

Abstract
Aims
MEDI0382 is a balanced glucagon‐like peptide‐1/glucagon receptor dual agonist under development for the treatment of type 2 diabetes mellitus and non‐alcoholic steatohepatitis. The primary objective was to assess the safety of MEDI0382 in healthy subjects.
Methods
In this placebo‐controlled, double‐blind, Phase 1 study, healthy subjects (aged 18–45 years) were randomized (3:1) to receive a single subcutaneous dose of MEDI0382 or placebo after ≥8 h of fasting. The study consisted of six cohorts that received study drug at 5 μg, 10 μg, 30 μg, 100 μg, 150 μg or 300 μg. The primary objective was safety and tolerability. Secondary endpoints included assessments of pharmacokinetics and immunogenicity. All subjects were followed for up to 28 days.
Results
A total of 36 subjects received MEDI0382 (n = 6 per cohort) and 12 subjects received placebo (n = 2 per cohort). Treatment‐emergent adverse events (TEAEs) occurred more frequently with MEDI0382 vs. placebo, which was mostly due to an increased occurrence at MEDI0382 doses ≥150 μg. All TEAEs were mild or moderate in severity. The most common TEAEs were vomiting, nausea and dizziness. There appeared to be a dose‐dependent increase in heart rate with MEDI0382 treatment. MEDI0382 showed linear pharmacokinetic profile (time to maximum plasma concentration: 4.50–9.00 h; elimination half‐life: 9.54–12.07 h). No immunogenicity was observed in the study.
Conclusions
In this single‐dose, Phase 1 study in healthy subjects, the safety and pharmacokinetic profiles of MEDI0382 support once‐daily dosing and further clinical development of MEDI0382.
Keywords: diabetes, drug safety, pharmacokinetics–pharmacodynamics, Phase 1, randomized controlled trial
What is Already Known about this Subject
MEDI0382 is a balanced glucagon‐like peptide‐1 (GLP‐1)/glucagon receptor dual agonist.
MEDI0382 administration in mice and non‐human primates resulted in robust glycaemic control and weight loss.
What this Study Adds
In this first‐in‐human, single‐dose study in healthy subjects, the safety and pharmacokinetic profile of MEDI0382 support once‐daily dosing.
Administration of MEDI0382 was associated with a dose‐dependent reduction in glucose that was within the normal range and was not associated with a rise in blood glucose. This finding supports preclinical data suggesting that the glucagon and GLP‐1 activity in MEDI0382 is adequately balanced.
Further evaluation of MEDI0382 in patients with type 2 diabetes mellitus is warranted.
Introduction
Type 2 diabetes mellitus is characterized by insulin resistance, obesity and weight gain, which contribute to progressive beta‐cell failure, ultimately resulting in steadily worsening glycaemic control 1. Despite the variety of antihyperglycaemic agents approved for type 2 diabetes mellitus, none of them result in substantial weight loss at doses licensed for blood glucose reduction 2. Weight loss, therefore, remains a significant unmet medical need in these patients 2. Approximately 50% of patients with type 2 diabetes mellitus progress from oral monotherapy (usually metformin) for blood glucose level control to initiation of insulin within 10 years 3, often taking multiple oral combination therapies before initiating insulin. The use of insulin exacerbates weight gain, which can be as great as 6 kg in the first year after starting insulin therapy 4. The overall weight gain, pre‐ and post‐diagnosis of diabetes, can lead to increased insulin resistance, which is associated with hypertension, dyslipidaemia, and an increased risk of major adverse cardiovascular events, such as cardiovascular mortality, nonfatal myocardial infarction or nonfatal stroke 5, 6. Significant weight loss (>5%) is the optimal intervention to reduce insulin resistance 7, although this can only be achieved reliably at present through intensive dietary and lifestyle interventions, and/or bariatric surgery 8, 9.
MEDI0382 is a synthetic oxyntomodulin‐like peptide with balanced dual http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5194) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1136 receptor agonist activity 10 that is under development for the treatment of type 2 diabetes mellitus and non‐alcoholic steatohepatitis. In diet‐induced obesity mouse models and nonhuman primates, MEDI0382 led to robust reductions in body weight, glucose levels, fasting insulin levels and hepatic fat content 10. In the diet‐induced obesity mouse model, greater weight loss was observed with MEDI0382 at lower molar doses compared with the approved GLP‐1 agonist, liraglutide 10. Moreover, in contrast to liraglutide, weight loss did not plateau with MEDI0382 over the course of the study 10. This suggests that MEDI0382 could drive greater weight loss through dual agonism of GLP‐1 and glucagon receptors compared with GLP‐1 mono‐agonists. This is supported by data with other dual agonist peptides as reviewed by Sánchez‐Garrido et al. 11. Preclinical data also suggest that, at least in the species studied, the balance of activity at the GLP‐1 and glucagon receptors was considered to be optimal with regard to achieving both weight loss and glycaemic control 10. These effects are thought to be mediated through stimulation of glucose‐mediated insulin release (the incretin effect), delayed gastric emptying, and increased fatty acid oxidation 12, 13. GLP‐1 and glucagon receptor dual agonists may also exert central effects on appetite, and glucagon receptor agonism has been shown to drive increased energy expenditure in animal models and humans 12, 14. Further, appetite suppression and increased energy expenditure in humans have been demonstrated with the use of oxyntomodulin, a weak GLP‐1/glucagon receptor dual agonist 15. Here, we report the results from a first‐in‐human, Phase 1, single‐dose study of MEDI0382 in healthy subjects.
Methods
Study design
This randomized, placebo‐controlled, double‐blinded study, evaluated the safety, tolerability, pharmacokinetic and pharmacodynamic properties of single ascending doses of MEDI0382 vs. placebo, and was conducted at a single site in Berlin, Germany. The study protocol and amendments to the protocol were approved by the Berlin independent ethics committee (identification code/approval number: 14/0576‐EK13). The study was conducted in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonisation Guidance for Good Clinical Practice, and all applicable local laws and requirements. All subjects provided written informed consent prior to initiating any study procedures. The trial was registered at ClinicalTrials.gov, NCT02394314.
The study was originally planned to evaluate the safety, tolerability, pharmacokinetic properties and immunogenicity of MEDI0382 up to single ascending doses of 2000 μg, with eight subjects per cohort and a total of eight or fewer cohorts (Figure 1). However, the 300 μg dose was not tolerated, as evidenced by events of significant vomiting (described in the Results section), and the dose was de‐escalated to 150 μg for the final cohort. Thus, contrary to the initial plan, the study included six dosing cohorts for MEDI0382 at doses of 5 μg, 10 μg, 30 μg, 100 μg, 150 μg and 300 μg (Figure 1).
Figure 1.

Planned and actual study doses and cohort population sizes
Study subjects
Eligible subjects were healthy volunteers, male or female (those who were not lactating and were not of childbearing potential), aged 18–45 years, with a body mass index (BMI) between ≥22 kg m−2 and ≤30 kg m−2, and body weight ≥70 kg. The BMI and weight criteria were chosen to approximate as closely as possible to a population of patients with type 2 diabetes mellitus, while minimizing the possibility for concomitant medical conditions. Subjects were required to have venous access suitable for multiple cannulations. Current or previous use (within 28 days of screening) of GLP‐1 receptor agonists or systemic corticosteroids were not allowed. Use of any products licensed for control of body weight or appetite 7 days prior to screening were prohibited. Subjects with a history or presence of any condition that in the opinion of the investigator would interfere with the study were excluded.
Randomization and masking
Eligible subjects were randomized (3:1) to receive MEDI0382 or placebo using an interactive voice‐ or web‐response system. Investigational products were supplied to the study site in vials of identical appearance in blinded coded kits. As such, the pharmacist, the subject, the investigator, staff and the Contract Research Organization personnel responsible for managing and monitoring the study were blinded to the treatment received. The study sponsor was not blinded to treatment allocation.
Procedures and materials
Subjects received a subcutaneous dose of the study drug within 24 h of randomization and following an overnight fast of ≥8 h. Subjects remained in the study facility for timed assessments and safety monitoring (including vital signs, pulse, blood pressure, cardiac telemetry and blood sample collections) until discharge from the unit on Day 3 post‐dose to allow for extended monitoring of cardiovascular effects. Outpatient visits were scheduled for Days 4, 7 and 28 post‐dose. A sentinel dosing approach was planned for each cohort. Two subjects in a given cohort were dosed on Day 1, and the randomization schedule ensured that these consisted of one active and one placebo subject. A time lag of 48 h occurred before additional subjects in the cohort were dosed and prior to dose escalations. Dosing was proposed to continue based on a lack of significant safety findings in the first two subjects who were dosed. The decision on whether to escalate the dose was based on review of the totality of unblinded safety and pharmacokinetic data by the dose‐escalation committee. The dose‐escalation committee consisted of the senior physician in charge of the MedImmune cardiovascular drug development group, the physician lead for MEDI0382, the clinical pharmacology lead for the study, and the principal investigator from the site. Dose escalation was not to proceed if moderate or severe adverse events (AEs) that were considered by the investigator to be related to the study drug occurred in >50% of MEDI0382‐treated subjects in a cohort, or if any serious AE considered by the investigator to be related to MEDI0382 occurred in a MEDI0382‐treated subject in a cohort.
MEDI0382 was provided as a sterile liquid drug product (nominal concentration of 2 mg ml−1). Double‐blind placebo, and open‐label diluent were provided in 50 mM sodium phosphate buffer containing 1.85% weight per volume of propylene glycol, pH level of 7.8, intended for subcutaneous administration. MEDI0382 was supplied by MedImmune Ltd (Cambridge, UK) 10. All doses were prepared by a treatment‐blinded pharmacist.
Outcome measures
The primary endpoint was the safety of MEDI0382, as assessed by summary measures of treatment‐emergent adverse events (TEAEs). Safety assessments also included electrocardiograms (ECGs), routine laboratory assessments, venous blood glucose and vital signs.
The secondary objectives were the pharmacokinetics and immunogenicity of MEDI0382. Pharmacokinetic analyses in ethylenediaminetetraacetic acid plasma were conducted using a liquid chromatography mass spectrometry method consisting of SCIEX API 5500 MS (SCIEX, Framingham, MA, USA) and Waters Acquity UPLC (Waters Corporation, Milford, MA, USA) systems. The assay method utilized an acetonitrile:water extraction (85:15 v/v) and a stable isotope‐labelled version of MEDI0382, resulting in a validated method with a calibration range of 0.4–400 ng ml−1 MEDI0382 in neat plasma. All instrument control, data collection, peak area integration and storage were performed using SCIEX Analyst version 1.6.2 (SCIEX). The data and statistical analysis comply with recommendations on experimental design and analysis in pharmacology 16.
Standard hospital meals, as supplied by the Charité hospitals group, were given to all subjects. The exploratory endpoint of the effect of study drug administration on food intake was estimated by the study nurse who assessed the proportion of food remaining when the trays were returned at the end of the meal. The exploratory endpoint of glucose levels around mealtimes following study drug administration was assessed using venous blood glucose sampling.
Statistical analysis
The sample size of eight subjects per cohort (up to eight cohorts and 64 subjects total) for this single ascending‐dose study was based on conventional study designs for first‐in‐human tolerance studies to obtain adequate safety, tolerability and pharmacokinetic data, and immunogenicity. All analyses were descriptive in nature; therefore, no formal hypothesis test‐driven sample size calculations were performed.
All summaries, except for analyses of pharmacokinetic properties, used the as‐treated population that included subjects who received investigational product, and were analysed as such. The pharmacokinetic population included all subjects who received any amount of investigational product and had at least one pharmacokinetic sample taken that was above the lower limit of quantitation.
Safety was monitored from the time of informed consent through Day 28. TEAEs were assessed by severity and relationship to the investigational product. TEAEs were coded by the most updated version of the Medical Dictionary for Regulatory Activities (MedDRA), and the type, incidence, severity and relationship to study investigational product was summarized by MedDRA system organ class and preferred term. Specific AEs were counted once for each subject for calculating percentages. In addition, if the same AE occurred multiple times within a particular subject, the highest severity and level of relationship observed was reported. The adverse events of special interest (AESIs) were clinically significant arrhythmia, vomiting and hepatic function abnormality (defined as any increase in alanine aminotransferase or aspartate aminotransferase to at least three times the upper limit of normal, and concurrent increase in bilirubin to at least twice the upper limit of normal). GI tolerability and arrhythmia were selected as an AESI because of the known profile of GLP‐1 or glucagon receptor agonists. As is typical for early‐stage drug development, liver events were collected as AESIs. For the MEDI0382 group, TEAEs were summarized by each dosing cohort and by all cohorts combined. For the placebo group, TEAEs were summarized by all cohorts combined. The clinical laboratory parameters and other safety evaluations were summarized with descriptive statistics.
Pharmacokinetic parameters were assessed in subjects receiving MEDI0382. These included the maximum observed plasma concentration (C max); time to C max (t max); apparent terminal elimination half‐life (t 1/2; obtained as the ratio of ln2/λz, where λz is the terminal phase rate constant estimated by linear regression analysis of the log transformed concentration–time data); area under the concentration–time curve (AUC) to the last quantifiable concentration (AUC0–last; determined using the linear trapezoidal rule for increasing concentrations and the logarithmic trapezoidal rule for decreasing concentrations); and AUC extrapolated to infinity (AUC0–∞) was calculated where data permitted, as the sum of AUC0–t and C t/λz, where C t is the observed plasma concentration obtained from the log‐linear regression analysis of the last quantifiable time‐point and λz is the terminal phase rate constant. A maximum of 20% of extrapolation on the AUC0–∞ was considered acceptable. Missing data were not imputed. Summary statistics were calculated for each of these parameters. Actual time of sampling, rather than nominal (planned) sampling time, was used to derive pharmacokinetic parameters using noncompartmental model 200–202 for extravascular administration using Phoenix WinNonlin 6.3 (Pharsight Corporation, Mountain View, CA, USA).
Immunogenicity of MEDI0382 was assessed on Days 1 (pre‐dose), 7 and 28, and at early discontinuation. A screening assay, in the form of a traditional ligand‐binding ‘bridging’ assay using electrochemiluminescence, was used to determine antidrug antibody‐positive samples. Any positive samples were confirmed as either positive or negative with respect to MEDI0382. Cross‐reactivity of antidrug antibody‐positive samples to GLP‐1 and glucagon may also have been assessed in the confirmatory assay, should the sample confirm a specific response for MEDI0382. Titre evaluation was performed on samples that were confirmed positive for antidrug antibody. The number and percentage of subjects in each treatment group showing an immunological response to MEDI0382 were summarized by study visit, for the as‐treated population and by cohort. All valid assay results from subjects who received any investigational product were included in immunogenicity summaries. Study discontinuation blood samples were summarized at the closest nominal time‐point that did not already have a value. Descriptive statistics were generated for the levels of glucose by treatment group at each specified time‐point.
Overall, the placebo groups were pooled together across all cohorts for analyses and presentation of results. Categorical data were summarized by the number and percentage of subjects in each category. Continuous variables were summarized by descriptive statistics, including mean, standard deviation, median, minimum and maximum. Baseline values were defined as the last valid assessment prior to the first administration of investigational product. All analyses were based on available data, and missing data were not imputed. Data analyses were conducted using the SAS® System version 9.3 or higher (SAS Institute Inc., Cary, NC, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 17, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18.
Results
Between 26 February 2015 and 12 August 2015, 48 subjects were randomized to receive MEDI0382 (n = 36; six subjects per cohort) or placebo (n = 12; two subjects per cohort). All subjects completed the study as planned and were included in the as‐treated population (Figure 2). A total of 33 of 36 subjects who received MEDI0382 had evaluable post‐dose pharmacokinetic data and were included in the pharmacokinetic population; only three subjects from the 5 μg cohort could not be included because of a lack of evaluable post‐dose pharmacokinetic data. Demographic characteristics were generally balanced across treatment groups. The mean age of subjects in the as‐treated population was 32.3 years, all subjects were male, and the majority were white (93.8%; Table 1).
Figure 2.
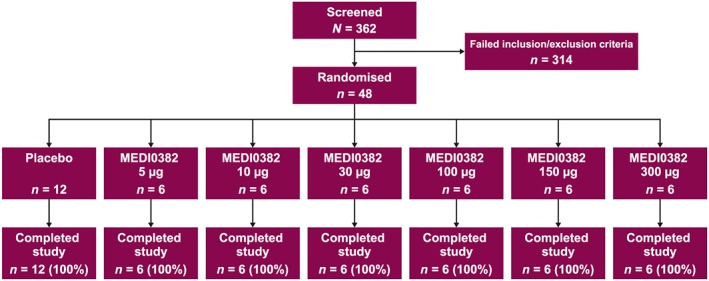
Subject disposition
Table 1.
Subject demographics and baseline characteristics
| MEDI0382 (n = 6 per cohort) | Pooled MEDI0382 (n = 36) | Pooled placebo (n = 12) | Total population (N = 48) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 5 μg | 10 μg | 30 μg | 100 μg | 150 μg | 300 μg | ||||
| Mean age, years (SD) | 37.3 (6.4) | 31.8 (6.8) | 33.2 (10.4) | 27.8 (4.3) | 34.5 (6.5) | 28.3 (5.9) | 32.2 (7.3) | 32.8 (9.1) | 32.3 (7.7) |
| Male, n (%) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 36 (100) | 12 (100) | 48 (100) |
| Race, n (%) | |||||||||
| White | 6 (100) | 5 (83.3) | 6 (100) | 6 (100) | 5 (83.3) | 6 (100) | 34 (94.4) | 11 (91.7) | 45 (93.8) |
| Other | 0 | 1 (16.7) | 0 | 0 | 1 (16.7) | 0 | 2 (5.6) | 1 (8.3) | 3 (6.3) |
| Mean BMI, kg m −2 (SD) | 27.6 (2.3) | 24.5 (1.6) | 24.7 (1.9) | 26.1 (1.6) | 26.4 (2.2) | 27.4 (2.8) | 26.1 (2.3) | 25.7 (2.4) | 26.0 (2.3) |
BMI, body mass index; SD, standard deviation
Overall, the incidence of TEAEs was higher in the MEDI0382 groups compared with the placebo group, and was highest in the 150 μg and 300 μg groups (Table 2). The majority of TEAEs were considered related to the investigational product, and related TEAEs were also more frequent in the highest MEDI0382 dose groups. At the preferred‐term level, the most frequent TEAEs were vomiting and nausea (Table 2). The onset of these TEAEs approximated to the t max (3–4 h post‐dose) and subsided approximately 12 h post‐dose. There was a total of 40 vomiting episodes for the 10 subjects who experienced vomiting. Of these subjects, 30 episodes were from five subjects at the MEDI0382 300 μg dose. With the exception of one event of vomiting in the MEDI0382 150 μg group, all events were considered related to the investigational product. All events resolved at the time of reporting and no event of vomiting led to withdrawal from the study or was classed as serious. However, because of the number of clinically significant events of vomiting observed at the 300 μg dose, the dose of MEDI0382 was de‐escalated to 150 μg for the final cohort.
Table 2.
Summary of safety and TEAEs by MedDRA, SOC and PT
| MEDI0382 (n = 6 per cohort) | Pooled MEDI0382 (n = 36) | Pooled placebo (n = 12) | ||||||
|---|---|---|---|---|---|---|---|---|
| 5 μg | 10 μg | 30 μg | 100 μg | 150 μg | 300 μg | |||
| TEAEs, n (%) | 2 (33.3) | 3 (50.0) | 4 (66.7) | 2 (33.3) | 5 (83.3) | 5 (83.3) | 21 (58.3) | 2 (16.7) |
| TEAEs by SOC and PT, n (%) | ||||||||
| Cardiac disorders | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 1 (16.7) | 3 (8.3) | 0 |
| Arrhythmia | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 1 (2.8) | 0 |
| AV block, second degree | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (2.8) | 0 |
| Ventricular extrasystoles | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (2.8) | 0 |
| GI disorders | 0 | 1 (16.7) | 0 | 1 (16.7) | 5 (83.3) | 5 (83.3) | 12 (33.3) | 1 (8.3) |
| Abdominal distension | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | 2 (5.6) | 0 |
| Abdominal pain | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (2.8) | 0 |
| Diarrhoea | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (2.8) | 0 |
| Nausea | 0 | 0 | 0 | 1 (16.7) | 4 (66.7) | 3 (50.0) | 8 (22.2) | 1 (8.3) |
| Vomiting | 0 | 0 | 0 | 1 (16.7) | 4 (66.7) | 5 (83.3) | 10 (27.8) | 0 |
| General disorders and administration site conditions | 0 | 0 | 1 (16.7) | 0 | 0 | 1 (16.7) | 2 (5.6) | 0 |
| Application site erosion | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.8) | 0 |
| Device‐associated injury | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (2.8) | 0 |
| Immune system disorders | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.8) | 0 |
| Seasonal allergies | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.8) | 0 |
| Infections and infestations | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 1 (2.8) | 1 (8.3) |
| Nasopharyngitis | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 1 (2.8) | 1 (8.3) |
| Injury, poisoning, and procedural complications | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.8) |
| Procedural dizziness | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.8) |
| Musculoskeletal and connective tissue disorders | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.8) | 0 |
| Musculoskeletal chest pain | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.8) | 0 |
| Nervous system disorders | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 2 (33.3) | 1 (16.7) | 7 (19.4) | 0 |
| Dizziness | 0 | 1 (16.7) | 1 (16.7) | 0 | 2 (33.3) | 1 (16.7) | 5 (13.9) | 0 |
| Headache | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 | 4 (11.1) | 0 |
| Respiratory, thoracic, and mediastinal disorders | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (2.8) | 0 |
| Oropharyngeal pain | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (2.8) | 0 |
AV, atrioventricular; GI, gastrointestinal; MedDRA, Medical Dictionary for Regulatory Activities; PT, preferred term; SOC, systems organ class; TEAE, treatment‐emergent adverse event
All TEAEs were grade 1 (mild) or grade 2 (moderate) in severity. There was one serious AE in the MEDI0382 10 μg group (atrioventricular block, second degree) that was considered unrelated to the investigational product and did not result in withdrawal from the study. There were no deaths or TEAEs that led to withdrawal from the study and, overall, there were no serious AEs related to the investigational product. No hepatic function abnormalities (an AESI) were observed, and no abnormalities in clinical laboratory or vital sign parameters were reported as TEAEs. Increased heart rate (Figure 3) and increased systolic blood pressure were observed with the MEDI0382 300 μg group. No trends were observed in vital sign parameters for any other treatment group.
Figure 3.
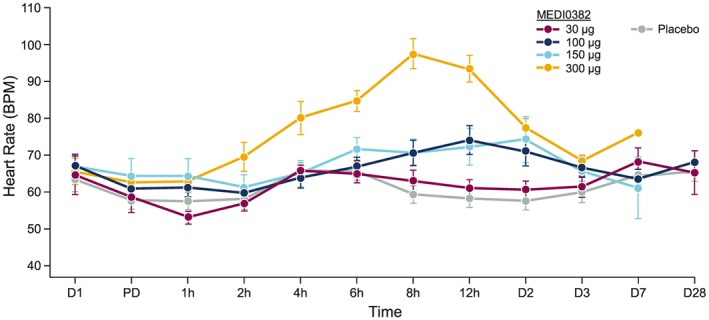
Effect of study drug treatment on heart rate (mean ± SEM) BPM, beats per minute; D, Day; h, hour; PD, pre‐dose
Plasma concentrations of MEDI0382 were measurable until 16–24 h in all subjects in the MEDI0382 5 μg and 10 μg groups. Plasma concentrations of MEDI0382 were measurable until 48 h in all subjects in the MEDI0382 100 μg and 300 μg groups. No subject had quantifiable plasma concentrations of MEDI0382 at 72 h post‐dose. The t max was estimated to be between 4.50 and 9.00 h, with t 1/2 estimated to be between 9.54 and 12.07 h (Table 3). MEDI0382 was associated with linear pharmacokinetic profiles for AUC0–∞ and C max (Figure 4). No subjects tested positive for anti‐drug antibodies following administration of MEDI0382 (data not shown).
Table 3.
Pharmacokinetic parameters of MEDI0382 (all data are geometric mean [95% CI], unless otherwise specified)
| Parameter | MEDI0382 | |||||
|---|---|---|---|---|---|---|
| 5 μg (n = 3) | 10 μg (n = 6) | 30 μg (n = 6) | 100 μg (n = 6) | 150 μg (n = 6) | 300 μg (n = 6) | |
| C max , ng ml −1 | 0.47 (0.44–0.50) | 0.80 (0.62–1.03) | 3.00 (2.59–3.47) | 6.19 (4.29–8.92) | 8.97 (6.20–12.97) | 20.98 (16.21–27.14) |
| t max , h a | 7.00 (5.88–10.00) | 6.94 (4.00–13.50) | 9.00 (4.50–10.02) | 7.53 (4.50–10.00) | 9.00 (4.00–12.00) | 4.50 (4.00–5.88) |
| AUC 0–last , ng.h ml −1 | 1.96 (0.58–6.64) | 9.97 (7.01–14.17) | 51.99 (44.55–60.67) | 137.33 (106.15–177.67) | 194.60 (162.84–232.56) | 364.47 (298.50–445.02) |
| AUC 0–∞ , ng.h ml −1 | NC | NC | 63.10b(51.49–77.33) | 150.03 (118.44–190.04) | 209.03 (178.45–244.86) | 379.12 (310.26–463.25) |
| t ½ , h | NC | NC | 11.43a (8.42–15.50) | 12.07 (9.58–15.20) | 10.97 (8.60–13.99) | 9.54 (8.63–10.56) |
Median (range)
n = 4
AUC0–last, area under the concentration–time curve from time zero to the last quantifiable concentration; AUC0–∞, area under the concentration–time curve from time zero to infinity; C max, maximum observed plasma concentration; NC, not calculated; t 1/2, apparent terminal elimination half‐life; t max, time to maximum observed plasma concentration
Figure 4.
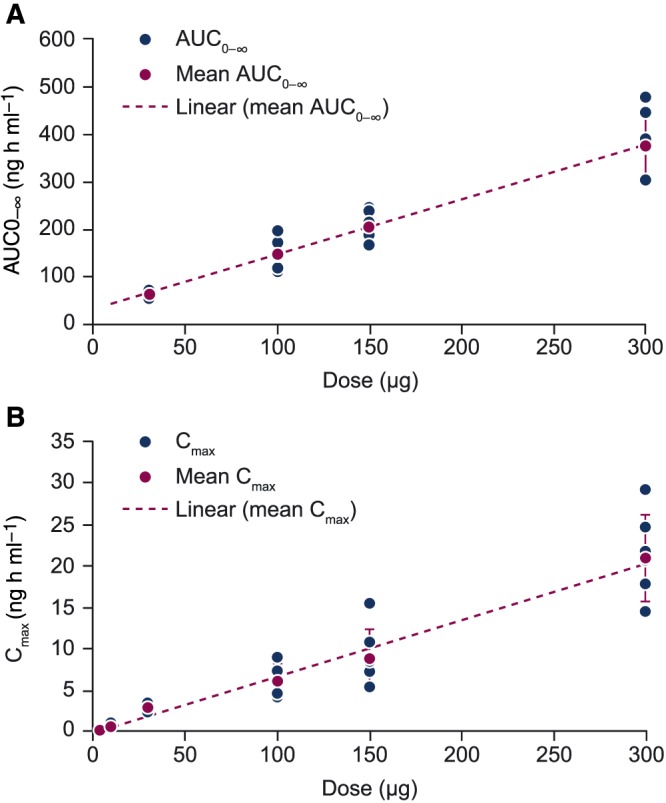
Dose proportionality of AUC0–∞ (A) and C max (B) observed with MEDI0382 treatment AUC0–∞, area under the concentration–time curve from time zero to infinity; C max, maximum observed plasma concentration
A trend indicating a dose‐dependent effect on glucose levels was observed even at low doses, especially after breakfast (2.0–4.5‐h intervals post‐dose) and dinner (13.0–14.5‐h intervals post‐dose; Figure 5). A dose‐dependent reduction in food intake was also observed following single doses of MEDI0382 (Table 4).
Figure 5.
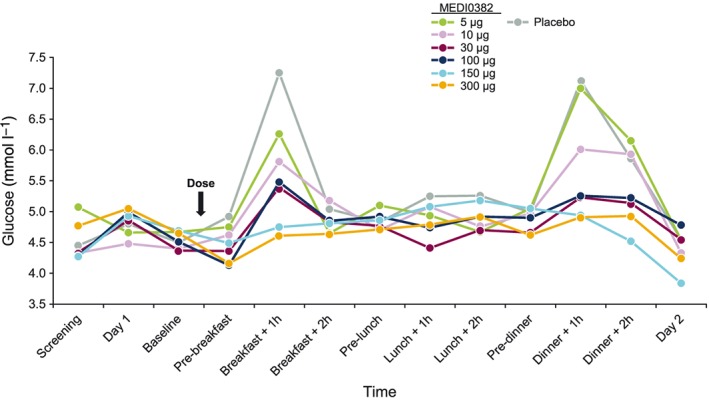
Effect of study drug treatment and food intake on median glucose levels over time
Table 4.
Mean percentage reduction in food intake and median percentage of peak glucose levels following single doses of study drug treatment
| Parameter, % | MEDI0382 | Pooled placebo | |||||
|---|---|---|---|---|---|---|---|
| 5 μg | 10 μg | 30 μg | 100 μg | 150 μg | 300 μg | ||
| Reduction in food intake | 0 | 0 | 4.1 | 17.8 | 40.0 | 76.6 | 0 |
| Median peak glucose level a | 86 | 80 | 74 | 76 | 66 | 64 | 100 |
After breakfast
Discussion
In this single‐dose, Phase 1 study, MEDI0382 was generally well tolerated at doses ≤100 μg in healthy subjects. MEDI0382 showed a linear pharmacokinetic profile and no subjects tested positive for anti‐drug antibodies. There was a trend towards dose‐dependent reduction in glucose levels with MEDI0382 even at the lowest dose of 10 μg. Moreover, a stepwise reduction in food intake was observed with MEDI0382 compared with placebo.
Increases in oxyntomodulin, a naturally occurring GLP‐1/glucagon receptor dual agonist, and GLP‐1 are often observed following bariatric surgery 18, 19. These increases in endogenous incretin levels are generally associated with rapid improvements in the control of blood glucose levels and marked weight reduction 15, 20. Subcutaneous delivery of oxyntomodulin in overweight and obese subjects is associated with weight loss over a 4‐week period 21. Because of its short half‐life, however, native oxyntomodulin does not represent an appropriate therapeutic option for the treatment of type 2 diabetes mellitus and obesity 22.
MEDI0382 is designed as a once‐daily, synthetically produced oxyntomodulin‐like peptide. By mimicking incretin changes observed after bariatric surgery, it is anticipated that GLP‐1/glucagon receptor dual agonists, such as MEDI0382, will result in superior weight reduction and glucose level control compared with GLP‐1 mono‐agonist therapy 11. As a step in this direction, this first‐in‐human study evaluated the safety, tolerability and pharmacokinetic properties of a single subcutaneous dose of MEDI0382 in healthy subjects.
With regard to safety, a higher proportion of TEAEs were observed in subjects receiving MEDI0382 compared with placebo. This difference in the proportion of TEAEs was largely driven by events of the system organ class of gastrointestinal disorders and nervous system disorders observed in the MEDI0382 group. Importantly, no event of vomiting led to withdrawal from the study or was classified as serious. Because significant increases in pulse rate have been observed with GLP‐1 agonists after single‐dose administration, including studies in healthy volunteers, as well as nausea and vomiting 23, 24, 25, these events were identified as AESIs 26. A rise in systolic blood pressure was observed at the 300 μg dose. Although direct effects on blood pressure have been observed with administration of high‐dose glucagon (3 mg), the relative glucagon agonist activity of a single 300 μg dose of MEDI0382 is unlikely to lead to a clinically significant rise in systolic blood pressure. The authors hypothesize that at least some of the rise in systolic blood pressure observed here may be due to profuse vomiting, driving anxiety in the highest dose cohort. In general, GLP‐1 agonists have not been shown to increase blood pressure 27.
Even though this was a first‐in‐human study only evaluating single doses, a pharmacological effect on both plasma glucose and food intake was reported. Effects on mealtime glucose excursions were observed with doses as low as 10 μg, while there were also significant effects on food intake from 100 μg. Both observations are suggestive of the high potency of MEDI0382 in humans, supporting the pre‐clinical evidence 10. The observed glucose reduction also suggests that the effects of glucagon receptor agonism with MEDI0382, do not lead to hyperglycaemia.
After administration of increasing single subcutaneous doses (5 μg, 10 μg, 30 μg, 100 μg, 150 μg and 300 μg), MEDI0382 appeared to be moderately absorbed with a median peak plasma concentration observed between 4.50 and 9.00 h post‐dose. The median t 1/2 of approximately 9.54 and 12.07 h could not be estimated accurately in the lowest MEDI0382 dose groups (5 μg and 10 μg) but was comparable across the other dose groups (30–300 μg).
As is common with approved GLP‐1 mono‐agonists 28, 29, tachyphylaxis, a reduction in gastrointestinal effects with multiple dosing, is expected with repeat administration of MEDI0382. Furthermore, dose titrations of GLP‐1 mono‐agonists such as semaglutide, have been shown to improve tolerability 30. Gastrointestinal effects of MEDI0382 at a single dose of ≤150 μg occurred without any major changes in pulse or blood pressure. Thus, a ‘tolerability window’ exists in which MEDI0382 can be safely administered to subjects with type 2 diabetes mellitus at a dose expected to improve glycaemic control and drive weight reduction, without a negative impact on the cardiovascular risk profile. Such doses could also be used as a starting point for dose up‐titrations. The results from this study indicate that commencing dosing of MEDI0382 up to 150 μg is acceptable with respect to initiating a multi‐dose clinical study. This conclusion is further supported by the pharmacokinetic and pharmacodynamic data observed in this study.
Overall, these data were consistent with a predictable pharmacokinetic profile amenable to once‐daily dosing, minimal accumulation of the compound in plasma after repeat dose, and steady‐state achievement within 4–7 days. Exploratory endpoints suggested that a lowering of blood glucose levels and a reduction in food intake were observed after administration of a single dose of MEDI0382 to healthy volunteers. A limitation to this study is that the method used to assess food intake (proportion of serving returned to the nurse after a meal) may be subject to bias.
In conclusion, the safety and pharmacokinetic profile of MEDI0382 support a once‐daily dosing regimen at doses less than 150 μg, followed by an up‐titration to higher doses. Multiple dosing studies will further inform the profile of MEDI0382 with respect to optimal titration regimen and the relative clinical effects of GLP‐1 and glucagon agonism in patients with type 2 diabetes mellitus.
Competing Interests
All authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors and have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author). P.D.A., M.P., and L.J. are employees of Medimmune, shareholders in AstraZeneca, and inventors on a patent application (WO2017153575 A1) related to this work. S.K. was an employee of Charité Research Organisation GmBH at the time of the study and received institutional funding. M.P. has nothing to disclose. W.P. was an employee of MedImmune at the time of the study. C.R. and B.H. are employees of MedImmune and shareholders of AstraZeneca.
We would like to thank Lan‐Feng Tsai of MedImmune for assisting with statistical analyses. Dr V. Ruvini Jayasinghe and Dr Suhaida Selamat of Oxford PharmaGenesis, Inc., Newtown, PA, USA, provided editorial assistance, which was contracted and funded by MedImmune. P.D.A. takes full responsibility for the contents of this article. This study was funded by MedImmune. Part of this study was previously presented at the American Diabetes Association Annual Meeting, New Orleans, LA, USA, on 10–14 June 2016.
Contributors
All authors contributed to drafting this manuscript by providing critical revisions and important intellectual content, and all approved the final version for submission. P.D.A was the medical monitor of this study. P.D.A., W.P., C.R. and M.P. were involved in the conception and design of the study.S.K. and M.P. were involved in the acquisition of data. P.D.A., S.K., C.R., L.J. and W.P. were involved in data analysis and interpretation. S.K. was the principal investigator.
Ambery, P. D. , Klammt, S. , Posch, M. G. , Petrone, M. , Pu, W. , Rondinone, C. , Jermutus, L. , and Hirshberg, B. (2018) MEDI0382, a GLP‐1/glucagon receptor dual agonist, meets safety and tolerability endpoints in a single‐dose, healthy‐subject, randomized, Phase 1 study. Br J Clin Pharmacol, 84: 2325–2335. 10.1111/bcp.13688.
References
- 1. Stumvoll M, Goldstein BJ, van Haeften TW. Type 2 diabetes: principles of pathogenesis and therapy. Lancet 2005; 365: 1333–1346. [DOI] [PubMed] [Google Scholar]
- 2. Baretić M, Troskot R. How to fight obesity with antidiabetic drugs: targeting gut or kidney? Minerva Endocrinol 2015; 40: 71–83. [PubMed] [Google Scholar]
- 3. Turner RC, Cull CA, Frighi V, Holman RR. Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: progressive requirement for multiple therapies (UKPDS 49). UK Prospective Diabetes Study (UKPDS) Group. JAMA 1999; 281: 2005–2012. [DOI] [PubMed] [Google Scholar]
- 4. Douek IF, Allen SE, Ewings P, Gale EA, Bingley PJ, Metformin Trial Group . Continuing metformin when starting insulin in patients with type 2 diabetes: a double‐blind randomized placebo‐controlled trial. Diabet Med 2005; 22: 634–640. [DOI] [PubMed] [Google Scholar]
- 5. Purnell JQ, Hokanson JE, Marcovina SM, Steffes MW, Cleary PA, Brunzell JD. Effect of excessive weight gain with intensive therapy of type 1 diabetes on lipid levels and blood pressure: results from the DCCT. Diabetes Control and Complications Trial. JAMA 1998; 280: 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wilcox R, Kupfer S, Erdmann E, PROactive Study investigators . Effects of pioglitazone on major adverse cardiovascular events in high‐risk patients with type 2 diabetes: results from PROspective pioglitAzone Clinical Trial In macro Vascular Events (PROactive 10). Am Heart J 2008; 155: 712–717. [DOI] [PubMed] [Google Scholar]
- 7. American Diabetes Association . Standards of medical care in diabetes – 2017. Diabetes Care 2017; 40 (Suppl. 1): S1–S135.27979885 [Google Scholar]
- 8. Grams J, Garvey WT. Weight loss and the prevention and treatment of type 2 diabetes using lifestyle therapy, pharmacotherapy, and bariatric surgery: mechanisms of action. Curr Obes Rep 2015; 4: 287–302. [DOI] [PubMed] [Google Scholar]
- 9. Astrup A, Finer N. Redefining type 2 diabetes: 'diabesity' or 'obesity dependent diabetes mellitus'? Obes Rev 2000; 1: 57–59. [DOI] [PubMed] [Google Scholar]
- 10. Henderson SJ, Konkar A, Hornigold DC, Trevaskis JL, Jackson R, Fredin MF, et al Robust anti‐obesity and metabolic effects of a dual GLP‐1/glucagon receptor peptide agonist in rodents and non‐human primates. Diabetes Obes Metab 2016; 18: 1176–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sánchez‐Garrido MA, Brandt SJ, Clemmensen C, Müller TD, DiMarchi RD, Tschöp MH. GLP‐1/glucagon receptor co‐agonism for treatment of obesity. Diabetologia 2017; 60: 1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lynch AM, Pathak N, Flatt YE, Gault VA, O'Harte FP, Irwin N, et al Comparison of stability, cellular, glucose‐lowering and appetite suppressing effects of oxyntomodulin analogues modified at the N‐terminus. Eur J Pharmacol 2014; 743: 69–78. [DOI] [PubMed] [Google Scholar]
- 13. Meier JJ, Gethmann A, Gotze O, Gallwitz B, Holst JJ, Schmidt WE, et al Glucagon‐like peptide 1 abolishes the postprandial rise in triglyceride concentrations and lowers levels of non‐esterified fatty acids in humans. Diabetologia 2006; 49: 452–458. [DOI] [PubMed] [Google Scholar]
- 14. Habegger KM, Stemmer K, Cheng C, Müller TD, Heppner KM, Ottaway N, et al Fibroblast growth factor 21 mediates specific glucagon actions. Diabetes 2013; 62: 1453–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wynne K, Park AJ, Small CJ, Meeran K, Ghatei MA, Frost GS, et al Oxyntomodulin increases energy expenditure in addition to decreasing energy intake in overweight and obese humans: a randomised controlled trial. Int J Obes (Lond) 2006; 30: 1729–1736. [DOI] [PubMed] [Google Scholar]
- 16. Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, et al Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 2015; 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wewer Albrechtsen NJ, Hornburg D, Albrechtsen R, Svendsen B, Toräng S, Jepsen SL, et al Oxyntomodulin identified as a marker of type 2 diabetes and gastric bypass surgery by mass‐spectrometry based profiling of human plasma. EBioMedicine 2016; 7: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ionut V, Burch M, Youdim A, Bergman RN. Gastrointestinal hormones and bariatric surgery‐induced weight loss. Obesity (Silver Spring) 2013; 21: 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drucker DJ, Habener JF, Holst JJ. Discovery, characterization, and clinical development of the glucagon‐like peptides. J Clin Invest 2017; 127: 4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wynne K, Park AJ, Small CJ, Patterson M, Ellis SM, Murphy KG, et al Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double‐blind, randomized, controlled trial. Diabetes 2005; 54: 2390–2395. [DOI] [PubMed] [Google Scholar]
- 22. Bhat VK, Kerr BD, Flatt PR, Gault VA. A novel GIP‐oxyntomodulin hybrid peptide acting through GIP, glucagon and GLP‐1 receptors exhibits weight reducing and anti‐diabetic properties. Biochem Pharmacol 2013; 85: 1655–1662. [DOI] [PubMed] [Google Scholar]
- 23. Mendis B, Simpson E, MacDonald I, Mansell P. Investigation of the haemodynamic effects of exenatide in healthy male subjects. Br J Clin Pharmacol 2012; 74: 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ellero C, Han J, Bhavsar S, Cirincione BB, Deyoung MB, Gray AL, et al Prophylactic use of anti‐emetic medications reduced nausea and vomiting associated with exenatide treatment: a retrospective analysis of an open‐label, parallel‐group, single‐dose study in healthy subjects. Diabet Med 2010; 27: 1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nauck M, Frid A, Hermansen K, Shah NS, Tankova T, Mitha IH, et al Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: the LEAD (liraglutide effect and action in diabetes)‐2 study. Diabetes Care 2009; 32: 84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Agersø H, Jensen LB, Elbrønd B, Rolan P, Zdravkovic M. The pharmacokinetics, pharmacodynamics, safety and tolerability of NN2211, a new long‐acting GLP‐1 derivative, in healthy men. Diabetologia 2002; 45: 195–202. [DOI] [PubMed] [Google Scholar]
- 27. Drucker DJ. The cardiovascular biology of glucagon‐like peptide‐1. Cell Metab 2016; 24: 15–30. [DOI] [PubMed] [Google Scholar]
- 28. Tong J, D'Alessio D. Give the receptor a brake: slowing gastric emptying by GLP‐1. Diabetes 2014; 63: 407–409. [DOI] [PubMed] [Google Scholar]
- 29. Umapathysivam MM, Lee MY, Jones KL, Annink CE, Cousins CE, Trahair LG, et al Comparative effects of prolonged and intermittent stimulation of the glucagon‐like peptide 1 receptor on gastric emptying and glycemia. Diabetes 2014; 63: 785–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nauck MA, Petrie JR, Sesti G, Mannucci E, Courreges JP, Lindegaard ML, et al A phase 2, randomized, dose‐finding study of the novel once‐weekly human GLP‐1 analog, semaglutide, compared with placebo and open‐label liraglutide in patients with type 2 diabetes. Diabetes Care 2016; 39: 231–241. [DOI] [PubMed] [Google Scholar]