

Abstract
Aims
Tildrakizumab, an interleukin (IL)‐23 inhibitor, is indicated for the treatment of moderate to severe chronic plaque psoriasis. Although tildrakizumab is not metabolized by, and does not alter, cytochrome P450 (CYP) expression in vitro, clinically significant pharmacokinetic effects through changes in systemic inflammation, which alters CYP metabolism, have been well documented. At the time of study conduct, the effect of modulation of inflammation/cytokines, including IL‐23 inhibition with tildrakizumab, on CYP metabolism, and therefore the potential for disease–drug interactions, in psoriasis patients was unknown. We therefore assessed whether tildrakizumab alters CYP metabolism in subjects with moderate to severe psoriasis.
Methods
This was an open‐label, fixed‐sequence, two‐period trial. In Period 1 (Day 1), subjects received an oral CYP probe cocktail of up to five drugs (midazolam 2 mg [3A4], caffeine 200 mg [1A2], warfarin 10 mg [2C9], omeprazole 40 mg [2C19] and dextromethorphan 30 mg [2D6]), followed by a 7‐day washout. In Period 2, subjects received tildrakizumab 200 mg subcutaneously on Days 1 and 29 and a second CYP probe cocktail on Day 57. Substrate or metabolite pharmacokinetics, safety and changes in Psoriasis Severity Area Index (PASI), interleukin‐6 (IL‐6) and high‐sensitivity C‐reactive protein (hs‐CRP), were assessed.
Results
Twenty subjects (13 men, 7 women) were enrolled. Tildrakizumab had no clinically relevant effect on the pharmacokinetics of any of the probe substrates tested. On Day 57 of Period 2, the median percentage decrease from baseline in PASI score following tildrakizumab was ~93%. There were no clinically relevant changes in IL‐6 or hs‐CRP. Treatment with tildrakizumab was generally well tolerated.
Conclusion
In subjects with moderate to severe psoriasis, tildrakizumab 200 mg did not have a discernible effect on CYP metabolism. The potential for clinically significant drug–drug interactions (DDIs) with tildrakizumab in patients with psoriasis is low. The difference in the occurrence of DDIs seen with anti‐inflammatory agents in rheumatoid arthritis patients compared with psoriasis patients may be due to the much greater extent of systemic inflammation in rheumatoid arthritis as compared to psoriasis.
Keywords: cytochrome P450, disease–drug interaction, high‐affinity humanized IgG1k antibody, interleukin‐23, psoriasis, tildrakizumab
What is Already Known about this Subject
Psoriasis patients are typically treated with multiple concomitant therapies.
There are no publications examining disease–drug interactions with immunomodulator therapy in psoriasis.
We assessed whether the IL‐23 inhibitor tildrakizumab alters CYP metabolism and explored its effects on safety, changes in Psoriasis Area and Severity Index (PASI) and circulating inflammatory markers.
What this Study Adds
In subjects with moderate to severe psoriasis, treatment with tildrakizumab 200 mg resulted in a clinically meaningful reduction in median PASI scores, but did not have a clinically meaningful effect on CYP metabolism.
The potential for clinically meaningful disease–drug interactions with tildrakizumab in psoriasis patients is low.
Introduction
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=8093 (MK‐3222) is a high‐affinity, humanized IgG1/κ antibody that binds selectively to the unique p19 sub‐unit of interleukin‐23 (IL‐23) and is indicated for the treatment of moderate to severe chronic plaque psoriasis.
Psoriasis patients often have comorbidities 1 and therefore are typically treated with multiple concomitant therapies. The possibility of drug–drug interactions (DDIs) is an important clinical consideration in these patients. Monoclonal antibodies (mAbs) like tildrakizumab are not metabolized by cytochrome P450 (CYP) enzymes, nor do they generally have an off‐target direct inductive or inhibitory effect on metabolic enzymes 2. However, during chronic inflammation, specific pro‐inflammatory cytokines (i.e., TNFα, IL‐2, IL‐1β, IL‐6) may suppress the expression of CYP enzymes 3, 4, 5, 6, 7, 8. Treatment with an anti‐inflammatory agent may directly or indirectly reduce the level of inflammatory cytokines, increasing CYP enzyme expression and metabolism, which could potentially alter exposure to co‐administered drugs that are metabolized by CYP enzymes. Clinically significant DDIs through the anti‐inflammatory effects of anti‐inflammatory mAbs have been reported in patients with rheumatoid arthritis 4. Furthermore, increased inflammation associated with acute or chronic illness has been associated with increased exposure and/or reduced clearance of drugs 9, 10, 11. There are also robust data demonstrating that the administration of cytokines or cytokine modulators can affect CYP expression and/or drug interactions 12, 13, 14, 15, 16, 17. These data clearly demonstrate that modulation of systemic inflammation has the potential to significantly affect CYP metabolism.
While modulation of cytokines in severe psoriasis has been observed 18, to our knowledge, DDIs as a result of treatment with immunomodulators in psoriasis have not been reported in the literature. Initially, a preclinical assessment of the potential for an interaction was undertaken. The in vitro effect of IL‐23 on human hepatocytes was examined in the presence and absence of Kupffer cells (specialized macrophages located in the liver). Exposure of hepatocyte:Kupffer cell co‐cultures to IL‐23 resulted in minimal inflammatory cytokine release which was similar to that of untreated controls and also minimal suppression of CYP3A4 activity, suggesting the lack of effect of IL‐23 on hepatocytes 19. While in vitro studies were performed, they were generally considered to have limited value because of the difficulty in making quantitative projections of clinical effects 20. Thus, the present clinical trial was driven by the paucity of data surrounding immunomodulator therapy and CYP metabolism in the psoriasis population and the limited translatability of preclinical evaluations to the clinic. This trial determined the effect of tildrakizumab on CYP metabolism and explored the drug's effects on cytokines associated with systemic inflammation in patients with psoriasis.
Methods
Subjects
Male and female subjects aged between 18 and 65 with a diagnosis of moderate to severe psoriasis vulgaris (affected body surface area (BSA) ≥10%, Psoriasis Area Severity Index (PASI) score ≥12) for at least 6 months and a body mass index (BMI) ≤32 kg m−2 were eligible for enrolment. Major exclusion criteria included a history of clinically significant diseases or abnormalities (including hepatitis B, hepatitis C, and HIV), an INR of >1.2, use of a systemic immunosuppressive agent or other systemic agents to treat psoriasis (prednisone, PUVA, phototherapy) within 4 weeks of treatment, receipt of topical treatment for psoriasis within 2 weeks of treatment, receipt of a live vaccine (s) within 1 month of treatment, or any contraindication to receiving the drugs included in the probe cocktail. During the trial, subjects were to refrain from use of drugs known to be CYP inhibitors or inducers or from systemic or topical psoriasis therapy.
Trial design
The study design is summarized in Figure 1. The trial protocol (PN009) was approved by the appropriate ethics review committees (Comitetul National de Expertiza Etica a Studiului Clinic [National Committee for Ethical Expertise of Clinical Trials], Chisinau, Moldova; Independent Local Ethics Committee, Tbilisi, Georgia). The trial was conducted in accordance with the guidelines on Good Clinical Practice and with the ethical standards for human experimentation established by the Declaration of Helsinki. All subjects provided written informed consent prior to participating in the trial. The trial was conducted at two trial centres, located in Tbilisi, Georgia and Chisinau, Moldova, between 18 February 2015 and 29 February 2016.
Figure 1.
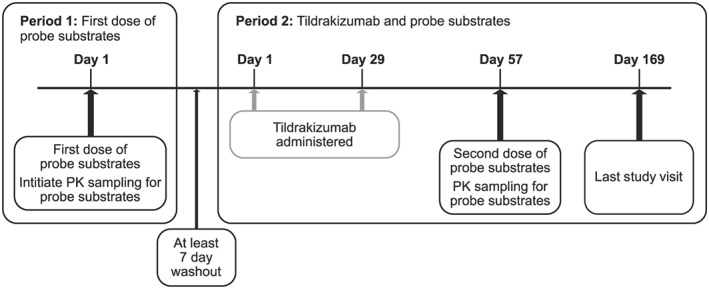
Study design
This was a fixed‐sequence, two‐period, parallel‐group, two‐site, open‐label, multiple‐dose trial of tildrakizumab in subjects with moderate to severe psoriasis. Twenty subjects were enrolled. All subjects were to receive a single oral dose of up to five CYP probe substrates (commercial sources of CYP probe substrates are provided in the Supporting Information Data S1) as a cocktail (http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6853 10 mg (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1326 probe) 21 + vitamin K 10 mg, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3342 2 mg (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1337 probe) 22, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6953 30 mg (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1329 probe) 23, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4279 40 mg (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=262 probe) 24, and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=407 200 mg (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1319 probe) 24) on Day 1 of Period 1. Subjects participating at the Moldova trial site received all five CYP probe substrates. Subjects participating at the Georgia trial site (n = 4) were only administered CYP probes that could be sourced locally within Georgia (omeprazole, caffeine, warfarin + vitamin K). Vitamin K was included in the cocktail as a precaution to offset any potential anticoagulant effects of warfarin. There was at least a 7‐day washout between Period 1 and Period 2.
On Day 1 and Day 29 of Period 2, subjects were administered tildrakizumab 200 mg subcutaneously (SC). On Day 57 of Period 2, all subjects received a single dose of the probe cocktail. Blood samples to evaluate the pharmacokinetics (PK) of the probe substrates were collected on Day 1 of Period 1 and Day 57 of Period 2 following administration of the probe cocktail. Blood samples were collected before dosing, and at 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h after dosing for caffeine and paraxanthine; 3, 6, 12, 24, 48, 72 and 96 h after dosing for S‐warfarin and 7‐hydroxywarfarin; 0.5, 1, 1.5, 2, 2.5, 3, 4, 6 and 8 h after dosing for omeprazole and 5‐hydroxyomperazole; 1, 2, 3, 4, 6, 8, 12, 24, 48 and 72 h after dosing for dextromethorphan and dextrorphan; and 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6 and 9 h after dosing for midazolam and 1‐hydroxymidazolam. Blood samples for measurement of serum concentrations of high‐sensitivity C‐reactive protein (hs‐CRP) and interleukin‐6 (IL‐6) were obtained prior to the administration of the CYP probes on Day 1 of Period 1 and Day 57 of Period 2. In each treatment period, subjects fasted from all food and drink except water for at least 8 h prior to dosing of the probe cocktail (there were no meal/drink restrictions regarding dosing of tildrakizumab) and 4 h post dose. Subjects received the probe cocktail with 480 ml of water (240 ml for midazolam, which was administered 3 h prior to the other probes, and 240 ml for the remaining cocktail), with water restricted 1 h prior to and 1 h after probe cocktail administration. Due to potential interference of food content in the DDI assessment, the following foods were prohibited for 1 week prior to receiving the probe cocktail in each period through to the last PK sample of the probe cocktail in each period: vegetables from the mustard green family (which contain high levels of glucosinolates, which are known to induce a number of CYP isozymes, including CYP1A1 and CYP1A2 25) and charbroiled meats (which contain high levels of polycyclic aromatic hydrocarbons, which are known to induce CYP1A2 26). Additionally, caffeine/caffeinated beverages and theobromine‐containing products were not to have been consumed from 48 h before through to completion of probe (only) PK sampling in each period due to potential contamination with the dosed caffeine probe in the bioanalytical assay.
Pharmacokinetic assessments
Serum concentrations of probe substrates and key metabolites were assessed by validated assays incorporating extraction followed by detection via liquid chromatography or high performance liquid chromatography (HPLC) separation and mass spectrometry (MS)/MS detection. Plasma concentrations of omeprazole, 5‐hydroxyomeprazole, S‐warfarin, 7‐hydroxywarfarin, caffeine and paraxanthine were determined by PPD (Middleton, WI). Plasma concentrations of midazolam, 1‐hydroxymidazolam, dextromethorphan and dextrorphan were determined by Inventiv Health Clinique Inc. (Quebec, Canada).
Pharmacokinetic parameter values were calculated using Phoenix WinNonlin (Version 6.3) (Cetera, Princeton, NJ, USA). Pharmacokinetic variables of interest include: area under the concentration–time curve from time zero to infinity (AUC0–∞), maximum concentration (C max), time to maximum concentration (T max) and apparent terminal half‐life (t 1/2).
For all probe substrates, with the exception of omeprazole, parent drug AUC0–∞ was the primary endpoint 18, 21, 22, 23, 24. For omeprazole, the 5‐hydroxyomeprazole AUC0–∞, to omeprazole AUC0–∞, ratio was the primary endpoint 24. As a secondary endpoint, the metabolite to parent AUC0–∞ geometric mean ratio (GMR) for each of the CYP substrate probes was estimated. These metabolite to parent endpoints provide an additional measure of any change in CYP activity 23, 24. The secondary endpoint for the CYP2C19 probe was the AUC0–∞ GMR (omeprazole after treatment with tildrakizumab/omeprazole prior to tildrakizumab treatment).
Pharmacodynamic assessments
Pharmacodynamic endpoints included PASI assessments 27 and serum concentrations of hs‐CRP and IL‐6. Plasma levels of hs‐CRP were assayed using a high sensitivity latex turbidimetric immunoassay (LLOQ = 0.1 mg l−1; Roche Diagnostics, Basel, Switzerland, and Wako Chemicals GmbH, Neuss, Germany), which was performed by local laboratories (Eurolab, Chisinau, Moldova [Wako Chemicals assay]; Scientific Research Institute of Clinical Medicine, Tbilisi, Georgia [Roche Diagnostics assay]). Plasma levels of IL‐6 were assayed using the Quantikine High Sensitivity Human IL‐6 Immunoassay (a solid phase ELISA; LLOQ = 0.108 pg ml−1; R&D Systems, Inc., Minneapolis, MN, USA), which was performed by PPD (Middleton, WI, USA).
Genotyping assessments
One blood sample for determination of the subject's CYP genotype (CYP2C9, CYP2C19 and CYP2D6) was collected on Day 1, Period 1. DNA was extracted and clinically validated assays were used to identify carriers of the following alleles: CYP2C19, *2, *3, *4, *5, *6, *8, and *17; CYP2C9 *2, *3; and CYP2D6 *2 *3 *4 *5 *6 *7 *8 *9 *10 *11, *12, *14, *15, *17, *41, *1XN, *2XN, *4XN, *10XN, *17XN, and *41XN at Labcorp (Research Triangle Park, NC, USA). If the previously mentioned alleles were not detected, it was assumed that the allele was *1. Any subject having two of the following alleles was considered a poor metabolizer for the given cytochrome P450: CYP2C9 *2 and *3; CYP2C19 *2‐*8; CYP2D6 *3, *4, *5, *6, *7, *8 *11, *15, *4XN. Poor metabolizer status was used to exclude subjects from some analyses, as described below in the statistical analysis section.
Safety assessments
The safety and tolerability of tildrakizumab were assessed throughout the trial by repeated clinical evaluations, including vital signs, physical examinations, 12‐lead ECGs and standard laboratory safety tests (haematology, chemistry and urinalysis). Subjects were also monitored for adverse events (AEs) throughout the trial.
Statistical analysis
Individual AUC0–∞ values were natural log‐transformed and analysed with a linear mixed effect model with fixed effect term for treatment. An unstructured covariance matrix was used to allow for unequal treatment variances and to model the correlation between the two treatment measurements within each subject via the REPEATED statement in SAS PROC MIXED. Kenward and Roger's method was used to calculate the denominator degrees of freedom for the fixed effects (DDFM = KR). A 90% confidence interval (CI) was constructed for the difference in least squares (LS) means on the log scale for AUC0–∞. These confidence limits were then exponentiated to obtain a CI for the true AUC0–∞GMR (probe analyte + tildrakizumab/probe analyte). LS geometric means and 95% CIs were also obtained for each treatment. AUC0–∞ and C max of each of the following probe analytes, omeprazole, 1‐hydroxymidazolam/midazolam ratio, 7‐hydroxywarfarin/S‐warfarin ratio, dextrorphan/dextromethorphan ratio and paraxanthine/caffeine ratio, were analysed using the model described above.
For each independent probe substrate, the primary statistical analysis excluded subjects who were phenotypically defined as CYP2C9, CYP2D6 or CYP2C19 poor metabolizers as determined by genotype. One subject was defined as a CYP2D6 poor metabolizer and was excluded from the statistical analysis related to the probe substrate dextromethorphan and its metabolite. Similarly, a CYP2C9 poor metabolizer was excluded from the statistical analysis related to the probe substrate, S‐warfarin and its metabolite.
A sample size of 16 (α = 0.05) provided a 99% probability that the 90% confidence interval (CI) of the true GMR (probe with tildrakizumab/probe) will be contained in the interval [0.5, 2.0]. Probe AUC0–∞ changes of this magnitude would likely be considered clinically significant. As the study duration was long, additional subjects were enrolled to ensure adequate power in the event of subject discontinuations.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 28, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 29.
Results
A total of 20 subjects enrolled in this trial, of which three discontinued (one due to an adverse event [see Safety section below for further details on this subject] and two due to withdrawal of consent). The demographics and baseline characteristics of the 20 enrolled subjects are summarized in Table 1.
Table 1.
Demographics and baseline disease characteristics
| Parameter | Subjects (N = 20) |
|---|---|
| Mean age (range), yrs | 40.2 (22–63) |
| Male, n (%) | 13 (65.0) |
| Race, n (%) | |
| White | 20 (100.0) |
| Mean ± SD body mass index, kg m −2 | 28 ± 3 |
| Psoriasis Area Severity Index (PASI) score | |
| Mean ± SD | 20 ± 8 |
| Median | 17 |
| Range | 12–42 |
SD, standard deviation
Pharmacogenetics
A total of 20 subjects were genotyped and the results are shown in Table 2. Data for three subjects and their CYP2D6 genotype were not obtained. While the number of subjects genotyped is small, the frequency of the observed alleles/haplotypes is generally consistent with that expected in a primarily white, Caucasian population. Based on the assumptions that, *1 genotype/haplotype is a functional allele, and that where CYP2D6 genotypes were not obtained (three subjects) that these subjects were not CYP2D6 poor metabolizers, two poor metabolizers were predicted based on their respective genotypes and the algorithm used for classification; one CYP2D6 and one CYP2C9.
Table 2.
Subject listing of genotype–phenotype data
| Subject | Gene | Genotype | Inferred phenotypic status |
|---|---|---|---|
| 1 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 1 | CYP2C9 | *1/*2 | Not a poor metabolizer |
| 1 | CYP2D6 | *1/*4 | Extensive metabolizer |
| 2 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 2 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 2 | CYP2D6 | *4/*4 | Poor metabolizer |
| 3 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 3 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 3 | CYP2D6 | *1/*35 | Extensive metabolizer |
| 4 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 4 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 4 | CYP2D6 | (*4/*10)XN | Intermediate metabolizer |
| 5 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 5 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 5 | CYP2D6 | *1/*1 | Extensive metabolizer |
| 6 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 6 | CYP2C9 | *1/*3 | Not a poor metabolizer |
| 6 | CYP2D6 | *1/*35 | Extensive metabolizer |
| 7 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 7 | CYP2C9 | *1/*2 | Not a poor metabolizer |
| 7 | CYP2D6 | *6/*35 | Extensive metabolizer |
| 8 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 8 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 8 | CYP2D6 | *1/*2 | Extensive metabolizer |
| 9 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 9 | CYP2C9 | *1/*2 | Not a poor metabolizer |
| 10 | CYP2C9 | *1/*2 | Not a poor metabolizer |
| 10 | CYP2D6 | *1/*41 | Extensive metabolizer |
| 10 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 11 | CYP2D6 | *1/*1 | Extensive metabolizer |
| 11 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 11 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 12 | CYP2D6 | *1/*4 | Extensive metabolizer |
| 12 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 12 | CYP2C19 | *1/*2 | Intermediate metabolizer |
| 13 | CYP2D6 | *1/*2 | Extensive metabolizer |
| 13 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 13 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 14 | CYP2D6 | *1/*1 | Extensive metabolizer |
| 14 | CYP2C9 | *1/*3 | Not a poor metabolizer |
| 14 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 15 | CYP2C9 | *1/*2 | Not a poor metabolizer |
| 15 | CYP2D6 | *1/*4 | Extensive metabolizer |
| 15 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 16 | CYP2D6 | *1/*1 | Extensive metabolizer |
| 16 | CYP2C9 | *1/*2 | Not a poor metabolizer |
| 16 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 17 | CYP2C19 | *1/*17 | Ultra‐rapid metabolizer |
| 17 | CYP2C9 | *1/*2 | Not a poor metabolizer |
| 17 | CYP2D6 | (*1/*4)XN | Extensive metabolizer |
| 18 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 18 | CYP2C9 | *2/*3 | Poor metabolizer |
| 18 | CYP2D6 | *1/*4 | Extensive metabolizer |
| 19 | CYP2C19 | *17/*17 | Ultra‐rapid metabolizer |
| 19 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 19 | CYP2D6 | *1/*41 | Extensive metabolizer |
| 20 | CYP2C9 | *1/*1 | Not a poor metabolizer |
| 20 | CYP2C19 | *1/*1 | Extensive metabolizer |
| 20 | CYP2D6 | *1/*4 | Extensive metabolizer |
Pharmacokinetics
Overall, treatment with two doses of tildrakizumab 200 mg SC did not have a clinically meaningful effect on the AUC0–∞ or C max of any of the CYP probe substrates tested (Table 3; Figure 2). These results are consistent with the GMR results for the metabolite to parent AUC0–∞ and C max (Table 4).
Table 3.
Statistical comparison of the serum PK for CYP probe substrates following the administration of a single oral dose of probe cocktail as probes alone, and following treatment with two subcutaneous doses of tildrakizumab 200 mg
| Compound | PK | CYP probe alone (Period 1, Day 1) | CYP probe + tildrakizumab (Period 2, Day 57) | GMR (90% CI) for CYP probe + tildrakizumab/CYP probe alone | ||
|---|---|---|---|---|---|---|
| n | GM (95% CI) | n | GM (95% CI) | |||
| Midazolam | AUC0–∞ (hr*ng ml−1)a | 16 | 24.6 (19.8, 30.7) | 15 | 27.3 (23.4, 32.0) | 1.11 (0.94, 1.32) |
| C max (ng ml−1)a | 9.15 (7.51, 11.2) | 9.66 (8.04, 11.6) | 1.06 (0.86, 1.29) | |||
| T max (h)b | 0.50 (0.50, 1.00) | 0.50 (0.50, 1.00) | ||||
| t 1/2 (h)c | 3.46 (29.8) | 3.15 (31.1) | ||||
| S‐Warfarin | AUC0‐∞ (hr*ng ml−1)a | 19 | 15 600 (13 200, 18 400) | 17 | 16 700 (13 900, 20 100) | 1.07 (0.98, 1.17) |
| C max (ng ml−1)a | 381 (348, 418) | 377 (344, 414) | 0.99 (0.95, 1.03) | |||
| T max (h)b | 3.00 (3.00, 3.00) | 3.00 (2.98, 3.00) | ||||
| t 1/2 (h)c | 32.4 (25.8) | 34.8 (22.4) | ||||
| 5‐hydroxyomeprazole/Omeprazole | AUC0–∞ (hr*ng ml−1)a | 16 | 0.591 (0.435, 0.802) | 15 | 0.567 (0.394, 0.817) | 0.96 (0.77, 1.19) |
| C max (ng ml−1)a | 20 | 0.419 (0.324, 0.540) | 18 | 0.415 (0.315, 0.547) | 0.99 (0.85, 1.15) | |
| Dextromethorphan | AUC0–∞ (hr*ng ml−1)a | 15 | 6.85 (3.81, 12.3) | 14 | 8.24 (4.51, 15.1) | 1.20 (1.00, 1.45) |
| C max (ng ml−1)a | 0.872 (0.494, 1.54) | 1.02 (0.573, 1.82) | 1.17 (0.96, 1.43) | |||
| T max (h)b | 2.00 (1.98, 3.00) | 1.99 (1.98, 3.00) | ||||
| t 1/2 (h)c | 7.85 (54.6) | 7.41 (31.0) | ||||
| Caffeine | AUC0–∞ (hr*ng ml−1)a | 19 | 33 800 (27 400, 41 800) | 17 | 38 500 (31 800, 46 600) | 1.14 (1.01, 1.28) |
| C max (ng ml−1)a | 4520 (3980, 5130) | 4340 (3830, 4920) | 0.96 (0.88, 1.05) | |||
| T max (h)b | 0.98 (0.48, 1.98) | 0.98 (0.48, 2.00) | ||||
| t 1/2 (h)c | 5.07 (37.0) | 5.93 (42.3) | ||||
Back‐transformed least squares mean and confidence interval from linear mixed effects model performed on natural log‐transformed values
Median (min, max) reported for T max
Geometric mean and percent geometric CV reported for apparent terminal t1/2
CYP, cytochrome P450; GM, geometric least‐squares mean; GMR, geometric least‐squares mean ratio; CI, confidence interval
Midazolam alone: A single oral dose of 2 mg midazolam
S‐warfarin alone: A single oral dose of 10 mg S‐warfarin
Dextromethorphan alone: A single oral dose of 30 mg dextromethorphan
Caffeine alone: A single oral dose of 200 mg caffeine
Figure 2.
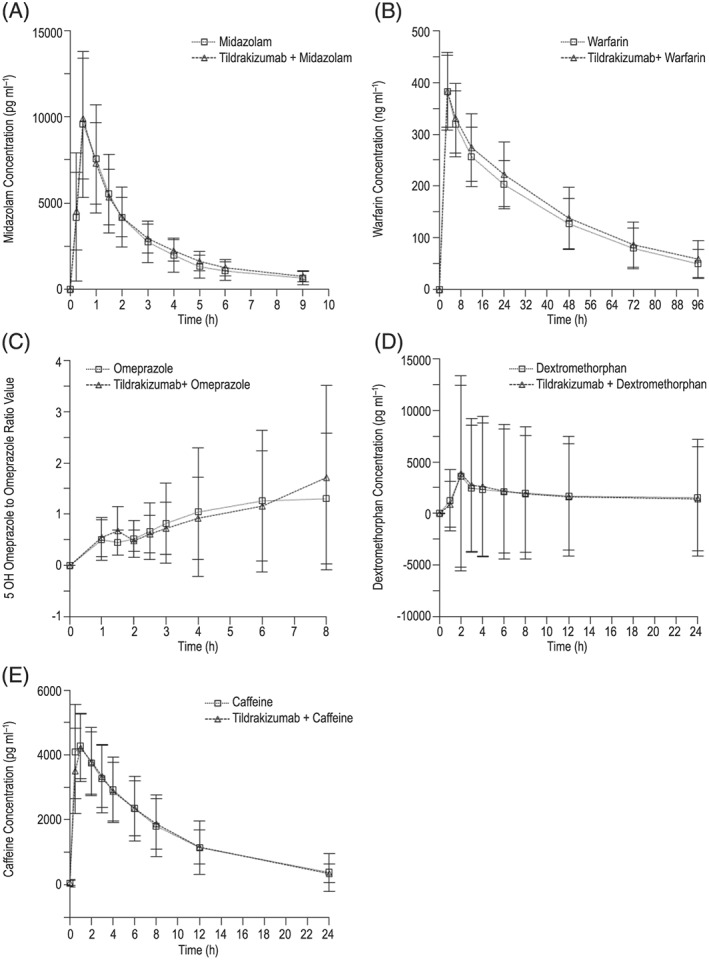
Mean (SD) serum concentration–time profiles of cytochrome P450 (CYP) probe substrates (midazolam, warfarin, dextromethorphan, caffeine) and mean (SD) CYP probe substrate metabolite to parent (5‐hydroxyomeprazole/omeprazole) serum concentration ratio–time profile following oral single‐dose administration of CYP probe substrates (A) midazolam 2 mg [CYP3A4 probe]; (B) S‐warfarin 10 mg + vitamin K 10 mg [CYP2C9 probe]; (C) omeprazole 40 mg [CYP2C19 probe]; (D) dextromethorphan 30 mg [CYP2D6 probe]; (E) caffeine 200 mg [CYP1A2 probe]) with or without coadministration of multiple doses of tildrakizumab SC (200 mg on Days 1 and 29) in subjects with moderate to severe psoriasis
Table 4.
Statistical comparison of the serum PK for the CYP probe substrate metabolite to parent ratios following the administration of a single oral dose of probe cocktail as probes alone, and following treatment with two subcutaneous doses of tildrakizumab 200 mg
| Compound | PK | CYP probe alone (Period 1, Day 1) | CYP probe + tildrakizumab (Period 2, Day 57) | GMR (90% CI) for CYP probe + tildrakizumab/CYP probe alone | ||
|---|---|---|---|---|---|---|
| n | GM (95% CI) | n | GM (95% CI) | |||
| 1‐Hydroxymidazolam/Midazolam | AUC0–∞ (hr*ng ml−1)a | 16 | 0.285 (0.248, 0.326) | 15 | 0.250 (0.201, 0.310) | 0.88 (0.73, 1.05) |
| C max (ng ml−1)a | 0.331 (0.287, 0.382) | 0.290 (0.233, 0.361) | 0.88 (0.73, 1.05) | |||
| 7‐Hydroxywarfarin/ S‐warfarin | AUC0–∞ (hr*ng ml−1)a | 12 | 0.209 (0.131, 0.333) | 11 | 0.211 (0.150, 0.297) | 1.01 (0.59, 1.73) |
| C max (ng ml−1)a | 19 | 0.101 (0.0861, 0.119) | 17 | 0.107 (0.0914, 0.125) | 1.06 (0.92, 1.22) | |
| Omeprazole | AUC0–∞ (hr*ng ml−1)a | 17 | 1440 (1020, 2040) | 15 | 1510 (1030, 2230) | 1.05 (0.83, 1.32) |
| C max (ng ml−1)a | 20 | 651 (489, 866) | 18 | 565 (464, 688) | 0.87 (0.69, 1.10) | |
| T max (h)b | 2.00 (1.50, 6.00) | 2.00 (1.00, 8.00) | ||||
| t 1/2 (h)c | 17 | 1.18 (43.9) | 15 | 1.06 (40.1) | ||
| Dextrorphan/Dextromethorphan | AUC0–∞ (hr*ng ml−1)a | 15 | 2.41 (1.45, 4.01) | 14 | 2.29 (1.31, 4.01) | 0.95 (0.81, 1.11) |
| C max (ng ml−1)a | 4.33 (2.73, 6.87) | 4.24 (2.53, 7.11) | 0.98 (0.82, 1.16) | |||
| Paraxanthine/Caffeine | AUC0–∞ (hr*ng ml−1)a | 11 | 1.03 (0.899, 1.17) | 8 | 1.08 (0.833, 1.40) | 1.05 (0.85, 1.30) |
| C max (ng ml−1)a | 19 | 0.393 (0.339, 0.456) | 17 | 0.440 (0.372, 0.520) | 1.12 (0.98, 1.27) | |
Back‐transformed least squares mean and confidence interval from linear mixed effects model performed on natural log‐transformed values
Median (min, max) reported for T max
Geometric mean and percent geometric CV reported for apparent terminal t 1/2
CYP, cytochrome P450; GM, geometric least‐squares mean; GMR, geometric least‐squares mean ratio; CI, confidence interval
Midazolam alone: A single oral dose of 2 mg midazolam
S‐warfarin alone: A single oral dose of 10 mg S‐warfarin
Dextromethorphan alone: A single oral dose of 30 mg dextromethorphan
Caffeine alone: A single oral dose of 200 mg caffeine
Pharmacodynamics
Following treatment with two doses of tildrakizumab 200 mg SC, median PASI decreased markedly from 16.6 on Day 1 to 1.20 on Day 57, representing a ~93% change from baseline (Table 5). The majority of the baseline hs‐CRP and IL‐6 values were within the normal range (Table 5).
Table 5.
Pre‐study, Day 57, and median percentage change from baseline at Day 57 measures for PASI, IL‐6, and hs‐CRP
| Parameter | Pre‐study | Period 2, Day 57 | Median (range) percentage change from baseline at Period 2, Day 57 | ||
|---|---|---|---|---|---|
| n | Median (range) | n | Median (range) | ||
| PASI | 20 | 16.60 (12.40 to 42.00) | 18 | 1.20 (0.00 to 6.20) | −92.65% (−100.00 to −69.00) |
| IL‐6 (pg ml −1 ) | 20 | 1.15 (0.44 to 25.50) | 18 | 1.14 (0.36 to 3.04) | −20.5% (−89.9 to 164.4) |
| hs‐CRP (mg l −1 ) | 20 | 3.18 (0.32 to 19.13 | 18 | 3.90 (1.27 to 15.63) | 68.4% (−85.3 to 296.9) |
PASI, Psoriasis Area Severity Index; IL‐6, interleukin‐6; hs‐CRP, high‐sensitivity C‐reactive protein
Three subjects had IL‐6 concentrations that were just below the lower normal limit of 0.5 pg ml−1 at screening and were subsequently within the normal range following treatment with tildrakizumab. While results were variable, the majority of subjects had a decrease in IL‐6 concentrations following treatment with tildrakizumab with a median (range) percentage change of −20.50 (−89.88, 164.39).
With regard to hs‐CRP, four subjects had hs‐CRP values that were above the normal range at screening and were subsequently within the normal range post‐treatment with tildrakizumab. Eleven subjects had hs‐CRP values that were within the normal range at screening and were above the normal range following treatment with tildrakizumab. The majority of subjects had an increase in hs‐CRP following treatment with tildrakizumab, with a median (range) percentage change from baseline of 68.42 (−85.31, 296.88).
Safety
Fourteen of the 20 subjects (70.0%) reported a total of 30 AEs, nine of which were considered by the investigator to be related to tildrakizumab. The majority of AEs were mild in severity.
One subject was discontinued due to a serious AE of infected dermal cyst, which required surgical drainage and was considered by the investigator to be related to tildrakizumab. Following drainage of the cyst and treatment with antibiotics, the wound healed completely.
The most common AE reported was hyperglycaemia (six subjects); however, none of these AEs was considered related to treatment. The majority of the enrolled subjects were overweight or obese and had hyperglycaemia documented at screening and/or prior to tildrakizumab treatment.
Though there were instances of increases in AST, ALT, and bilirubin that were considered AEs, these changes were generally mild and transient.
Discussion
Tildrakizumab is an IL‐23p19 monoclonal antibody indicated for the treatment of chronic plaque psoriasis. Tildrakizumab was demonstrated to improve significantly the signs and symptoms of psoriasis and was generally well tolerated 30. This trial was conducted to assess the potential effect of tildrakizumab on CYP metabolism through modulation of inflammation. Biologics such as tildrakizumab do not undergo disposition pathways relevant to small molecules. The lack of overlapping pathways is expected to limit the potential for PK‐based DDIs between these therapeutics. However, increased inflammation has been associated with increased exposure and/or reduced clearance of drugs 9, 10, 11. Literature demonstrate that the administration of cytokines or cytokine modulators affects CYP expression and/or drug interactions 3, 4, 5, 6, 7, 8, 12, 13, 14, 15, 16, 17, 31. These data clearly demonstrate that modulation of systemic inflammation has the potential to significantly affect CYP metabolism 3, 4, 5, 6, 7, 8. As previously mentioned, while modulation of cytokines in severe psoriasis has been observed 18, to our knowledge, DDIs as a result of treatment with immunomodulators in psoriasis have not been reported in the literature.
A preclinical evaluation was also considered when evaluating the potential for tildrakizumab to modulate CYP expression. A co‐culture model of hepatocytes and Kupffer cells was designed to assess a potential indirect cytokine impact on hepatocytes through stimulation of Kupffer cell‐mediated cytokine release. Exposure of hepatocyte:Kupffer cell co‐cultures to IL‐23 resulted in inflammatory cytokine (IL‐8, TNF‐α and INF‐γ) levels that were similar to those of untreated controls and only minimal suppression of CYP3A4 activity, suggesting the lack of effect of IL‐23 on hepatocytes 19. The lack of effect of IL‐23 may be attributed to the absence of, or less than detectable, mRNA expression of IL‐23R in the hepatocyte‐Kupffer cell co‐cultures as measured by RT‐PCR, thereby limiting IL‐23 from directly interacting with these cells 19. Independent studies have also observed that IL‐23 and IL‐12 do not alter expression or activity of multiple P450 enzymes (CYP2B6, CYP2C9, CYP2C19 and CYP3A4) in human hepatocytes 32. As IL‐23 does not appear to affect hepatic P450 expression, clinical drug–drug interactions through P450 pathways are unlikely; however, in vitro or animal studies may have limited value in prediction of clinical effects 20.
Thus, this trial evaluated the potential for tildrakizumab, as an immunomodulator, to perpetrate drug–drug interactions in subjects with moderate to severe psoriasis. A well‐established, validated approach known as the Cooperstown 5 + 1 cocktail was used to evaluate simultaneously the potential for several CYP‐based drug interactions 33. The dose and dose regimen of the orally administered CYP probe substrates (caffeine 200 mg, of warfarin 10 mg + vitamin K oral 10 mg, omeprazole 40 mg, dextromethorphan oral 30 mg, and midazolam 2 mg) are supported by published cocktail studies 22, 24, 33, 34, 35, 36, 37, 38, 39.
The results of this trial demonstrated that tildrakizumab did not have a clinically meaningful effect on the metabolism of any of the CYP probe substrates evaluated. The AUC0–∞ GMR (probe + tildrakizumab/tildrakizumab alone) and 90% CIs for midazolam, dextromethorphan, caffeine, S‐warfarin and 5‐hydroxyomeprazole/omeprazole were not meaningfully altered with mean changes ranging from a 4% decrease to 20% increase. Further, the results for parent drug were consistent with GMRs for the metabolite to parent AUC0–∞. These results suggest a very low potential for an interaction between tildrakizumab and commonly coadministered drugs in the psoriasis population and across therapeutic classes that are metabolized by any of the major CYP isoenzymes including CYP1A2, CYP2C9, CYP2C19, CYP2D6 and CYP3A4. Increases in inflammation have also been shown to decrease expression of transporters 5, 40, 41, hence theoretically the risk of transporter‐mediated DDIs is possible. However, given the lower systemic levels of inflammatory cytokines observed in the psoriasis population (as opposed to other autoimmune diseases such as rheumatoid arthritis), the potential for a change in transporter expression following treatment with tildrakizumab in this population is unlikely.
To ensure that the potential for tildrakizumab to perpetrate a disease–drug interaction was adequately tested, a substantial effect of tildrakizumab on PASI was anticipated. For example, in the Phase 1 study in patients with moderate to severe psoriasis given tildrakizumab IV, results showed that at the 3 mg kg−1 (n = 15) and 10 mg kg−1 (n = 14) doses, the placebo‐corrected mean (90% confidence interval [CI]) percentage change from baseline in PASI score was −56.2 (−71.3, −41.0) and −69.1 (−84.5, −53.6) on Day 56, following tildrakizumab administration on Days 1 and 28 (data on file). Though these were not the maximal efficacy effects observed during the study, based on the expected time‐lag for suppression of inflammation and clearing of the skin (i.e., normalized keratinocytes require 28 days to migrate from basal layer to cornified layer of the epidermis), these data would suggest a significant reduction of inflammation at this time point. Therefore, any effect of treatment of inflammation on expression of CYP enzymes would be expected to be evident by Day 57 of Period 2 30. To confirm anti‐IL‐23 activity, PASI response was evaluated at Day 57, in addition to the evaluation of post‐treatment CYP metabolism activity. All subjects with pre‐ and post‐treatment PASI data demonstrated a marked decrease in PASI score. The median percentage decrease in PASI score from baseline was approximately 92%, indicating that the evaluation of CYP metabolism was conducted under conditions of meaningful anti‐IL‐23 activity.
The lack of a disease–drug interaction in this study is in contrast to what has been observed in a disease–drug interaction study with tocilizumab, an mAb to IL‐6R, in subjects with rheumatoid arthritis 4. In the tocilizumab study, the exposure of the CYP3A4 substrate simvastatin, was reduced by approximately 57% following treatment with tocilizumab, which was likely the result of decreased systemic inflammation and increased CYP3A4 expression and metabolism. The difference between the trials may be due to the magnitude of systemic inflammation in rheumatoid arthritis as compared to psoriasis. In the tocilizumab study, the mean baseline IL‐6 and hs‐CRP concentrations were approximately 50 pg ml−1 and 40 to 50 mg l−1, respectively. Conversely, in the present study, the mean baseline IL‐6 and hs‐CRP concentrations were approximately 2.6 pg ml−1 and 4.36 mg l−1, respectively. Data reported for IL‐6 serum concentrations in patients with psoriasis are variable 42. The relative lack of abnormally high IL‐6 baseline values observed in the present trial are consistent with a meta‐analysis of 78 studies 18 evaluating markers of systemic inflammation in psoriasis, which demonstrated significant, but only modest, increases in inflammatory parameters such as IL‐6 when compared to healthy subjects. The meta‐analysis reported a standard mean difference (95% CI) of 1.32 (0.83, 1.81) for IL‐6 18. The baseline hs‐CRP concentrations observed in the present study were consistent with those observed for hs‐CRP and CRP in other psoriasis studies 43, 44. Furthermore, the trend towards an increase in hs‐CRP was modest and may have been associated with variability and is not likely of clinical relevance. As the sample size of this study was driven by the evaluation of clinically relevant changes in PK endpoints, a limitation of this study is that it was likely too small to detect small or modest changes in markers of systemic inflammation as a result of variability in these parameters and the magnitude of the increase observed in the psoriasis population 18. However, these data suggest that CYP metabolism may not be as dramatically modulated by inflammation in psoriasis when compared to a disease such as rheumatoid arthritis, which is associated with marked elevations in markers of systemic inflammation. These results are consistent with the US prescribing information for the recently approved anti‐IL23 IL‐23p19 monoclonal antibody, guselkumab 45. However, one subject receiving guselkumab was determined to have an approximately three‐fold change in the substrate for CYP2D6 (dextromethorphan); these data have yet to be published and therefore cannot be discussed further.
The present study had the limitation of small sample size, which only allowed for the detection of larger changes that would likely be considered clinically relevant if tildrakizumab was coadministered with drugs metabolized by the CYP enzymes evaluated in the current study (i.e., 50% reduction or two‐fold increase in exposure of the CYP probes evaluated).
In conclusion, for subjects in this trial with moderate to severe psoriasis, treatment with the IL‐23 inhibitor tildrakizumab resulted in a clinically meaningful reduction in median PASI scores (~93% median decrease compared to baseline), but did not have a clinically meaningful effect on the metabolism of the CYP probe substrates tested. The potential for clinically meaningful DDIs with tildrakizumab in psoriasis patients appears to be low. The difference in the occurrence of disease–drug interactions seen with anti‐inflammatory agents in rheumatoid arthritis patients compared with psoriasis patients may be due to the much greater extent of systemic inflammation in rheumatoid arthritis as compared to psoriasis.
Competing Interests
S.K., A.H., D.M., V.L., P.M.S., C.B.‐S., X.S.G., A.C., M.M. and M.I. are employees of, and may hold stock and/or stock options in, Merck & Co., Inc., Kenilworth, NJ, USA. I.B. and L.M. (Principal Investigators) are employees of ARENSIA Exploratory Medicine LLC, which was contracted by Merck & Co., Inc., Kenilworth, NJ, USA to conduct this trial. This trial was funded by Merck & Co., Inc., Kenilworth, NJ, USA.
The authors give sincere thanks and appreciation to the subjects and staff who participated in this trial. The authors wish to thank Alan Meehan and Amy Johnson‐Levonas (both of Merck & Co., Inc., Kenilworth, NJ, USA) for writing assistance. The authors also wish to thank Jennifer Pawlowski (Merck & Co., Inc., Kenilworth, NJ, USA) for assistance with preparing this paper for publication.
Supporting information
Data S1 Commercial Sources of Cytochrome P450 Probe Substrates Used in Cooperstown 5 + 1 Cocktail
Khalilieh, S. , Hussain, A. , Montgomery, D. , Levine, V. , Shaw, P. M. , Bodrug, I. , Mekokishvili, L. , Bailey‐Smith, C. , Glasgow, X. S. , Cheng, A. , Martinho, M. , and Iwamoto, M. (2018) Effect of tildrakizumab (MK‐3222), a high affinity, selective anti‐IL23p19 monoclonal antibody, on cytochrome P450 metabolism in subjects with moderate to severe psoriasis. Br J Clin Pharmacol, 84: 2292–2302. 10.1111/bcp.13670.
References
- 1. Gottlieb AB, Chao C, Dann F. Psoriasis comorbidities. J Dermatolog Treat 2008; 19: 5–21. [DOI] [PubMed] [Google Scholar]
- 2. Zhou H, Mascelli MA. Mechanisms of monoclonal antibody–drug interactions. Annu Rev Pharmacol Toxicol 2011; 51: 359–372. [DOI] [PubMed] [Google Scholar]
- 3. Zhou H, Davis HM. Risk‐based strategy for the assessment of pharmacokinetic drug–drug interactions for therapeutic monoclonal antibodies. Drug Discov Today 2009; 14: 891–898. [DOI] [PubMed] [Google Scholar]
- 4. Schmitt C, Kuhn B, Zhang X, Kivitz AJ, Grange S. Disease–drug–drug interaction involving tocilizumab and simvastatin in patients with rheumatoid arthritis. Clin Pharmacol Ther 2011; 89: 735–740. [DOI] [PubMed] [Google Scholar]
- 5. Aitken AE, Richardson TA, Morgan ET. Regulation of drug‐metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol 2006; 46: 123–149. [DOI] [PubMed] [Google Scholar]
- 6. Renton KW. Cytochrome P450 regulation and drug biotransformation during inflammation and infection. Curr Drug Metab 2004; 5: 235–243. [DOI] [PubMed] [Google Scholar]
- 7. Seitz K, Zhou H. Pharmacokinetic drug–drug interaction potentials for therapeutic monoclonal antibodies: reality check. J Clin Pharmacol 2007; 47: 1104–1118. [DOI] [PubMed] [Google Scholar]
- 8. Dickmann LJ, Patel SK, Rock DA, Wienkers LC, Slatter JG. Effects of interleukin‐6 (IL‐6) and an anti‐IL‐6 monoclonal antibody on drug‐metabolizing enzymes in human hepatocyte culture. Drug Metab Dispos 2011; 39: 1415–1422. [DOI] [PubMed] [Google Scholar]
- 9. Chang KC, Bell TD, Lauer BA, Chai H. Altered theophylline pharmacokinetics during acute respiratory viral illness. Lancet 1978; 1: 132–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frye RF, Schneider VM, Frye CS, Feldman AM. Plasma levels of TNF‐alpha and IL‐6 are inversely related to cytochrome P450‐dependent drug metabolism in patients with congestive heart failure. J Card Fail 2002; 8: 315–319. [DOI] [PubMed] [Google Scholar]
- 11. Glassman AH, Johnson LL, Giardina EG, Walsh BT, Roose SP, Cooper TB, et al The use of imipramine in depressed patients with congestive heart failure. JAMA 1983; 250: 1997–2001. [PubMed] [Google Scholar]
- 12. Williams SJ, Baird‐Lambert JA, Farrell GC. Inhibition of theophylline metabolism by interferon. Lancet 1987; 330: 939–941. [DOI] [PubMed] [Google Scholar]
- 13. Okuno H, Takasu M, Kano H, Seki T, Shiozaki Y, Inoue K. Depression of drug‐metabolizing activity in the human liver by interferon‐beta. Hepatology 1993; 17: 65–69. [PubMed] [Google Scholar]
- 14. Elkahwaji J, Robin MA, Berson A, Tinel M, Lettéron P, Labbe G, et al Decrease in hepatic cytochrome P450 after interleukin‐2 immunotherapy. Biochem Pharmacol 1999; 57: 951–954. [DOI] [PubMed] [Google Scholar]
- 15. Chen YL, Le Vraux V, Leneveu A, Dreyfus F, Stheneur A, Florentin I, et al Acute‐phase response, interleukin‐6, and alteration of cyclosporine pharmacokinetics. Clin Pharmacol Ther 1994; 55: 649–660. [DOI] [PubMed] [Google Scholar]
- 16. Sifontis NM, Benedetti E, Vasquez EM. Clinically significant drug interaction between basiliximab and tacrolimus in renal transplant recipients. Transplant Proc 2002; 34: 1730–1732. [DOI] [PubMed] [Google Scholar]
- 17. Vasquez EM, Pollak R. OKT3 therapy increases cyclosporine blood levels. Clin Transplant 1997; 11: 38–41. [PubMed] [Google Scholar]
- 18. Dowlatshahi EA, van der Voort EA, Arends LR, Nijsten T. Markers of systemic inflammation in psoriasis: a systematic review and meta‐analysis. Br J Dermatol 2013; 169: 266–282. [DOI] [PubMed] [Google Scholar]
- 19. Nguyen TV, Ukairo O, Khetani SR, McVay M, Kanchagar C, Seghezzi W, et al Establishment of a hepatocyte‐Kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab Dispos 2015; 43: 774–785. [DOI] [PubMed] [Google Scholar]
- 20. Huang S‐M, Zhao H, Lee J‐I, Reynolds K, Zhang L, Temple R, et al Therapeutic protein–drug interactions and implications for drug development. Clin Pharmacol Ther 2010; 87: 497–503. [DOI] [PubMed] [Google Scholar]
- 21. Dumond JB, Vourvahis M, Rezk NL, Patterson KB, Tien HC, White N, et al A phenotype‐genotype approach to predicting CYP450 and P‐glycoprotein drug interactions with the mixed inhibitor/inducer tipranavir/ritonavir. Clin Pharmacol Ther 2010; 87: 735–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Doroshyenko O, Rokitta D, Zadoyan G, Klement S, Schlafke S, Dienel A, et al Drug cocktail interaction study on the effect of the orally administered lavender oil preparation silexan on cytochrome P450 enzymes in healthy volunteers. Drug Metab Dispos 2013; 41: 987–993. [DOI] [PubMed] [Google Scholar]
- 23. Borges S, Li L, Hamman MA, Jones DR, Hall SD, Gorski JC. Dextromethorphan to dextrorphan urinary metabolic ratio does not reflect dextromethorphan oral clearance. Drug Metab Dispos 2005; 33: 1052–1055. [DOI] [PubMed] [Google Scholar]
- 24. Johnson BM, Song IH, Adkison KK, Borland J, Fang L, Lou Y, et al Evaluation of the drug interaction potential of aplaviroc, a novel human immunodeficiency virus entry inhibitor, using a modified Cooperstown 5+1 cocktail. J Clin Pharmacol 2006; 46: 577–587. [DOI] [PubMed] [Google Scholar]
- 25. Vang O, Jensen H, Autrup H. Induction of cytochrome P‐450IA1, IA2, IIB1, IIB2 and IIE1 by broccoli in rat liver and colon. Chem‐Biol Interactions 1991; 78: 85–96. [DOI] [PubMed] [Google Scholar]
- 26. Fontana RJ, Lown KS, Paine MF, Fortlage L, Santella RM, Felton JS, et al Effects of a chargrilled meat diet on expression of CYP3A, CYP1A, and P‐ glycoprotein levels in healthy volunteers. Gastroenterol 1999; 117: 89–98. [DOI] [PubMed] [Google Scholar]
- 27. Fredriksson T, Pettersson U. Severe psoriasis – oral therapy with a new retinoid. Dermatologica 1978; 157: 238–244. [DOI] [PubMed] [Google Scholar]
- 28. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res 2018; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Petersn JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2018; 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kopp T, Riedl E, Bangert C, Bowman EP, Greisenegger E, Horowitz A, et al Clinical improvement in psoriasis with specific targeting of interleukin‐23. Nature 2015; 521: 222–226. [DOI] [PubMed] [Google Scholar]
- 31. Lee J‐I, Zhang L, Men AY, Kenna LA, Huang S‐M. CYP‐mediated therapeutic protein–drug interactions – clinical findings, proposed mechanisms and regulatory implications. Clin Pharmacokinet 2010; 49: 295–310. [DOI] [PubMed] [Google Scholar]
- 32. Dallas S, Chattopadhyay S, Sensenhauser C, Batheja A, Singer M, Silva J. Interleukins‐12 and ‐23 do not alter expression or activity of multiple cytochrome P450 enzymes in cryopreserved human hepatocytes. Drug Metab Dispos 2013; 41: 689–693. [DOI] [PubMed] [Google Scholar]
- 33. Chainuvati S, Nafziger AN, Leeder JS, Gaedigk A, Kearns GL, Sellers E, et al Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, and xanthine oxidase activities with the "Cooperstown 5+1 cocktail". Clin Pharmacol Ther 2003; 74: 437–447. [DOI] [PubMed] [Google Scholar]
- 34. Turnpault S, Brian W, Van Horn R, Santoni A, Poitiers F, Donazzolo Y, et al Pharmacokinetic assessment of a five‐probe cocktail for CYPs 1A2, 2C9, 2C19, 2D6 and 3A. Br J Clin Pharmacol 2009; 68: 928–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shelepova T, Nafziger AN, Victory J, Kashuba ADM, Rowland E, Zhang Y, et al Effect of triphasic oral contraceptive on drug‐metabolizing enzyme activity as measured by the validated Cooperstown 5+1 cocktail. J Clin Pharmacol 2005; 45: 1413–1421. [DOI] [PubMed] [Google Scholar]
- 36. Wexler D, Courtnet R, Richards W, Banfield C, Lim J, Laughlin M. Effect of posaconazole on cytochrome P450 enzymes: a randomized, open‐label, two‐way crossover study. Eur J Pharma Sci 2004; 21: 645–653. [DOI] [PubMed] [Google Scholar]
- 37. Streetman DS, Bleakley JF, Kim JS, Nafziger AN, Leeder JS, Gaedigk A, et al Combined phenotypic assessment of CYP1A2, CYP2C19, CYP2D6, CYP3A, N‐acetlytransferase‐2, and xanthine oxidase with the "Cooperstown cocktail". Clin Pharmacol Ther 2000; 68: 375–383. [DOI] [PubMed] [Google Scholar]
- 38. Gorski JC, Hall SD, Becker P, Affrime MB, Cutler DL, Haehner‐Daniels B. In vivo effects of interleukin‐10 on cytochrome P450 activity. Clin Pharmacol Ther 2000; 67: 32–43. [DOI] [PubMed] [Google Scholar]
- 39. Ma JD, Nafziger AN, Villano SA, Gaedigk A, Bertino JS Jr. Maribavir pharmacokinetics and the effects of multiple‐dose maribavir on cytochrome P450 (CYP) 1A2, CYP 2C9, CYP 2C19, CYP 2D6, CYP 3A, N‐acetyltransferase‐2, and xanthine oxidase activities in healthy adults. Antimicrob Agents Chemother 2006; 50: 1130–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petrovic V, Teng S, Piquette‐Miller M. Regulation of drug transporters during infection and inflammation. Mol Interv 2007; 7: 99–111. [DOI] [PubMed] [Google Scholar]
- 41. Zhou H, Meibohm B, eds. Drug–Drug Interactions for Therapeutic Biologics. Hoboken, NJ: John Wiley & Sons, 2013. [Google Scholar]
- 42. Pietrzak AT, Zalewska A, Chodorowska G, Krasowska D, Michalak‐Stoma A, Nockowski P, et al Cytokines and anticytokines in psoriasis. Clin Chim Acta 2008; 394: 7–21. [DOI] [PubMed] [Google Scholar]
- 43. Kanelleas A, Liapi C, Katoulis A, Stavropoulos P, Avgerinou G, Georgala S, et al The role of inflammatory markers in assessing disease severity and response to treatment in patients with psoriasis treated with etanercept. Clin Exp Dermatol 2011; 36: 845–850. [DOI] [PubMed] [Google Scholar]
- 44. Strober B, Teller C, Yamauchi P, Miller JL, Hooper M, Yang YC, et al Effects of etanercept on C‐reactive protein levels in psoriasis and psoriatic arthritis. Br J Dermatol 2008; 159: 322–330. [DOI] [PubMed] [Google Scholar]
- 45. US prescribing information for TREMFYA™ (guselkumab) injection, for subcutaneous use . 2017. Available at https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761061s000lbl.pdf (last accessed 22 September 2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Commercial Sources of Cytochrome P450 Probe Substrates Used in Cooperstown 5 + 1 Cocktail