

Abstract
Aims
The oncostatin M (OSM) pathway drives fibrosis, inflammation and vasculopathy, and is a potential therapeutic target for inflammatory and fibrotic diseases. The aim of this first‐time‐in‐human experimental medicine study was to assess the safety, tolerability, pharmacokinetics and target engagement of single subcutaneous doses of GSK2330811, an anti‐OSM monoclonal antibody, in healthy subjects.
Methods
This was a phase I, randomized, double‐blind, placebo‐controlled, single‐dose escalation, first‐time‐in‐human study of subcutaneously administered GSK2330811 in healthy adults (NCT02386436). Safety and tolerability, GSK2330811 pharmacokinetic profile, OSM levels in blood and skin, and the potential for antidrug antibody formation were assessed. The in vivo affinity of GSK2330811 for OSM and target engagement in serum and skin blister fluid (obtained via a skin suction blister model) were estimated using target‐mediated drug disposition (TMDD) models in combination with compartmental and physiology‐based pharmacokinetic (PBPK) models.
Results
Thirty subjects were randomized to receive GSK2330811 and 10 to placebo in this completed study. GSK2330811 demonstrated a favourable safety profile in healthy subjects; no adverse events were serious or led to withdrawal. There were no clinically relevant trends in change from baseline in laboratory values, with the exception of a reversible dose‐dependent reduction in platelet count. GSK2330811 exhibited linear pharmacokinetics over the dose range 0.1–6 mg kg–1. The estimated in vivo affinity (nM) of GSK2330811 for OSM was 0.568 [95% confidence interval (CI) 0.455, 0.710] in the compartmental with TMDD model and 0.629 (95% CI 0.494, 0.802) using the minimal PBPK with TMDD model.
Conclusions
Single subcutaneous doses of GSK2330811 were well tolerated in healthy subjects. GSK2330811 demonstrated sufficient affinity to achieve target engagement in systemic circulation and target skin tissue, supporting the progression of GSK2330811 clinical development.
Keywords: affinity, first‐time‐in‐human, GSK2330811, oncostatin M, target engagement
What is Already Known about this Subject
Oncostatin M (OSM) is a member of the glycoprotein 130/interleukin‐6 cytokine family that acts on a broad range of cell types to elicit pleiotropic effects, including cell differentiation and proliferation, and inflammatory mediator release.
Preclinical studies have suggested an involvement of OSM in the pathogenesis of a range of inflammatory and fibrotic diseases.
What this Study Adds
Single subcutaneous administrations of GSK2330811, an anti‐OSM monoclonal antibody, were well tolerated and showed pharmacokinetic characteristics typical of an immunoglobulin G1 monoclonal antibody in healthy subjects.
GSK2330811 had sufficient affinity to achieve high target engagement in systemic circulation and target skin tissue, supporting the progression of GSK2330811 clinical development.
The skin suction blister model is a useful technique for assessing drug pharmacokinetic and target engagement in the skin compartment.
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5035 is a pleiotropic member of the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2317 cytokine family that also includes http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4998, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=5016 and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4995 1. It is produced by leukocytes, including macrophages, activated T cells and neutrophils, and acts primarily via OSM receptors on a broad range of cell types, including chondrocytes, fibroblasts, keratinocytes and endothelial cells 1, 2, to elicit diverse biological functions 3. Depending on the context, its functions include: activation of endothelium; induction of the acute phase response; induction of cellular proliferation and/or differentiation of cell types such as fibroblasts, epithelial cells and keratinocytes; modulation of erythropoiesis and megakaryopoiesis; inflammatory mediator release; and promotion of wound healing 4, 5, 6. OSM is implicated in a broad range of inflammatory and fibrotic diseases, including inflammatory bowel disease, liver fibrosis, idiopathic pulmonary fibrosis and systemic sclerosis (SSc) 4, 7, 8, 9, an autoimmune disease characterized by fibrosis of the skin and internal organs 10. As such, OSM is an important therapeutic target.
GSK2330811 is a humanized immunoglobulin G1 kappa (IgG1κ) monoclonal antibody (mAb) that functionally blocks human OSM from binding to the gp130 receptor. It is currently in development for the treatment of SSc and other immune‐mediated diseases. Compared with GSK315234, an anti‐OSM mAb previously developed for the treatment of rheumatoid arthritis and discontinued owing to a lack of efficacy that is probably associated with poor binding affinity 11, GSK2330811 has superior affinity for human OSM, with an approximately 10‐fold increased binding affinity, as measured in vitro using surface plasmon resonance with drug capture and solution‐phase affinity methodology (six‐fold and 20‐fold, respectively; data on file).
This first‐time‐in‐human study was an experimental medicine study that assessed the safety and tolerability of GSK2330811 in healthy subjects. It was also prospectively designed to determine whether the improved in vitro affinity of GSK2330811, compared with GSK315234, for OSM led to improved affinity in vivo.
A skin suction blister model was used to analyze GSK2330811 levels and target engagement of GSK2330811 to OSM in the skin compartment, a target tissue for inflammatory and fibrotic diseases such as SSc. Skin suction blisters are a minimally invasive method for studying target engagement in skin 12 and have been applied successfully to cytokine measurements in previous studies 13. This model involved separating the epidermis from the dermis at the lamina lucida through the application of prolonged negative pressure 14. In vivo antibody affinity was determined using compartmental and physiology‐based pharmacokinetic (PBPK) with target‐mediated drug disposition (TMDD) models.
Methods
Study design
This was a phase I, first‐time‐in‐human, randomized, double‐blind (sponsor open), placebo‐controlled, single‐centre, single‐dose escalation study of subcutaneously (SC) administered GSK2330811 in healthy subjects (NCT02386436; GSK study number: 201246). A 30‐day screening phase was followed by an 8‐day inpatient dosing and monitoring period and a 105–133‐day follow‐up phase (Figure S1). A total of nine outpatient visits were scheduled over 76 days plus a follow‐up visit on day 105, and on day 133 for cohorts 4 and 5 only. Enrolment was planned as 40 subjects in five sequential cohorts (eight per cohort) in a 3:1 (GSK2330811 : placebo) ratio.
The study was conducted at a single centre (GlaxoSmithKline Clinical Unit Cambridge, Addenbrooke's Hospital, Cambridge, UK) in accordance with the ethical principles of the Declaration of Helsinki and International Council for Harmonisation – Good Clinical Practice (GCP), and the applicable subject privacy requirements. The ethics committee was the National Research Ethics Service Committee East of England, Cambridge Central. All subjects provided written informed consent.
Subject randomization was carried out using validated in‐house software. Subjects and site personnel, with the exception of the pharmacy team who prepared treatments, remained blinded to treatment allocation.
Investigational treatment
Subjects received the study medication according to their body weight and were injected in the abdomen. GSK2330811 was administered as single ascending SC doses from 0.1 mg kg–1 to 6.0 mg kg–1: cohort 1 (0.1 mg kg−1), cohort 2 (0.3 mg kg–1), cohort 3 (1 mg kg–1), cohort 4 (3 mg kg−1) and cohort 5 (6 mg kg–1). In order to limit the volume of each injection to a maximum of 1.2 ml, cohorts 1–3 received one injection of 1 ml, cohort 4 received three injections of 1 ml each, and cohort 5 received four injections of 1.2 ml each. Multiple injections were administered immediately after each other.
Dose levels were selected based on pharmacokinetic (PK)/pharmacodynamic (PD) predictions and preclinical data. The lowest administered dose of 0.1 mg kg–1 corresponded with the minimal anticipated biological effect level 15, with a maximum predicted PD inhibition of 41%, according to human PK/PD predictions in the best‐case scenario. The highest planned dose of 6 mg kg–1 was expected to provide full target engagement in serum (defined as >90%) lasting 14–40 days, with lower target engagement levels (<90%) predicted to be achieved in tissue compartments, including skin. The predicted highest exposures, measured by the peak plasma concentration (Cmax) and area under the curve (AUC), were almost 100‐fold below the safety margin provided by the toxicology study.
A sentinel group (n = 2) in each cohort was randomized to receive GSK2330811 or placebo; dosing of the remainder of the cohort proceeded if no safety concerns were identified up to and including 5 days postdose. Dose escalation in subsequent cohorts was based on safety data during a minimum of 28 days postdose in ≥5 subjects who received GSK2330811, accumulated safety and tolerability data from previous cohorts and all available PK data.
Study population
Subjects eligible for the study were healthy adults, including men and women of nonreproductive potential, 18–65 years of age, with body weight ≤100 kg (≤80 kg for cohort 5; in order to limit the number of injections required for 6 mg kg–1 of GSK2330811 to four injections of 1.2 ml) and body mass index (BMI) 18.5–29.9 kg m–2. Subjects with abnormal clinical chemistry values, including platelet or haemoglobin values below the lower limit of normal, a history of gastrointestinal bleeding disorders or donation of >500 ml of blood within 56 days prior to dosing, were excluded, as were those with a history of haematological disease or acquired platelet disorders and coagulation disorders, or of opportunistic infection or serious, active or unresolved infection. Paracetamol at doses of ≤ 2 g day–1 was permitted at any time during the study; other concomitant medications were considered on a case‐by‐case basis.
Endpoints and assessments
The primary objective of the study was to evaluate the safety and tolerability of single SC doses of GSK2330811 in healthy subjects. Primary endpoints were the frequency of adverse events (AEs) and serious AEs (SAEs), and changes in clinical laboratory evaluations, vital signs and 12‐lead electrocardiograms (ECGs). Blood samples for safety assessments (haematology and clinical chemistry) were collected during the inpatient monitoring, at each visit during the outpatient monitoring and at follow‐up. Secondary objectives were to evaluate the PK profile (endpoints: plasma concentrations of GSK2330811 and derived PK parameters) and assess the potential for antidrug antibody formation (endpoints: incidence, specificity and titres of anti‐GSK2330811 antibodies) following single SC doses of GSK2330811 in healthy subjects. Exploratory objectives were included to explore GSK2330811 PD and the PK/PD relationship in the blood (endpoints: serum levels of free and total OSM) and GSK2330811 PK, PD and the PK/PD relationship in the skin in healthy subjects (endpoints: skin blister fluid levels of GSK2330811, and of free and total OSM). These exploratory endpoints were used to assess target engagement, based on free and total OSM levels in serum and skin blister fluid.
PK plasma and PD serum samples were collected via an indwelling cannula or by direct venepuncture, predose and at 8 h, 24 h, 48 h, 96 h and 144 h, and on day 10, day 14, day 21, day 28, day 42, day 56, day 84, day 105 and, for cohorts 4 and 5 only, day 133 postdose.
Suction blisters were raised by applying prolonged negative pressure using a VP25 or VP28 suction pump (Eschmann, Lancing, West Sussex, UK) attached to a suction chamber placed over unblemished skin on the volar surface of the left and right forearm (one site per visit) on days −1, 7 and 42, similar to the method described by Akbar et al. 14. The negative pressure was applied for 4 h until a single unilocular blister was formed. Skin blister fluid was collected 4 h postblister induction and briefly centrifuged to remove cellular contents. Serum samples for immunogenicity assays were collected at predose, day 14, day 28 and day 105 (follow‐up visit). The suction blister procedure was only performed for subjects in the higher dose cohorts (3–5) in which target engagement in the skin was anticipated to be measurable.
PK samples were analyzed for GSK2330811 concentrations in plasma and skin blister fluid using a qualified analytical method based on sample dilution followed by immunoassay analysis. The lower limit of quantification (LLQ) for GSK2330811 was 100 ng ml–1, using a 40 μl aliquot of plasma or a 10 μl aliquot of blister fluid diluted 25‐fold into assay buffer, with a higher limit of quantification (HLQ) of 5000 ng ml–1. Further details are provided in the Supplementary Methods.
PD (free OSM and total OSM) samples were measured using a validated ligand‐binding assay. The LLQ for both free and total OSM (free and GSK2330811 bound) was 1.45 pg ml–1, with an HLQ of 2500 pg ml–1. Further details are provided in the Supplementary Methods.
PK/PD analysis
Actual blood sampling times were used to determine individual PK parameters. The concentration–time results for GSK2330811 were used to assess Cmax, AUC, apparent clearance, steady‐state apparent volume of distribution, time of maximum plasma concentration and terminal half‐life. Values below the LLQ of the PK assay (5% of data, mainly in the 0.1 mg kg–1 cohort at the latest time points) were not included in the analysis. Parameters were determined via noncompartmental PK analysis using WinNonlin Phoenix version 6.4 (Certara USA, Inc., Princeton, NJ).
Plasma PK and serum OSM data above the LLQ of the assays were used to evaluate the relationship between PK and target engagement using nonlinear mixed‐effect modelling. A one‐compartment PK with TMDD model 16, 17 (Figure 1) was developed using the observed GSK2330811 plasma concentration and total OSM (free plus complex) serum concentration to assess the in vivo affinity of GSK2330811 for OSM. Free target was assumed to be synthesized at a zero‐order rate and degraded at a first‐order rate. Free OSM levels in serum and skin blister fluid at baseline were used to estimate target turnover parameters. A quasi‐steady‐state solution was considered among drug, free target and drug–OSM complex 18. In addition, GSK2330811 and OSM concentrations in blister fluid were used together with plasma and serum data to develop a minimal PBPK (mPBPK) with TMDD model 19 (Figure 1). Physiological parameters of the mPBPK model were fixed to their physiological value 19. Drug absorption rate constant, plasma clearance and interstitial fluid volume were estimated from the data. Parameters describing the target turnover, drug–target binding and complex clearance were estimated from the data. The percentage of target engagement (TE%) at every time point was computed as TE (t) = [baseline free OSM – free OSM (t)]/baseline free OSM * 100. Predicted free levels were used as measured values and were all below the LLQ of the assay after drug administration. Model selection was based on general goodness‐of‐fit criteria, including diagnostic goodness‐of‐fit plots, reduction in minimum value of objective function, precision of parameter estimates, convergence of the minimization algorithm and visual predictive check for the final PK/PD model. Model parameters were estimated using the Monte Carlo importance sampling (IMP) estimation method with the software NONMEM (version 7.2, ICON Development Solutions, Ellicott City, MD, USA) and Perl‐speaks‐NONMEM (version 3.4.2) 20. The differential equations describing the one‐compartment model and the mPBPK model are provided in the Supplementary Methods.
Figure 1.
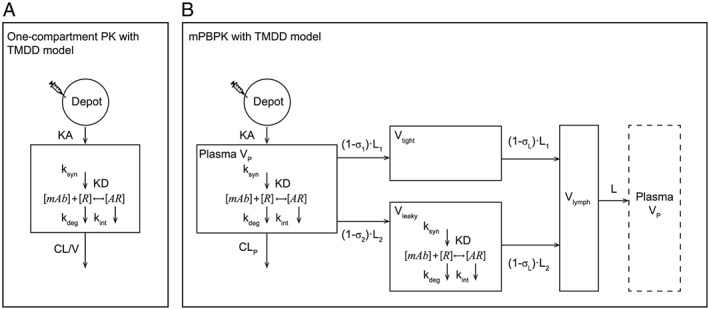
Block scheme of the one‐compartment model with TMDD in plasma (A) and the mPBPK model including TMDD in plasma and leaky tissue (B). AR, antibody–target complex; CL, clearance; CLp, plasma clearance; KA, absorption constant; KD, dissociation constant; kdeg, degradation rate constant; Kint, antibody–target complex degradation rate constant; ksyn, biosynthesis rate constant; L, lymph flow; σ,vascular reflection coefficient; mAb, monoclonal antibody; mPBPK, minimal physiology‐based pharmacokinetic; PK, pharmacokinetics; R, target; TMDD, target‐mediated drug disposition; V, volume; Vp, plasma volume
Sample size and statistical analysis
Sample size was based primarily on feasibility. Unless otherwise stated, all analyses were carried out in the all‐subjects population, comprising all randomized subjects who received a dose of study treatment. The PK population comprised all subjects for whom a PK sample was obtained and analyzed.
Nomenclature of target and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 21, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 22.
Results
Study population
The first patient was enrolled on 21 April 2015 and the last subject completed the study on 11 April 2016. In total, 89 subjects were screened and 41 were randomized; 40 were included in the all‐subjects population (one subject was withdrawn prior to dosing at the investigator's discretion) (Figure S2). All dose escalations occurred as planned in the protocol.
All randomized subjects were male except for one female subject in cohort 4. Mean body weight was lower in cohort 5 (69.5 kg) compared with the other cohorts (78.5–85.4 kg) due to the lower body weight inclusion criterion for this group (≤80 kg vs. ≤100 kg). Mean BMI (23.7 kg m–2 vs. 24.6–26.7 kg m–2) and age (36.8 vs. 41.5–47.7 years) were also lower in cohort 5 than in the other cohorts.
Safety and tolerability
All subjects except one in each of the GSK2330811 0.3 mg kg–1 and 1 mg kg–1 groups reported ≥1 AE. Nasopharyngitis and headache were the most commonly reported (Table 1); oropharyngeal pain was reported at higher dose levels only (≥3 mg kg–1). Two reports of injection site bruising (placebo group and 0.3 mg kg–1 group) and one of injection site pain (0.1 mg kg–1 group) were suspected by the investigator to be related to the study drug. Skin suction blisters were well tolerated and no AEs related to blister healing were reported. There were no deaths, nonfatal SAEs, or AEs leading to withdrawal from the study.
Table 1.
AEs by category and individual events with ≥2 subjects in any group (all‐subjects population)
| GSK2330811 | ||||||
|---|---|---|---|---|---|---|
| Placebo N = 10 | 0.1 mg kg–1 N = 6 | 0.3 mg kg–1 N = 6 | 1 mg kg–1 N = 6 | 3 mg kg–1 N = 6 | 6 mg kg–1 N = 6 | |
| Any AE, n (%) | 10 (100) | 6 (100) | 5 (83) | 5 (83) | 6 (100) | 6 (100) |
| Infections and infestations, n (%) | 7 (70) | 1 (17) | 4 (67) | 2 (33) | 1 (17) | 3 (50) |
| Nasopharyngitis | 6 (60) | 0 (0) | 2 (33) | 2 (33) | 1 (17) | 1 (17) |
| Folliculitis | 0 (0) | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) |
| Nervous system disorders, n (%) | 3 (30) | 2 (33) | 1 (17) | 0 (0) | 3 (50) | 5 (83) |
| Headache | 3 (30) | 2 (33) | 1 (17) | 0 (0) | 2 (33) | 2 (33) |
| Sinus headache | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (33) |
| Injury, poisoning and procedural complications, n (%) | 3 (30) | 4 (67) | 1 (17) | 2 (33) | 0 (0) | 1 (17) |
| Scratch | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Soft tissue injury | 0 (0) | 2 (33) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Respiratory, thoracic and mediastinal disorders, n (%) | 0 (0) | 1 (17) | 1 (17) | 1 (17) | 4 (67) | 2 (33) |
| Oropharyngeal pain | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (33) | 2 (33) |
| Skin and subcutaneous tissue disorders, n (%) | 3 (30) | 1 (17) | 0 (0) | 1 (17) | 2 (33) | 1 (17) |
| Dermatitis contact | 3 (30) | 0 (0) | 0 (0) | 1 (17) | 0 (0) | 0 (0) |
| Gastrointestinal disorders, n (%) | 2 (20) | 1 (17) | 1 (17) | 0 (0) | 1 (17) | 1 (17) |
| General disorders and administration site conditions, n (%) | 1 (10) | 1 (17) | 1 (17) | 1 (17) | 0 (0) | 2 (33) |
| Musculoskeletal and connective tissue disorders, n (%) | 3 (30) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (50) |
AE, adverse event.
No treatment‐emergent occurrences of antidrug antibodies were reported. Treatment‐unrelated pre‐existing antibodies, detected in three subjects, were low titre and were not associated with any specific AEs, inhibition of target engagement or alteration in PK.
Liver function tests showed no clinically significant elevation of liver enzymes; no subjects recorded alanine aminotransferase (ALT) >2 times the upper limit of normal and no dose–response relationship was observed in subjects with Grade 1 ALT elevations. There were no clinically significant changes in other clinical chemistry laboratory parameters (haematology results are detailed below), vital signs or ECGs which were accompanied by any signs and symptoms.
Platelet count and blood cell parameters
A reversible dose‐dependent reduction in platelet count was seen after single doses of GSK2330811 in the three highest dose groups (1 mg kg–1, 3 mg kg–1 and 6 mg kg–1) (Figure 2A). Median time to platelet count nadir was similar, with a range of 20 to 26 days in these groups (Table S1). Platelet count had recovered to the normal range by day 42 in all subjects. The number of subjects with graded Common Terminology Criteria for Adverse Events version 4.03 (CTCAE) events of decreased platelet count rose 23 with increasing dose level. One subject in the 6 mg kg–1 group had a Grade 2 CTCAE (73 GI l–1 [where GI = 109] on day 18; Table S1).
Figure 2.
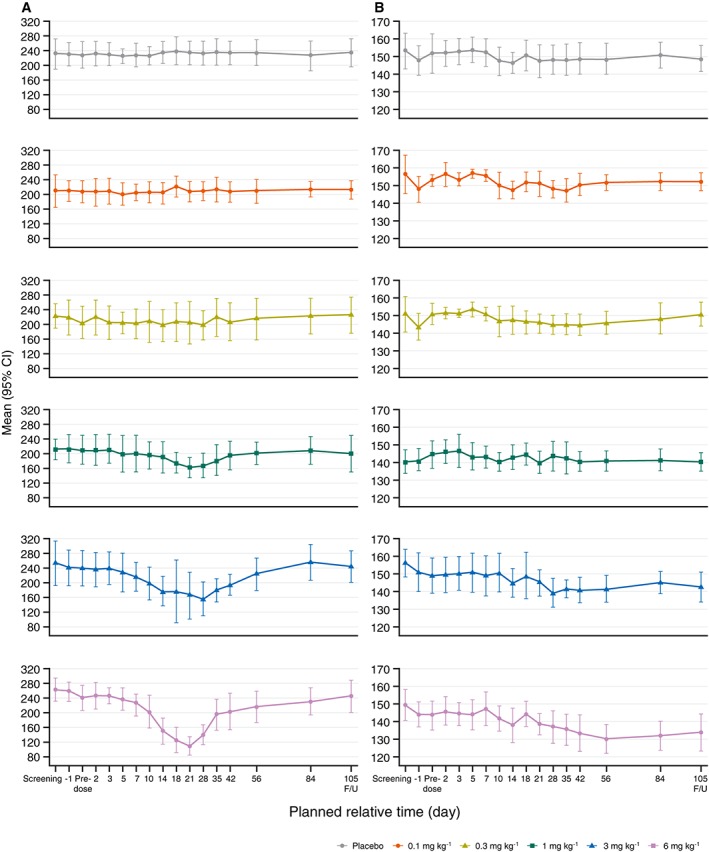
Mean platelet count (GI l−1) (A) and mean haemoglobin (g l−1) (B) to first follow‐up visit by GSK2330811 treatment group (all‐subjects population). CI, confidence interval; FU, Follow‐up
A reduction in haemoglobin values was observed in the 6 mg kg–1 group: this was maximal at day 56 (Figure 2B). In all subjects, values had returned to the normal range by day 133. Four subjects in the highest dose group had Grade 1 CTCAEs of anaemia; no subjects had Grade 2 or higher anaemia. Changes in red blood cell counts followed similar trends to changes in haemoglobin levels. An initial decrease in reticulocyte count starting from day 10 was seen in the 3 mg kg–1 and 6 mg kg–1 groups, followed by a reticulocytosis starting from day 21 to day 28 (Figure S3).
Only one CTCAE Grade 3 laboratory abnormality was observed (lymphopenia). This occurred in a subject receiving GSK2330811 3 mg kg–1 who recorded a lymphocyte count of 0.42 GI l–1 on day 10. The subsequent lymphocyte counts were 0.93 GI l–1 on day 14 and 1.83 GI l–1 on day 18 (normal range, 1.2–3.65 GI l–1). Three additional subjects (one in each of the placebo, GSK2330811 1 mg kg–1 and GSK2330811 3 mg kg–1 groups) recorded a transient Grade 2 neutropenia. No dose‐related trends were observed in either lymphocyte or neutrophil numbers or in total white cell count.
PK
GSK2330811 exhibited approximately linear PK over 0.1 mg kg–1 to 6 mg kg–1 following a single SC administration (Figure 3), and PK parameters determined with noncompartmental analysis were consistent with an IgG1 antibody against a soluble target (Table 2). Following single SC administration, the median time to reach the maximum observed plasma concentration ranged from 119.77 h to 264.18 h. GSK2330811 was eliminated from the circulation with a geometric mean terminal half‐life ranging from 462.1 h to 609.2 h.
Figure 3.
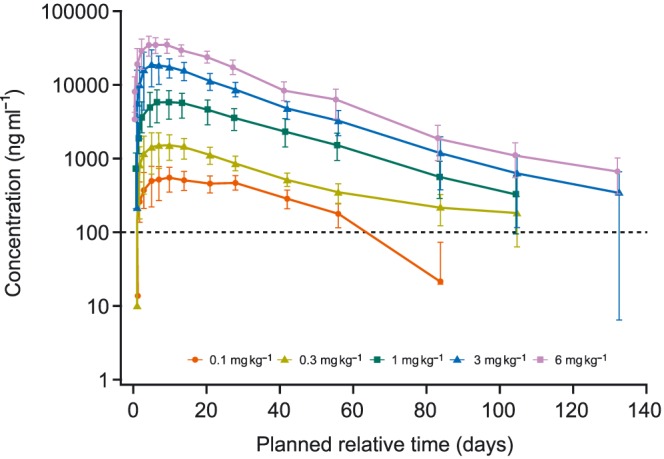
Mean (±SD) GSK2330811 concentration by treatment group in plasma (PK population). LLQ, lower limit of quantification; PD, pharmacodynamic; PK, pharmacokinetic; SD, standard deviation. Note: GSK2330811 concentration values lower than the LLQ were not included in the noncompartmental analysis and PK/PD modelling analysis
Table 2.
GSK2330811 plasma PK parameters determined with noncompartmental analysis (PK population)
| PK parameter | GSK2330811 | ||||
|---|---|---|---|---|---|
| 0.1 mg kg–1 N = 6 | 0.3 mg kg–1 N = 6 | 1 mg kg–1 N = 6 | 3 mg kg–1 N = 6 | 6 mg kg–1 N = 6 | |
| AUC0–t, h*μg ml–1 | 510 (31.3) | 1427 (16.3) | 5099 (41.7) | 14 321 (21.6) | 27 187 (22.7) |
| AUC0–inf, h*μg ml–1 | 615 (26.2) | 1605 (21.6) | 5316 (42.5) | 14 656 (21.6) | 27 681 (23.3) |
| Cmax, μg ml–1 | 0.6 (32.3) | 1.5 (42.7) | 5.7 (43.5) | 19.0 (42.7) | 36.4 (22.9) |
| Vss/F, ml kg–1 | 108.44 (22.8) | 164.30 (27.5) | 127.19 (33.4) | 136.46 (42.5) | 163.23 (24.0) |
| CL/f, ml h–1 kg–1 | 0.1625 (26.2) | 0.1870 (21.6) | 0.1881 (42.5) | 0.2047 (21.6) | 0.2168 (23.3) |
| tmax, h a | 264.18 (96.0, 650.0) | 216.28 (96.0, 482.0) | 178.56 (142.3, 337.7) | 180.66 (24.0, 309.8) | 119.77 (96.0, 213.2) |
| t½, h | 462.6 (15.2) | 609.2 (13.7)b | 468.6 (15.5) | 462.1 (43.5) | 522.0 (17.0) |
Median (range) for tmax only, all other PK parameters are geometric mean (%CVb);
n = 3, as t½ could not be identified unambiguously for three subjects
AUC0–inf, area under the concentration–time curve from zero to infinity; AUC0–t, area under the concentration–time curve from zero hours to time t; Cmax, maximum observed concentration; CL/f, apparent clearance; CVb, between‐subject coefficient of variation; PK, pharmacokinetic; t½, terminal phase half‐life; tmax, time to reach the maximum observed plasma concentration; Vss/F, steady‐state volume of distribution/bioavailability
The observed concentration–time profiles of GSK2330811 were adequately described by a one‐compartmental PK model with first‐order absorption and first‐order elimination (Figure 4). Comparable fitting and prediction of PK data were obtained with the mPBPK model (data not shown). The impact of age and weight as covariates was tested. No correlation was found; however, this could have been due to the limited number of subjects, typical of a first‐time‐in‐human study. The GSK2330811 ratio between mean skin blister fluid (Table 3) and plasma concentration ranged from 19% to 45%.
Figure 4.
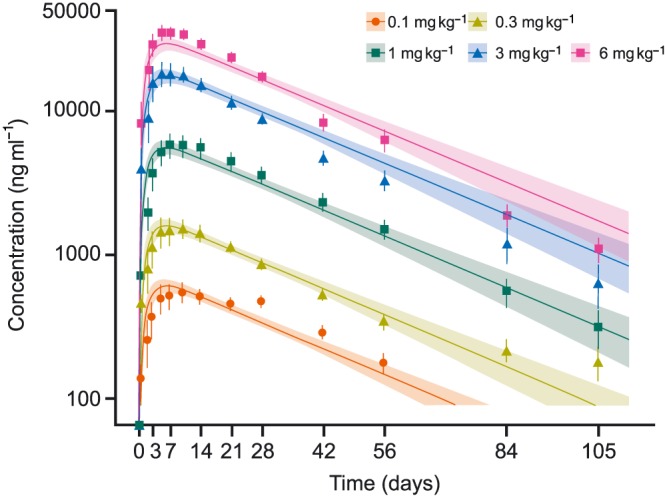
Mean (±SE) observed vs. mean predicted total GSK2330811 concentration in plasma by treatment group (PK population). PK, pharmacokinetic; SE, standard error
Table 3.
Summary of blister fluid concentration (PK population)
| GSK2330811 | |||
|---|---|---|---|
|
1 mg kg–1
N = 6 |
3 mg kg–1
N = 6 |
6 mg kg–1
N = 6 |
|
| Day 7, ng ml–1, mean (SD) | 1115.0 (383.36) | 5479.7 (2821.99) | 9438.2 (2855.00) |
| Day 42, ng ml–1, mean (SD) | 442.2 (159.0) | 2113.8 (985.70) | 2951.3 (1384.51) |
PK, pharmacokinetic; SD, standard deviation.
Target engagement and in vivo affinity assessment
All free OSM levels in both serum and skin blister fluid were below the LLQ after drug administration, indicating substantial OSM inhibition. Baseline total OSM levels were, in general, consistent among dose groups, with mean values ranging from 9.1 pg ml–1 to 14.1 pg ml–1 in serum, and from 11.7 pg ml–1 to 19.2 pg ml–1 in skin blister fluid, with the placebo group showing a similar concentration profile over time (data not shown). Total OSM levels increased rapidly after GSK2330811 administration, achieving a saturation level at doses ≥3 mg kg–1 (approximately 80‐fold in serum and 26‐fold in skin blister fluid), with a prolonged saturation observed at 6 mg kg–1. Observed vs. predicted mean total OSM is shown in Figure 5A for serum and 5B for skin blister fluid.
Figure 5.
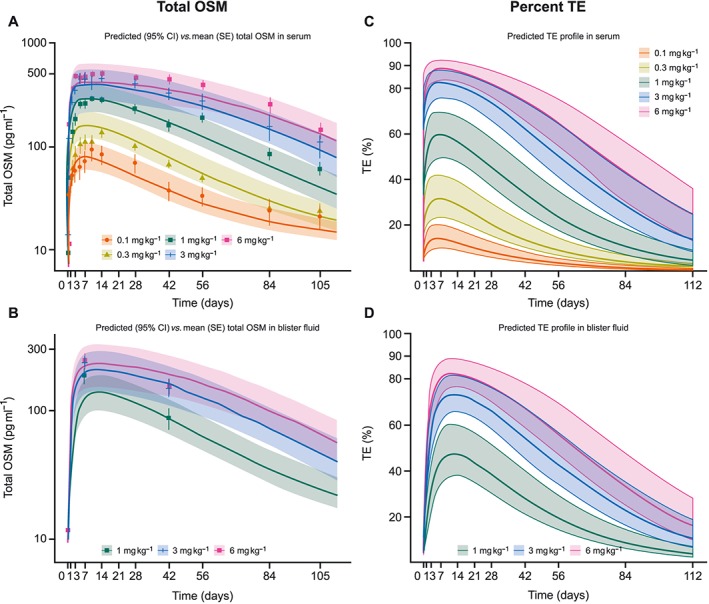
Mean observed vs. mean predicted total OSM concentration by treatment group in serum (A) and blister fluid (B); and mean predicted % TE in serum (C) and blister fluid (D) (PK population). CI, confidence interval; OSM, oncostatin M; PK, pharmacokinetic; SE, standard error; TE, target engagement
The estimated in vivo affinity of GSK2330811 to OSM was 0.568 nM [95% confidence interval (CI) 0.455, 0.710] in the one‐compartment PK with TMDD model. A rapid free OSM degradation rate constant of 2.05 h−1 (95% CI 1.62, 2.59) was estimated. Similar results were obtained using the combined mPBPK and TMDD model [in vivo affinity of GSK2330811 to OSM: 0.629 nM (95% CI 0.494, 0.802); free OSM target turnover rate: 1.90 h−1 (95% CI 1.45, 2.50)].
Target engagement was assessed using model predictions as measured free OSM levels were below the LLQ. Following a single 6 mg kg–1 SC administration of GSK2330811, target engagement predictions from the physiologically based PK model (mPBPK) were approximately 90% in serum (Figure 5C) and >80% in skin (Figure 5D). Model‐predicted free OSM concentrations were estimated above the LLQ of the assay (Figure S4). This was inconsistent with free OSM observations that were below the LLQ after dosing administration.
Discussion
This first‐time‐in‐human study demonstrated a favourable safety and tolerability profile of GSK2330811 in healthy subjects. There were no deaths, nonfatal SAEs, or AEs leading to withdrawal from the study. Three AEs were suspected by the blinded investigator to be drug related – two of injection site bruising (one in each of the placebo and GSK2330811 0.3 mg kg–1 groups) and one of injection site pain (in the GSK2330811 0.1 mg kg–1 group). No treatment‐related anti‐GSK2330811 antibodies were detected in any subjects. There were no clinically relevant trends in change from baseline in laboratory values, vital signs or ECGs, with the exception of changes in platelet counts.
A reversible reduction in platelet count was observed at the higher (1–6 mg kg–1) dose levels. This was expected, as the previous anti‐OSM antibody (GSK315234) demonstrated a modest dose‐dependent effect on platelet count 11, although platelet reduction with GSK315234 was not considered clinically significant. In addition, OSM‐deficient mice exhibit anaemia and thrombocytopenia 24. OSM is known to regulate haematopoiesis via stimulation of bone marrow stromal cells and haematopoietic progenitors 25, 26. A reduction in haemoglobin and red blood cell parameters was also seen at the 6 mg kg–1 dose level, with evidence of recovery beginning during the study period and reticulocytosis starting from day 21 to 28, providing early evidence of compensatory mechanisms. Dose‐related reductions in peripheral platelet counts have also been observed with treatment with anti‐IL‐6 agents, including tocilizumab 27, 28, but in contrast to IL‐6 inhibition, inhibition of OSM did not lead to a dose‐related reduction in neutrophil counts in the present study.
PK parameters estimated using modelling for GSK2330811 were consistent with an IgG1 antibody against a soluble target 29, with a typical apparent distribution volume of 11.5 l and typical apparent systemic clearance of 14.1 ml h–1. GSK2330811 PK were approximately linear over the dose range of 0.1 mg kg–1 to 6 mg kg–1, and the mean terminal half‐life was estimated at approximately 24 days.
Understanding the complex pharmacology and interdependent PK and PD properties of mAbs is important in improving their clinical success 30. For mAbs against soluble targets such as cytokines, in many cases drug PK will not be affected by binding to the target, but rather the kinetics of the target will be affected by the drug. Ligands such as cytokines often have very low baseline levels and short half‐lives in the range of minutes, while the mAbs targeting them often have longer half‐lives in the range of weeks and are administered in high molar excess. Binding of a mAb to a ligand with rapid turnover usually results in significant accumulation of mAb–ligand complex above baseline levels of the ligand, and in low free‐ligand concentrations 31, 32 often below the LLQ of the assay. However, it is of central relevance to determine the reduction in the free ligand due to binding to the target, as it is a direct measurement of target engagement and its magnitude and duration theoretically drive the mAb efficacy. When free ligand cannot be measured directly owing to assay sensitivity, PK/PD modelling approaches using total target data (unbound and bound) can be used to assess target engagement.
The present study was prospectively designed to determine if the improved in vitro affinity of GSK2330811 led to improved affinity in vivo and to measure target engagement in skin, a target tissue in SSc. Saturation of total OSM was observed after dosing with 3 mg kg–1 and 6 mg kg–1, with total OSM concentration achieving up to an 80‐fold accumulation with respect to baseline values. Comparable accumulation levels had been observed with GSK315234 after dosing with 30 mg kg–1. Furthermore, total OSM accumulation levels obtained after dosing with 1 mg kg–1 GSK2330811 were comparable with levels obtained after dosing with 10 mg kg–1 GSK315234 (data on file). These results showed an improved in vivo affinity consistent with 10‐fold increased binding in vitro affinity. PK/PD models using drug PK and total OSM data were developed to assess the in vivo affinity of GSK2330811 for OSM. The one‐compartment PK with TMDD model gave an estimated in vivo affinity of GSK2330811 for OSM of approximately 0.6 nM, with an OSM half‐life of approximately 30 min (half‐life was computed as the ratio between the natural logarithm of 2 and the estimated OSM degradation rate constant). A similar estimate of in vivo GSK2330811 to target OSM affinity and target turnover was achieved from a mPBPK model including both serum and skin blister fluid data.
PKPD models development and validation were based only on total OSM and total GSK2330811 data. Predicted free OSM was not in agreement with the below LLQ measurements for the free target. Discrepancy between free and total target data is not unusual for therapeutic proteins binding to soluble target. As widely reported in the literature, measuring the concentration of a free soluble target in the presence of a capturing drug is challenging, and total data are considered more reliable 32, 33.
According to the model predictions, GSK2330811 achieved approximately 90% and 80% target engagement in serum and skin blister fluid, respectively, following a single 6 mg kg–1 GSK2330811 SC administration. Model predictions after repeat dosing can be used to inform dose selection in subsequent studies.
The skin blister model allowed measurement of OSM and GSK2330811 levels in skin interstitial fluid and provided information on target engagement within the skin. GSK2330811 was successfully detected in skin blister fluid at concentrations ranging from 19% to 45% of those observed in plasma, in line with the results of Dragatin et al. 34. The skin blisters were well tolerated, with no AEs related to blister healing reported. These findings, alongside another study using the skin blister model 35, support the use of a dermal suction blister model as a minimally invasive technique for measuring PK and assessing target engagement of cytokines in the skin compartment, and highlight its potential utility in future clinical studies for indications with skin involvement.
The limitations of the present study included the fact that GSK2330811 was assessed as a single dose and in a healthy study population; further assessment of GSK2330811 distribution and skin OSM levels following repeat dosing and in a patient population are required. Due to the parallel study design of the study, within‐subject variability to each ascending dose level could not be studied.
In conclusion, single SC doses of 0.1–6.0 mg kg–1 GSK2330811 were well tolerated in healthy subjects. GSK2330811 showed linear PK over this dose range, and a mean terminal half‐life estimated at approximately 24 days with sufficient affinity to achieve target engagement in the systemic circulation and target skin tissue. Together with the favourable safety and tolerability profile, this finding supports the progression of GSK2330811 clinical development in inflammatory and fibrotic disease. A proof‐of‐mechanism study in patients with diffuse cutaneous SSc is now under way (NCT03041025).
Competing Interests
All authors are employees of GSK and hold stock or stock options. J.R., S.Z., M.F., C.Z. and N.W. hold a GSK patent.
The authors thank Dr Subramanya Kumar, Karen McGlashan, Sharon Crosby and the biomarker laboratory and pharmacy staff at the GlaxoSmithKline (GSK) Clinical Unit, Cambridge, UK; Professor Caroline Savage and Dr Shaun Flint of the Immunoinflammation Department at GSK, and staff in the GSK Drug Metabolism and Pharmacokinetics and Clinical Immunology Departments. The authors acknowledge medical writing support provided by Clare Slater and Katy Tucker of Fishawack Indicia Ltd, UK, funded by GSK.
Contributors
K.E. and D.F. made substantial contributions to the conception and design of the work; acquisition, analysis and interpretation of the data; as well as drafting and revising the work. J.R., S.Z., K.N. and N.W. made substantial contributions to the conception and design of the work, analysis and interpretation of the data, as well as drafting and revising the work. S.‐A.R. and M.F. made substantial contributions to the conception and design of the work, as well as drafting and revising the work. C.Z. made substantial contributions to the analysis and interpretation of the data, as well as drafting and revising the work. All authors have revised the work critically for important intellectual content and have provided final approval of the version to be published. Authors give their agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supporting information
Figure S1 Study design
Figure S2 Subject disposition
Figure S3 Mean reticulocytes (TI l–1) to first follow‐up visit, by GSK2330811 treatment group (all‐subjects population)
Figure S4 Predicted (95% confidence interval) vs. baseline free oncostatin M in serum (A) and skin blister fluid (B)
Table S1 Overview of platelet counts (all‐subjects population)
Supplementary Materials Supplementary Methods
Reid, J. , Zamuner, S. , Edwards, K. , Rumley, S.‐A. , Nevin, K. , Feeney, M. , Zecchin, C. , Fernando, D. , and Wisniacki, N. (2018) In vivo affinity and target engagement in skin and blood in a first‐time‐in‐human study of an anti‐oncostatin M monoclonal antibody. Br J Clin Pharmacol, 84: 2280–2291. 10.1111/bcp.13669.
References
- 1. Mosley B, De Imus C, Friend D, Boiani N, Thoma B, Park LS, et al Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM‐specific receptor activation. J Biol Chem 1996; 271: 32635–32643. [DOI] [PubMed] [Google Scholar]
- 2. Gearing DP, Comeau MR, Friend DJ, Gimpel SD, Thut CJ, McGourty J, et al The IL‐6 signal transducer, gp130: an oncostatin M receptor and affinity converter for the LIF receptor. Science 1992; 255: 1434–1437. [DOI] [PubMed] [Google Scholar]
- 3. Hermanns HM. Oncostatin M and interleukin‐31: cytokines, receptors, signal transduction and physiology. Cytokine Growth Factor Rev 2015; 26: 545–558. [DOI] [PubMed] [Google Scholar]
- 4. Mozaffarian A, Brewer AW, Trueblood ES, Luzina IG, Todd NW, Atamas SP, et al Mechanisms of oncostatin M‐induced pulmonary inflammation and fibrosis. J Immunol 2008; 181: 7243–7253. [DOI] [PubMed] [Google Scholar]
- 5. Richards CD. The enigmatic cytokine oncostatin m and roles in disease. ISRN Inflamm 2013; 2013: 512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tanaka M, Hirabayashi Y, Sekiguchi T, Inoue T, Katsuki M, Miyajima A. Targeted disruption of oncostatin M receptor results in altered hematopoiesis. Blood 2003; 102: 3154–3162. [DOI] [PubMed] [Google Scholar]
- 7. West NR, Hegazy AN, Owens BMJ, Bullers SJ, Linggi B, Buonocore S, et al Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor‐neutralizing therapy in patients with inflammatory bowel disease. Nat Med 2017; 23: 579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsuda M, Tsurusaki S, Miyata N, Saijou E, Okochi H, Miyajima A, et al Oncostatin M causes liver fibrosis by regulating cooperation between hepatic stellate cells and macrophages in mice. Hepatology 2018; 67: 296–312. [DOI] [PubMed] [Google Scholar]
- 9. Feeney M, Syed F, Khan K, Shiwen X, Sully K, Trinder S, et al Oncostatin M as a potential molecular target in systemic sclerosis. Arthritis Rheumatol 2015; 67 (Suppl. 10): 1–4046. [Google Scholar]
- 10. Allanore Y, Simms R, Distler O, Trojanowska M, Pope J, Denton CP, et al Systemic sclerosis. Nat Rev Dis Primers 2015; 1: 15002. [DOI] [PubMed] [Google Scholar]
- 11. Choy EH, Bendit M, McAleer D, Liu F, Feeney M, Brett S, et al Safety, tolerability, pharmacokinetics and pharmacodynamics of an anti‐oncostatin M monoclonal antibody in rheumatoid arthritis: results from phase II randomized, placebo‐controlled trials. Arthritis Res Ther 2013; 15: R132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kuhns DB, DeCarlo E, Hawk DM, Gallin JI. Dynamics of the cellular and humoral components of the inflammatory response elicited in skin blisters in humans. J Clin Invest 1992; 89: 1734–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clark KE, Lopez H, Abdi BA, Guerra SG, Shiwen X, Khan K, et al Multiplex cytokine analysis of dermal interstitial blister fluid defines local disease mechanisms in systemic sclerosis. Arthritis Res Ther 2015; 17: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akbar AN, Reed JR, Lacy KE, Jackson SE, Vukmanovic‐Stejic M, Rustin MH. Investigation of the cutaneous response to recall antigen in humans in vivo . Clin Exp Immunol 2013; 173: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Committee for Medicinal Products for Human Use . Guideline on strategies to identify and mitigate risks for first‐in‐human clinical trials with investigational medicinal products. 2007. Available at https://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf (last accessed 25 June 2018). [DOI] [PMC free article] [PubMed]
- 16. Aston PJ, Derks G, Raji A, Agoram BM, van der Graaf PH. Mathematical analysis of the pharmacokinetic‐pharmacodynamic (PKPD) behaviour of monoclonal antibodies: predicting in vivo potency. J Theor Biol 2011; 281: 113–121. [DOI] [PubMed] [Google Scholar]
- 17. Mager DE, Jusko WJ. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J Pharmacokinet Pharmacodyn 2001; 28: 507–532. [DOI] [PubMed] [Google Scholar]
- 18. Gibiansky L, Gibiansky E. Target‐mediated drug disposition model and its approximations for antibody‐drug conjugates. J Pharmacokinet Pharmacodyn 2014; 41: 35–47. [DOI] [PubMed] [Google Scholar]
- 19. Cao Y, Jusko WJ. Incorporating target‐mediated drug disposition in a minimal physiologically‐based pharmacokinetic model for monoclonal antibodies. J Pharmacokinet Pharmacodyn 2014; 41: 375–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN)‐‐a Perl module for NONMEM related programming. Computer methods and programs in biomedicine 2004; 75: 85–94. [DOI] [PubMed] [Google Scholar]
- 21. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 2017; 174 (Suppl. 1): S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. National Institutes of Health . Common terminology criteria for adverse events (CTCAE) v4.03. 2010. Available at https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf (last accessed 25 June 2018).
- 24. Minehata K, Takeuchi M, Hirabayashi Y, Inoue T, Donovan PJ, Tanaka M, et al Oncostatin m maintains the hematopoietic microenvironment and retains hematopoietic progenitors in the bone marrow. Int J Hematol 2006; 84: 319–327. [DOI] [PubMed] [Google Scholar]
- 25. Wallace PM, MacMaster JF, Rillema JR, Peng J, Burstein SA, Shoyab M. Thrombocytopoietic properties of oncostatin M. Blood 1995; 86: 1310–1315. [PubMed] [Google Scholar]
- 26. Miyajima A, Kinoshita T, Tanaka M, Kamiya A, Mukouyama Y, Hara T. Role of Oncostatin M in hematopoiesis and liver development. Cytokine Growth Factor Rev 2000; 11: 177–183. [DOI] [PubMed] [Google Scholar]
- 27. Gibiansky L, Frey N. Linking interleukin‐6 receptor blockade with tocilizumab and its hematological effects using a modeling approach. J Pharmacokinet Pharmacodyn 2012; 39: 5–16. [DOI] [PubMed] [Google Scholar]
- 28. Roche Registration Ltd . RoActemra (toclizumab) Summary of Product Characteristics. 2017. Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/000955/WC500054890.pdf (last accessed 25 June 2018).
- 29. Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci 2004; 93: 2645–2668. [DOI] [PubMed] [Google Scholar]
- 30. Kamath AV. Translational pharmacokinetics and pharmacodynamics of monoclonal antibodies. Drug Discov Today Technol 2016; 21–22: 75–83. [DOI] [PubMed] [Google Scholar]
- 31. Davda JP, Hansen RJ. Properties of a general PK/PD model of antibody‐ligand interactions for therapeutic antibodies that bind to soluble endogenous targets. MAbs 2010; 2: 576–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee JW, Kelley M, King LE, Yang J, Salimi‐Moosavi H, Tang MT, et al Bioanalytical approaches to quantify ʻtotalʼ and ʻfreeʼ therapeutic antibodies and their targets: technical challenges and PK/PD applications over the course of drug development. AAPS J 2011; 13: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang C, Prueksaritanont T. Theoretical analysis of interplay of therapeutic protein drug and circulating soluble target: temporal profiles of 'free' and 'total' drug and target. Pharm Res 2011; 28: 2447–2457. [DOI] [PubMed] [Google Scholar]
- 34. Dragatin C, Polus F, Bodenlenz M, Calonder C, Aigner B, Tiffner KI, et al Secukinumab distributes into dermal interstitial fluid of psoriasis patients as demonstrated by open flow microperfusion. Exp Dermatol 2016; 25: 157–159. [DOI] [PubMed] [Google Scholar]
- 35. Bouma G, Zamuner S, Hicks K, Want A, Oliveira J, Choudhury A, et al CCL20 neutralization by a monoclonal antibody in healthy subjects selectively inhibits recruitment of CCR6+ cells in an experimental suction blister. Br J Clin Pharmacol 2017; 83: 1976–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Study design
Figure S2 Subject disposition
Figure S3 Mean reticulocytes (TI l–1) to first follow‐up visit, by GSK2330811 treatment group (all‐subjects population)
Figure S4 Predicted (95% confidence interval) vs. baseline free oncostatin M in serum (A) and skin blister fluid (B)
Table S1 Overview of platelet counts (all‐subjects population)
Supplementary Materials Supplementary Methods