Sjögren C (2004) Postreplicative recruitment of cohesin to double‐strand breaks is required for DNA repair. Mol Cell 16: 1003–1015 [DOI] [PubMed] [Google Scholar]
- Ström L, Karlsson C, Lindroos HB, Wedahl S, Katou Y, Shirahige K, Sjögren C (2007) Postreplicative formation of cohesion is required for repair and induced by a single DNA break. Science 317: 242–245 [DOI] [PubMed] [Google Scholar]
- Tateishi S, Niwa H, Miyazaki J, Fujimoto S, Inoue H, Yamaizumi M (2003) Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol Cell Biol 23: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tercero JA, Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557 [DOI] [PubMed] [Google Scholar]
- Tittel‐Elmer M, Lengronne A, Davidson MB, Bacal J, François P, Hohl M, Petrini JHJ, Pasero P, Cobb JA (2012) Cohesin association to replication sites depends on Rad50 and promotes fork restart. Mol Cell 48: 98–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich HD, Jentsch S (2000) Two RING finger proteins mediate cooperation between ubiquitin‐conjugating enzymes in DNA repair. EMBO J 19: 3388–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I, Hajdú I, Fátyol K, Szakál B, Blastyák A, Bermudez V, Hurwitz J, Prakash L, Prakash S, Haracska L (2006) Human SHPRH is a ubiquitin ligase for Mms2‐Ubc13‐dependent polyubiquitylation of proliferating cell nuclear antigen. Proc Natl Acad Sci USA 103: 18107–18112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unk I, Hajdú I, Fátyol K, Hurwitz J, Yoon JH, Prakash L, Prakash S, Haracska L (2008) Human HLTF functions as a ubiquitin ligase for proliferating cell nuclear antigen polyubiquitination. Proc Natl Acad Sci USA 105: 3768–3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanoli F, Fumasoni M, Szakal B, Maloisel L, Branzei D (2010) Replication and recombination factors contributing to recombination‐dependent bypass of DNA lesions by template switch. PLoS Genet 6: e1001205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez MV, Rojas V, Tercero JA (2008) Multiple pathways cooperate to facilitate DNA replication fork progression through alkylated DNA. DNA Repair 7: 1693–1704 [DOI] [PubMed] [Google Scholar]
- Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Shibahara K, Stillman B (2000) PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 408: 221–222 [DOI] [PubMed] [Google Scholar]
- Zhang H, Lawrence CW (2005) The error‐free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister‐strand recombination. Proc Natl Acad Sci USA 102: 15954–15959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuin J, Franke V, van Ijcken WF, van der Sloot A, Krantz ID, van der Reijden MI, Nakato R, Lenhard B, Wendt KS (2014) A cohesin‐independent role for NIPBL at promoters provides insights in CdLS. PLoS Genet 10: e1004153 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 2C
Source Data for Figure 3D