

Abstract
Multidrug-resistant gonorrhea has become an urgent issue for global public health. As the causative agent of gonorrhea, Neisseria gonorrhoeae, has been progressively developing resistance to nearly all prescribed antimicrobial drugs, monitoring its antimicrobial resistance on a broader scale has become a crucial agenda for effective antibiotic stewardship. Unfortunately, gold standard antimicrobial susceptibility testing (AST) relies on time and labor-intensive phenotypic assays, which lag behind the current diagnostic workflow for N. gonorrhoeae identification based on nucleic acid amplification tests (NAAT). Newer assay technologies based on NAAT can rapidly identify N. gonorrhoeae from clinical specimen but fundamentally lack the capacity to provide phenotypic AST information. Herein, we propose a direct-quantitative PCR (direct-qPCR) assay that enables pathogen-specific identification and phenotypic AST via quantitative measurement of N. gonorrhoeae growth directly from a liquid medium without any sample preprocessing. The assay has an analytical sensitivity of 102 CFU/mL and is highly specific to N. gonorrhoeae in the presence of urogenital flora and clinical swab eluent. We tested seven N. gonorrhoeae strains against three antibiotic agents, penicillin, tetracycline, and ciprofloxacin, and achieved 95.2% category agreement and 85.7% essential agreement with the FDA-approved E-test. The assay presented in this work has the unique ability to identify N. gonorrhoeae and provide phenotypic AST directly from the liquid medium with cell densities as low as 102 CFU/mL, demonstrating an accelerated, sensitive, and scalable workflow for performing both identification and AST of N. gonorrhoeae.
Keywords: Neisseria gonorrhoeae, direct PCR, phenotypic antimicrobial susceptibility testing, pathogen identification, growth measurement, antimicrobial resistance, penicillin, ciprofloxacin, tetracycline
Graphical Abstract

Antimicrobial-resistant gonorrhea has been marked by the World Health Organization and the Centers for Disease Control and Prevention (CDC) as a top-tier antimicrobial resistance threat.1–3 In 2006, five recommended treatment options for gonorrhea were available including penicillin, sulfonamides, tetracyclines, fluoroquinolones, and the early generations of cephalosporins. However, because of the rapid development of antimicrobial resistance in reported cases of N. gonorrhoeae over the past decade,3 the CDC’s treatment recommendation has now become limited to ceftriaxone and azithromycin.4 More alarmingly, resistance of N. gonorrhoeae to ceftriaxone and azithromycin is also being reported as an emerging threat.3,5–7 Both of these antimicrobials are currently used in the most effective regimen for treating gonorrhea,1,4 and the emergence of resistance to ceftriaxone and azithromycin highlights the urgent need for timely and accurate surveillance of gonococcal antimicrobial susceptibility to guide clinical practice. Unexpectedly, a growing shift in gonorrhea diagnostic resources toward the faster and more sensitive NAAT tests8–10 has also limited the development of capacity for N. gonorrhoeae culture and phenotypic antimicrobial susceptibility testing (AST) in clinical laboratories, further compounding the challenges.
The CDC initiated the Gonococcal Isolate Surveillance Project (GISP) with the intention of monitoring antimicrobial resistance trends of N. gonorrhoeae and to provide guidance for clinical practice.11,12 However, the use of GISP as an effective surveillance tool is limited by its sample size. Currently, less than 2% of all reported cases of gonorrhea are sampled,1 limited in part by the time-consuming and subjective diagnostic procedures utilizing manual agar plate-based techniques for susceptibility determination. To facilitate effective antimicrobial resistance surveillance in gonorrhea, there is a critical need for a timely and objective phenotypic assay that can accommodate the recent shift toward NAAT in clinical diagnostics. To this end, many investigators have focused on genotypic approaches to profiling the antimicrobial susceptibility of N. gonorrhoeae by detecting the presence or mutation of resistant genes.10,13,14 However, genotypic characterization does not always match phenotypic susceptibility testing.3 Additionally, the continued emergence of previously unidentified mechanisms for antimicrobial resistance13 challenges the use of gene panels as an effective surveillance tool for accurately predicting phenotypic susceptibility.
A recent study has demonstrated the potential of a hybrid approach to phenotypic AST by utilizing molecular assays to measure bacterial growth. Athamanolap and co-workers recently developed an assay for combined identification and AST by targeting the 16S rRNA gene in a panel of urinary tract pathogens including Escherichia coli, Enterococcus faecalis, Proteus mirabilis, and Staphylococcus aureus via PCR and high-resolution melt analysis.14 Although this approach uniquely enables both species-level identification and phenotypic drug resistance in a single reaction, a high level of agreement with established methods for minimum inhibitory concentration (MIC) determination is necessary to demonstrate clinical utility. An essential component to facilitate this in gonorrhea diagnostics is a robust liquid growth medium for N. gonorrhoeae culture directly from clinical swab specimen, capable of growth under low inoculum and in the presence of nongonococcal flora. Furthermore, the workflow requirement for DNA isolation prior to PCR mitigates the potential gain in diagnostic speed and throughput that could be achieved by this approach.
Several innovations in recent years have emerged as potential solutions to these challenges. A potential technique to surmount the workflow constraint of routine DNA purification in NAAT is direct PCR, in which crude nucleic acid-containing samples are directly added to PCR reagent for detection.15–17 Direct PCR methods utilize inhibitor-resistant polymerases to perform DNA/RNA amplification directly from raw samples, such as blood, swabs, and body fluids, without additional DNA extraction or purification. Furthermore, significant efforts have been made to develop new liquid media that permit dense growth of N. gonorrhoeae.18,19 Of particular note is the Graver-Wade (GW) medium, which is a fully defined, protein-free medium with a demonstrated ability to support the growth of various strains of N. gonorrhoeae from low inoculum.18
Herein, we present a direct-qPCR assay for integrated identification, and MIC determination of N. gonorrhoeae. We have identified an inhibitor-resistant polymerase Hemo KlenTaq that is compatible with GW medium for developing a direct-PCR assay. Our assay targets an N. gonorrhoeae-specific genetic marker opa20,21 to accurately measure the growth of N. gonorrhoeae without additional DNA extraction and purification. Our direct-qPCR assay can specifically detect as low as 102 CFU/mL of N. gonorrhoeae in the presence of urogenital flora. By incorporating a dose–response model to interpret quantitative PCR data, we can accurately determine the antimicrobial susceptibility of N. gonorrhoeae within an hour of incubation under AST condition. To evaluate our assay, we tested two reference strains and five clinical isolates against three antimicrobials and compared the accuracy of our assay with the FDA approved commercial E-tests. Our AST results have 94.5% category agreement and 85.7% essential agreement with E-tests.
RESULTS
Assay Overview
The assay workflow consists of a two-step process (Figure 1). First, clinical samples (e.g., urethral or penile-meatal swabs) can be directly eluted in GW medium and treated with different concentrations of antimicrobials. All the antimicrobial-treated samples are incubated at 37 °C with 5% CO2 under constant agitation. After brief cultivation of 1–4 h, aliquots of each sample are added directly to PCR mixture containing a real-time reporter dye. Afterward, quantitative PCR is performed to measure the amount of N. gonorrhoeae-specific nucleic acid marker, the opa gene. Following data processing via dose–response modeling, a comprehensive antibiogram of the isolated N. gonorrhoeae is generated. The entire process involves only two steps of liquid handling and after a few hours of broth cultivation, then providing a rapid and facile method of combined identification and AST for N. gonorrhoeae.
Figure 1.

Overview of the direct-qPCR assay for N. gonorrhoeae identification and AST. In this streamlined workflow, N. gonorrhoeae cells are inoculated in GW medium. The inoculated medium is aliquoted for each test condition and treated with different antimicrobials as needed. A direct-qPCR assay is performed using a real-time PCR instrument, followed by a built-in melting curve analysis step to verify amplicon specificity. Phenotypic responses to tested antimicrobial conditions (as a function of genomic DNA replication) are determined by the difference in Cq between test reaction and antimicrobial-free control reaction (ΔCq).
Quantitation of N. gonorrhoeae via Direct-qPCR Assay
The direct-qPCR assay enables quantitation of N. gonorrhoeae cells directly from culture medium without additional DNA extraction and purification. We use an inhibitor-resistant polymerase, Hemo KlenTaq, to perform the qPCR. The qPCR reagent exhibits a high level of tolerance to the GW medium (Figure S1) to ensure sensitivity and quantitation capabilities of the direct-qPCR method. Comparison of the direct-qPCR approach to other common cell lysis and DNA extraction techniques, such as mechanical lysis and enzymatic lysis via lysozyme and proteinase K, showed no substantial differences in assay performance as indicated by comparable Cq (Figure S2). In contrast to other species-agnostic methods for AST, assay specificity to N. gonorrhoeae in the direct-qPCR assay is ensured by the use of opa gene-specific primers. While the downside of using the inhibitor-resistant polymerase HemoKlenTaq is nonspecific amplification may occur, this issue can be resolved using melting curve analysis performed postamplification to further assess specificity of the PCR reaction (Figure S3). We were able to quantify N. gonorrhoeae cells ranging from 102 CFU/mL (2 CFU/reaction) to 107 CFU/mL (2 × 105 CFU/reaction) (Figure 2A). The PCR efficiency is about 100% based on the slope of the linear regression line (Figure 2B), and the high coefficient of determination (R2 = 0.997) indicates excellent correlation between the measured amount of the opa target and the number of N. gonorrhoeae cells in GW medium.
Figure 2.

Use of direct-qPCR assay for quantitation of N. gonorrhoeae in GW medium. (A) Direct-qPCR quantifies cells ranging from 107 CFU/mL to 102 CFU/mL. Cq values were obtained using regression fit determined by the BioRad CFX Manager v3.1. Amplification curves are highly consistent across technical replicates, although some stochastic effects were observed when only a few target molecules (102 CFU/mL) are present in the sample. (B) Linear fit of the quantitative Cq values using Origin Pro 2017 with instrumental weighting. PCR efficiency of approximately 100% was obtained from the slope value of the linear fit.
Growth Measurement via Direct-qPCR
We used the quantitative capability of our direct-qPCR assay to measure the growth of N. gonorrhoeae in GW medium (Figure S4A). Cq is a base-2 logarithmic quantity, with a fold-increase in cell density represented by how much the measured value has decreased from Cq at time-zero. A lower Cq indicates a higher number of cells, where one Cq decrease corresponds to a two-fold increase in cell density. As such, the baseline-subtracted quantity −ΔCq can be used to quantify the fold-change in bacterial density on a log scale. Using this representation, doubling time can be calculated by taking the slope of the −ΔCq-versus-cultivation time plot. In the example of gonococcal strains used in this study, −ΔCq increases approximately linearly as a function of cultivation time with a doubling time of approximately 1 h, indicating exponential growth (Figure S4B). It should be noted that quantification based on absolute fluorescence signal is not a reliable measure of DNA product quantity due to confounding factors including variations in the optical densities of sample and the intensities of the excitation source. The use of Cq to determine DNA quantity enables sample- and instrument-agnostic method of measuring growth.
An important feature of our assay is the ability to detect phenotypic growth with a shorter duration of cultivation, enabled by the high degree of precision and sensitivity inherent to qPCR. To demonstrate this, we used the reference strain ATCC 49226 as a model and cultivated the cells in GW medium in the presence or absence of 0.06 μg/mL ciprofloxacin. Then we performed a side-by-side comparison of ΔCq measurement by direct-qPCR and the conventional OD measurement to monitor the growth for 17 h. Cells without the antimicrobial can proliferate in the GW medium, whereas cells challenged with the antimicrobial show no growth. Using the direct-qPCR method, we are able to differentiate between growth and nongrowth as early as 1 h of cultivation (Figure 3A), indicating that the assay time for gonorrhea AST can be as short as 1 h. In contrast, the OD-based method requires more than 9 h cultivation to obtain signal above background (Figure 3B). Moreover, compared with the OD method, our direct-qPCR assay exhibits higher dynamic range and smaller deviation.
Figure 3.

Growth of ATCC 49226 as measured via direct-qPCR and OD. One-hundred-thousand N. gonorrhoeae cells were inoculated into 10 mL of GW medium with (black points) and without (red points) 0.06 μL/mL of ciprofloxacin. Samples (300 μL) were taken every hour to perform the tests. All Cq values measured were normalized by subtracting each measured value with Cq at 0 h. (A) Direct-qPCR method exhibits higher quantification range and it differentiates the growth of drug-treated and nontreated cells in 1 h cultivation. (B) OD method shows a comparatively limited quantification capability with a narrower dynamic range. It differentiates the growth of drug-treated and nontreated cells in 9 h cultivation. As a point of comparison, 24-h incubation is the typical time frame cited for plate-based cultivation.11
By targeting the gonorrhea-specific gene opa, we are able to specifically measure the growth of N. gonorrhoeae in complex sample matrix, even in the presence of other microbial species. We added 104 CFU/mL of one organism from a panel of nine common urinary tract pathogens and commensals into the N. gonorrhoeae culture and cocultured each organism with N. gonorrhoeae. We did not observe significant inhibition to N. gonorrhoeae growth during a 6-h cultivation (Figure 4A). To further demonstrate our assay’s capability of working with clinical sample matrix, we tested five N. gonorrhoeae-negative vaginal swabs. We collected all the cells and cell debris from the swabs and added them into N. gonorrhoeae culture. During a 6-h cultivation, no noticeable inhibition of N. gonorrhoeae growth was observed (Figure 4B).
Figure 4.

Growth measurement of N. gonorrhoeae in complex sample matrices. (A) N. gonorrhoeae was spiked with one of nine urinary tract pathogens and commensals at a concentration of 104 CFU/mL and cocultured. All samples were cultivated at 37 °C for 6 h and a 10 μL aliquot of each sample was collected at 1 h interval for direct-qPCR analysis. No significant interference of N. gonorrhoeae growth is observed. (B) Five individual N. gonorrhoeae negative vaginal swabs were eluted in 500 μL of GW. Then each eluent was diluted 10-fold with GW medium and final concentration of 104 CFU/mL of N. gonorrhoeae cells was inoculated. The samples were cultivated at 37 °C for 6 h, and a 10 μL aliquot of each sample was collected at 1 h interval for direct-qPCR analysis. The growth of N. gonorrhoeae is not significantly affected by the contents eluted from the vaginal swabs.
Objective Determination of AST Using Dose–Response Model
To demonstrate objective and quantitative AST via MIC determination using our assay, we challenged the reference strain ATCC 49226 with three common antimicrobials penicillin, tetracycline, and ciprofloxacin. We cultured the cells with different concentration of the drugs for 4 h and then measured the growth using our assay. The Cq of each sample was obtained via direct-qPCR. Using cell quantitation based on ΔCq, we linearized the quantity using the function 2−ΔCq and normalized the quantity in a 0–100 scale to represent cell viability. Using this scale, the antimicrobial condition resulting in the largest amount of growth as measured by ΔCq represents maximal viability, while the condition resulting in the smallest ΔCq represents minimal viability. Lastly, to overcome the limited precision of traditional AST methods that use doubling dilution series to generate censored discrete data [22], we applied the dose–response model to analyze the relationship between antimicrobial concentration and cell viability. The model enables us to estimate the effective concentration at which cell viability is 80% of maximum (EC80) and to predict the resistance level of a given antimicrobial (Figure 5). The EC80 values derived in this manner are in full concordance with the CLSI quality control range (indicated as a shaded box in Figure 5). The dose–response model enables the use of direct-qPCR assay result to quantitatively assess antimicrobial susceptibility of N. gonorrhoeae.
Figure 5.
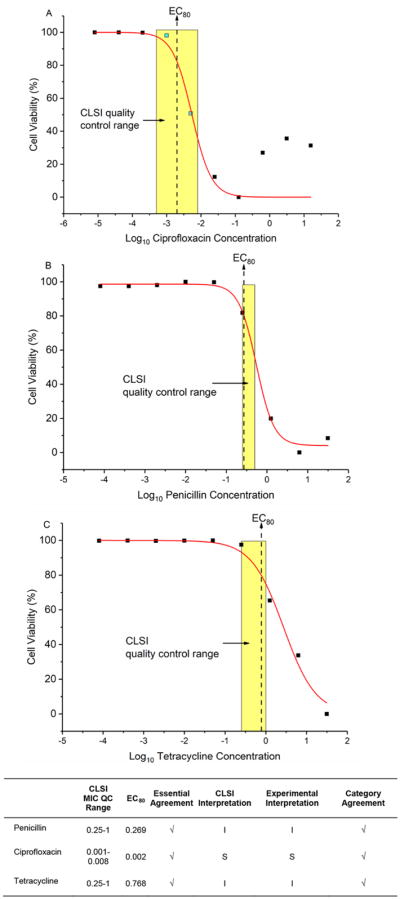
Dose–response modeling of ATCC 49226 phenotypic responses under the treatment of ciprofloxacin, penicillin, and tetracycline via direct-qPCR. A sigmoidal dose–response curve was fitted to the 2−ΔCq values using the function (1), where f(x) is normalized 2−ΔCq(in the range of 0–100%), x is the base-10 logarithm of antimicrobial concentration in μg/mL, A1 is the upper asymptote, A2 is the lower asymptote, log(x0) is the center of the curve, and p is the hill slope, respectively. The EC80 is the antimicrobial concentration at which the inhibitory effect is 80%. Dose ranges indicated by yellow shades are quality control MIC ranges reported by CLSI. Our predicted AST results (EC80) are in full concordance with the CLSI quality control references.
Analytical Validation of qPCR-Based Quantitative Antimicrobial Susceptibility Testing for N. gonorrhoeae
The direct-qPCR assay offers accurate and quantitative assessment of N. gonorrhoeae antimicrobial susceptibility. To further evaluate our method, we tested two reference strains (ATCC 49226 and ATCC 43069) plus five clinical isolates against three antimicrobials penicillin, tetracycline, and ciprofloxacin. We cultured each strain with different concentrations of the antimicrobials and measured the growth by using our direct-qPCR assay. We employed the dose–response model to quantitatively represent the antimicrobial susceptibility of a given strain against a given drug (Table S1). We calculated EC10, EC20, EC50, EC80, and EC90 values from the dose–response fittings and identified EC80 as the most accurate parameter to predict MIC (Table S2). To compare our assay with the conventional E-test, we plotted the EC80 values generated via our assay and the MIC obtained via E-test (Figure 6). In this comparative analysis, we analyzed the essential agreement (Figure 6A), defined as the prediction of MIC using the test assay within ± one two-fold dilution of the MIC determined by a gold standard test. We also analyzed categorical agreement (Figure 6B), defined as the accurate interpretation of susceptibility based on MIC value ranges in three discrete categories (resistant, intermediate, and susceptible). Our method shows 85.7% essential agreement (Figure 6A) and 94.5% categorical agreement (Figure 6B) with the gold standard method E-test, demonstrating the potential of our assay to deliver accurate and objective clinical interpretation for antimicrobial resistance of N. gonorrhoeae.
Figure 6.

Comparative analysis of our assay with the gold standard E-test. (A) EC80 values of our assay are plotted against the MIC values of E-test to evaluate the essential agreement of our assay. The diagonal of the plot represents 100% agreement between EC80 and the E-test MIC. The region between dash lines is ± one two-fold dilution of variation from the diagonal of the plot, indicating essential agreement between EC80 and MIC. The data points outside the range of essential agreement are marked in red. Our assay’s essential agreement of with E-test is 85.7%. (B) Categorical antimicrobial susceptibility R (Resistant), I (Intermediate), or S (Susceptible) was determined using the CLSI reference. Our assay’s categorical interpretation is plotted against E-test’s interpretation. The green boxes in the diagonal of the plot indicate 100% category agreement between EC80 and MIC. Minor discrepancy is defined if E-test result is R or S and our EC80 result is I, or the E-test result is I and our EC80 result is R or S. Major discrepancy is defined if the E-test result is S and our EC80 result is R; very major discrepancy is defined if E-test result is R and our EC80 result is S. Only 4.8% of minor discrepancy (3 out of 21 strain/antimicrobial combinations) and no major or very major discrepancies were observed.
DISCUSSION
NAATs have become the gold standard for pathogen identification in sexually transmitted infections because they are substantially faster and more sensitive than bacterial culture.8–10 While this has simplified diagnostic workflow and accelerated time-to-result, it has also resulted in a reduction of gonococcal culture in clinical laboratories, limiting the capacity to perform AST testing, and antimicrobial susceptibility surveillance. Given the limitation of genotypic AST methods and the complexity of mechanisms leading to antimicrobial resistance, bacterial growth-based phenotypic approaches will continue to serve as the gold standard for performing AST. On a fundamental level, the assay presented in this work fills the critical gap in a NAAT-based clinical diagnostic workflow by incorporating a growth-based phenotypic component enabled by direct-qPCR.
This work also highlights the importance of the AST medium as the key enabling component in a phenotypic assay. The high level of assay compatibility enabled by the protein-free medium is essential to quantitative growth measurement required for accurate MIC determination. In the context of emerging technologies for AST, techniques such as direct cell counting,22 cytological profiling,23 and cellular metabolite measuring24,25 all share broth microdilution as the common enabling technology. Given the ubiquity of this component, active investigation toward the development of robust growth medium for cultivation of N. gonorrhoeae in more challenging incubation conditions is warranted. Specifically, decoupling gonococcal AST incubation step from the requirement for a CO2-controlled incubator would enable the implementation of the direct-qPCR workflow directly in many of the automated NAAT systems in use today.
Our assay builds on innovations exemplified by two key contributions in literature. A recent work by Foerster et al. demonstrated the use of a resazurin-based microdilution assay for MIC determination of N. gonorrhoeae in 6 h.25 A key innovation of this work is the demonstration of the GW medium as applied to the broth microdilution AST for N. gonorrhoeae. While the work by Foerster et al. was applicable only to clinical isolates due to the lack of specificity of the resazurin indicator, our work introduces an additional layer of diagnostic capacity by enabling gonorrhea-specific identification via direct-qPCR, which allows for the measurement of gonorrhea-specific growth in more complex sample matrices and in the presence of nongonococcal flora. Another recent work by Athamanolap et al. demonstrated the use of ΔCq measurement and high-resolution melt analysis as a way of identifying and testing antimicrobial susceptibility in urinary tract pathogens.14 While the preceding work highlights the methodology of obtaining ΔCq for drug susceptibility testing, our work presented here drastically simplifies the assay workflow for N. gonorrhoeae detection by incorporating direct-PCR and N. gonorrhoeae-specific primers. The high level of correlation demonstrated against a gold standard comparator is a unique contribution highlighting the potential utility of the direct-qPCR assay for clinical applications. In this study, we applied the direct-PCR assay to test N. gonorrhoeae strains against three previously implemented antibiotic agents, penicillin, tetracycline, and ciprofloxacin, with only 4 h of drug exposure, demonstrating the potential utility to guide antibiotic selection for individualized treatment. The assay is also applicable for testing azithromycin and ceftriaxone (Tables S1 and S2) and can be implemented for monitoring antibiotic resistance trend of this currently recommended dual therapy.
The direct-qPCR approach to AST is not without technical limitations. Albeit only for a few hours, the need for an antibacterial exposure step prior to qPCR-based detection requires incubation conditions identical to standard culture-based methods. In light of purely genetic marker-based tests in the market for genotypic AST, our proposed approach will take a longer amount of time to complete. We anticipate that some of these limitations could be addressed over time by optimizing various components of the assay. For example, implementing AST incubation upstream of direct-qPCR assay may be more easily achieved on a conventional heat block using liquid medium formulations capable of sustaining growth of N. gonorrhoeae in absence of external CO2 source. Additionally, utilizing immediate-early genes in place of opa gene may improve the sensitivity of direct-qPCR assay and help to further reduce the required AST incubation time.
Developing and validating new assay technologies is essential to advancing the standard of care in clinical microbiology. Innovations in gonorrhea AST are especially needed to keep pace with the improvement in diagnostic turnaround time enabled by NAAT. The direct-qPCR assay described in this work overcomes a key limitation of a NAAT-based workflow for gonorrhea diagnosis and provides a method for effective surveillance of antimicrobial resistance of N. gonorrhoeae. Additional studies are needed to evaluate the use of direct-qPCR AST in clinical specimen and to assess the feasibility of implementing such workflow in an automated instrument for increased throughput.
MATERIALS AND METHODS
N. gonorrhoeae Strains
Two reference strains ATCC 43069 and ATCC 49226 were purchased from the American Type Culture Collection (ATCC, VA USA). Five NG clinical isolates, 16–01–02, 16–01–15, BAL-Control-1, BAL-625, and BAL-660, were provided by Dr. Jonathan Zenilman from Johns Hopkins University. These isolates were collected in Baltimore, MD as part of routine surveillance projects in 2005 (BAL-625, BAL-660, and BAL-Control-1) and in 2016 (16–01–02, 16–01–15). Each strain was first plated on a Chocolate II Agar plate (Becton Dickinson) at 37 °C and 5% CO2 overnight. Isolated colonies from each plate were suspended in GW broth and then mixed with 20% glycerol (v/v; Sigma-Aldrich, St. Louis, MO), aliquoted, frozen, and stored at −80 °C until use. At time of use, each aliquot was thawed and platted on Chocolate II Agar (BBL, Becton Dickinson). The concentration of all N. gonorrhoeae suspensions was matched to 1 × 107 CFU/mL using 0.5 McFarland standard on the Nanodrop spectrometer (Thermo Fisher Scientific, MA, USA) and verified via plating and colony enumeration.
Assay Reagents
Hemo KlenTaq polymerase and Hemo KlenTaq Reaction Buffer were purchased from New England Biolabs (Ipswich, MA USA). Primers were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA, USA). Deoxynucleotide (dNTP) Solution Mix was purchased from Thermo Fisher Scientific (Waltham, MA, USA). EvaGreen Dye was purchased from Biotium Inc. (Fremont, CA, USA). All components for GW medium (medium 199, dextrose, ammonium bicarbonate, sodium acetate trihydrate, L-glutamine, spermidine, L-arginine, hypoxanthine, uracil, oxaloacetate, thiamine hydrochloride, L-ornithine, nicotinamide adenine dinucleotide, and sodium D,L-lactate) were purchased from MilliporeSigma (St. Louis, MO, USA). All components were weighed or measured by volume, reconstituted in working concentration, and adjusted to pH 6.8 at room temperature as described by Wade and Graver.18 All antimicrobials used in this study (penicillin, tetracycline, and ciprofloxacin) were purchased from MilliporeSigma. All E-test strips were purchased from BioMérieux (Marcy-I’Étoile, France).
AST and Culture Protocol
For each growth measurement test, fresh cultures were prepared by thawing and plating the N. gonorrhoeae cells on Chocolate II Agar at 37 °C and 5% CO2 overnight. Single colonies were then collected and plated under the same condition to passage the cells once before use. Multiple colonies were collected from the subculture plates and suspended in GW broth. The cell solution was passed through a cell strainer with a mesh size of 100 μm and was adjusted to 0.5 McFarland standard (equivalent to OD600 of 0.1). The cell samples were diluted to 1 × 104 CFU/mL. Stock solutions of antimicrobials were prepared according to the manufacturer’s instructions. All antimicrobials were diluted in the 1 × 104 CFU/mL cell solutions to reach desired working concentrations, which were determined according to the CLSI resistance breakpoints.26 All broth cultures were prepared in 50 mL conical tubes and agitated at 180 rpm (rpm) on an orbital shaker during incubation. Broth cultures were incubated at 37 °C and 5% CO2. Samples were collected before and after cultivation for measurement via direct-qPCR.
Direct-qPCR Protocol
Hemo KlenTaq PCR mixture was prepared according to the manufacturer’s instructions. Two microliters of broth was added directly to the PCR mixture for a final volume of 20 μL for each reaction. The qPCR was performed using a CFX96 Touch Real-time PCR Detection System (Bio-Rad Laboratories) and analyzed using the accompanying stock software, CFX Manager. For all qPCR assays, the qPCR cycling conditions were as follows: 10 min at 95 °C, followed by 35 cycles of 95 °C for 30 s, 30 s at the annealing temperature 55 °C, and a 30-s extension step at 72 °C. The PCR primers used in this study have been previously validated for sensitivity and specificity to N. gonorrhoeae.20
Dose–Response Modeling and Statistical Analysis
Quantitation cycle (Cq) values were obtained from the BioRad CFX Manager software using the regression determination mode. A lower Cq correlates with higher number of molecular targets in a sample (in this work, the opa gene). The mean Cq of each sample prior to cultivation is designated as Cq0. ΔCq is calculated by subtracting Cq0 from the measured Cq of the samples tested after cultivation under test conditions. Cell growth measurement for each antimicrobial condition were linearized using the base-2 exponential function 2−ΔCq and normalized on a 0–100 scale to represent cell viability. The effect of each antimicrobial on the different bacterial strains was quantified using the dose–response model:25
| (1) |
where f(x) is normalized 2−ΔCq, x is the base-10 logarithm of the antimicrobial concentration in μg/mL, A2 is the upper asymptote (representing 100% cell viability), A1 is the lower asymptote (representing 0% cell viability), and p is the hill slope, and log(x0) denotes the value of x at the center of the curve. Effective concentrations at different percentage of response (ECF) were calculated using the following equation:
| (2) |
where F is the response (cell viability) in a 0–100 scale. We set the upper asymptote to 100 and the lower asymptote to 0 when performing curve fitting to ensure that the primary sigmoidal feature was captured for each experiment performed. In this work, we used EC80 to determine the MIC values for quantifying antimicrobial susceptibility. Dose–response curves were generated using OriginPro 2017 SR2 software (Origin-Lab Corp., Northampton, MA, USA).
Supplementary Material
Acknowledgments
We would like to thank our funding source the National Institutes of Health (R01AI117032, R01AI137272, U54EB007958) for supporting this work.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfec-dis.8b00104.
Compatibility of GW medium and direct-qPCR; comparison of direct-qPCR with other common cell lysis and DNA extraction techniques; melting curve analysis post PCR; characterization of N. gonorrhoeae growth in GW medium; experimental EC80 and E-test MIC; antimicrobial concentrations at different levels of response of all dose–response fittings (PDF)
References
- 1.Bolan GA, Sparling PF, Wasserheit JN. The emerging threat of untreatable gonococcal infection. N Engl J Med. 2012;366(6):485–487. doi: 10.1056/NEJMp1112456. [DOI] [PubMed] [Google Scholar]
- 2.Unemo M, Shafer WM. Antibiotic resistance in Neisseria gonorrhoeae: origin, evolution, and lessons learned for the future. Ann N Y Acad Sci. 2011;1230:E19–28. doi: 10.1111/j.1749-6632.2011.06215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Unemo M, Del Rio C, Shafer WM. Antimicrobial Resistance Expressed by Neisseria gonorrhoeae: A Major Global Public Health Problem in the 21st Century. Microbiol Spectr. 2016;4(3):213. doi: 10.1128/microbiolspec.EI10-0009-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Workowski KA, Bolan GA. Prevention, Sexually transmitted diseases treatment guidelines, 2015. MMWR Recomm Rep. 2015;64(RR-03):1–137. [PMC free article] [PubMed] [Google Scholar]
- 5.Ohnishi M, Golparian D, Shimuta K, Saika T, Hoshina S, Iwasaku K, Nakayama S, Kitawaki J, Unemo M. Is Neisseria gonorrhoeae initiating a future era of untreatable gonorrhea?: detailed characterization of the first strain with high-level resistance to ceftriaxone. Antimicrob Agents Chemother. 2011;55(7):3538–3545. doi: 10.1128/AAC.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unemo M, Golparian D, Hellmark B. First three Neisseria gonorrhoeae isolates with high-level resistance to azithromycin in Sweden: a threat to currently available dual-antimicrobial regimens for treatment of gonorrhea? Antimicrob Agents Chemother. 2014;58(1):624–625. doi: 10.1128/AAC.02093-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Unemo M, Shafer WM. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin Microbiol Rev. 2014;27(3):587–613. doi: 10.1128/CMR.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer HM, Mallinson H, Wood RL, Herring AJ. Evaluation of the specificities of five DNA amplification methods for the detection of Neisseria gonorrhoeae. J Clin Microbiol. 2003;41(2):835–837. doi: 10.1128/JCM.41.2.835-837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tabrizi SN, Unemo M, Limnios AE, Hogan TR, Hjelmevoll SO, Garland SM, Tapsall J. Evaluation of six commercial nucleic acid amplification tests for detection of Neisseria gonorrhoeae and other Neisseria species. J Clin Microbiol. 2011;49(10):3610–3615. doi: 10.1128/JCM.01217-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goire N, Sloots TP, Nissen MD, Whiley DM. Protocol for the molecular detection of antibiotic resistance mechanisms in Neisseria gonorrhoeae. Methods Mol Biol. 2012;903:319–328. doi: 10.1007/978-1-61779-937-2_22. [DOI] [PubMed] [Google Scholar]
- 11.Kirkcaldy RD, Kidd S, Weinstock HS, Papp JR, Bolan GA. Trends in antimicrobial resistance in Neisseria gonorrhoeae in the USA: the Gonococcal Isolate Surveillance Project (GISP), January 2006–June 2012. Sex Transm Infect. 2013;89(Suppl4):iv5–10. doi: 10.1136/sextrans-2013-051162. [DOI] [PubMed] [Google Scholar]
- 12.Schwarcz SK, Zenilman JM, Schnell D, Knapp JS, Hook EW, 3rd, Thompson S, Judson FN, Holmes KK. National surveillance of antimicrobial resistance in Neisseria gonorrhoeae. The Gonococcal Isolate Surveillance Project. JAMA, J Am Med Assoc. 1990;264(11):1413–1417. [PubMed] [Google Scholar]
- 13.Unemo M, Golparian D, Nicholas R, Ohnishi M, Gallay A, Sednaoui P. High-level cefixime- and ceftriaxone-resistant Neisseria gonorrhoeae in France: novel penA mosaic allele in a successful international clone causes treatment failure. Antimicrob Agents Chemother. 2012;56(3):1273–1280. doi: 10.1128/AAC.05760-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Athamanolap P, Hsieh K, Chen L, Yang S, Wang TH. Integrated Bacterial Identification and Antimicrobial Susceptibility Testing Using PCR and High-Resolution Melt. Anal Chem. 2017;89(21):11529–11536. doi: 10.1021/acs.analchem.7b02809. [DOI] [PubMed] [Google Scholar]
- 15.Cascella R, Strafella C, Ragazzo M, Zampatti S, Borgiani P, Gambardella S, Pirazzoli A, Novelli G, Giardina E. Direct PCR: a new pharmacogenetic approach for the inexpensive testing of HLA-B*57:01. Pharmacogenomics J. 2015;15(2):196–200. doi: 10.1038/tpj.2014.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kricke S, Mhaldien L, Fernandes R, Villanueva C, Shaw A, Veys P, Adams S. Chimerism Analysis in the Pediatric Setting: Direct PCR from Bone Marrow, Whole Blood, and Cell Fractions. J Mol Diagn. 2018;20(3):381–388. doi: 10.1016/j.jmoldx.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 17.McPherson AS, Dhungyel OP, Whittington RJ. Detection and Serogrouping of Dichelobacter nodosus Infection by Use of Direct PCR from Lesion Swabs To Support Outbreak-Specific Vaccination for Virulent Footrot in Sheep. J Clin Microbiol. 2018;56(4):e01730-17. doi: 10.1128/JCM.01730-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wade JJ, Graver MA. A fully defined, clear and protein-free liquid medium permitting dense growth of Neisseria gonorrhoeae from very low inocula. FEMS Microbiol Lett. 2007;273(1):35–37. doi: 10.1111/j.1574-6968.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- 19.Takei M, Yamaguchi Y, Fukuda H, Yasuda M, Deguchi T. Cultivation of Neisseria gonorrhoeae in liquid media and determination of its in vitro susceptibilities to quinolones. J Clin Microbiol. 2005;43(9):4321–4327. doi: 10.1128/JCM.43.9.4321-4327.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tabrizi SN, Chen S, Tapsall J, Garland SM. Evaluation of opa-based real-time PCR for detection of Neisseria gonorrhoeae. Sex Transm Dis. 2005;32(3):199–202. doi: 10.1097/01.olq.0000154495.24519.bf. [DOI] [PubMed] [Google Scholar]
- 21.Geraats-Peters CW, Brouwers M, Schneeberger PM, van der Zanden AG, Bruisten SM, Weers-Pothoff G, Boel CH, van den Brule AJ, Harmsen HG, Hermans MH. Specific and sensitive detection of Neisseria gonorrhoeae in clinical specimens by real-time PCR. J Clin Microbiol. 2005;43(11):5653–5659. doi: 10.1128/JCM.43.11.5653-5659.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Broeren MA, Maas Y, Retera E, Arents NL. Antimicrobial susceptibility testing in 90 min by bacterial cell count monitoring. Clin Microbiol Infect. 2013;19(3):286–291. doi: 10.1111/j.1469-0691.2012.03800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quach DT, Sakoulas G, Nizet V, Pogliano J, Pogliano K. Bacterial Cytological Profiling (BCP) as a Rapid and Accurate Antimicrobial Susceptibility Testing Method for Staphylococcus aureus. E Bio Medicine. 2016;4:95–103. doi: 10.1016/j.ebiom.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saint-Ruf C, Crussard S, Franceschi C, Orenga S, Ouattara J, Ramjeet M, Surre J, Matic I. Antibiotic Susceptibility Testing of the Gram-Negative Bacteria Based on Flow Cytometry. Front Microbiol. 2016;7:1121. doi: 10.3389/fmicb.2016.01121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foerster S, Desilvestro V, Hathaway LJ, Althaus CL, Unemo M. A new rapid resazurin-based microdilution assay for antimicrobial susceptibility testing of Neisseria gonorrhoeae. J Antimicrob Chemother. 2017;72:1961–1968. doi: 10.1093/jac/dkx113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weinstein MP, Limbago B, Patel JB, Mathers AJ, Campeau S, Mazzulli T, Eliopoulos GM, Patel R, Galas MF, Richter SS, Humphries RM, Satlin M, Jenkins SG, Swenson JM, JSL, Zimmer BL. M100 Performance Standards for Antimicrobial Susceptibility Testing. CLSI; 2018. https://clsi.org/standards/products/microbiology/documents/m100/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.