

Abstract
Aims
Trastuzumab is a humanized monoclonal antibody that binds the human epidermal growth factor receptor 2 (HER2) oncoprotein and is an effective therapy for HER2‐overexpressing breast cancer. MYL‐1401O is a trastuzumab biosimilar. Here, we report results from a phase 1 study that investigated bioequivalence among MYL‐1401O, reference EU‐trastuzumab and US‐trastuzumab.
Methods
This single‐centre, randomized, double‐blind, three‐arm, parallel‐group, phase 1 study was conducted in healthy adult male volunteers. Subjects were randomized 1:1:1 to receive a single 8 mg kg−1 dose of MYL‐1401O, EU‐trastuzumab or US‐trastuzumab as a 90‐min intravenous infusion. The primary objective was to assess PK similarity among all three products. Primary endpoints assessed were peak serum concentration (Cmax), area under the serum concentration–time curve from time of dosing to time of last quantifiable concentration and from time of dosing to infinity. Secondary endpoints included time of Cmax, elimination rate constant, half‐life, safety and immunogenicity.
Results
Of 132 subjects enrolled (44/treatment), 120 (MYL‐1401O, n = 42; EU‐trastuzumab, n = 41; US‐trastuzumab, n = 37) were included in the PK analysis. The 90% confidence intervals of the ratios of geometric means for the primary endpoints were bounded within the predefined bioequivalence criterion of 80–125%. Secondary endpoints time of Cmax, elimination rate constant and half‐life were similar among groups. All treatment‐emergent adverse events were mild or moderate, similar across groups and no serious adverse events were reported. No treatment‐related antidrug antibodies were detected.
Conclusions
MYL‐1401O was well tolerated and demonstrated PK and safety profiles similar to EU‐trastuzumab and US‐trastuzumab in healthy volunteers (ClinicalTrials.gov, NCT02594761).
Keywords: bioequivalence, breast cancer, cancer, pharmacokinetics, phase 1
What is Already Known about this Subject
The monoclonal antibody trastuzumab is an effective therapy for HER2‐overexpressing breast cancer
Biosimilars of existing therapies can decrease costs and improve patient access
MYL‐1401O is a trastuzumab biosimilar and this manuscript presents data from a phase 1 pharmacokinetic trial evaluating its bioequivalence with reference trastuzumab
What this Study Adds
In this population of healthy male subjects, MYL‐1401O demonstrated pharmacokinetic bioequivalence to reference trastuzumab; additionally, safety and tolerability results were consistent with previous trastuzumab studies
These data, in combination with data from the phase 3 HERITAGE study, support the biosimilarity between MYL‐1401O and trastuzumab
Introduction
Monoclonal antibodies (mAbs) have been successfully established as monotherapy, or combination therapies, in the first‐ and second‐line setting, and as palliative therapy for solid tumours and haematological malignancies 1, 2, 3. These antibodies can target the host immune response or modify specific intracellular pathways, reducing tumour proliferation itself, increasing the efficacy of radiation or chemotherapies, or both 1.
Approximately 15–20% of invasive breast cancers overexpress the oncoprotein human epidermal growth factor receptor 2 (HER2), which is normally present in limited amounts on the outer surface of the cell membrane in various tissues 4, 5. HER2 is unique in that it can form ligand‐independent homodimers and overexpression of HER2 can lead to its activation 6, 7. Trastuzumab is a humanized immunoglobulin G1 (IgG1) mAb directed against HER2; blocking receptor activity leads to selective inhibition of the downstream HER2 pathway 1, which arrests the cell cycle in G1 and induces antibody‐dependent cellular toxicity 8. Trastuzumab has been evaluated in female patients with metastatic breast cancer; in a phase 3 trial, compared with chemotherapy alone, chemotherapy in combination with trastuzumab improved survival and response in HER2‐positive metastatic breast and gastric cancers 9, 10, 11, 12. Trastuzumab is approved for the treatment of HER2‐overexpressing breast cancer and metastatic gastric or gastroesophageal junction adenocarcinoma 10.
Despite significant therapeutic improvement, biologic therapies such as mAbs are costly, limiting access across the globe 13, 14. Biosimilars, developed as copies of existing biologics, have been available in European markets for roughly a decade. The European Medicines Agency (EMA) approved the first mAb infliximab biosimilars in 2013, Remsima (Celltrion, Incheon, Republic of Korea) and Inflectra (Pfizer, New York, NY, USA) 15. In 2016, the US Food and Drug Administration (FDA) approved its first two mAb biosimilars: Inflectra, an infliximab biosimilar, and Amjevita (Amgen, Thousand Oaks, CA, USA), an adalimumab biosimilar 16. Both agencies have continued to approve biosimilars, and as patents expire on existing biologics and new regulations are implemented, biosimilars will become more readily available worldwide 13, 17, 18. Biosimilars are not characterized in the same way as traditional small molecules because of the molecular complexity and multifaceted production process of biologics; therefore, the FDA and the EMA have established guidance for the development and approval of biosimilars. The FDA defines biosimilarity to mean “the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components,” with “no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity, and potency of the product” 19. As most biologics are often used in combination with chemotherapy and have a variety of indications, demonstrating bioequivalence with mAbs is complicated 20.
MYL‐1401O is the first FDA‐approved biosimilar for the treatment of breast or stomach cancer and the second FDA‐approved biosimilar for the treatment of cancer 21. It encodes an IgG1 mAb that is 100% identical in amino acid sequence to the heavy chain and light chain of the trastuzumab sequence. Physicochemical analyses and previous studies, including the phase 3 HERITAGE study, have demonstrated the similarity of MYL‐1401O to European‐sourced trastuzumab (EU‐trastuzumab) and US‐sourced trastuzumab (US‐trastuzumab) 9, 12, 22, 23, 24. This manuscript presents data from a phase 1 pharmacokinetic (PK) trial evaluating the bioequivalence of MYL‐1401O with both EU‐trastuzumab and US‐trastuzumab, and of EU‐trastuzumab with US‐trastuzumab.
Methods
Study design
This phase 1, single‐centre, randomized, double‐blind, three‐arm, parallel‐group, 10‐week study was performed between 8 August 2013 and 27 February 2014, in healthy male adults (NCT02594761). The study was conducted using a parallel‐group design because a crossover design was deemed impractical owing to the long treatment periods and washout interval, which can last as long as 7 months 10. Drugs used in this study were MYL‐1401O (Biocon Ltd, Bangalore, India, for Mylan Inc, Canonsburg, PA), reference EU‐trastuzumab (Herceptin, Roche Diagnostics GmbH, Mannheim, Germany) and reference US‐trastuzumab (Herceptin, Genentech, Inc, South San Francisco, CA).
Subjects were randomized on a 1:1:1 basis per protocol to receive MYL‐1401O, EU‐trastuzumab or US‐trastuzumab (Fig. 1). Subjects received a single 8 mg kg−1 intravenous infusion per body weight of test or study drug reconstituted and diluted in 250 ml of saline over 90 min. The dose of 8 mg kg−1 was chosen because it is the clinically approved loading dose of trastuzumab for the every‐3‐weeks regimen (i.e. initial dose of 8 mg kg−1 followed by 6 mg kg−1 every 3 weeks); this is in agreement with authorities for assessment of PK, safety and immunogenicity in healthy human volunteers 10. Subjects were excluded if they had a history of any significant disease, used any medication 7 days before start of study or participated in a clinical trial within 30 days of start of study. Healthy male subjects aged 18–55 years who weighed >50 kg with body mass index 18–30 kg m–2 inclusive, had no known cardiac history (nonhypertensive, left ventricular ejection fraction >50%), understood study procedures, agreed to participate and were willing to give informed consent were eligible.
Figure 1.
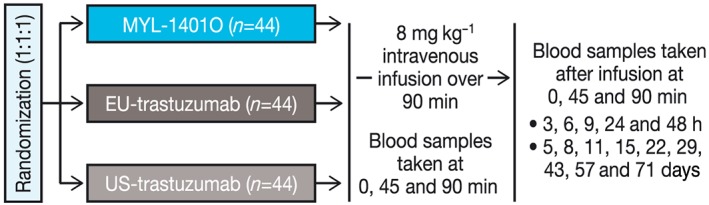
Study design. Healthy males were randomized to receive a 90‐min infusion of 8 mg kg−1 of MYL‐1401O, EU‐trastuzumab or US‐trastuzumab. Blood samples were collected as shown
The study was approved by Alpha Institutional Review Board (San Clemente, CA) and conducted in accordance with the guidelines set forth by the EMA, the International Council for Harmonization of Guidelines for Good Clinical Practice (E6) and the US Code of Federal Regulations Guidelines for Good Clinical Practice (21 CFR Parts 50 and 56) regarding the treatment of human subjects in a study. All subjects who agreed to participate in the study were required to provide the investigators with properly documented and fully executed informed consent before initiation of any study‐specific activities.
PK endpoints
Blood samples (3.5 ml each) were collected immediately before dose administration (0 h); at 45 and 90 min after the start of infusion; postdose at 3, 6, 9, 24 and 48 h relative to the start of infusion; and on days 5, 8, 11, 15, 22, 29, 43, 57 and 71. Serum samples were assayed for trastuzumab concentration using a validated enzyme‐linked immunosorbent assay (ELISA) method (described below). Three similarity assessments were performed: (i) MYL‐1401O and EU‐trastuzumab; (ii) MYL‐1401O and US‐trastuzumab; and (iii) EU‐trastuzumab and US‐trastuzumab. The primary PK endpoints were peak serum concentration of trastuzumab (Cmax), area under the serum concentration–time curve from the time of dosing to the time of last quantifiable concentration (AUC0‐last) and AUC from the time of dosing to infinity (AUC0‐∞). Secondary endpoints included time of Cmax (tmax), elimination rate constant (λz) and half‐life (t½).
ELISA
A validated sandwich ELISA format was used with a monoclonal anti‐idiotype antibody against trastuzumab. Calibration standards and quality controls were prepared in neat pooled human serum. The concentration of MYL‐1401O, EU‐trastuzumab or US‐trastuzumab in samples was back‐calculated from the calibration curve. The lower limit of quantitation of the validated assay was 75 ng ml−1 in neat serum. Analytical studies were performed at Covance Laboratories (Harrogate, UK).
Safety and tolerability
Safety and tolerability assessments included adverse event (AE) collection, vital signs, cardiac monitoring, clinical laboratory assessments [including C‐reactive protein (CRP) and immunoglobulins], local tolerability assessments (according to the phlebitis scale), physical examination and immunogenicity. Any clinically significant observations outside the range of normal were recorded as AEs. A treatment‐emergent AE (TEAE) was defined as any new AE or worsening of an existing condition after treatment administration up to and including the end‐of‐study visit (day 71).
Blood and urine samples were collected at screening and study exit for clinical laboratory evaluations of haematology, urinalysis, clinical chemistry and serology. Cardiac parameters were measured by 12‐lead electrocardiogram (ECG) and echocardiography using left ventricular ejection fraction. A 12‐lead ECG was obtained and evaluated at screening, predose (baseline), and at 90 min (before end of infusion) and 3 h postdose (relative to the start of infusion), and on day 71 (or at early termination). Twelve‐lead ECGs were collected in the supine position after the subject had been quietly resting for 10 min. Echocardiography was measured at screening, day 15 and day 71 (or at early termination).
Measurements of CRP, a marker of acute inflammatory response, were evaluated at screening and at days 1 (predose and 3 h postdose), 2, 3, 8 and 71 (or at early termination).
Immunogenicity testing
Measurements of IgA, IgG and IgM were evaluated at days 1, 8 and 71 (or at early termination). Immunogenicity samples were taken at predose (day 1) and at the last PK sample time point (day 71 or early termination) and measured with a bridging immunoassay format using the Meso Scale Discovery electrochemiluminescence (ECL) detection technology (Rockville, MD, USA). Samples were analysed as recommended in a multitiered fashion 25, 26, 27. Sample ECL signals that were greater than the cut‐off (established during validation at a 5% false‐positive rate) were identified as positive in this screening assay and then tested in a confirmatory test where the percentage inhibition of ECL signal in the presence of excess drug was determined and compared with a confirmatory cut‐off (0.1% false‐positive rate). Confirmed positive samples were then evaluated in the titration tier where samples were serially diluted using 1.5‐fold dilution steps until the ECL signal dropped below the screening assay cut‐off. The last dilution (including the minimum required dilution) that yielded an ECL signal above the cut‐off was the reported titre. Results were also reported as the log10 of the dilution. The assays for antidrug antibody (ADA) detection were fully validated before use in the evaluation of study samples. During validation, the assays were demonstrated to detect 50 ng ml–1 of ADA (as the surrogate positive control) in the presence of 100 μg ml–1 of trastuzumab. During study sample analysis, the drug tolerance limit was not exceeded for any of the immunogenicity samples. Analytical studies were performed at Covance Laboratories.
Statistical analysis
Single‐dose PK parameters for trastuzumab were calculated using noncompartmental techniques. Statistical analyses were performed on PK parameters using the general linear models procedure (PROC GLM) of SAS software (SAS Institute, Cary, NC, USA). The postdosing drug concentration values were used for the calculation. The Cmax and tmax were determined from the observed serum concentration‐time profile over the sampling time interval. The λz was determined by linear regression of the terminal linear phase of the log serum concentration–time profile. The AUC0‐last was the sum of the linear trapezoidal estimation of the areas from the time of dosing to the time of the last quantifiable concentration. The AUC0‐∞ was calculated as AUC0‐∞ = AUC0‐last + LQC / λz where LQC is the last quantifiable concentration; t½ was calculated as t½ = 0.693 / λz. The hypothesis of nonequivalent bioavailability between any two study drugs was to be rejected if each 90% confidence interval (CI) for two one‐sided tests (α = 0.05) for Cmax, AUC0‐last and AUC0‐∞ was in the 80–125% range for the natural log‐transformed data. Single‐dose PK parameters (natural log‐transformed parameter CI) for trastuzumab were analysed using analysis of variance (ANOVA).
Determination of sample size
On the basis of a previous crossover PK study, a sample size of 132 subjects (44 per treatment group) was deemed sufficient to provide at least 82% power overall, assuming intersubject variability ≤25%, a true geometric mean ratio (GMR) between 0.92 and 1.08 and ≥ 90% of subjects completing the study. The power calculation was for a 90% CI (for two one‐sided α = 0.05 tests) of the GMR in the range of 80–125%.
Results
Subject demographics
A total of 132 subjects (44 per treatment group) were enrolled per protocol in the study. The mean age was 32.9 years and the mean body mass index was 25.5 kg m−2. Baseline characteristics were generally similar across all treatment groups (Table 1). Of the subjects enrolled, 121 completed the clinical portion of the study (MYL‐1401O, n = 42; EU‐trastuzumab, n = 41; US‐trastuzumab, n = 38) and 11 withdrew consent and discontinued (MYL‐1401O, n = 2; EU‐trastuzumab, n = 3; US‐trastuzumab, n = 6; Supplemental Figure). No subjects withdrew because of TEAEs and all subjects who withdrew consent did so for personal reasons, including work‐schedule conflicts and family emergencies. One subject in the US‐trastuzumab group did not receive the correct amount of dose in infusion because of a dose preparation error and was discontinued from bioanalytical analysis by the sponsor. Thus, 120 subjects were included in the PK analysis.
Table 1.
Baseline characteristics
| Parameter, mean (SD) | MYL‐1401O (n = 44) | EU‐trastuzumab (n = 44) | US‐trastuzumab (n = 44) |
|---|---|---|---|
| Age, y | 31.9 (9.5) | 33.0 (10.0) | 33.9 (9.9) |
| Weight, kg | 82.0 (10.9) | 79.2 (11.1) | 79.2 (12.2) |
| Height, cm | 177.8 (7.8) | 176.6 (8.4) | 177.2 (7.3) |
| BMI, kg m −2 | 25.9 (2.8) | 25.3 (2.7) | 25.1 (2.6) |
BMI, body mass index; SD, standard deviation
PK analysis
The trastuzumab serum concentration–time profile was similar across all three treatment groups (Fig. 2). The mean (coefficient of variation [%CV]) for Cmax in the MYL‐1401O, EU‐trastuzumab and US‐trastuzumab groups was 207.1 μg ml−1 (12.44), 199.4 μg ml−1 (13.69) and 205.8 μg ml−1 (15.67), respectively (Table 2). The mean (%CV) for AUC0‐last in the MYL‐1401O, EU‐trastuzumab and US‐trastuzumab groups was 49 704 μg h ml−1 (16.64), 51 642 μg h ml−1 (19.82) and 51 887 μg h ml−1 (14.44), respectively (Table 2). The mean (%CV) for AUC0‐∞ in the MYL‐1401O, EU‐trastuzumab and US‐trastuzumab groups was 49 899 μg h ml−1 (16.93), 51 902 μg h ml−1 (20.01) and 52 259 μg h ml−1 (14.36), respectively (Table 2). Results from ANOVA comparing the primary endpoints of Cmax, AUC0‐last and AUC0‐∞ across all three treatment groups showed that the 90% CIs of the ratios of geometric means ranged between 89.16% and 109.41% and were within the predefined bioequivalence interval of 80–125%. Mean (%CV) for the secondary PK endpoints of tmax (2.880 h [54.83], 3.028 h [118.2] and 2.625 h [53.37]), λz (0.0046 h−1 [22.80], 0.0044 h−1 [27.14] and 0.0042 h−1 [23.45]), and t½ (160.0 h [28.39], 173.8 h [32.92] and 176.4 h [29.85]) was also similar across the MYL‐1401O, EU‐trastuzumab and US‐trastuzumab groups, respectively (Table 2). The ANOVA for secondary PK endpoints under the null hypothesis of no difference resulted in P values of 0.7614 for tmax, 0.2497 for λz and 0.3139 for t½, demonstrating that there was no statistically significant difference among groups for these parameters.
Figure 2.
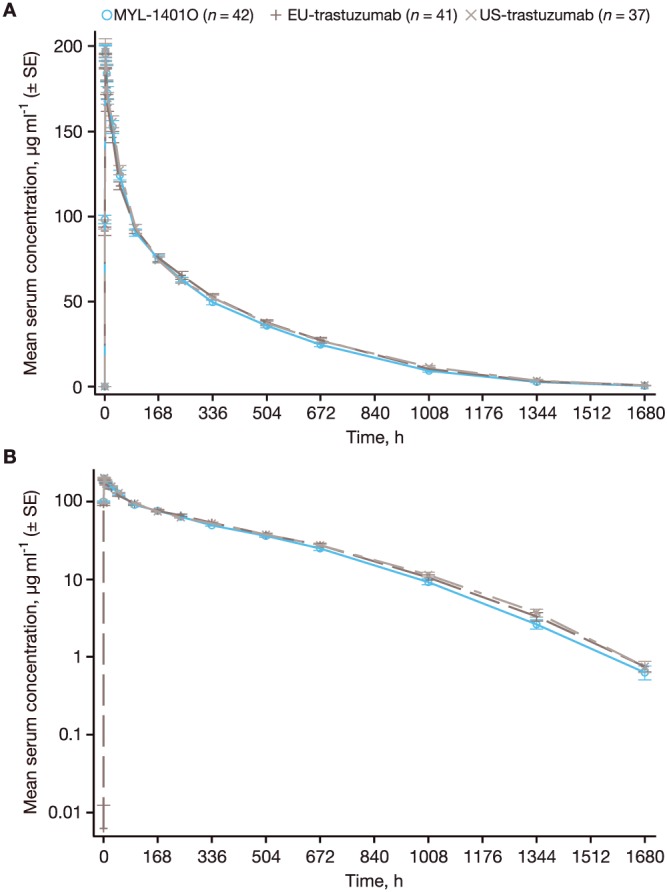
Mean (± standard error) serum concentrations of MYL‐1401O, EU‐trastuzumab and US‐trastuzumab over the course of the study. (A) Linear scale. (B) Semi‐log scale. SE, standard error
Table 2.
Summary of pharmacokinetic parameters in serum (PK analysis set)
| MYL‐1401O/EU‐trastuzumab | MYL‐1401O/US‐trastuzumab | ||||||
|---|---|---|---|---|---|---|---|
| Parameter, mean (%CV) | MYL‐1401O (n = 42) | EU‐trastuzumab (n = 41) | US‐trastuzumab (n = 37) | LS mean ratio | 90% CIa | LS mean ratio | 90% CIa |
| Cmax, μg ml −1 | 207.1 (12.44) | 199.4 (13.69) | 205.8 (15.67) | 1.04 | 98.80–109.41 | 1.01 | 95.81–106.39 |
| AUC 0‐last , μg h ml −1 | 49 704 (16.64) | 51 642 (19.82) | 51 887 (14.44) | 0.97 | 90.90–102.91 | 0.95 | 89.54–101.71 |
| AUC 0‐∞ , μg h ml −1 | 49 899 (16.93) | 51 902 (20.01) | 52 259 (14.36) | 0.97 | 90.76–102.84 | 0.95 | 89.16–101.36 |
| tmax, h | 2.880 (54.83) | 3.028 (118.2) | 2.625 (53.37) | — | — | — | — |
| λz, h −1 | 0.0046 (22.80) | 0.0044 (27.14) | 0.0042 (23.45) | — | — | — | — |
| t ½ , h | 160.0 (28.39) | 173.8 (32.92) | 176.4 (29.85) | — | — | — | — |
AUC0‐∞, area under the serum concentration‐time curve from the time of dosing to infinity; AUC0‐t, area under the serum concentration‐time curve from the time of dosing to the time of last quantifiable concentration; CI, confidence interval; Cmax, peak serum concentration; %CV, coefficient of variation; LS, least squares; PK, pharmacokinetic; t½, half‐life; tmax, time of Cmax; λz, elimination rate constant
Natural log‐transformed parameter was used
Safety and tolerability
Of the 132 subjects who received study medication, 83 experienced a total of 227 TEAEs (MYL‐1401O, 31 subjects with 91 TEAEs; EU‐trastuzumab, 28 subjects with 80 TEAEs; US‐trastuzumab, 24 subjects with 56 TEAEs). All TEAEs were mild or moderate in severity, and no serious AEs were reported. No subjects withdrew because of TEAEs. The most frequently reported AE in all groups was headache (MYL‐1401O, 27.3%; EU‐trastuzumab, 29.5%; US‐trastuzumab, 22.7%; Table 3). Back pain and influenza‐like illness were more frequent in the MYL‐1401O group compared with the reference groups (MYL‐1401O, 15.9% and 11.4%; EU‐trastuzumab, 4.5% and 4.5%; US‐trastuzumab, 2.3% and 0, respectively; Table 3). Chills were more frequent in the EU‐trastuzumab group (25.0%) compared with the MYL‐1401O group (4.5%) and the US‐trastuzumab group (4.5%). The most frequent treatment‐related TEAEs (≥5% of subjects) in the MYL‐1401O group were headache (20.5%), influenza‐like illness (11.4%), back pain (11.4%), fatigue (6.8%) and nausea (6.8%).
Table 3.
Summary of most frequently reported treatment‐emergent adverse events (≥5% of subjects) by treatment
| Adverse event (preferred term), n (%) | MYL‐1401O (n = 44) | EU‐trastuzumab (n = 44) | US‐trastuzumab (n = 44) |
|---|---|---|---|
| Headache | 12 (27.3) | 13 (29.5) | 10 (22.7) |
| Back pain | 7 (15.9) | 2 (4.5) | 1 (2.3) |
| Influenza‐like illness | 5 (11.4) | 2 (4.5) | 0 |
| Upper respiratory tract infection | 4 (9.1) | 4 (9.1) | 2 (4.5) |
| Nausea | 4 (9.1) | 2 (4.5) | 4 (9.1) |
| Pain in extremity | 3 (6.8) | 2 (4.5) | 1 (2.3) |
| Dizziness | 3 (6.8) | 3 (6.8) | 3 (6.8) |
| Fatigue | 3 (6.8) | 1 (2.3) | 2 (4.5) |
| Chills | 2 (4.5) | 11 (25.0) | 2 (4.5) |
| Dry lips | 1 (2.3) | 3 (6.8) | 0 |
| Vomiting | 0 | 3 (6.8) | 1 (2.3) |
An increase in CRP (usually within the normal range) was observed in all subjects within the first 24 h after administration, which returned to each subject's baseline by day 8. No differences were found in cardiac parameters among treatment groups, measured either by ECG or echocardiography. There were no clinically significant changes in vital signs observed during the study. Infusions were well tolerated and local reactions observed were minimal. Three instances of erythema were noted at the infusion site (two of these subjects received MYL‐1401O, one received US‐trastuzumab) and there were two reports of pain (both subjects received EU‐trastuzumab) with erythema or oedema (grade 2 on the phlebitis scale) 90 min postdose, which were resolved within 3 h postdose.
Immunogenicity
The number of subjects with antidrug antibodies (ADA) was comparable among treatment groups (Table 4). Of 252 samples analysed from 126 subjects, six were confirmed positive against MYL‐1401O (n = 3; two samples at predose, one at day 71), EU‐trastuzumab (n = 1; one sample at predose) and US‐trastuzumab (n = 2; one sample at predose, one at day 71). Of the four subjects who were ADA positive at predose, two remained ADA positive at day 71. The change in titre observed for these two subjects was within the precision boundaries of the assay; the change in titre was not significant from baseline, indicating that there were no treatment‐induced or treatment‐boosted ADA‐positive subjects in the study. Serum IgA, IgG and IgM evaluations indicated no evidence of immunogenicity, and analysis of covariance (ANCOVA) indicated a similar response among the three treatment groups (data not shown).
Table 4.
Summary of antidrug antibodies (ADA) samples by treatment
| Visit | Result | MYL‐1401O (n = 43) | EU‐trastuzumab (n = 43) | US‐trastuzumab (n = 40) |
|---|---|---|---|---|
| Day 1 (before dosing), n (%) | ADA negative | 41 (95.3) | 42 (97.7) | 39 (97.5) |
| ADA positive | 2 (4.7) | 1 (2.3) | 1 (2.5) | |
| Day 71 (last PK sample), n (%) | ADA negative | 42 (97.7) | 42 (100) | 39 (97.5) |
| ADA positive | 1 (2.3) | 0 | 1 (2.5) |
PK, pharmacokinetic
Discussion
Bioequivalence was demonstrated in this single‐dose, phase 1 study of MYL‐1401O, EU‐trastuzumab and US‐trastuzumab administered as an 8‐mg kg−1 intravenous infusion. When comparing the primary endpoints across all three treatment groups, ANOVA results showed that the 90% CIs of the ratios of geometric means for these PK parameters ranged between 89.96% and 109.82% and were all within the predefined bioequivalence interval of 80–125% for the natural log‐transformed data for each of the comparisons. Secondary PK endpoints of tmax, λz and t½ were also similar across all treatment groups. MYL‐1401O, EU‐trastuzumab and US‐trastuzumab had similar safety profiles: there was no evidence of immunogenicity, no serious AEs were reported, all TEAEs were mild or moderate in severity, and local reactions were infrequent and mild, indicating that all three products were well tolerated in this population of healthy volunteers.
The PK parameters reported in this study were comparable with those previously reported for other trastuzumab biosimilars and the marketed trastuzumab product when administered as a single intravenous dose of 6 mg kg−1 8, 28, 29. A previous trial compared a trastuzumab biosimilar, FTMB (Synthon Biopharmaceuticals, Nijmegen, The Netherlands), to reference trastuzumab in healthy males and reported a half‐life of ~6.0 days for FTMB and ~6.8 days for reference trastuzumab 10. An open‐label, two‐part, phase 1/1b study (NCT00800436) using EU‐trastuzumab reported half‐lives of ~10.6 days in healthy males and ~10.2 days in female patients with HER2‐positive early breast cancer 29. The phase 1 REFLECTIONS trial randomized healthy male subjects to PF‐05280014 (the Pfizer trastuzumab biosimilar), EU‐trastuzumab or US‐trastuzumab and found the half‐life of these compounds to be ~8.9, ~9.2 and ~8.8 days, respectively 8. Compared with those trials, this study reports shorter half‐lives for MYL‐1401O (~6.7 days), EU‐trastuzumab (~7.2 days) and US‐trastuzumab (~7.4 days) when administered as an 8‐mg kg−1 intravenous infusion. The reason for the comparatively shorter half‐lives is not known; however, in single‐dose studies of MYL‐1401O, we noted higher clearances when drug concentrations tapered to very low levels where receptor binding desaturates. Although still within normal range, we did note increases in CRP in the current study, and CRP has been shown to correlate positively with mAb clearance 30. Values for Cmax, AUC0‐last and AUC0‐∞ for MYL‐1401O were higher in this study compared with those for FTMB and PF‐05280014; the values for reference EU‐trastuzumab and US‐trastuzumab in this trial were also higher compared with those for reference trastuzumab in the other trials. The values reported among MYL‐1401O and reference trastuzumab within this study, and within the FTMB bioequivalence study, the phase 1/1b study and the REFLECTIONS trial, are all similar 8, 10, 29.
MYL‐1401O, EU‐trastuzumab and US‐trastuzumab were well tolerated, with no serious AEs reported in any treatment group. Cardiac safety, CRP, ECG and echocardiography were performed throughout the study and all measures were found to be within normal ranges, with no differences observed among treatment groups. In innovator clinical trials, trastuzumab demonstrated a low risk (1.4%–15%) of causing subclinical or clinical cardiac failure, which was reversible in approximately 50% of affected patients 10. The highest incidence of cardiomyopathy occurs when trastuzumab is administered with an anthracycline, and guidelines for use of trastuzumab indicate that cardiac function should be tested every 3 months during and upon completion of trastuzumab, and every 6 months for at least 2 years after completion of trastuzumab as a component of adjuvant therapy 10. As the present investigation was a single‐dose study, a decline in cardiac function was not necessarily expected nor seen.
Overall, the most frequently reported AE in this trial was headache, reported by an average of 26.5% of subjects; this is consistent with 28.6% of subjects who experienced headache in the phase 1 REFLECTIONS trial of another trastuzumab biosimilar and the safety profile of reference trastuzumab 8, 10. The second most frequently reported AE was back pain, reported in the range of 2.3–15.9% of subjects across treatment groups; AEs of back pain were observed in 22% of patients in a trial of US‐trastuzumab for the treatment of metastatic breast cancer 10. While higher rates of back pain and influenza‐like symptoms occurred in the MYL‐1401O group relative to the reference compound groups, the number of subjects exposed to each treatment was small (n = 44) and, accordingly, the number of subjects experiencing these TEAEs was small. The present study was not designed or powered to compare differences in reported TEAEs; in contrast, the phase 3 trial comparing MYL‐1401O with EU‐trastuzumab and US‐trastuzumab was designed to assess differences in these safety profiles 12. In the phase 3 study, neither back pain nor influenza‐like symptoms were reported in >5% of patients (n = 493), suggesting that these TEAEs are not expected to affect a substantial proportion of patients 12. Acute or delayed anaphylactic reactions did not develop in subjects who were ADA positive, indicating that there was no product‐specific immunogenicity. Overall, safety and tolerability of MYL‐1401O and reference trastuzumab observed in this study were consistent with results from previous studies of trastuzumab 8, 10, 23, 28, 29; all AEs were mild or moderate with no serious AEs observed, indicating that these products were well tolerated.
Conclusions
This study demonstrated that MYL‐1401O, a trastuzumab biosimilar, is bioequivalent to both EU‐trastuzumab and US‐trastuzumab when administered as a single‐dose 8 mg kg–1 intravenous infusion over 90 min in healthy male subjects. Results from ANOVA showed that the 90% CIs of the ratios of geometric means for primary PK parameters were within the predefined bioequivalence interval of 80–125%. Additionally, EU‐trastuzumab and US‐trastuzumab were demonstrated to be bioequivalent to each other. Overall, MYL‐1401O, EU‐trastuzumab and US‐trastuzumab were well tolerated, and no significant safety issues emerged.
Competing Interests
C.F.W. is a consultant/advisory board member for Mylan Inc. A.V. has nothing to disclose. T.E.L., A.S., M.S.L., A.B., P.G., C.D. and E.J.P. are paid employees of Mylan Inc and may hold stock with the company. N.S. is a paid employee of Biocon Research Ltd and may hold stock with the company. M.B and R.S. were employees of Mylan Inc at the time of analysis and may hold stock with the company.
This study was supported by Mylan Inc, Canonsburg, PA, USA, and Biocon Ltd, Bangalore, India. Editorial assistance was provided under the direction of the authors by Ali Rosenberg, PhD, William Turner, PhD, and Jennifer Rossi, MA, ELS, MedThink SciCom, with support from Mylan Inc.
Supporting information
Figure S1 Subject flow chart. aSubject was discontinued by sponsor from bioanalytical analysis because he did not receive the correct amount of dose in infusion because of a dose preparation error
Waller, C. F. , Vutikullird, A. , Lawrence, T. E. , Shaw, A. , Liu, M. S. , Baczkowski, M. , Sharma, R. , Barve, A. , Goyal, P. , Donnelly, C. , Sengupta, N. , and Pennella, E. J. (2018) A pharmacokinetics phase 1 bioequivalence study of the trastuzumab biosimilar MYL‐1401O vs. EU‐trastuzumab and US‐trastuzumab. Br J Clin Pharmacol, 84: 2336–2343. 10.1111/bcp.13689.
References
- 1. Eisenbeis AM, Grau SJ. Monoclonal antibodies and Fc fragments for treating solid tumors. Biol Targets Ther 2012; 6: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bellati F, Napoletano C, Gasparri ML, Visconti V, Zizzari IG, Ruscito I, et al Monoclonal antibodies in gynecological cancer: a critical point of view. Clin Dev Immunol 2011; 2011: 890758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharkey RM, Goldenberg DM. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J Clin 2006; 56: 226–243. [DOI] [PubMed] [Google Scholar]
- 4. Iqbal N, Iqbal N. Human epidermal growth factor receptor 2 (HER2) in cancers: overexpression and therapeutic implications. Mol Biol Int 2014; 2014: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burstein HJ. The distinctive nature of HER2‐positive breast cancers. N Engl J Med 2005; 353: 1652–1654. [DOI] [PubMed] [Google Scholar]
- 6. Cohen BD, Kiener PA, Green JM, Foy L, Fell HP, Zhang K. The relationship between human epidermal growth‐like factor receptor expression and cellular transformation in NIH3T3 cells. J Biol Chem 1996; 271: 30897–30903. [DOI] [PubMed] [Google Scholar]
- 7. Keshamouni VG, Mattingly RR, Reddy KB. Mechanism of 17‐β‐estradiol‐induced Erk1/2 activation in breast cancer cells: a role for HER2 and PKC‐δ. J Biol Chem 2002; 277: 22558–22565. [DOI] [PubMed] [Google Scholar]
- 8. Yin D, Barker KB, Li R, Meng X, Reich SD, Ricart AD, et al A randomized phase 1 pharmacokinetic trial comparing the potential biosimilar PF‐05280014 with trastuzumab in healthy volunteers (REFLECTIONS B327‐01). Br J Clin Pharmacol 2014; 78: 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eiermann W. Trastuzumab combined with chemotherapy for the treatment of HER2‐positive metastatic breast cancer: pivotal trial data. Ann Oncol 2001; 12 (Suppl. 1): S57–S62. [PubMed] [Google Scholar]
- 10. Herceptin [package insert]. South San Francisco, CA: Genentech, Inc, 2017. [Google Scholar]
- 11. Gunturu KS, Woo Y, Beaubier N, Remotti HE, Saif MW. Gastric cancer and trastuzumab: first biologic therapy in gastric cancer. Ther Adv Med Oncol 2013; 5: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rugo HS, Barve A, Waller CF, Hernandez‐Bronchud M, Herson J, Yuan J, et al Effect of a proposed trastuzumab biosimilar compared with trastuzumab on overall response rate in patients with ERBB2 (HER2)‐positive metastatic breast cancer: a randomized clinical trial. JAMA 2017; 317: 37–47. [DOI] [PubMed] [Google Scholar]
- 13. Rugo HS, Linton KM, Cervi P, Rosenberg JA, Jacobs I. A clinician's guide to biosimilars in oncology. Cancer Treat Rev 2016; 46: 73–79. [DOI] [PubMed] [Google Scholar]
- 14. Chopra R, Lopes G. Improving access to cancer treatments: the role of biosimilars. J Glob Oncol 2017; 3: 596–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. European Medicines Agency . European public assessment reports (EPAR): biosimilars [document on the internet]. c1995–2017. Available at http://www.ema.europa.eu/ema/index.jsp?searchType=name&mid=WC0b01ac058001d124&searchGenericType=biosimilars&keyword=Enter+keywords&alreadyLoaded=true&curl=pages%2Fmedicines%2Flanding%2Fepar_search.jsp&status=Authorised&searchTab=searchByAuthType&pageNo=2 (last accessed 25 May 2017).
- 16. US Food and Drug Administration . Purple book: lists of licensed biological products with reference product exclusivity and biosimilarity or interchangeability evaluations [document on the internet]. Silver Spring, MD; no date. Available at https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/ucm411418.htm (last accessed 23 May 2017).
- 17. Al‐Sabbagh A, Olech E, McClellan JE, Kirchhoff CF. Development of biosimilars. Semin Arthritis Rheum 2016; 45 (Suppl. 5): S11–S18. [DOI] [PubMed] [Google Scholar]
- 18. Abi‐Raad R, Smith BR. Biosimilar biologics: never identical but close enough. Transfusion 2015; 55: 229–231. [DOI] [PubMed] [Google Scholar]
- 19. US Food and Drug Administration . Scientific considerations in demonstrating biosimilarity to a reference product guidance for industry [document on the internet]. Silver Spring, MD; 2015. Available at https://www.fda.gov/downloads/drugs/guidances/ucm291128.pdf (last accessed 12 September 2017).
- 20. Schellekens H, Smolen JS, Dicato M, Rifkin RM. Safety and efficacy of biosimilars in oncology. Lancet Oncol 2017; 17: e502–e509. [DOI] [PubMed] [Google Scholar]
- 21. US Food and Drug Administration . FDA approves first biosimilar for the treatment of certain breast and stomach cancers [document on the internet]. Silver Spring, MD; 2017. Available at https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm587378.htm (last accessed 12 December 2017).
- 22. Chtioui H, Vallotton L, Audran R, Dao K, Rothuizen L, Winterfeld U. A bioequivalence study for Hercules, a biosimilar trastuzumab candidate in development. Poster presented at: Pharmacology 2015; December 15–17, 2015; London, UK.
- 23. Rugo H, Barve A, Waller CF, Bronchud M, Herson J, Yuan J, et al Heritage, a phase III safety and efficacy trial of the proposed trastuzumab biosimilar, MYL‐1401O vs trastuzumab. Ann Oncol 2016; 27 (suppl 6): vi552–vi587. [Google Scholar]
- 24. Waller CF, Vutikullird A, Lawrence TE, Shaw A, Liu MS, Baczkowski M, et al A pharmacokinetics (PK) bioequivalence trial of proposed trastuzumab biosimilar, MYL‐1401O (A) vs EU‐Herceptin (B) and US‐Herceptin (C). J Clin Oncol 2016; 34 (Suppl. 15): 583. [Google Scholar]
- 25. US Food and Drug Administration . Immunogenicity assessment for therapeutic protein products guidance for industry [document on the internet]. Silver Spring, MD; 2014. Available at https://www.fda.gov/downloads/drugs/guidances/ucm338856.pdf (last accessed 13 April 2018).
- 26. Mire‐Sluis AR, Barrett YC, Devanarayan V, Koren E, Liu H, Maia M, et al Recommendations for the design and optimization of immunoassays used in the detection of host antibodies against biotechnology products. J Immunol Methods 2004; 289: 1–16. [DOI] [PubMed] [Google Scholar]
- 27. US Food and Drug Administration . Assay development and validation for immunogenicity testing of therapeutic protein products guidance for industry [document on the internet]. Silver Spring, MD; 2016. Available at https://www.fda.gov/downloads/Drugs/Guidances/UCM192750.pdf (last accessed 13 April 2018).
- 28. Wisman LA, De Cock EP, Reijers JA, Kamerling IM, Van Os SH, de Kam ML, et al A phase I dose‐escalation and bioequivalence study of a trastuzumab biosimilar in healthy male volunteers. Clin Drug Investig 2014; 34: 887–894. [DOI] [PubMed] [Google Scholar]
- 29. Wynne C, Harvey V, Schwabe C, Waaka D, McIntyre C, Bittner B. Comparison of subcutaneous and intravenous administration of trastuzumab: a phase I/Ib trial in healthy male volunteers and patients with HER2‐positive breast cancer. J Clin Pharmacol 2013; 53: 192–201. [DOI] [PubMed] [Google Scholar]
- 30. Ordás I, Mould DR, Feagan BG, Sandborn WJ. Anti‐TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics‐based dosing paradigms. Clin Pharmacol Ther 2012; 91: 635–646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Subject flow chart. aSubject was discontinued by sponsor from bioanalytical analysis because he did not receive the correct amount of dose in infusion because of a dose preparation error