

Abstract
Excessive complement activation contributes significantly to the pathogeneses of various diseases. Currently, significant developmental research efforts aim to identify complement inhibitors with therapeutic uses have led to the approval of one inhibitor for clinical use. However, most existing complement inhibitors are based on monoclonal antibodies, which have many drawbacks such as high costs and limited administration options. With this report, we establish an inexpensive, cell imaging-based high-throughput assay for the large-scale screening of potential small molecule complement inhibitors. Using this assay, we screened a library containing 3115 bioactive chemical compounds and identified cisplatin and pyridostatin as two new complement inhibitors in addition to nafamostat mesylate, a compound with known complement inhibitory activity. We further demonstrated that cisplatin and pyridostatin inhibit C5 convertases in the classical pathway of complement activation but have no effects on the alternative pathway of complement activation. In summary, this work has established a simple, large-scale, high-throughput assay for screening novel complement inhibitors and discovered previously unknown complement activation inhibitory activities for cisplatin and pyridostatin.
Introduction
Complement, a key component of innate immunity, serves as the first defense system against infection(Ricklin et al., 2010). Complement functions through one of three major activation pathways: classical, lectin, or alternative. The classical pathway is activated after antibody-antigen complexes bind with C1, the lectin pathway is initiated when certain exposed mannose or other sugar moieties bind to mannose-binding lectin (MBL) in serum. The alternative complement activation pathway is triggered by the spontaneous hydrolysis of the internal thioester bond of C3, subsequent binding of C3b to factor B, and activation of factor B by factor D. Following these initiation processes, the C3 convertases C4b2a for both the classical and lectin pathway, and C3bBb for the alternative pathway, form and subsequently cleave C3 into the small C3a and large C3b fragments. Subsequently, C3a, an anaphylatoxin, is released into the fluid phase while C3b is deposited on the cell surface at the complement activation site to facilitate phagocytosis and the formation of C5 convertase (C4b2a3b for the classical and lectin pathway, and C3bBb3b for the alternative pathway). C5 convertase then activates C5 to release C5a, another anaphylatoxin, as well as to generate the large C5b fragment, which initiates the assembly of membrane attack complexes (MAC, a.k.a., C5b–9). The released anaphylatoxins promote inflammation by recruiting and activating leukocytes to killing the invading pathogens, additionally, the formed MACs cause osmotic cell damage or even lysis of the target cells.
However, activated complement must be tightly controlled to maintain homeostasis. Notably, the integral involvement of excessive complement activation has been identified in the pathogenesis of many human disorders, including cancer(Hajishengallis et al., 2017), age-related macular degeneration (AMD)(Edwards et al., 2005; Hageman et al., 2005; Klein et al., 2005), ischemia/reperfusion-induced injury(Danobeitia et al., 2014), and transplant rejection(de Cordoba et al., 2012). Consequently, intensive developmental research has focused on biologicals with complement activation inhibitory activities as novel therapeutic agents(Ricklin and Lambris, 2016). One such monoclonal antibody (mAb), eculizumab, which is an anticomplement component 5 (C5) mAb, has been approved for the treatment of paroxysmal nocturnal hemoglobinuria (PNH)(DeZern and Brodsky, 2015), atypical hemolytic uremic syndrome (aHUS)(Mache et al., 2009), and myasthenia gravis(Farmakidis et al., 2018) with an revenue of approximately $3 billion in 2017 alone. Other complement inhibitors, such as anti-C5a(Riedemann et al., 2017) and anti-MASP-2 mAbs, are being investigated in various stages of clinical trials for the treatment of various diseases including hidradenitis suppurativa(Kanni et al., 2018), IgA nephropathy, and thrombotic microangiopathy, with promising results(Ricklin et al., 2018).
As demonstrated above, most currently investigated complement inhibitors are mAbs, which are effective but also have some disadvantages such as a high cost (~$500,000/patient/year for eculizumab)(Coyle et al., 2014), limited routes of administration, and potential antigenicity. Accordingly, interest has increased in therapeutics based on novel small-molecule complement inhibitors, which feature many benefits, including relatively low costs and multiple administration routes, e.g., oral and subcutaneous. However, the requirement for robust large-scale, high-throughput screening assays for the identification of promising lead compounds represents the first major hurdle to the further modification, evaluation, and development of small-molecule complement inhibitors.
Current hemolysis-based complement assays represent certain challenges for high throughput screening. Time-consuming centrifugation and supernatant transfer for optical density analysis as well as associated cost increase may hinder successful workflow development and screening process. Imaging-based assays may be considered as an alternative high throughput-friendly approach. In this report, we describe a low-cost, cell imaging-based, high throughput screening system for the rapid and efficient identification of potential complement inhibitors. Using this system, we screened a small molecule library and identified three leading compounds; of these, only one had been previously identified as a complement inhibitor.
Methods and reagents
Chemical Compounds
The Collection of Biologically Active Molecules (Collection3115) was compiled from the LOPAC library (Sigma, St. Louis, MO, USA) and Bioactive Compound Library (Selleck Chemicals, Houston, TX, USA). A total of 3115 mechanistically annotated and partially redundant compounds were subjected to screening. All compounds were dissolved in DMSO to a stock concentration of 10 mM. The final DMSO concentration did not exceed 0.1 % in the screening assay or during hit validation, and the negative and positive controls contained the same vehicle percentage. Upon hit identification, all compounds were retested as 10 mM stock solutions purchased from Selleck Chemicals.
Cell imaging-based high-throughput screening
To prepare antibody-sensitized sheep erythrocytes (EshA) for the screening, 1ml of sheep erythrocytes (Hemostat Laboratories, Dixon, CA, USA) were washed with GVB-E for twice and suspended with 8ml of GVB-E. 10μl of rabbit anti-sheep erythrocytes serum (MP Biomedicals, Santa Ana, CA, USA) was added. After incubation of 30min in 37°C and another 30min on ice, the antibody-sensitized erythrocytes were washed and re-suspended. For high-throughput screening, 384-well plates (#3707; Corning, Corning, NY, USA) containing final test concentrations (10 μM) of each chemical were prepared using a Janus liquid handling platform (Perkin Elmer, Waltham, MA, USA) equipped with a 50-nL pin transfer tool (V&P Scientific, San Diego, CA, USA). A total of 20 μL of phosphate-buffered saline (PBS) supplemented with 0.15 mM Ca2+ and 1 mM Mg2+ (PBS+CaMg) were dispensed into each well of the plates using an automated dispenser (MultiFlo FX; Biotek, Winooski, VT, USA). Subsequently, 20 μL of 2.5% normal human serum (NHS) in PBS+CaMg were added to the 20 μL of PBS+CaMg buffer with EshA in 10 μL of the PBS+CaMg buffer. After a 40-min incubation at ambient temperature (22ºC), 20 μL of 2.5% glutaraldehyde were added to the wells to stop the reaction and preserve cell morphology. In each plate, two columns (32 wells) containing vehicle only (i.e., without NHS) and two columns containing NHS only were included as positive and negative controls, respectively.
Automated cell imaging and analysis.
An Operetta high-content imaging system with a 20X objective (PerkinElmer) was used for cell imaging. EshA were imaged using the digital-phase contrast mode of the Operetta system at 3–6 hr after the addition of glutaraldehyde. Image analyses and calculations were performed using Acapella and Harmony 4.1 software packages (PerkinElmer). Intact EshA were identified based on the cell size (from 6 to 45 μm2, shape (roundness >0.8), and phase-contrast signal intensity (>185 units). Three non-consecutive fields per well were imaged, analyzed, and averaged; this corresponded to approximately 104 EshA in control wells without NHS. The average number of intact EshA per field were determined for each well, and the percent inhibition was calculated using the following equation:
where CMPD is the number of intact EshA per field in wells treated with compounds, CONTROL_no_NHS is the number in control wells without NHS, and CONTROL_NHS is the number in control wells with NHS.
Assay quality and screening plate data quality were determined based on Z’ factor calculations performed as described(Zhang et al., 1999).
Classical and alternative complement pathway inhibition assay
Classical complement pathway activation was assessed using EshA in Gelatin Veronal Buffer supplemented with Ca2+ and Mg2+ (GVB++; 10 mM Barbital, 145 mM NaCl, 0.1% Gelatin, 0.5 mM MgCl2, 0.15 mM CaCl2, pH 7.3 ± 0.15; Boston BioProducts, Ashland, MA, USA) (Morgan, 2000). In brief, approximately 5 × 106 EshA were incubated with serially diluted NHS (20% to 2.5%) and different titrations (0–50 μM) of cisplatin or pyridostatin in GVB++ at 37ºC for 10 min. For negative controls, 5 mM EDTA was added to the buffer to inhibit complement activation. For the alternative pathway, rabbit erythrocytes (Erabb) and Gelatin Veronal Buffer supplemented with Mg2+ & EGTA (GVB-Mg-EGTA; 10 mM Barbital, 145 mM NaCl, 0.1% Gelatin, 0.5 mM MgCl2, 10 mM EGTA, pH 7.4 ± 0.2; Boston BioProducts,) were used (Morgan, 2000). All the other conditions were the same as that in the classical pathway. After incubation, the erythocytes were centrifuged after the incubation, and the optical density (OD) at 414 nm (OD414) was measured in each 80 μl aliquots of supernatants containing released hemoglobin and used to calculate the percentage of hemolysis as follows: Hemolysis rate (%) = [(A - B)/(C - B)] × 100%, where A is the OD414 of tested samples, B is the OD414 of negative controls with EDTA, and C is the OD414 of maximum hemolysis induced by H2O.
Classical complement pathway convertase assays
The effects of cisplatin and pyridostatin on the classical pathway C3 and C5 convertases were evaluated using EshA, human C3 or C5-depleted serum (ComplementTech, Tyler, TX, USA), guinea pig serum (MP Biomedicals, Solon, OH, USA), and PBS+CaMg buffer according to a published protocol(Blom et al., 2014; Okroj et al., 2012). Briefly, EshA were incubated at 37ºC for 5 min with 5% C3 or C5-depleted serum in 100 μl of GVB++ in the presence of various dosages of the compounds (0, 1.25, 2.5, 5,10 μM). In the absence of C3 or C5, C3 (C4b2a) or C5 convertase (C4b2a3b) assemble on the EshA without further cascade initiation. After washing, 100 μl of 20 mM EDTA-GVB containing 3% guinea pig serum was added to each tube to initiate EshA lysis via pre-assembled convertases. The samples were incubated for 20 min at 37ºC and centrifuged, and the OD414 of the supernatants were determined. Hemolysis rates were calculated using the equation described in the previous section.
Results
Hemolytic complement assay development
The EshA -based complement activity assay described herein was designed and implemented in a 384-well plate format. Baseline reads of a plate containing just buffer no added compound demonstrated a robust signal-to-noise ratio and excellent well-to-well uniformity (Fig. 1). After incubating EshA with complement (NHS), the numbers of intact cells decreased dramatically (20-fold relative to untreated cells) as a result of complement-mediated hemolysis (Fig. 1A-D), and the associated Z’ factor(Zhang et al., 1999) exceeded 0.65. This result was routinely reproduced upon repeated testing. Furthermore, the addition of EDTA, a known inhibitor of complement activation, increased the number of intact EshA in a concentration-dependent manner (Fig. 1E). The addition of glutaraldehyde effectively stopped the reaction and preserved cell morphology for more than 72 hours, while both the Z’ factor and the signal-to-background ratio remained static throughout the stability test ((Supplementary Table 2). This excellent stability and robust performance in a 384-well plate format indicate that our assay could be used for automated screening. A correlation analysis further demonstrated the good reproducibility of screening results between combinations of paired sets of small molecules (Fig. 1F), with a correlation coefficient (R2) of >0.9 in a comparison of two independent experiments.
Figure 1.
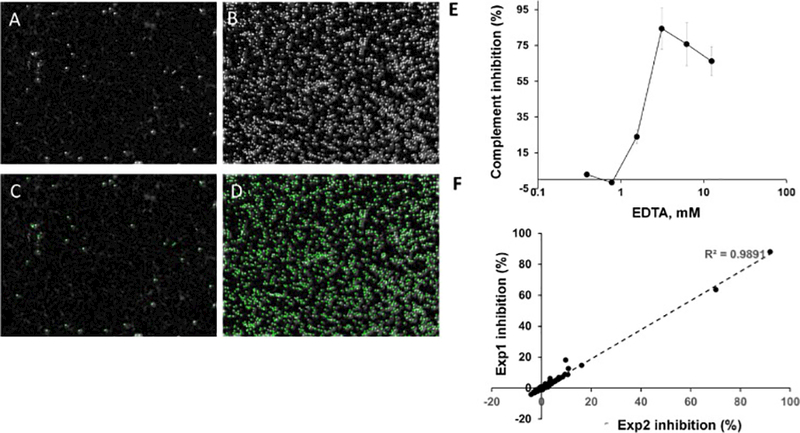
Evaluation of complement activity using automated cell imaging and image analysis.
A-D, Representative images of wells with EshA in the presence of absence of complement. EshA were incubated in wells of a 384 well plate the presence (A) or absence (B) of complement, then the numbers of intact red blood cells were analyzed by the Operetta image system. A, B, Digital phase-contrast images. C, D, Automated image analysis. Intact EshA are outlined in green. E, Complement inhibitory effect of EDTA as determined by imaging and image analysis. F, Correlation between the inhibition values obtained from two independent experiments with a set of 353 small molecules. EXP, experiment.
Quantitative high-throughput screening of the Collection 3115 library
The Collection 3115 library was screened in high-throughput screening mode using the above-described complement activity assay. All library compounds were tested at a single concentration of 10 μM. The assay performance remained robust and the Z’ factor remained high (Z’ >0.6, data not shown) throughout the screening process. Using the criterion of an inhibitory response of ≥50% to identify active hit candidates, an analysis of the cumulative library response revealed that very few compounds (n = 5) demonstrated significant inhibitory activity (Fig. 2). After two hit candidates were found to represent the same compound (cisplatin), the list of potential hit candidates was reduced to four compounds: nafamostat, cisplatin, cDPCP, and pyridostatin.
Figure 2.
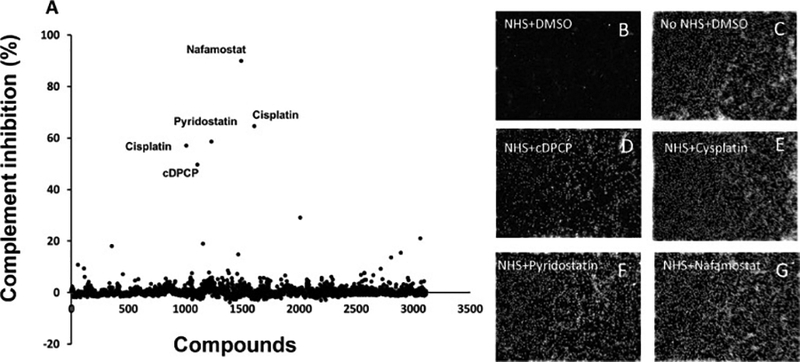
Screening of 3115 individual small molecules and identification of several potential complement inhibitors.
A, The complement inhibitory activities of 3115 compounds are plotted relative to the negative control. Compound IDs are listed on the X-axis, while complement inhibition (%) is shown along the Y-axis. Compound names are listed for the small molecules with the strongest inhibitory activities. B-G, representative digital phase-contrast images of control wells (B-C) and wells containing treated red blood cells (D-G). NHS, normal human serum.
Characterization of the hit compounds
Further experiments demonstrated that all four identified compounds inhibited complement-mediated hemolysis in a concentration-dependent manner (Fig. 3). However, only three compounds exhibited inhibitory activity levels exceeding 50% at concentrations <10 μM: nafamostat (IC50 = 0.89 μM), cisplatin (IC50 = 3.96 μM), and pyridostatin (IC50 = 7.57 μM).
Figure 3.
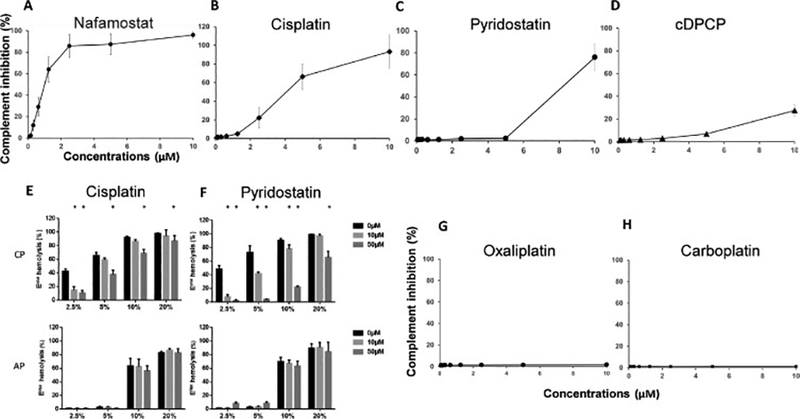
Further analysis of the hit compounds and others in a concentration-dependent manner.
A, Nafamostat mesylate, a known complement inhibitor, was independently identified using our screening method. B-D, Our screening method identified three new complement activation inhibitors—cisplatin, pyridostatin, and cDPCP. All three compounds inhibit complement activation in a concentration-dependent manner. E-F, Cisplatin and pyridostatin inhibit the classical pathway (CP) of complement activation in a sensitized sheep erythrocyte (EshA)-based hemolysis assay (upper panels), but not in the alternative pathway (AP) in a rabbit erythrocyte (Erabb)-based hemolysis assay (lower panels). G-H. Two other platinum-containing agents, oxaliplatin and carboplatin, do not inhibit complement activation. All experiments were performed in duplicate, and the results are shown as mean ± standard deviations from one representative experiment. NHS, normal human serum.
Because our screening assay used EshA and thus evaluated complement activation through the classical pathway, we used an Erabb–based hemolysis assay to determine whether cisplatin and/or pyridostatin could also inhibit complement activation via the alternative pathway. However, we found that although both compounds inhibited the complement-mediated hemolysis of EshA (classical pathway), they had no inhibitory effect on the hemolysis of Erabb (alternative pathway) even at concentrations as high as 50 μM (Fig. 3E, F, lower panel).
Complement activation inhibitory activity is not common to all platinum-containing agents
Our initial screening of the Collection 3115 library revealed that cisplatin and cDPCP, two of four included platinum-containing molecules, exhibited inhibitory activity toward classical pathway complement activation-mediated hemolysis. Accordingly, we considered that platinum might be essential to the observed complement inhibitory activity. However, additional experiments revealed that oxaliplatin (Weiss and Christian, 1993) and carboplatin(Lee et al., 1983), the other two platinum-containing compounds, did not exhibit any inhibitory activity against complement-mediated hemolysis, even at concentrations as high as 10 μM (Fig. 3G, H).
Inhibition of the classical pathway C3 and C5 convertase
We next explored the potential mechanism by which the newly identified inhibitors of classical pathway complement-mediated hemolysis inhibit complement activation at the level of C3 and/or C5 convertases. Using an assay involving the surface preassembly of C3 or C5 convertase on EshA, we demonstrated that although cisplatin slightly augmented C3 convertase activity at 10 μM, (Fig. 4A) it strongly inhibited C5 convertase activity at concentrations as low as 1.25 μM (Fig. 4C). In addition, while pyridostatin had no significant effect on C3 convertase activity even at a concentration as high as 10 μM (Fig. 4B), it inhibited C5 convertase at concentrations as low as 1.25 μM (Fig. 4D)
Figure 4.
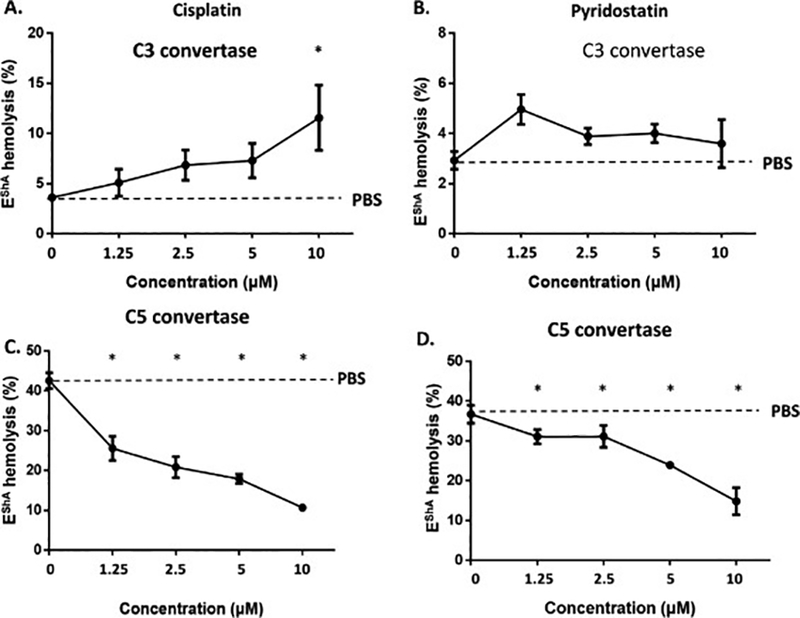
Classical pathway C3 and C5 convertase inhibition assay
The inhibitory activities of cisplatin and pyridostatin against the C3 and C5 convertases in the classical pathway of complement activation were evaluated in an antibody-sensitized sheep erythrocyte (EshA) hemolysis assay. A, Effects of cisplatin on C3 convertase. B, Effects of pyridostatin on C3 convertase. C, Effects of cisplatin on C5 convertase. D, Effects of pyridostatin on C5 convertase. Equivalent volumes of PBS were used as controls (PBS). *p <0.05.
Discussion
In this report, we describe a robust cell imaging-based assay for the high-throughput screening of complement inhibitors. Using this assay, we screened more than 3000 bioactive small molecule compounds and identified two new complement inhibitors, cisplatin and pyridostatin, as well as nafamostat, a previously reported complement inhibitor(Fujii and Hitomi, 1981). These two newly identified compounds do not appear to inhibit the alternative pathway of complement activation, but were found to target C5 convertase in the classical pathway.
Large-scale screening is commonly used to identify lead compounds for further modifications and improvements during drug discovery and development. Such screening procedures require minimal human interference and are preferably conducted by machines in an automatic and relatively inexpensive manner. In this work, we developed an assay based on standard 384-well plates, with machine-based handling of reagents, samples, and the final imaging analysis. These specifications allowed this assay to be used for high-throughput screening. Indeed, our system could be used to easily screen more than 3000 compounds for potential complement inhibitors within one day, at an approximate reagent cost of $0.01 per sample. Thus, our cell imaging-based assay is valuable for the large-scale screening of potential lead compounds during the development of novel complement inhibitors as drug candidates.
While developing our screening assay, we used EshA as target cells in measurements of complement (i.e., MAC) inhibitory activities because the antibody-antigen complexes formed on the surfaces of EshA activate complement in the NHS via the classical pathway and thus initiate MAC formation and cell lysis(Morgan, 2000). This reaction can be effectively stopped by the addition of glutaraldehyde, which denatures all proteins and fixes the EshA for later quantitative imaging analyses of intact cells. This step is particularly useful during large-scale screening experiments wherein plates must be stored prior to image analysis.
Our screening process identified only 3 compounds with apparent complement inhibitory activity from a library of 3115 bioactive small molecules; in other words, our assay appears to be highly specific. The inclusion of nafamostat, a previously reported complement inhibitor, among the three “hits” further validated our assay and suggested that the identified compounds are highly likely to exhibit complement inhibitory activity. Indeed, subsequent confirmatory experiments demonstrated that all the newly identified compounds inhibited complement activation (i.e., complement-mediated hemolysis) in a dose-dependent manner. Based on the success of this assay, we predict that rabbit RBCs could be used similarly for the hight-hroughput screening of inhibitors of the alternative pathway of complement activation.
Eculizumab, a complement inhibitor approved for the clinical treatment of PNH and aHUS, inhibits MAC formation at the C5 step of the complement activation cascade while leaving the C3 step intact(Kaplan, 2002). In our C3/C5 convertase assays, we found that both newly identified complement inhibitors, cisplatin and pyridostatin, inhibited C5 convertase rather than C3 convertase. Furthermore, the C5 convertase inhibitory activities of the 2 hit compounds appeared to be specific to the classical pathway, as we did not observe any inhibitory effects of either compound on the complement-mediated lysis of Erabb , which is mediated by the alternative pathway of complement activation. As noted above, the classical and alternative pathway use different C5 convertases (C4b2a3b v.s. C3bBb3b). Accordingly, this difference might explain why cisplatin and pyridostatin target the C5 convertase of one pathway but not the other.
Interestingly, both novel hit compounds identified during our screen are tumoricidal agents. Currently, the precise role of complement in cancer remains elusive. Activated complement induces direct MAC-mediated tumor cell killing(Taylor and Lindorfer, 2014) and the recruitment and activation of other leukocytes to further damage the tumor cells. However, activated complement, especially in the local tumor microenvironment, has also been reported to promote tumor cell survival directly by signaling through C3aR and/or C5aR on tumor cells(Ajona et al., 2017), and indirectly by recruiting immunosuppressive myeloid-derived suppressor cells(Markiewski et al., 2008).
Cisplatin is a chemotherapy drug commonly used to treat various types of cancer, including ovarian and cervical cancers(Wichtowski et al., 2017). Mechanistically, it kills cancer cells by crosslinking the purine bases on DNA strands to interfere with cell proliferation, with the eventual outcomes of cell apoptosis and death. Cisplatin is administrated via i.v. infusion, and is fully exposed to complement in the blood. Therefore, cisplatin might still exert complement-related biological effects in patients even though its complement inhibitory activity appears to be weaker than that of nafamostat. As described above, local complement activation in the tumor microenvironment may promote tumor cell survival(Pio et al., 2014). Possibly, cisplatin might accumulate within this microenvironment at sufficiently high concentrations to exert complement inhibitory activity. This is a particularly interesting concept because complement inhibition is currently considered a promising approach to cancer treatment (Pio et al., 2013). Further studies are warranted to address the potential involvement of this newly discovered complement inhibitory activity in the mechanisms by which cisplatin mediates its tumoricidal effects.
Pyridostatin, the other hit compound identified during our screen, is a G-quadruplex-stabilizing small molecule with telomerase inhibitory activity(McLuckie et al., 2013). Accordingly, exposure to this drug induces cell senescence and apoptosis. Currently, pyridostatin is under development as a novel tumoricidal drug. Our new discovery of the weak but detectable complement inhibitory activity of pyridostatin might shed further light on the documented tumoricidal effects of this drug.
In summary, we have developed a cell imaging-based assay for the large-scale, high-throughput screening of potential complement inhibitors and subsequently used this assay to screen a library of >3000 bioactive small molecules. In addition to nafamostat, a known complement inhibitor, our screening newly identified two tumoricidal agents, cisplatin and pyridostatin, with inhibitory activity against the classical but not the alternative pathway of complement activation. These results validate our newly developed assay for the large-scale screening of potential complement inhibitors, and suggest that complement inhibition might contribute to the established tumoricidal effects of cisplatin and pyridostatin.
Supplementary Material
Acknowledgment:
This work was supported in part by NIH grant DK10358 (to F.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
References:
- Ajona D, Ortiz-Espinosa S and Pio R, 2017. Complement anaphylatoxins C3a and C5a: Emerging roles in cancer progression and treatment. Semin Cell Dev Biol. [DOI] [PubMed] [Google Scholar]
- Blom AM, Volokhina EB, Fransson V, Stromberg P, Berghard L, Viktorelius M, Mollnes TE, Lopez-Trascasa M, van den Heuvel LP, Goodship TH, Marchbank KJ and Okroj M, 2014. A novel method for direct measurement of complement convertases activity in human serum. Clin Exp Immunol 178, 142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle D, Cheung MC and Evans GA, 2014. Opportunity cost of funding drugs for rare diseases: the cost-effectiveness of eculizumab in paroxysmal nocturnal hemoglobinuria. Med Decis Making 34, 1016–29. [DOI] [PubMed] [Google Scholar]
- Danobeitia JS, Djamali A and Fernandez LA, 2014. The role of complement in the pathogenesis of renal ischemia-reperfusion injury and fibrosis. Fibrogenesis Tissue Repair 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Cordoba SR, Tortajada A, Harris CL and Morgan BP, 2012. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology 217, 1034–46. [DOI] [PubMed] [Google Scholar]
- DeZern AE and Brodsky RA, 2015. Paroxysmal Nocturnal Hemoglobinuria: A Complement-Mediated Hemolytic Anemia. Hematol Oncol Clin North Am 29, 479–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C and Farrer LA, 2005. Complement factor H polymorphism and age-related macular degeneration. Science 308, 421–4. [DOI] [PubMed] [Google Scholar]
- Farmakidis C, Pasnoor M, Dimachkie MM and Barohn RJ, 2018. Treatment of Myasthenia Gravis. Neurol Clin 36, 311–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S and Hitomi Y, 1981. New synthetic inhibitors of C1r, C1 esterase, thrombin, plasmin, kallikrein and trypsin. Biochim Biophys Acta 661, 342–5. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M and Allikmets R, 2005. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A 102, 7227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Reis ES, Mastellos DC, Ricklin D and Lambris JD, 2017. Novel mechanisms and functions of complement. Nat Immunol 18, 1288–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanni T, Zenker O, Habel M, Riedemann N and Giamarellos-Bourboulis EJ, 2018. Complement activation in hidradenitis suppurativa: a new pathway of pathogenesis? Br J Dermatol. [DOI] [PubMed] [Google Scholar]
- Kaplan M, 2002. Eculizumab (Alexion). Curr Opin Investig Drugs 3, 1017–23. [PubMed] [Google Scholar]
- Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C and Hoh J, 2005. Complement factor H polymorphism in age-related macular degeneration. Science 308, 385–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FH, Canetta R, Issell BF and Lenaz L, 1983. New platinum complexes in clinical trials. Cancer Treat Rev 10, 39–51. [DOI] [PubMed] [Google Scholar]
- Mache CJ, Acham-Roschitz B, Fremeaux-Bacchi V, Kirschfink M, Zipfel PF, Roedl S, Vester U and Ring E, 2009. Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol 4, 1312–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markiewski MM, DeAngelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G and Lambris JD, 2008. Modulation of the antitumor immune response by complement. Nat Immunol 9, 1225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLuckie KI, Di Antonio M, Zecchini H, Xian J, Caldas C, Krippendorff BF, Tannahill D, Lowe C and Balasubramanian S, 2013. G-quadruplex DNA as a molecular target for induced synthetic lethality in cancer cells. J Am Chem Soc 135, 9640–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan BP, 2000. Complement methods and protocols. Humana Press, Totowa, N.J. [Google Scholar]
- Okroj M, Holmquist E, King BC and Blom AM, 2012. Functional analyses of complement convertases using C3 and C5-depleted sera. PLoS One 7, e47245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pio R, Ajona D and Lambris JD, 2013. Complement inhibition in cancer therapy. Semin Immunol 25, 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pio R, Corrales L and Lambris JD, 2014. The role of complement in tumor growth. Advances in experimental medicine and biology 772, 229–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K and Lambris JD, 2010. Complement: a key system for immune surveillance and homeostasis. Nat Immunol 11, 785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D and Lambris JD, 2016. New milestones ahead in complement-targeted therapy. Semin Immunol 28, 208–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricklin D, Mastellos DC, Reis ES and Lambris JD, 2018. The renaissance of complement therapeutics. Nat Rev Nephrol 14, 26–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedemann NC, Habel M, Ziereisen J, Hermann M, Schneider C, Wehling C, Kirschfink M, Kentouche K and Guo R, 2017. Controlling the anaphylatoxin C5a in diseases requires a specifically targeted inhibition. Clin Immunol 180, 25–32. [DOI] [PubMed] [Google Scholar]
- Taylor RP and Lindorfer MA, 2014. The role of complement in mAb-based therapies of cancer. Methods 65, 18–27. [DOI] [PubMed] [Google Scholar]
- Weiss RB and Christian MC, 1993. New cisplatin analogues in development. A review. Drugs 46, 360–377. [DOI] [PubMed] [Google Scholar]
- Wichtowski M, Murawa D, Kulcenty K and Zaleska K, 2017. Electrochemotherapy in Breast Cancer – Discussion of the Method and Literature Review. Breast Care (Basel) 12, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD and Oldenburg KR, 1999. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.