

Abstract
Phosphorylation is one of the most frequent post-translational modifications on proteins. It regulates many cellular processes by modulation of phosphorylation on protein structure and dynamics. However, the mechanism of phosphorylation-induced conformational changes of proteins is still poorly understood. Here, we report a computational study of three representative groups of tyrosine in ADP-ribosylhydrolase 1, serine in BTG2, and serine in Sp100C by using six molecular dynamics (MD) simulations and quantum chemical calculations. Added phosphorylation was found to disrupt hydrogen bond (H-bond), and increase new weak interactions (H-bond and hydrophobic interaction) during MD simulations, leading to conformational changes. Quantum chemical calculations further indicate that the phosphorylation on tyrosine, threonine and serine could decrease the optical band gap energy (Egap) energy, which can trigger electronic transitions to form or disrupt interactions easily. Our results provide an atomic and electronic description of how phosphorylation facilitates conformational and dynamic changes in proteins, which may be useful for studying protein function and protein design.
Keywords: Post-translational modification, Phosphorylation, Molecular dynamics simulation, Quantum chemical calculation, Conformational changes
INTRODUCTION
Post-translational modifications (PTMs) play critical roles in regulating cellular activities and functions.1,2 In particular, one of the most extensive PTMs is phosphorylation, which regulates various cellular processes, such as differentiation, growth, metabolism, apoptosis, cellular transport, and signal transduction.3,4 In prokaryotic and eukaryotic proteins, phosphorylation can occur on serine, threonine, tyrosine, histidine5, arginine or lysine6. The addition of a phosphate (PO4) to a polar group of an amino acid in phosphorylation can turn a portion of a protein more hydrophilic, and hence change local conformation/dynamics and induce protein’s activity. However, the underlying molecular mechanism of phosphorylation at the atomic level is poorly understood. Characterization of a phosphorylation site at the atomic level is difficult. Possible experimental techniques include mass spectrometry, fluorescence immunoassays, microscale thermophoresis, FRET, TRF, fluorescence polarization, fluorescence-quenching, mobility shift, bead-based detection, and cell-based formats,7,8 but all of them are costly and time consuming.
Molecular dynamics (MD) simulation and quantum chemical calculations provide powerful tools to probe molecular mechanisms with atomic and dynamic details9–13. For example, they can help determine the effects of mutations on protein function and stability, and study enzyme reactions. Recently, computational methods have been used to model phosphorylation networks and study the dynamic effects of phosphorylation.14–20 Michel et al analyzed the impact of Ser17 phosphorylation on the dynamics of the oncoprotein MDM2 using enhanced sampling molecular dynamics simulations. The study indicated that the phosphorylated residues stabilize a “closed” state, with respect to non-phosphorylated types20. Cannon applied molecular dynamics to study the impact of Thr74 phosphorylation on structural alterations leading to PP1 activation.14 The result showed that frequency of I2 Tyr149 displacement from the PP1 active site was significantly increased upon I2 Thr74 phosphorylation14. Chiappori et al.16 pointed out that the phosphorylation of serine’s at the dimer interface of αβ-Crystallin induced the formation of hexamers (the active state of αβ-Crystallin) in the MD simulations. Nevertheless, there is lack of computational studies on general phosphorylation effects at the atomic level, especially using quantum mechanics.
In this study, we aimed to computationally study the phosphorylation mechanism using three representative systems, i.e., the phosphorylation on tyrosine (human ADP-ribosylhydrolase 1, ARH1),21,22 and serine (human BTG223 and human nuclear antigen Sp100C24) based on MD and quantum chemical calculations. Our computational studies led to a mechanistic insight into three different systems. We have investigated the electronic transitions in the phosphorylated residues. In particular, using MD simulation methods, we have tested how the phosphorylation of specific residues (tyrosine and serine) induce conformational switches and thus affect protein activities. Our results provide an atomic and electronic description of the biochemical and dynamic effects of phosphorylation on proteins.
MATERIALS AND METHODS
Quantum chemical calculations.
The quantum chemical calculations were carried out using the B3LYP function 25–28 as implemented in the Gaussian 09 program at the 6–31 G* set.25–28 Solvation effects were evaluated by single-point calculations on the optimized geometries at the B3LYP 6–31G* level of theory as the geometry optimizations using the conductor-like polarized continuum model (C-PCM).28 In the calculations, ε = 4 was used for the surrounding solvent. Frequency calculations were performed to obtain free energy corrections at 298.15 K and 1 atm pressure. Multiwfn, a multifunctional program for wave function analysis of quantum chemical calculation results29 was used to analyze the weak interaction for serine, threonine and tyrosine versus phosphorylated serine, threonine and tyrosine (p-Ser, p-Thr and p-Tyr), respectively. The number of grids was set to 200 × 200 × 200 in three-dimensional space. The 5000 frames of trajectories were extracted to average the density. To analyze traditional H-bond occupancy, the angle and distance between the donor and acceptor were set to 35° and 3.5 Å, respectively, as thresholds.
Conventional MD simulations.
All the six MD (two 180 and four 100 ns) simulations were carried out with the NAMD 2.10b1 package30 using the CHARMM36 all-atom force field.31 These six simulations were all placed into a cubic box and solvated with TIP3P waters,32 which extended 15Å from the protein atoms. Before the energy minimization, we also added sodium chloride ions to electro-neutralize the system with the concentration of 0.15mol/L. Each MD simulation had three stages. At the first stage, the main chain of the protein and the heavy atoms of ligand were constrained to minimize the energy of the system in 20,000 steps, and another same process was performed without the constraints. At the second stage, a 1.0 ns MD simulation was applied to slowly heat the system from 0 K to 298.15 K. Finally, the unconstrained production MD simulation was performed to generate the data. The bonds involving hydrogen atoms were constrained by the SHAKE algorithm.33 The Particle Mesh Ewald summation algorithm was used to calculate the long-range electrostatic interactions.34 All production simulations were performed at constant pressure (1 atm) maintained by the Langevin piston method35 and optimum temperature (298.15 k) with a 2-fs time step.
Principal component analysis.
It has been shown that biologically significant concerted motions can be extracted from MD simulations using cross-correlation analysis, which is a measure of similarity between two series as a function of the lag of one relative to the other,36 and principal components analysis (PCA), which is also called essential dynamics or quasi-harmonic analysis.37–41 PCA of the covariance matrix identifies dominant low-frequency and large-scale motions along a trajectory generated by MD simulations, and it is used to represent the most relevant correlated motions with a new basis set directly reflecting the collective motions undergone in the system.37–41 Using this approach, we have identified the protein regions involved in the most relevant collective conformational changes of phosphorylation on tyrosine and serine of proteins.
Molecular docking.
AutoDock 4.2 was used to study the binding of different ligands to the active pocket of ARH3.42 This tool uses a semi-empirical free energy force field as the scoring function to evaluate the docked conformation solution. For the ligand, Gasteigere–Marsili partial charges were assigned and non-polar hydrogen atoms were merged.43 All torsion angles were allowed to rotate during docking. The Lamarckian genetic algorithm, and the pseudo-Solis and Wets methods were also applied for energy minimization using the default parameters.43 The size of the docking box is 26 *26*26 Å. There are 22 atom types in the docking. The following parameters are used: ga_num_evals =25,000,000, ga_run = 2, and ga_run = 50.
MM-GBSA caculations.
The binding free energies were calculated by using the molecular mechanics surface area continuum solvation (MM/GBSA) method with 10 ns using Amber 14 software44,45 with ARH1-N(omega)-ADP-D-ribosyl)-L-arginine and Sp100C-petipe. In the MM–PBSA method, the free energy of the protein-ligand binding, ΔGbind, is obtained from the difference between the free energies of protein-ligand complex (Gcomplex), and the unbound receptor/protein (Gprotein) and ligand (Gligand) as follows
| (1) |
where ∆Gcomplex, ∆Gprotein, and ∆Gligand are the free energies of the complex, the protein, and the ligand, respectively. Each free energy term in Eq. (1) was calculated with the terms of (a) the absolute free energy (for protein, ligand, and their complex) in gas phase (Egas), (b) the solvation free energy (Gsolvation), and (c) the entropy term (TS) using Eq. (1), whose thermodynamic cycle is illustrated in Figure s1. For each MD-simulated complex, we calculated the ΔGbind values for the 1000 snapshots of the MD 10 ns trajectory (one snapshot for each 2 ps during the last 2000 ps of the stable trajectory). The final ΔGbind value was the average of the calculated ΔGbind values from these snapshots.
RESULTS AND DISCUSSIONS
Protein preparation.
We used three representative systems in our study: the phosphorylation on tyrosine (ARH1, PDB code 3HFW) (see Figure S2),21,22 and serine (BTG2, PDB code 3DJU, see Figure S3)23 and human nuclear antigen Sp100C PDB code 5FB0).24 ARH1 has 357 residues (Table S1), and it has attracted attention because bacterial virulence factors, including diphtheria, cholera, and pertussis toxin, use it as part of their pathogenic mechanism.21,22 Human BTG2 is the prototypical member of the TOB family, containing 127 residues, and is known to be involved in cell growth, differentiation and DNA repair (Table S1).21 Among the speckled protein (Sp) family members (Sp100, Sp140, Sp110, and Sp140L), Sp100, with 183 residues, is a nuclear autoantigen first identified in patients with the autoimmune disease primary biliary cirrhosis24 The data related to the ligands (e.g. N(omega)-ADP-D-ribosyl)-L-arginine) was downloaded from the Chemspider database and used in Gaussian 09 software at the B3LYP 6–31 G* set.25
Quantum chemical calculations.
In order to explore the mechanism of the phosphorylation on Tyr, Ser and Thr, quantum chemical calculations were carried out for six compounds (Ser, p-Ser, Thr, p-Thr, Tyr, and p-Tyr). Egap represents the energy difference between HOMO orbit and LUMO orbit. It depends on all of the coordinates of the system, which provides a more efficient sampling method than a geometrical reaction coordinate to better reflect the activities of the compounds.46 Table 1 lists the Egap values in the six compounds. It can be seen that Egap in the phosphorylated residues are lower than that of non-phosphorylated Ser, Thr, and Tyr, which indicates that the electron transfer may occur more easily in the phosphorylated residues than non-phosphorylated ones. In other words, phosphorylation can facilitate forming or destroying various interactions, such as hydrogen bond (H-bond), salt bridges, and van der Waals (vdW) interactions. Table 1 also shows the ionization potential (IP) and electron affinity (EA) energy between the non-phosphorylated Ser/Thr/Tyr and the phosphorylated type. It can be seen that IP and EA in the phosphorylated residues are higher than those of non-phosphorylated Ser, Thr and Tyr, which suggests that phosphorylated residues can get electron more easily than the non-phosphorylated ones.
Table 1.
Egap,, IP, EA energies between the non-phosphorylated Ser/Tyr/Thr type and the phosphorylated type.
| Energy components (eV) |
Ser | p-Ser | Tyr | p-Tyr | Thr | p-Thr |
|---|---|---|---|---|---|---|
| Ionization potential (IP) | 6.685 | 7.318 | 5.782 | 6.371 | 6.489 | 7.082 |
| Electron affinity (EA) | 0.162 | 0.500 | 0.044 | 0.242 | 0.25 | 0.631 |
| Energy gap (Egap) | 7.185 | 7.156 | 6.129 | 5.826 | 7.12 | 6.832 |
Figure 1(a, c, e) shows LUMO orbits of the non-phosphorylated Ser, Tyr and Thr, while Figure 1(b, d, f) shows the LUMO orbits of the phosphorylated Ser, Tyr and Thr. It can be seen that the LUMO component of the oxygen atom in the side chain of non-phosphorylated Ser, Tyr or Thr is lower than that of the phosphorylated type. The lager LUMO component of the oxygen atom is, the more nucleophilic the site is. This indicates that the compounds become more active with phosphorylation. Since phosphorylated residues can get electron more easily and become chemically more active based on the calculated HOMO and LUMO gaps, it suggests a possible approach to engineer more efficient enzymes for certain types of chemical reactions by having phosphorylated residues in the binding pocket instead of non-phosphorylated versions of those residues.
Figure 1.
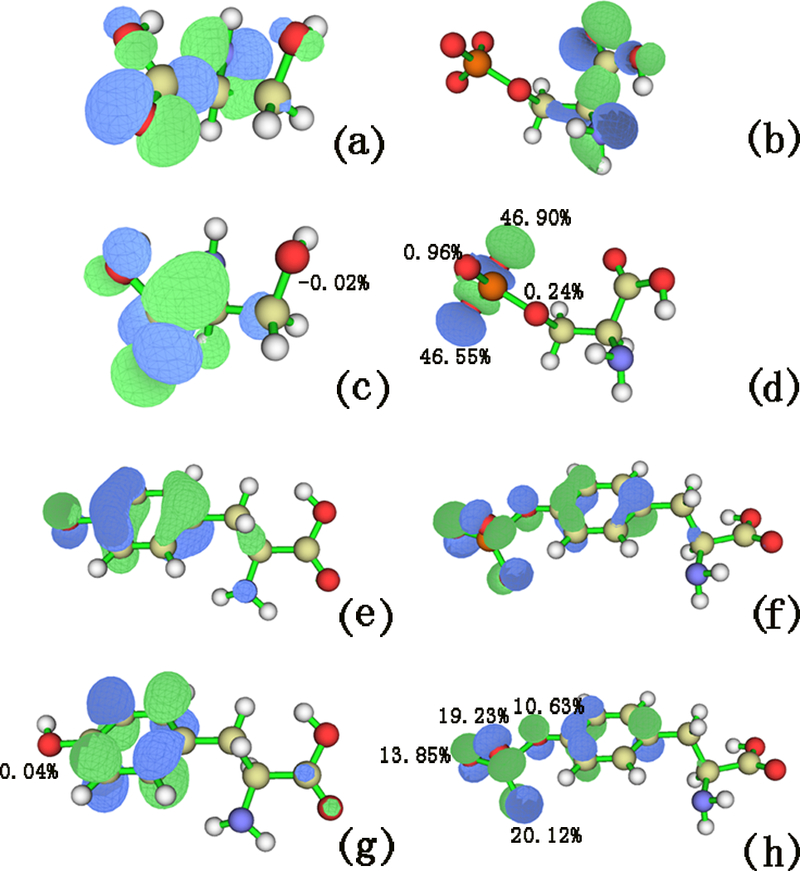
Electronic orbits. (a) LUMO orbit of serine, (b) LUMO orbit of phosphorylated serine, (c) LUMO orbit of tyrosine, (d) LUMO orbit of phosphorylated tyrosine, (e) LUMO orbit of threonine, (f) LUMO orbit of phosphorylated threonine.
The quantum interaction analysis can provide details on favorable and unfavorable interactions of compounds. It also complements analyses of H-bond, steric repulsion and vdW interaction.47 The average reduced density gradients (aRDG) of six compounds versus averaging effective density were calculated. The left and right graphs of Figure 2(a-d) indicate the attractive and steric repulsive effects, respectively. It shows that the H-bond interaction (left graph) in the phosphorylated type is larger than that of the non-phosphorylated type, which indicates the phosphorylated type forms new interactions more easily. The may be because the phosphorylated type provides more negative charge group and hence can be more interactive with other residues.
Figure 2.

Sign (lambda2) rho versus aRDG. (a) Ser, (b) p-Ser, (c) Tyr, (d) p-Tyr, (e) Thr, (f) p-Thr.
Conformational changes of ARH1 induced by tyrosine phosphorylation.
The human genome encodes three DRAG-related proteins designated ARH1, ARH2, and ARH3, which have 357, 354, and 363 amino acids, respectively.21,22 ARH1, whose 3D structure is shown in Figure S1a, like DRAG, specifically de-ADP-ribosylates proteins and mono-ADP-ribosylates on arginine residues.48,49 We docked the ligand at the active site with the phosphorylated type and non-phosphorylated type, respectively. Figure S1b indicates that His165, Ser270, Ser264, Gly130, Asp320, Gly101, Glu25, and Gly100 were the active residues for the ligand binding. The binuclear Mg2+ center is found in the active site containing residues Asp56, Asp55, Ser54, Asp15, Asp304, Glu25, Ser303, Asp302, Gly301, His299, leu27, and Trp24, as shown in Figure 3b. ARH1 has a unique feature among the family of glycohydrolases,50 as its ARH fold with 16 α-helices (shown in Figure S1c).51 From Figure S4 and Table S1, the sites of phosphorylations of ARH1 located at residues 4, 19, 20, and 205, are not in the active site while residues 19 and 20 are near the active residue Glu25. How can the phosphorylation of these residues induce the conformational changes of ARH1?
Figure 3.
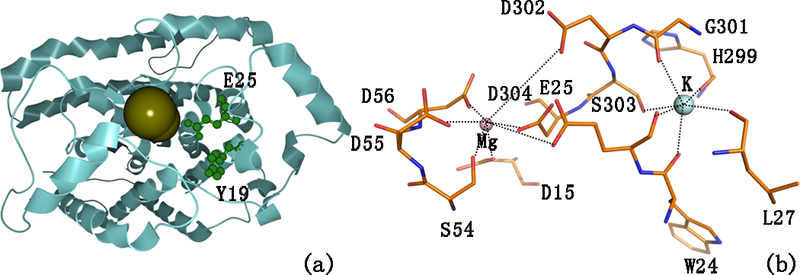
Modeling of tunnel and ion-substrate coordination. (a) Tunnel of the non-phosphorylated type Glu25 and Tyr19 located at the tunnel, and (b) coordination of Mg ions in the orthorhombic crystal form of hARH1 (PDB code. 3HFW). H-bonds are represented as dashed lines.
To explore the phosphorylation mechanics, 180 ns MD simulations were applied on the non-phosphorylated type and the phosphorylated type, respectively. The average values of RMSD of two systems are shown in Figure S5a, which indicates the two proteins reached equilibration around 120 ns. Hence, a time scale of 180 ns was set in this work to guarantee an equilibrium state for the two systems. Figure S5b shows the RMSF of residues 25 and 165 in the phosphorylated type are slightly higher than those of the non-phosphorylated type. The variation of the main chain torsion of Glu25 is changed drastically in the phosphorylated type during about 30–60 ns MD simulations (Figure 3a). The phosphorylation of ARH1 located at residues 4, 19, 20, and 205 makes Glu25 more disordered and hence destroy the H-bonds among Glu25, Gln28 and Tyr19 (Figure 3b).
We also used the CAVER 3.0 software52 for geometry-based analysis of tunnel in MD simulations, as shown in Figure 4a. Glu25 and Tyr19 are most frequent bottleneck in the ARH1 tunnel both in the non-phosphorylated type and in the phosphorylated type.
Figure 4.
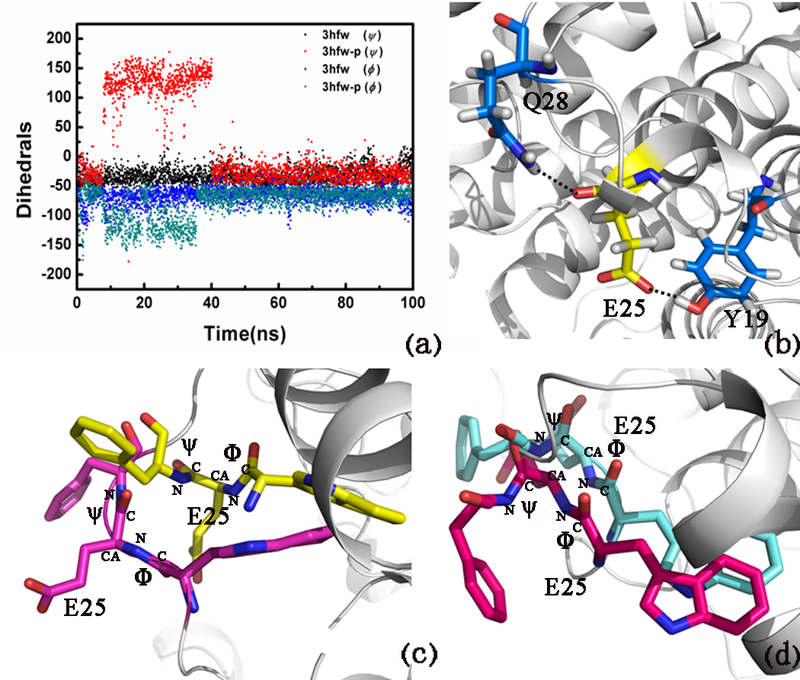
Modeling of ARH1. (a) Variation of the main-chain torsion of Glu25 with respect to simulation time during 100 ns, (b) H-bonds among Glu25, Gln28, and Tyr19 (only disappeared in the non-phosphorylated ARH1 in 30 ns), (c) Glu25 in the non-phosphorylated ARH1 (30 ns, shown in blue) and in the ARH1-P type (30 ns, shown in green), and (d) Glu25 in the non-phosphorylated ARH1 (60 ns, shown in yellow) and in the ARH1-P type (60 ns, shown in pink).
To further inspect the direction of the fluctuation in the two systems, we performed the free energy landscape (FEL) for all Cα atoms of the protein structure from the 180 ns trajectory. A lower relative free energy of the complex indicates a stronger conformational stability of the complex. The conformations of the phosphorylated type (Figure S7a) are also distributed more compactly than the non-phosphorylated type (Figure S7b), which indicates that the phosphorylated type is more stable than that of the non-phosphorylated type.
We further analyzed the H-bonds of different systems by calculating the occupancies of H-bonds during MD simulations (Tables 2 and 3). In the non-phosphorylated type, it can be seen that there are two H-bonds between Glu25 and Tyr19. While in the phosphorylated type, H-bonds between Glu25 and Tyr19 disappeared. From the two tables, the low H-bond occupancy was found in the non-phosphorylated type while the high H-bond occupancy was found in the phosphorylated type. The above analysis suggests that Glu25 are important residues in ligand binding, Mg2+ coordination, and binding (unbinding) pathway. The phosphorylation of four residues can disrupt the H-bond between Glu25 and Tyr19 and lead to more flexibility for Glu25, which may help the ligand binding and Mg2+ coordination, and thus can affect the catalytic efficiency of ARH1.
Table 2.
H-bond occupancies of the non-phosphorylated ARH1.
| 3HFW | Occupancy (%) | |
|---|---|---|
| TYR4 | TYR4:HH...O:LEU140 | 86.92 |
| TYR19 | TYR19:HN...O:ASP15 | 31.60 |
| TYR19:O...HN:GLY22 | 43.08 | |
| TYR19:O...OE1:GLU25 | 18.36 | |
| TYR19:O...OE2:GLU25 | 16.88 | |
| TYR20 | TYR20:HN...O:ALA16 | 11.20 |
| TYR20:O...HN:LYS23 | 26.48 | |
| TYR205 | TYR205:HH...OE2:GLU157 | 67.88 |
| TYR205:HN...O:GLU201 | 28.16 | |
| TYR205:HN...O:ALA202 | <10 | |
| TYR205:O...HE22:GLN208 | <10 | |
| TYR205:O...HN:GLN208 | <10 | |
| TYR205:O...HN:SER209 | 89.64 | |
| TYR205:O...HB:SER209 | 11.32 |
Table 3.
H-bond occupancies of the phosphorylated type.
| p-3HFW | Occupancy (%) | |
|---|---|---|
| TYR4 | TP4:O...HN:MET8 | 10.16 |
| TYR19 | TP19:HN...O:ASP15 | 77.56 |
| TP19:O...HN:GLY22 | 59.16 | |
| TP19:H11...O:GLY96 | 28.68 | |
| TP19:OH...HA:ARG97 | <10 | |
| TYR20 | TP20:HN...O:ALA16 | 55.72 |
| TP20:O...HN:LYS23 | 18.44 | |
| TYR205 | TP205:O9...HH22:ARG119 | <10 |
| TP205:H11...OE2:GLU157 | <10 | |
| TP205:H11...OE1:GLU157 | 58.12 | |
| TP205:HN...O:GLU201 | 11.48 | |
| TP205:HN...O:ALA202 | <10 | |
| TP205:O...HE21:GLN208 | <10 | |
| TP205:O...HN:GLN208 | <10 | |
| TP205:O...HN:SER209 | 75.16 | |
| TP205:O...HB:SER209 | 5.04 | |
| TP205:O11...HB1:SER209 | <10 |
Conformational changes of human BTG2 induced by serine phosphorylation.
As the prototypical member of an anti-proliferative family, BTG2 contains three highly conserved domains among various species, i.e., Box A (Y50–N71), Box B (L96–E115)53 and Box C (D116-P127) (Figure S2b). Box A (also named GR) and Box B appear to play key roles in anti-proliferative function and binding to a number of molecular targets.53 Two copies of an LXXLL motif in BTG2 are involved in the regulation of ERa-mediated activation: L1 (L42-L46) and L2 (L92-L96), referred to as the NR (nuclear receptor) box.54
The two LXXLL motifs are located on α-helices 2 and 5, respectively. Interestingly, these two motifs are located on opposite sides of BTG2 and provide hydrophilic surfaces, which might facilitate contact with nuclear receptors, while the hydrophobic residues are buried inside the core of the protein.23 Moreover, Box C is known to interact with PRMT1 in vitro.54 The structure is comprised of five α-helices and four β-strands that form two antiparallel β-sheets. Box A is composed of β strand 1, the short α-helix 3, part of the α-helix 2, and a connecting loop between them. Two antiparallel β-strands (2 and 3) form Box B, while Box C is composed of β strand 4 and the extended C-terminal loop. Figure S2c suggests that serine phosphorylation (residue 83 at BGT2) can lead to the protein surface contact change between the non-phosphorylated and phosphorylated types.
To explore the conformational changes of human BTG2 induced by serine phosphorylation, two 100 ns MD simulations were applied. According to RMSD analysis (Figure S6a), the two systems reached equilibration around 80 ns, and a time scale of 100 ns was therefore set in this work to guarantee an equilibrium state for the two systems. Figure S6b indicates that RMSDs around residues 20–50 and residues 80–100 in the phosphorylated type are slightly higher than that of the non-phosphorylated type. It is well known that the two LXXLL motifs L1 (L42-L46) and L2 (L92-L96) (see Figure 5c) are located in these domains. Time-dependent solvent-accessible surface area (SASA) was calculated for residues 92–96 from the simulations. Figure 5(a-b) shows that Leu95 in the phosphorylated type has larger SASA than that of the non-phosphorylated type. The larger SASA of residue is, the more hydrophobicity of residue is. The increased hydrophobicity of Leu95 may facilitate contact with nuclear receptors.
Figure 5.
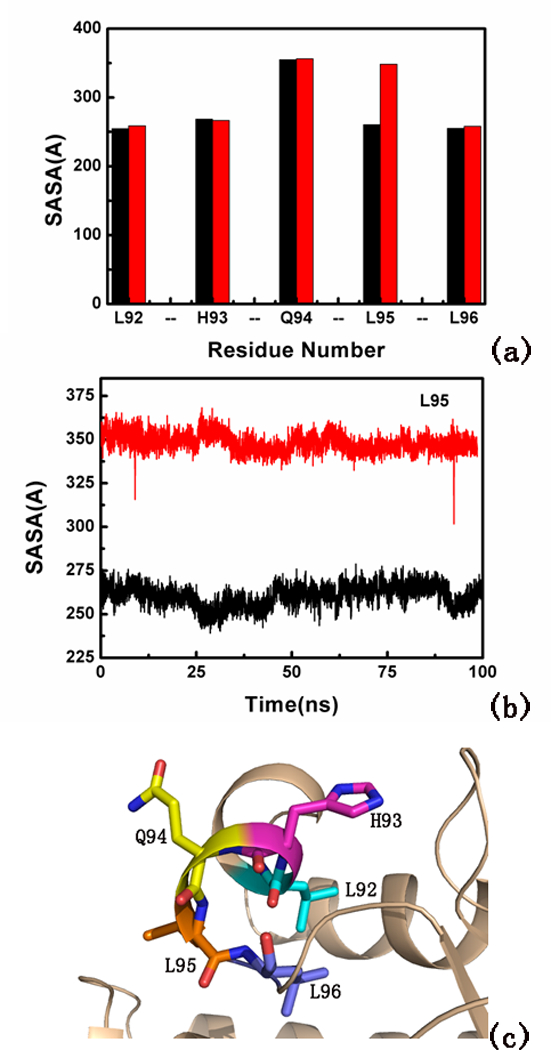
Simulation of the active site of BGT2. (a) Total SASA of residues 92–96 and (b) total SASA of Leu95 during 100 ns MD. Red represents the phosphorylated type, and black represents the non-phosphorylated type. (c) Residues 92–96 at the hydrophobic pocket of BGT2.
We further analyzed the H-bonds of different systems during MD simulations. As shown in Figure 6(a-d), the distances between Ser83 and Ser79, and between Ser83 and Arg80 were unstable in the phosphorylated type during MD simulations, which indicates these H-bonds may disappear in the phosphorylated type. The H-bond interruption may help more hydrophobicity of residue 95 located next to the α-helix of BTG2, which may facilitate contact with nuclear receptors.
Figure 6.
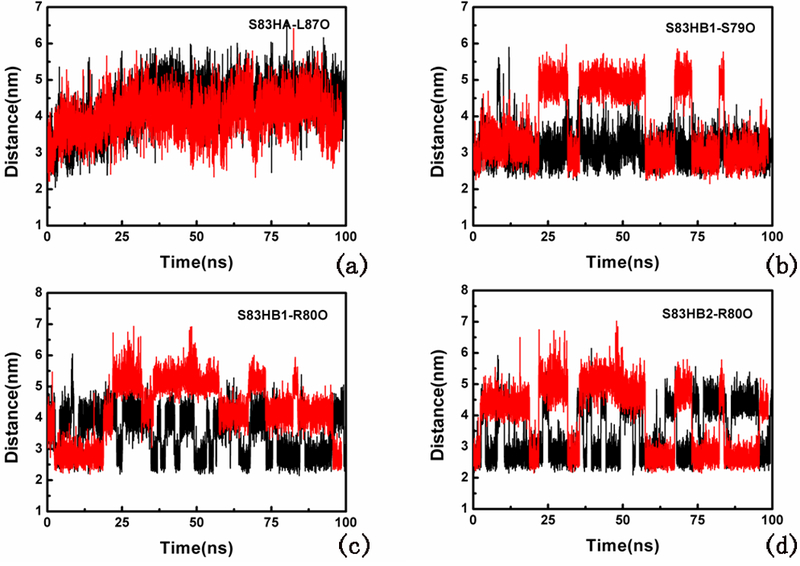
H-bond distance between (a) Ser83 HA and Leu87 O, (b) Ser83 HB1 and Ser79 O, (c) Ser83 HB1 and Arg80 O, and (d) Ser83 HB2 and Arg80 O. Red represents the phosphorylated type, and black represents the non-phosphorylated type.
Conformational changes of human nuclear antigen Sp100C induced by serine phosphorylation.
The speckled protein (Sp) family members (Sp100, Sp140, Sp110, and Sp140L) constitute a class of multinodular nuclear proteins that play key roles in intrinsic immunity and transcriptional regulation.24 Mutagenesis assays were performed on Asp696, Asn701, Asn703, and Asp718, which led to a binding loss of 7–330-fold activity.24 Deletion of residues 696–700 caused a 25-fold binding loss.24 To explore the conformational changes of human Sp100C induced by serine phosphorylation, two 100 ns MD simulations were performed. As shown in Figure 7a, time-dependent solvent-accessible surface area (SASA) was also calculated for residue 696 from the simulations. After a period of 100 ns, the SASA of Asp696 in the phosphorylated type is smaller than that of the non-phosphorylated type. The larger SASA of residue is, the more hydrophobicity of residue is. Figure 7(b-c) indicates that the main chain torsions of Asp696 are changed drastically in the phosphorylated type, which may induce the ligand binding.
Figure 7.
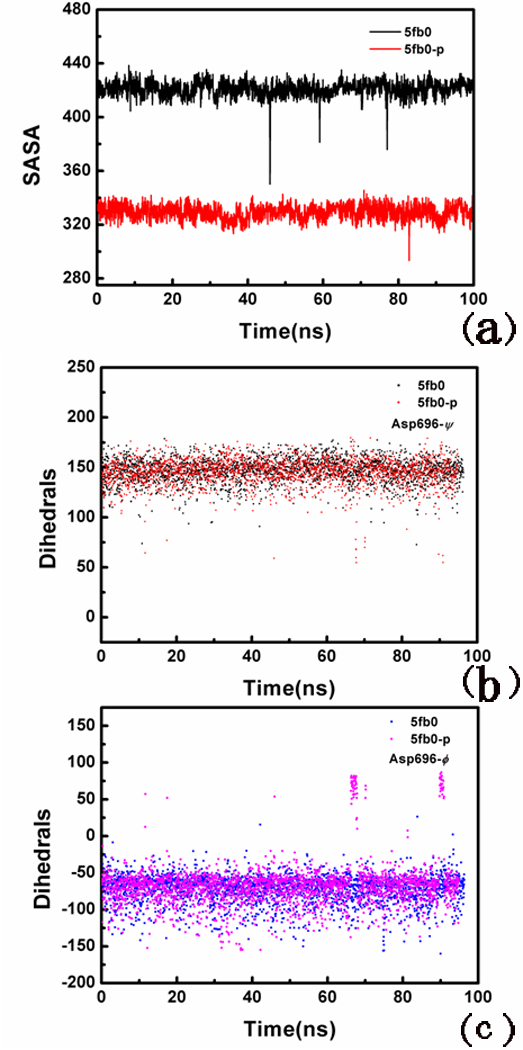
Modeling of Asp696 in Sp100C. (a) Total SASA of residue Asp696, (b) variation of the main chain torsion of Asp696 (psi) with respect to simulation time during 100 ns, and (c) variation of the main chain torsion of Asp696 with respect to simulation time during 100 ns.
The binding free energy from the MM/GBSA methodology can provide a semi-quantitative estimate of ligand (inhibitor) affinity with enzyme. Table 4 shows the binding free energies and their components for H3. The binding free energies (ΔGbind) of the H3 with the non-phosphorylated and phosphorylated types were both negative values, indicating that these complexes were energetically favorable. The two binding free energies were also compared, and the H3 phosphorylated type (−32.79 Kcal/mol) was lower in energy than H3 with the non-phosphorylated type (−29.64 Kcal/mol). This result suggests that the H3 with the phosphorylated type has a higher binding energy. For each component of MM/GBSA binding free energies, electrostatic energies (ΔEele) contribute to total energies to a greater extent than vdW energies (ΔEvdW) in the two complexes. Hence, electrostatic energies interaction was observed in the dominant position in the interaction of the H3 with both non-phosphorylated and phosphorylated types. These results are consistent with the observations in MD trajectories.
Table 4.
Free energy of binding of H3 with the non-phosphorylated and phosphorylated types (Kcal/mol).
| ΔEele | ΔEvdw | ΔGsol_np | ΔGsol_polar | ΔGpolar | ΔGnonpolar | ΔGbind | |
|---|---|---|---|---|---|---|---|
| Sp100C | −70.40 | −59.87 | −8.19 | 108.81 | 38.41 | −68.05 | −29.64 |
| p-Sp100C | −103.14 | −57.54 | −8.20 | 136.09 | 32.95 | −65.74 | −32.79 |
To sum up, it can be concluded that phosphorylation can change amino acid properties “on the fly”. These changes can facilitate function and dynamics of protein. However, the mechanisms of the changes may vary: some alter local conformations (dihedral angels); some lead to hydrophobicity changes; some make atomic groups chemically more active; and some alter H-bonds. The diverse mechanisms of phosphorylation can increase proteomic diversity,55 where one protein after the translation process without mutation can deliver diverse gene functions.
CONCLUSIONS
In conclusion, we have demonstrated that the phosphorylation on tyrosine, threonine and serine can decrease the Egap energy, which can lead to the electronic transitions and forming new interactions easily by quantum chemical calculations. Meanwhile, six 100 ns MD simulations were performed to explore the possible conformational changes of proteins induced by tyrosine or serine phosphorylation. Our results indicate that phosphorylation (1) can make some main chain of residues flip in the MD simulation and hence the residue may discord, (2) may disrupt some improper H-bond, and (3) may increase the SASA of some residue, which is useful to ligand binding. Added phosphorylation may disrupt an improper interaction and increase a new weak interaction during MD simulations, replaced with a new interaction leading to a more stable conformation. Our results provide some theoretical clues for further studies of phosphorylation mechanisms.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by National Institutes of Health grant R01-GM100701. We like to thank the anonymous reviewer for the helpful comments and suggestions.
REFERENCES
- (1).Balmant KM; Zhang T; Chen S Protein Phosphorylation and redox modification in stomatal guard cells. Front. Physiol 2016, 7 (Pt 1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Behar J; Yaniv Y Dynamics of PKA phosphorylation and gain-of-function in cardiac pacemaker cells: a computational model analysis. Ajp Heart & Circulatory Physiology 2016. [DOI] [PubMed]
- (3).Ahmad MF; Raman B; Ramakrishna T; Rao CM Effect of phosphorylation on αB-crystallin: differences in stability, subunit exchange and chaperone activity of homo and mixed oligomers of αB-Crystallin and its phosphorylation-mimicking mutant. J. Mol. Biol 2008, 375 (4), 1040–1051. [DOI] [PubMed] [Google Scholar]
- (4).Chiappori F; Mattiazzi L; Milanesi L; Merelli I A novel molecular dynamics approach to evaluate the effect of phosphorylation on multimeric protein interface: the αB-Crystallin case study. Bmc Bioinformatics 2016, 17 (4), 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Srivastava S;1,2,3 Panda S;1,2,3 Li Z;1,2,3 Fuhs SR;4 Hunter T;4 Thiele DJ;5,6 Hubbard SR;1,3 Skolnik EY 1,2,3 Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. Elife 2016, DOI: 10.7554/eLife.16093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Matthews HR Protein kinases and phosphatases that act on histidine, lysine, or arginine residues in eukaryotic proteins: a possible regulator of the mitogen-activated protein kinase cascade. Pharmacol Ther 1995, 67(3), 323–50. [DOI] [PubMed] [Google Scholar]
- (7).Gerber SA; Rush J; Stemman O; Kirschner MW; Gygi SP Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. USA 2003, 100 (12), 6940–6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Olive DM; Quantitative methods for the analysis of protein phosphorylation in drug development. Expert Review of Proteomics 2014, 1 (3), 327–341. [DOI] [PubMed] [Google Scholar]
- (9).Gao J Correlation between postranslational modification and intrinsic disorder in ptotein. Pacific Symposium on Biocomputing, 2012, 94–103. [PMC free article] [PubMed]
- (10).Aldridge BB; Burke JM; Lauffenburger DA; Sorger PK Physicochemical modelling of cell signalling pathways. Nature Cell Biology 2006, 8 (8), 1195–1203. [DOI] [PubMed] [Google Scholar]
- (11).Sathe C; Girdhar A; Leburton J; Schulten K Electronic detection of dsDNA transition from helical to zipper conformation using graphene nanopores. Nanotechnology, 2014, 25, 445105–445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Chandler D; Strümpfer J; Sener M; Scheuring S; Schulten K Light harvesting by lamellar chromatophores in Rhodospirillum photometricum. Biophys J 2014, 106, 2503–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Solov’yov IA; Domratcheva T; Shahi A;R;M; Schulten K Decrypting cryptochrome: Revealing the molecular identity of the photoactivation reaction. J Am Chem Soc 2012, 134, 18046–18052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cannon JF How phosphorylation activates the protein phosphatase-1 • inhibitor-2 complex. Bioch Biophys Acta 2013, 1834, 71–86. [DOI] [PubMed] [Google Scholar]
- (15).Kardos J; Kiss B; Micsonai A; Rovó P; Menyhárd DK; Kovács J; Váradi G; Tóth GK; Perczel A Phosphorylation as conformational switch from the native to amyloid state: Trp-cage as a protein aggregation model. J Phys. Chem. B 2015, 119 (7), 2946–2955. [DOI] [PubMed] [Google Scholar]
- (16).Chiappori F; Mattiazzi L; Milanesi L; Merelli I A novel molecular dynamics approach to evaluate the effect of phosphorylation on multimeric protein interface: the αB-Crystallin case study. BMC Bioinformatics 2016, 17(57), 226–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yonezawa Y Molecular dynamics study of the phosphorylation effect on the conformational states of the C-terminal domain of RNA polymerase II. J. Phys. Chem. B 2014, 118 (17), 4471–4478. [DOI] [PubMed] [Google Scholar]
- (18).Pisani P; Caporuscio F; Carlino L; Rastelli G Molecular dynamics simulations and classical multidimensional scaling unveil new metastable states in the conformational landscape of CDK2. Plos One 2016, 11. [DOI] [PMC free article] [PubMed]
- (19).Velazquez HA; Hamelberg D Dynamical role of phosphorylation on serine/threonine-proline Pin1 substrates from constant force molecular dynamics simulations. J. Chem. Phys 2015, 142 (7), 551–562. [DOI] [PubMed] [Google Scholar]
- (20).Bueren-Calabuig JA; Miche J Impact of Ser17 Phosphorylation on the Conformational Dynamics of the Oncoprotein MDM2. Biochemistry 2016, 55, 2500–2509. [DOI] [PubMed] [Google Scholar]
- (21).Moss J; Stanley SJ; Nightingale MS; Murtagh JJ; Monaco L,; Mishima K; Chen HC; Williamson KC; Tsai SC Molecular and immunological characterization of ADP-ribosylarginine hydrolases. J. Biol. Chem 1992, 267, 10481–10488. [PubMed] [Google Scholar]
- (22).Mueller-Dieckmann C; Koch-Nolte F The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation. Proc. Natl. Acad. Sci. USA 2006, 103 (41), 15026–15031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Morita M; Suzuki T; Bartlam M; Yamamoto T Crystal structures of human BTG2 and mouse TIS21 involved in suppression of CAF1 deadenylase activity. Nucleic Acids Research 2008, 36 (21), 6872–6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang X; Zhao; Xiong X; He Z; Li H Multifaceted histone H3 methylation and phosphorylation readout by the plant homeodomain finger of human nuclear antigen Sp100C. J.Biol.Chem 2016, 291, 12786–12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Jin HY; Zhou ZH; Wang D; Guan SS; Han WW Molecular dynamics simulations of acylpeptide hydrolase bound to chlorpyrifosmethyl oxon and dichlorvos. Int. J. Mol. Sci 2015, 16 (3), 6217–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wu RR; Rodgers MT Mechanisms and energetics for N-glycosidic bond cleavage of protonated adenine nucleosides: N3 protonation induces base rotation and enhances N-glycosidic bond stability. Phys. Chem. Chem. Phys 2016, 18(23), 16021–16032. [DOI] [PubMed] [Google Scholar]
- (27).Klapötke TM; Stierstorfer J; Weyrauther M; Witkowski TG Synthesis and Investigation of 2,6‐Bis(picrylamino)‐3,5‐dinitro‐pyridine (PYX) and Its Salts. Chemistry 2016, 22(25), 8619–8626. [DOI] [PubMed] [Google Scholar]
- (28).Paulson LO; Kaminský J; Anderson DT; Bouř P; Kubelka J Theoretical study of vibrationally averaged dipole moments for the ground and excited C═O stretching states of trans-formic acid 2010, 6 (3), 817–827. [DOI] [PubMed] [Google Scholar]
- (29).Lu T; Chen F Multiwfn: a multifunctional wavefunction analyzer. J. Chem. Theory Comput 2012, 33 (5), 580–592. [DOI] [PubMed] [Google Scholar]
- (30).Poudel KR; Dong Y; Yu H; Su A; Ho T; Liu Y; Schulten K; Bai J A time-course of orchestrated endophilin action in sensing, bending, and stabilizing curved membranes. Mol Biol Cell 2016, 27(13), 2119–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Best RB; Zhu X; Shim J; Lopes PE; Mittal J; Feig M; M. A Jr Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput 2012, 8 (9), 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Mark Pekka; Nilsson L Structure and dynamics of the TIP3P, SPC, and SPC/E water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar]
- (33).Kräutler V; Gunsteren WFV; Hünenberger PH A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem 2001, 22 (22), 501–508. [Google Scholar]
- (34).Kolafa J; Perram JW Cutoff frrors in the ewald summation formulae for point charge systems. Mol. Simul 1992, 9 (5), 351–368. [Google Scholar]
- (35).Feller SE; Zhang Y; Pastor RW; Brooks BR Constant pressure molecular dynamics simulation: The langevin piston method. J. Chem. Physic 1995, 103 (11), 4613–4621. [Google Scholar]
- (36).Ichiye T; Karplus M Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins Structure Function & Bioinformatics 1991, 11 (11), 205–217. [DOI] [PubMed] [Google Scholar]
- (37).Levy RM; Srinivasan AR; Olson WK; Mccammon JA Quasi-harmonic method for studying very low frequency modes in proteins. Biopolymers 1984, 23 (6), 1099–1112. [DOI] [PubMed] [Google Scholar]
- (38).García AE, Large-amplitude nolinear motions in proteins. Phys. Rev. Lett 1992, 68 (17), 2696–2699. [DOI] [PubMed] [Google Scholar]
- (39).Laberge M; Yonetani T Molecular dynamics simulations of hemoglobin A in different states and bound to DPG: effector-linked perturbation of tertiary conformations and HbA concerted dynamics. Biophys. J 2008, 94 (7), 2737–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Amadei A; Linssen ABM; Berendsen HJC Essential dynamics of proteins. Proteins-structure Function & Bioinformatics 1993, 17 (4), 412–425. [DOI] [PubMed] [Google Scholar]
- (41).Hayward S; And AK; Gō N Harmonic and anharmonic aspects in the dynamics of BPTI: a normal mode analysis and principal component analysis. Protein Science 1994, 3 (6), 936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Morris GM; Goodsell DS; Halliday RS; Huey R; Hart WE; Belew RK; Olson AJ Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- (43).Huey R; Morris GM; Olson AJ; Goodsell DS A semiempirical free energy force field with charge-based desolvation. J. Comput. Chem 2007, 28 (6), 1145–1152. [DOI] [PubMed] [Google Scholar]
- (44).Case DA; Cheatham III TE; Darden T; Gohlke H; Luo R; Merz KM; Onufriev A; Simmerling C; Wang B; Woods R The Amber biomolecular simulation programs. J. Computat. Chem 2005, 26, 1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Genheden S; Ryde U The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug. Discov 2015, 10(5), 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gouveia AF; Sczancoski JC; Ferrer MM; Lima AS; Santos MR; Li MS,; Santos RS; Longo E; Cavalcante LS Experimental and theoretical investigations of electronic structure and photoluminescence properties of β-Ag2 MoO4 microcrystals. Inorg. Chem 2014, 53(11), 5589–5599. [DOI] [PubMed] [Google Scholar]
- (47).Guardavaccaro D; Corrente G; Covone F; Micheli L; D’Agnano I; Starace G; Caruso M; Tirone F Arrest of G(1)-S progression by the p53-inducible gene PC3 is Rb dependent and relies on the inhibition of cyclin D1 transcription. Mol. Cell. Biol 2000, 20 (5), 1797–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Moss J; Stanley SJ; Nightingale MS; Murtagh JJ; Monaco L; Mishima K; Chen HC; Bliziotes MM; Tsai SC Molecular and immunological characterization of ADP-ribosylarginine hydrolases. J Biol Chem 1992, 10481–10488. [PubMed] [Google Scholar]
- (49).Oka S; Kato J; Moss J Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem 2006, 281 (2), 705–713. [DOI] [PubMed] [Google Scholar]
- (50).Davies GJ; Gloster TM; Henrissat B Recent structural insights into the expanding world of carbohydrate-active enzymes. Curr. Opin. Struct. Biol 2005, 15 (6), 637–644. [DOI] [PubMed] [Google Scholar]
- (51).Kanyo ZF; Scolnick LR; Ash DE; Christianson DW Structure of a unique binuclear manganese cluster in arginase. Nature 1996, 383 (6600), 554–557. [DOI] [PubMed] [Google Scholar]
- (52).Chovancova E; Pavelka A; Benes P; Strnad O; Brezovsky J; Kozlikova B; Gora A; Sustr V; Klvana M; Medek P CAVER 3.0: A tool for the analysis of transport pathways in dynamic protein structures. Plos. Comput. Biol 2012, 8 (10), 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Prévôt D; Morel AP; Voeltzel T; Rostan MC; Rimokh R; Magaud JP; Corbo L Relationships of the antiproliferative proteins BTG1 and BTG2 with CAF1, the human homolog of a component of the Yeast CCR4 transcriptional complex. J. Biol. Chem 2001, 276, 9640–9648. [DOI] [PubMed] [Google Scholar]
- (54).Revol V; Samarut C; Lukaszewicz A; Dehay C; Dumontet C; Magaud JP; Rouault JP Interaction of PRMT1 with BTG/TOB proteins in cell signalling: molecular analysis and functional aspects. Genes to Cells 2002, 7 (1), 29–39. [DOI] [PubMed] [Google Scholar]
- (55).Prabakaran S,; Lippens G; Steen H; Gunawardena J Post-translational modification: nature’s escape from genetic imprisonment and the basis for dynamic. Interdiscip Rev Syst Biol Med 2012, 4(6), 565–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.