

Abstract
Evidence is reviewed that behavioral training and neural injury can engage metaplastic processes that regulate adaptive potential. This issue is explored within a model system that examines how training affects the capacity to learn within the lower (lumbosacral) spinal cord. Response-contingent (controllable) stimulation applied caudal to a spinal transection induces a behavioral modification indicative of learning. This behavioral change is not observed in animals that receive stimulation in an uncontrollable manner. Exposure to uncontrollable stimulation also engages a process that disables spinal learning for 24-48 hrs. Controllable stimulation has the opposite effect; it engages a process that enables learning and prevents/reverses the learning deficit induced by uncontrollable stimulation. These observations suggest that a learning episode can impact the capacity to learn in future situations, providing an example of behavioral metaplasticity. The protective/restorative effect of controllable stimulation has been linked to an up-regulation of brain-derived neurotrophic factor (BDNF). The disruption of learning has been linked to the sensitization of pain (nociceptive) circuits, which is enabled by a reduction in GABA-dependent inhibition. After spinal cord injury (SCI), the co-transporter (KCC2) that regulates the outward flow of Cl- is down-regulated. This causes the intracellular concentration of Cl- to increase, reducing (and potentially reversing) the inward flow of Cl- through the GABA-A receptor. The shift in GABA function (ionic plasticity) increases neural excitability caudal to injury and sets the stage for nociceptive sensitization. The injury-induced shift in KCC2 is related to the loss of descending serotonergic (5HT) fibers that regulate plasticity within the spinal cord dorsal horn through the 5HT-1A receptor. Evidence is presented that these alterations in spinal plasticity impact pain in a brain-dependent task (place conditioning). The findings suggest that ionic plasticity can affect learning potential, shifting a neural circuit from dampened/hard-wired to excitable/plastic.
Keywords: Spinal cord injury, central sensitization, instrumental conditioning, BDNF, TNF, KCC2
Graphical abstract
1. Introduction
Learning in response to environmental stimulation can impact both the performance of a particular response and the capacity to learn when challenged by new environmental relations. The latter effect suggests that training can induce a lasting alteration (a form of plasticity) that impacts plastic potential, a phenomenon known as metaplasticity (Abraham, 2008, Hulme et al., 2013). The concept of metaplasticity emerged from studies examining factors that influence the development of long-term potentiation (LTP) and depression (LTD) using electrophysiological techniques (Abraham and Bear, 1996). In general, shifts in the development of LTP/LTD have been related to alterations in synaptic function, with a particular focus on the neurotransmitter glutamate and the degree to which this ligand elicits a response in the postsynaptic cell, mediated by the AMPA and NMDA receptors. This line of work has revealed that stimulation can induce a lasting modification that impacts the magnitude and direction of synaptic change (synaptic plasticity), and has linked these alterations to ligands that impact AMPA and NMDA receptor function (Abraham, 2008).
In the present review we take a more top-down approach to the topic of metaplasticity. We begin with a set of behavioral phenomena that reveal how a learning episode can impact future learning, demonstrating a form of learning to learn (behavioral metaplasticity) (Schmidt et al., 2013, Grau et al., 2014). After we unpack the criteria for invoking these concepts, we will illustrate how the observations apply to a simple form of spinally-mediated learning. We will show that neurons within the lower (lumbosacral) spinal cord are sensitive to behavioral relations and that this learning affects the capacity to learn when the system is challenged with new environmental events. We will unpack how concepts derived from the study of brain-dependent learning and memory, as well as from the pain literature, inform our understanding of spinal cord plasticity. Spinal learning depends upon a form of NMDA receptor (NMDAR) mediated plasticity and involve ligands known to affect LTD/LTP, such as brain derived neurotrophic factor (BDNF) and tumor necrosis factor (TNF) (Stellwagen and Malenka, 2006, Grau, 2014, Grau et al., 2014). Further work has revealed that plastic potential within the spinal cord is modulated by descending fibers that regulate the inhibitory action of the neurotransmitter GABA (Grau et al., 2016, Huang et al., 2016, Grau et al., 2017). We review how a shift in GABA-dependent inhibition (ionic plasticity) allows spinal learning and promotes the sensitization of pain (nociceptive) fibers. The work, we believe, challenges some long-standing beliefs—spinal circuits are not fixed and GABA does not, in adult animals, uniformly inhibit neural function. We suggest that the lessons learned from a detailed analysis of plasticity within the spinal cord may inform our views of how neural function is regulated more generally.
2. Metaplasticity
Abraham and Bear (1996) put forth the concept of metaplasticity to capture a set of electrophysiological observations. Here, the plasticity of interest concerns a synaptic modification, with an emphasis on factors (e.g., the trafficking of AMPARs) that affect the response elicited within the postsynaptic cell, that modulate the development of LTP and LTD. Interestingly, prior events can affect the development of this plasticity. Because the initiating event engages a lasting modification (a form of plasticity), that affects the development of synaptic plasticity in response to subsequent stimulation, the phenomenon was characterized as a kind of plasticity of plasticity (meta-plasticity) (Abraham, 2008). For example, a short burst of stimulation within area CA1 of the hippocampus produces a transient potentiation that decays within ten min. Yet, when a stronger stimulus is applied twenty min later, LTP is inhibited (Huang et al., 1992). Importantly, this was observed even though the initial electrophysiological response within the postsynaptic cell was unchanged at the time of testing. Rather, what was seemingly altered was the magnitude of synaptic modification produced by the postsynaptic activity (depolarization). This type of modification can be represented within a model that relates the development of LTP/LTD to postsynaptic neural activity (Bienenstock et al., 1982). In this framework, metaplasticity involves a shift in the relation between the postsynaptic response and the consequent synaptic modification (Fig. 1A) (Abraham, 2008). For example, treatment with a mGlu agonist or TNF can foster the development of LTP, producing a leftward shift in the function relating postsynaptic activity to alterations in synaptic efficacy. A shift of this type can emerge from either an intrinsic alteration within the stimulated circuit (homosynaptic metaplasticity) or through the activation of an additional (extrinsic) process that regulates plastic potential (heterosynaptic metaplasticity). By engaging an extrinsic process, heterosynaptic metaplasticity can impact neural plasticity in other circuits.
Fig. 1.
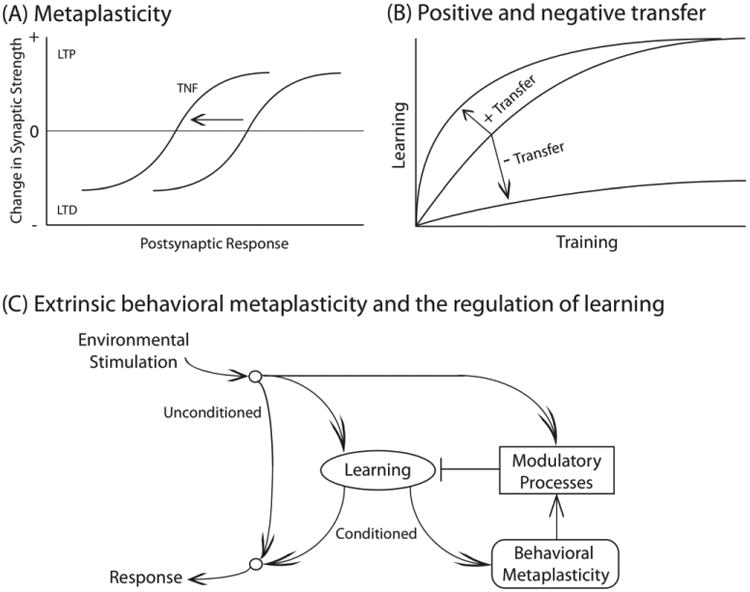
(A) The Bienenstock, Cooper and Murno (BCM) model links the induction of LTD and LTP to the postsynaptic response (Bienenstock et al., 1982). Within this framework, metaplastic alterations can be represented as a shift in the threshold (theta) for inducing LTD versus LTP. For example, a treatment (e.g., TNF) that promotes the development of LTP is hypothesized to shift the function to the left. (B) Behavioral metaplasticity impacts the rate of learning, producing either a facilitation [positive (+) transfer] or impairment [negative (-) transfer]. (C) A theoretical framework for representing an extrinsic form of behavioral metaplasticity. Environmental stimulation can engage unconditioned (unlearned) behavioral responses, learning, and modulatory process that regulate the rate of learning. Learning can also invoke a secondary process that represents a form of “learning to learn” (behavioral metaplasticity). This would induce a lasting modification that impacts future learning, both in the trained and other (extrinsic) neural circuits by regulating the activation of modulatory processes.
A defining feature of metaplasticity is that it has a lasting effect (Abraham and Bear, 1996). This is important because many forms of stimulation can engage an unlearned (unconditioned) response that acutely modulates neural plasticity but fades soon after the event has ended. Another key feature concerns the relation between the target effect (the change in the synaptic function) and the processes that underlies metaplasticity. At issue is whether or not the initiating event has a lasting effect simply because it induces a subtle modification in a common process (e.g., increasing the density of AMPARs in the active zone of the postsynaptic membrane). Additivity of this sort can be difficult to rule out in cases where the priming and test events involve the same (intrinsic) circuit. The issue is of less concern when the initiating event affects other (extrinsic) neural circuits. Elsewhere we have suggested another key feature of metaplasticity is reversibility (Grau et al., 2014). Are the cellular processes that underlie the learning still (potentially) functional? Treatments that have a lasting effect because they are cytotoxic, and thereby eliminate the capacity for learning, do not represent examples of metaplasticity. To demonstrate metaplasticity, the effect of our experimental treatment needs to be reversible (e.g., by a drug antagonist)
More recently, the concept of metaplasticity has been extended to a range of behavioral phenomena that impact the capacity to learn when the organism is challenged by new environmental events (Schmidt et al., 2013). It is assumed here that behavioral metaplasticity reflects an alteration in neuronal function (neural plasticity), that this may involve either a presynaptic or postsynaptic modification, and that non-neuronal cells (astrocytes and microglia) may play a role. In behavioral terms, the claim is that an initial learning episode can impact the capacity to learn in future situations, a kind of learning to learn (Sehgal et al., 2013, Grau et al., 2014). This can affect learning in two obvious ways (Fig. 1B): By promoting subsequent learning (positive transfer) or disrupting it (negative transfer).
Behavioral metaplasticity is assumed to have a lasting effect, where lasting is typically defined on the order of hours to weeks (Grau et al., 2014). As with cellular metaplasticity, care needs be taken to assure that the modification in the rate of learning is not simply due to a kind of additivity, wherein an alteration induced by the first learning episode simply adds to (or subtracts from) a common process when the organism is challenged to learn a second time. For example, we would not want to claim that learning to turn left in a T-maze for a food reward has a metaplastic effect because it interferes with later learning to turn right for reward. In this case, the motor response established by the first learning episode could simply interfere with learning to perform the new response. Rather, behavioral metaplasticity involves cases wherein the initial training allows for the abstraction of a new, qualitatively distinct form of learning—establishing a memory that impacts the capacity for learning when the organism is later challenged with new environmental events/tasks (Fig. 1C). Again, it is also important to show that the metaplastic modification is reversible and not due to factors such as cell death and/or injury.
3. The plastic spinal cord: A simple model system
In exploring whether neurons within the lumbosacral spinal cord can learn, we discovered that training can have a lasting effect on adaptive potential (Grau et al., 2006). To understand how our findings implicated a kind of behavioral metaplasticity, we need to introduce how spinal learning has been studied. Spinal neurons can support a range of basic learning phenomena, including single stimulus learning (habituation and sensitization), Pavlovian conditioning, and instrumental learning (Grau, 2014). Here, we will focus on just two kinds of learning: sensitization due to exposure to a noxious stimulus (Willis, 2001, Ji et al., 2003, Latremoliere and Woolf, 2009, Sandkuhler, 2009) and learning about response-outcome (instrumental) relations (Grau et al., 1998, Grau et al., 2006).
It was recognized decades ago that exposure to a noxious stimulus, which engages pain (nociceptive) fibers, can facilitate the performance of a spinal reflex after communication with the brain has been surgically interrupted by means of a rostral transection (Thompson and Spencer, 1966, Groves et al., 1969). More recently, research has shown that the activation of unmyelinated (C) nociceptive fibers can sensitize neurons within the dorsal horn of the spinal cord, a phenomenon known as central sensitization (Latremoliere and Woolf, 2009). Interestingly, the induction of central sensitization can enhance behavioral reactivity to non-noxious mechanical stimulation and transform how the stimulus is perceived, causing it to generate pain (Simone et al., 1989). This phenomenon is known as allodynia and is a hallmark of neuropathic pain. Neurobiological studies have linked centrally-mediated nociceptive sensitization to the development of LTP and NMDAR-mediated plasticity (Sandkuhler, 2009). From this view, the induction of central sensitization has a lasting effect because it lays down a kind of pain memory within the dorsal horn. Evidence suggests that the induction and maintenance of this memory depends upon signal pathways analogous to those involved in learning and memory within the brain (Ji et al., 2003).
The discovery that spinal neurons support NMDAR-mediated plasticity and the development of LTP implied a capacity to learn. Building on these insights, we explored the functional limits of this system, demonstrating it could support a range of Pavlovian phenomena (reviewed in Grau et al., 2014). Because the notion that spinal neurons may be sensitive to response-outcome (instrumental) relations had proven controversial, and because this type of learning seemed especially relevant to recovery after SCI, we focused our attention on this form of learning. Our first aim was to determine whether spinal neurons, isolated from the brain by means of a complete (thoracic) transection, are sensitive to response-outcome relations (Grau et al., 1998). By performing the injury at a remote site [second thoracic vertebra (T2)], we hoped that the injury per se would have a minimal effect on spinal function. Treatment and/or testing is typically initiated the next day. Using this procedure, we have found no evidence that a general process (e.g., spinal shock) interferes with learning (Grau et al., 1998, Grau et al., 2006). Indeed, as we will see below, injury appears to increase adaptive potential.
To elicit a behavioral response, shock is applied to one hindleg through electrodes implanted into the tibialis anterior muscle, which drives a flexion response. Leg position is monitored by taping a contact electrode to the rat's hindpaw. When the leg is relaxed, the electrode touches an underlying salt solution, which completes a circuit monitored by a computer. In our first experiment (Grau et al., 1998), one group of spinally transected rats (Master) was given controllable shock for 30 min. For rats in this group, shock was applied whenever the contact electrode touched the underlying solution. Master rats exhibited a progressive increase in flexion duration that reduced net shock exposure (Fig. 2A). Rats in another group were experimentally coupled (Yoked) to subjects in the master condition. Each rat in the yoked group received shock at the same time, and for the same duration, as its master partner. Because the yoked rat received the stimulation independent of leg position, the shock was uncontrollable. Yoked rats did not exhibit an increase in flexion duration, which suggests that the learning observed in the master rats depended upon the response (leg position)-outcome (shock) relation.
Fig. 2.
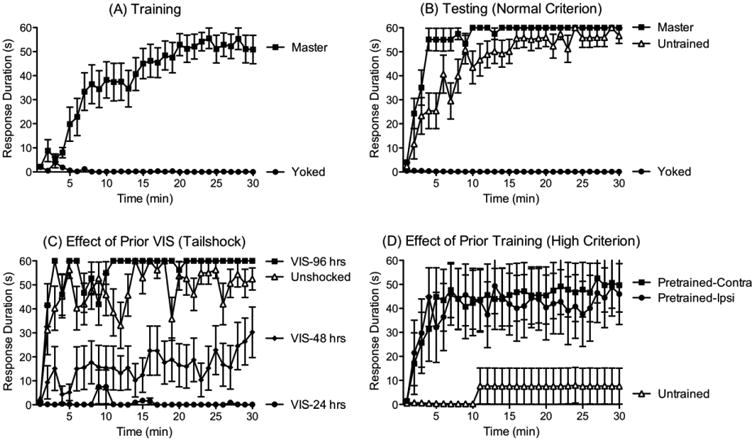
(A) Spinally transected rats that receive a noxious shock to one hind leg whenever the leg is extended (Master) exhibit a progressive increase in flexion (response) duration over 30 min of testing, which reduces net shock exposure (Grau et al., 1998). Rats that receive the same amount of noxious stimulation, but independent of leg position (Yoked), fail to learn. (B) Spinally transected rats that have previously undergone training with controllable stimulation (Master) learn faster (positive transfer) than untrained (naïve) controls (Grau et al., 1998). Rats that have previously received uncontrollable stimulation (Yoked) exhibit a learning impairment. Importantly, this impairment is not due to an inability to perform the target response; rats that have previously received uncontrollable stimulation exhibit a flexion response when shock is given, but this response-contingent stimulation does not induce an increase in flexion duration. (C) Exposure to variable intermittent tailshock (VIS) for 6 min (180 shocks) impairs learning for 48 hrs (Crown et al., 2002b). (D) Prior exposure to controllable shock (Pretrained) fosters learning when animals are tested with a higher response criterion (Crown et al., 2002a). Under these conditions, naïve (untrained) rats fail to learn. Pretrained rats are able to learn and this is true independent of whether they are tested on the same [ipsilateral (ipsi)] or opposite [contralateral (contra)] leg.
At the end of training, we re-equated response vigor and task difficulty across animals by adjusting the shock intensity used to elicit a flexion response and the depth of the contact electrode (used to monitor leg position). Animals were then tested animals for another 30 min with response-contingent (controllable) shock (Grau et al., 1998). We found that prior training with controllable stimulation fostered learning relative to the untrained controls (Fig. 2B). Interestingly, rats that had previously received the same amount of shock in an uncontrollable manner failed to learn. Further work showed that degrading response-outcome contiguity (by delaying the onset of shock) also disrupts learning (Grau et al., 1998).
Our results show that spinal cord neurons are sensitive to R-O relations. We have suggested that this learning depends upon the relation between an index of leg position (a proprioceptive signal) and shock onset, and that the system is biologically prepared to detect these relations (Grau et al., 2012). This is consistent with the view that proprioceptive input is integrated online to form a temporal-spatial “image” to guide limb dynamics (Edgerton et al., 2004). Our work is also consistent with the framework outlined by Windhorst (2007), who suggested that motor function can be adjusted through a form of back-propagation within the dendritic tree, mediated by NMDAR-dependent/Hebbian synaptic plasticity (Windhorst, 2007).
4. Experience induces a metaplastic-like change in the capacity to learn
We recognized that the learning phenomena described above could be intrinsic to the trained pathway (Crown et al., 2002a, Joynes et al., 2003). For example, exposure to uncontrollable stimulation might interfere with later learning simply because it induced a kind of motor fatigue, a peripheral modification that would undermine the performance of the target response. To examine how uncontrollable stimulation affects the capacity to learn, we developed a computer program that emulates the variable pattern of stimulation produced by a typical master rat (Crown et al., 2002b). This program presents brief (100 msec) shocks on a variable (0.2-3.8 s) schedule with a mean inter-stimulus interval (ISI) of 2 sec. We found that just 6 min of variable intermittent shock (VIS) impaired learning when rats were tested 24-48 hrs later (Fig. 2C). Importantly, VIS applied to one hind leg impaired learning when subjects were tested on the opposite (contralateral) leg (Joynes et al., 2003). Indeed, even VIS applied to the tail induced a learning impairment. The fact that stimulation to one dermatome affected learning when a distinct stimulus-response (S-R) circuit was tested suggests that VIS has a general (extrinsic) effect on adaptive potential within the lumbosacral spinal cord. To firmly establish that the learning impairment depended upon a central modification, we blocked the sensory input from one hind leg by cutting the sciatic nerve (Joynes et al., 2003). This eliminated the learning impairment induced by VIS applied to that leg. Likewise, inactivating spinal neurons by applying the anesthetic lidocaine [through an intrathecal (i.t.) catheter] prior to VIS blocked the learning impairment when rats were tested the next day. Interestingly, i.t. administration of a NMDAR antagonist, or a protein synthesis inhibitor, prior to VIS also blocked the induction of the learning impairment (Patton et al., 2004, Ferguson et al., 2006). Taken together, these observations suggest that the adverse effect of VIS on learning (i.e., maladaptive plasticity) is centrally mediated and depends upon an NMDAR-dependent process.
We performed a similar set of manipulations to confirm that adaptive response-outcome (instrumental) learning also depends upon neurons within the spinal cord (Crown et al., 2002a). Cutting the sciatic nerve prevented instrumental learning. Likewise, inactivating spinal cord neurons with lidocaine blocked learning. So too did i.t. administration of a NMDAR antagonist (Joynes et al., 2004). These findings imply that learning to perform an instrumental response depends upon a centrally-mediated NMDAR-dependent process
Next, we explored the facilitation of learning observed after instrumental conditioning. We reasoned that prior training might enable learning (positive transfer) when animals are tested using a higher response criterion. To increase task difficulty, we raised the level of the underlying solution, forcing animals to exhibit a stronger flexion response to terminate the shock (Crown et al., 2002a). This manipulation made the task so difficult that previously untrained animals could not learn (Fig. 2D). However, rats that had been previously trained were able to learn and this was true when animals were tested on the pretrained (ipsilateral) or opposite (contralateral) leg. The fact that instrumental training on one leg affects learning when animals are tested on the opposite leg again implies that a process was engaged within the spinal cord that has a general (extrinsic) effect on the capacity to learn.
Additional evidence that instrumental learning has a general effect on learning potential was obtained when we assessed the interaction between instrumental conditioning and the learning impairment. We found that prior exposure to controllable stimulation blocked the adverse effect of VIS applied to the tail (Crown and Grau, 2001). Importantly, the protective effect of instrumental training was evident when subjects were tested using the contralateral leg. We also assessed the effect of instrumental training given after the learning impairment was induced (Crown and Grau, 2001). To allow learning in VIS exposed animals, we pretreated rats with an opioid antagonist (naltrexone). Prior work had shown that administration of naltrexone blocks the expression, but not the induction, of the learning deficit (Joynes and Grau, 2004). By blocking the expression of the deficit, we were able to instrumentally train rats that had previously received VIS. We then tested rats the next day (after the drug had cleared the system) by applying response-contingent shock to the untreated (contralateral) leg. As usual, VIS to the tail induced a learning impairment. Rats that had received instrumental training after VIS did not exhibit a learning impairment.
Our observations yield a coherent story, demonstrating that exposure to uncontrollable stimulation induces a lasting learning impairment (Crown et al., 2002b). Controllable stimulation has the opposite effect—it enables learning and blocks the learning impairment (Crown and Grau, 2001, Grau et al., 2012). Both effects are lasting and involve a form of NMDAR-mediated plasticity (Joynes et al., 2004, Ferguson et al., 2006). We have also shown that uncontrollable, but not controllable, noxious stimulation impairs recovery after a contusion injury and fosters the development of chronic pain (Grau et al., 2004, Garraway, 2014, Grau et al., 2017).
4.1 Brain derived neurotrophic factor (BDNF) enables learning
Studies examining brain-mediated plasticity have shown that the neurotrophin BDNF can promote LTP, synaptic tagging, the trafficking of AMPARs, and memory consolidation, mediating a form of metaplasticity that generally enhances the development of synaptic plasticity (Gottmann et al., 2009, Waterhouse and Xu, 2009). This concept has been related to spinal cord function within the pain literature, where it has been shown that BDNF fosters the development of central sensitization (Coull et al., 2005a, Merighi et al., 2008b, Lu et al., 2009, Beggs and Salter, 2013). Other work has shown a benefit of locomotor training after SCI and related this effect to increased expression of BDNF (Gomez-Pinilla et al., 2002, Vaynman and Gomez-Pinilla, 2005, Boyce et al., 2007, Cote et al., 2011, Boyce et al., 2012, Boyce and Mendell, 2014). These findings led us to hypothesize that BDNF might be involved in the enabling effect of controllable stimulation. In collaboration with Gomez-Pinilla, we showed that training up-regulates BDNF mRNA expression, and other plasticity-related genes, that a protein (TrkB-IgG) that sequesters free BDNF blocks the enabling effect, and that pretreatment with BDNF substitutes for training to enable learning when rats are tested with a high response criterion (Gomez-Pinilla et al., 2007).
As noted above, instrumental conditioning has a protective effect that can block the induction of a learning deficit (Crown and Grau, 2001). We showed that this effect too is blocked by TrkB-IgG and that BDNF substitutes for training to restore the capacity to learn (Huie et al., 2012b). Conversely, the beneficial effect of training after uncontrollable stimulation is blocked by TrkB-IgG and treatment with BDNF can substitute for training to restore the capacity to learn. BDNF also blocks the learning impairment when given immediately before testing. Taken together, these findings show that training enables learning, and counters the development of the learning deficit, because it increases BDNF expression. Further work has linked the adverse effect that uncontrollable stimulation has on recovery after a contusion injury to the down-regulation of BDNF (Garraway et al., 2011).
4.2 Tumor necrosis factor (TNF) disables learning
Others have suggested that BDNF and the cytokine TNF can have opposing effects on synaptic plasticity (Stellwagen and Malenka, 2006). Given this, we explored whether the learning impairment involves TNF. We showed that exposure to uncontrollable stimulation induces a lasting (24 hr) increase in TNF protein, and that pretreatment with a TNF inhibitor blocked both the induction and expression of the learning impairment (Huie et al., 2012a). Furthermore, pretreatment with TNF produced a lasting learning impairment that was evident 24 hrs later, and the expression of this deficit was blocked by a TNF inhibitor administered prior to testing. Uncontrollable stimulation is also known to impair long-term recovery after a contusion injury and foster the development of chronic pain. These effects have been related to an up-regulation of TNF (Garraway, 2014).
Further research has shown that non-neuronal cells play an important role in the development of the learning impairment (Vichaya et al., 2009). Supporting this, activating glia with the toll-like receptor 4 (TLR4) agonist lipopolysaccharide (LPS) disrupted learning for 24 hrs and the induction of the deficit by LPS or VIS was blocked by a glial inhibitor (fluorocitrate). Fluorocitrate also blocked the induction of a learning impairment by TNF, and a TNF inhibitor blocked the expression of the learning deficit produced by LPS (Fig. 3).
Fig. 3.
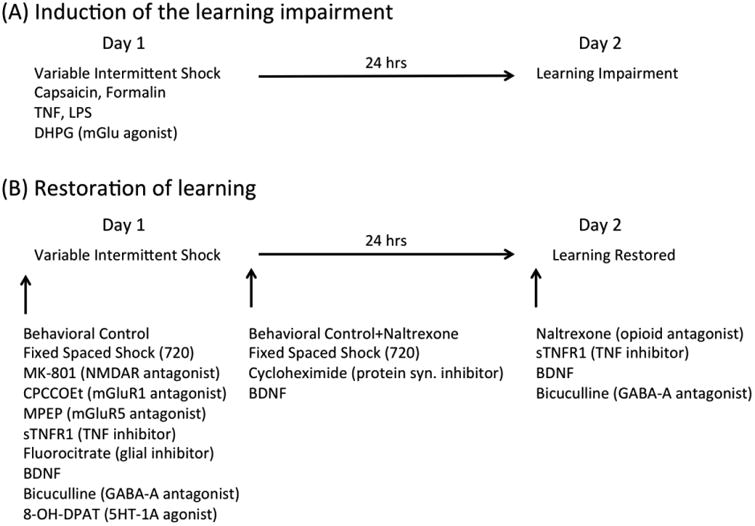
(A) A list of the treatments that have been shown to induce a lasting learning impairment. (B) A list of the treatments that have been shown to block the learning impairment induced by variable intermittent shock when given prior to shock treatment, immediately after shock treatment, or prior to testing.
Additional work has implicated other neurochemical processes linked to the induction of central sensitization, LTP, and metaplasticity, including the group I mGlu receptors (mGluR) (Neugebauer et al., 1999, Abraham, 2008, Grau et al., 2014). Pretreatment with drug antagonists that target the mGluR1 (CPCCOEt) or mGluR5 (MPEP) block the induction of the learning impairment, and administration of a group 1 mGluR agonist (DHPG) induces a lasting learning impairment (Ferguson et al., 2008). However, in contrast to the effect of TNF and LPS, we have not verified that the effect of DHPG treatment on learning can be reversed.
4.3 Temporal predictability impacts learning potential
We have shown that exposure to uncontrollable stimulation disables learning and that controllable stimulation has a restorative effect that can prevent and reverse the learning impairment. Other studies have shown that temporal predictability can also engage a metaplastic like effect (Baumbauer et al., 2009, Lee, 2015, 2016). This finding emerged from studies examining how the induction of the learning impairment is related to stimulus frequency (Baumbauer et al., 2008). We were motivated to explore this variable because it is known to regulate the induction of LTD and LTP, with low frequency (e.g., 1 Hz) stimulation favoring LTD whereas high frequency stimulation (e.g., 100 Hz) favors LTP (Malenka, 1994, Sandkuhler et al., 1997). Because it is more difficult to manipulate stimulus frequency when shocks occur in a variable manner, we gave rats intermittent shock at fixed (regular) intervals. Assuming that spinal neurons have no sense of time, we expected that this change in our procedure would have no effect. As predicted, 180 shocks (6 min of stimulation) induced a learning impairment independent of whether the stimuli occurred on variable time (VT) or fixed time (FT) schedule. But when shock number was increased 5 fold (to 900 shocks, presented at the same frequency), only VT shock induced a learning impairment. The fact that 180 FT shocks induces a learning impairment, while 900 does not, implies that presenting 720 additional shocks in a regular (temporally predictable) manner has a restorative effect. Given this, we asked whether 720 regularly spaced shocks given before 180 variably spaced shocks would prevent the induction of the learning impairment (Baumbauer et al., 2009). It did so, and this effect lasted 24 hrs. Further work revealed that the induction of this metaplastic effect depends upon the NMDAR and protein synthesis. In addition, the restorative effect of predictable stimulation was blocked by pretreatment with the BDNF sequestering antibody TrkB-IgG.
The key difference between FT and VT stimulation is that the FT schedule introduces a kind of temporal predictability. Given this, the fact that VT and FT stimulation have divergent effects implies that the spinal cord has a sense of time. Additional studies have reinforced this conclusion, demonstrating that processes within the spinal cord can abstract regularity across dermatomes, that this process shows a form of savings, and that the abstraction of regularity is related to the central pattern generator (CPG) that drives stepping behavior (Lee, 2015, 2016). For present purposes, what is most important is that both temporal predictability and behavioral control can induce a metaplastic effect that counters the development of the learning impairment and, in both cases, this effect depends upon BDNF.
5. Uncontrollable/unpredictable stimulation induces nociceptive sensitization
The studies reviewed above provide a straightforward account of how training affects learning within the spinal cord and the relation of these effects to NMDAR-mediated plasticity. It is assumed here that the learning is linked to a form of LTP that drives the motor response, leading to an increase in flexion duration that reduces net exposure to the nociceptive stimulus (Joynes et al., 2004). Uncontrollable/unpredictable stimulation appears to engage an opponentlike effect that inhibits adaptive plasticity. We posited that this could be mediated by a kind of LTD, or diffuse noxious inhibitory control (DNIC), that inhibits nociceptive activity (antinociception) and counters adaptive learning (Joynes et al., 2003). Indirect support for this hypothesis was obtained from pharmacological studies demonstrating that pretreatment with either an opioid or GABA antagonist blocks the expression of the learning impairment (Ferguson et al., 2003, Joynes and Grau, 2004).
In other studies, we had shown that exposure to a long continuous shock induces a form of spinally-mediated DNIC that inhibits behavioral reactivity to noxious stimulation (Meagher et al., 1990). If uncontrollable/unpredictable stimulation impairs adaptive learning because it induces a form of DNIC, then it too should have an antinociceptive effect. However, when we tested reactivity to a noxious thermal stimulus after continuous shock and VIS, we found that only continuous shock induced antinociception (Crown et al., 2002b). Moreover, while 6 min of continuous shock induced a robust antinociception, it did not induce a learning impairment. Furthermore, when continuous shock was applied concurrently with VIS, it blocked the induction of the learning impairment.
We also tested whether exposure to VIS affects reactivity to mechanical stimulation applied to the plantar surface of the hind paw using von Frey stimuli (Ferguson et al., 2006). To our surprise, VIS-treated animals were hyper-, not hypo-, responsive. Enhanced mechanical reactivity (EMR) is observed after a variety of treatments (e.g., peripheral application of the irritants formalin, caarageenan or capsaicin) known to sensitize nociceptive neurons within the dorsal horn of the spinal cord (Latremoliere and Woolf, 2009). As noted above, this centrally-mediated nociceptive sensitization depends upon a form of NMDAR-mediated plasticity (Ji et al., 2003). These parallels led us to an alternative hypothesis: VIS interferes with learning because it induces a form of central sensitization that saturates NMDAR-dependent plasticity (Moser and Moser, 1999, Ferguson et al., 2012b). If this is true, treatments that induce central sensitization should, like VIS, impair spinally-mediated instrumental learning. As predicted, peripheral application of the irritants carrageenan, formalin, or capsaicin induced a learning impairment (Ferguson et al., 2006, Hook et al., 2008, Baumbauer and Grau, 2011, Ferguson et al., 2012b). Furthermore, like VIS, exposure to capsaicin has a lasting effect that impairs learning when subjects are tested 24 hrs later, long after the capsaicin-induced EMR has faded (Hook et al., 2008). Like the learning impairment induced by VIS, training with controllable stimulation can prevent and reverse the capsaicin-induced learning deficit. More important from a translational perspective, behavioral control also prevents and reverses capsaicin-induced EMR. An extended exposure to regular (FT) stimulation had the same effect, blocking the capsaicin-induced EMR and learning impairment (Baumbauer and Grau, 2011, Baumbauer et al., 2012).
The idea that the learning impairment involves a form of central sensitization is also consistent with a number of neurobiological observations. For example, our data linking TNF and microglia activation to the induction of the learning deficit are consistent with findings in the pain literature implicating these processes in the development of central sensitization (Watkins et al., 1997, Milligan et al., 2003, Czeschik et al., 2008, Park et al., 2011). TNF may increase neural excitability by trafficking AMPARs to the synapse and increasing their permeability to Ca++ (Huie, 2015).
5.1 GABA drives nociceptive sensitization after injury
Our discovery that the learning impairment is related to the development of central sensitization raised a host of questions. One concerned the role of BDNF. We had suggested that controllable/predictable stimulation attenuates the learning impairment because it up-regulates BDNF (Gomez-Pinilla et al., 2007, Grau et al., 2012). Consistent with this hypothesis, i.t. BDNF can prevent and reverse the learning deficit (Huie et al., 2012b). This implies that, if the deficit reflects a form of central sensitization, i.t BDNF should also attenuate central sensitization. As a first test of this implication, spinally transected rats were exposed to VIS and tested using von Frey stimuli. As predicted, VIS led to EMR and this effect was blocked by pretreatment with BDNF (Huie et al., 2012b). The implication is that BDNF can attenuate central sensitization. This finding, however, ran counter to other studies demonstrating that BDNF typically has the opposite effect (Coull et al., 2005b, Merighi et al., 2008a, Latremoliere and Woolf, 2009, Lu et al., 2009, Biggs et al., 2010, Beggs and Salter, 2013).
Another problem stems from studies exploring the role of GABAergic neurons. GABA engages the ionotropic GABA-A receptor, which allows the anion Cl- to enter the cell (Medina et al 2014). This inward flow of Cl- has a hyper-polarizing (inhibitory) effect that dampens neural excitability within the spinal cord. Supporting this, blocking the GABA-A receptor with the GABA-A antagonist bicuculline in uninjured animals has a pronociceptive effect that enhances nociceptive signaling and behavioral reactivity (Roberts et al., 1986, Sivilotti and Woolf, 1994, Sorkin et al., 1998, Baba et al., 2003, Dougherty and Hochman, 2008). Based on these results, we would expect that pretreatment with a GABA-A antagonist would enhance neural excitability in response to VIS, which should (if anything) foster the development of the learning impairment in spinally transected rats. Contrary to our expectations, bicuculline blocked the induction of the deficit (Ferguson et al., 2003). Given our claim that the deficit reflects a form of central sensitization, this outcome seemed paradoxical because it implied that administration of a GABA-A antagonist will block VIS-induced EMR. To test this, spinally transected rats were administered bicuculline or its vehicle prior to receiving VIS to one hind leg (Huang, 2016). In the vehicle treated rats, VIS induced a bilateral EMR. Pretreatment with bicuculline eliminated this effect.
We explored the generality of these observations by testing the effect of bicuculline on the EMR elicited by a number of other treatments. Others have shown that i.t. application of LPS activates microglia and enhances reactivity to mechanical stimulation (Reeve et al., 2000, Young et al., 2007) and we reported above that LPS (i.t.) impairs spinal learning (Vichaya et al., 2009). LPS (i.t.) also induces EMR in spinally transected rats and this effect was blocked by bicuculline (Huang et al., 2016). Next, we assessed the effect of chemically activating C-fibers using capsaicin, a treatment that has been widely used to study central sensitization within the pain literature (Willis, 2001, Latremoliere and Woolf, 2009, Sandkuhler, 2009). As expected, application of capsaicin to one hind paw induced a robust bilateral EMR in spinally transected rats (Fig. 4B). This effect too was blocked by pretreatment with bicuculline. An alternative GABA-A antagonist (gabazine) had the same effect. We then assessed whether bicuculline would impact the maintenance of capsaicin-induced EMR. Transected rats had capsaicin applied to one paw and an hour later, after EMR had developed, received i.t. bicuculline (Huang, 2016). After central sensitization was induced, blocking the GABA-A receptor attenuated the resultant EMR.
Fig. 4.
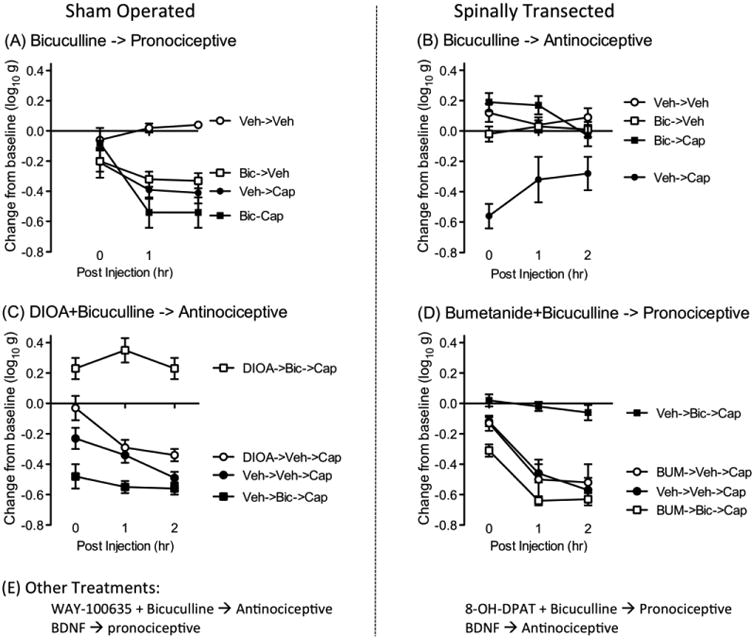
(A) Spinally transected rats that have been treated with the irritant capsaicin (Cap) applied to one hind paw exhibit enhanced mechanical reactivity (EMR) relative to the vehicle (Veh) controls (Huang et al., 2016). This effect is observed on both the treated and opposite leg. The figure depicts the change from baseline mechanical reactivity, averaged across the two legs, soon after capsaicin treatment (0) and 1 and 2 hrs later. In sham-operated rats, bicuculline (Bic) per se induced an EMR and potentiated the effect of capsaicin. (B) Capsaicin treatment also induced an EMR in spinally transected rats. This effect was blocked by bicuculline (Bic) (Huang et al., 2016). (C) Blocking the KCC2 channel with DIOA flipped how bicuculline affected capsaicin-induced EMR in sham operated rats (Huang et al., 2016). In the absence of DIOA, bicuculline enhanced capsaicin-induced EMR. After treatment with DIOA bicuculline had an antinociceptive effect that blocked capsaicin-induced EMR. (D) Blocking the NKCC1 channel with bumetanide (Bum) reversed the effect of bicuculline on capsaicin-induced EMR in spinally transected rats (Huang et al., 2016). In the absence of bumetanide, bicuculline had an antinociceptive effect that blocked capsaicin-induced EMR. After bumetanide treatment, bicuculline enhanced capsaicin-induced EMR. (E) Other treatments impact capsaicin-induced EMR. In sham operated rats, pretreatment (i.t.) with the 5HT-1A antagonist WAY-100635 causes bicuculline to have an antinociceptive effect that attenuates capsaicin-induced EMR. Conversely, in spinally transected rats, administration of a 5HT-1A agonist produces a state wherein bicuculline has a pronociceptive effect. BDNF affects capsaicin-induced EMR in opposite ways in sham-operated and spinally transected rats. In uninjured rats, it has a pronociceptive effect that enhances EMR. After SCI, it has an antinociceptive effect that attenuates VIS and capsaicin-induced EMR.
Research within the pain literature has shown that central sensitization is associated with the expression of the proto-oncogene c-fos and the phosphorylation of the protein extracellular-signal-regulated kinase (pERK) (Gao et al., 2010). To evaluate whether our experimental treatments affected these cellular indices, spinally transected rats were treated with bicuculline, or its vehicle, before capsaicin was applied to one paw (Huang, 2016). We again found that capsaicin induced a robust EMR that was blocked by bicuculline. Two hrs after capsaicin treatment, tissue was collected and we assessed c-fos and pERK expression within the dorsal horn. As expected, capsaicin induced the expression of c-fos and pERK. Pretreatment with bicuculline attenuated this effect.
Recognizing that our results ran counter to what has been reported in uninjured rats, we assessed the effect of bicuculline on nociceptive sensitization in rats that had undergone a sham surgery (Huang, 2016). As expected, capsaicin induced a robust EMR (Fig. 4A). Consistent with other studies, bicuculline alone enhanced behavioral reactivity in uninjured rats and, if anything, potentiated capsaicin-induced EMR. At the cellular level, capsaicin did not induce robust c-fos/pERK expression within the dorsal horn (relative to spinally transected rats) (also see (Castellanos et al., 2007)). This suggests that, in the absence of injury, supraspinal systems may mediate capsaicin-induced allodynia.
The results imply that SCI alters how GABA affects nociceptive circuits. To further explore this concept, we also tested the effect of a GABA-A agonist (muscimol). As reported by others, i.t. muscimol inhibited nociceptive reactivity and the development of nociceptive sensitization in uninjured rats (Huang et al., 2016). SCI eliminated both of these effects.
5.2 Ionic plasticity
Our findings indicate that SCI transforms how GABA affects the development of nociceptive sensitization. In uninjured animals, i.t. administration of a GABA-A antagonist enhances nociceptive responsiveness (Roberts et al., 1986, Sivilotti and Woolf, 1994, Sorkin et al 1998), which is consistent with the usual view—that GABA interneurons within the spinal cord inhibit neural excitability. But after SCI, pretreatment with a GABA-A antagonist blocks the development of nociceptive sensitization (Huang et al., 2016), which implies that GABA release is essential to the emergence of this effect—that GABA release drives the development of nociceptive sensitization. How could SCI so dramatically impact GABA function?
New findings suggest that this is related to the cellular processes that regulate the intracellular concentration of the anion Cl- (Kaila et al., 2014a). The GABA-A receptor functions as a ligand-gated ion channel. When activated, it allows Cl- to flow through the channel (Fig. 5A). The direction of Cl- flow depends upon the intracellular concentration of Cl-, which is controlled by two co-transporters, KCC2 and NKCC1, which regulate the outward and inward flow of Cl-, respectively (Cramer et al., 2008, Kahle et al., 2013, Kaila et al., 2014a, Medina et al., 2014). In uninjured adult animals, these co-transporters maintain a low concentration of Cl- within the cell. As a result, opening the channel allows Cl- to flow into the cell which exerts a hyperpolarizing (inhibitory) effect. Interestingly, NKCC1 emerges earlier in development than KCC2 (Ben-Ari, 2002, Rivera et al., 2005, Ben-Ari, 2014). Because NKCC1 allows Cl- to flow into the cell, there is a rise in the intracellular concentration of Cl- (Kaila et al., 2014a, Medina et al., 2014). Under these conditions, engaging the GABA-A receptor allows Cl- to flow out of the cell, which has a depolarizing (excitatory) effect (Fig. 5D). Consequently, early in development, GABA-dependent inhibition is lessened and neural excitability is enhanced, which could function to promote the adaptive wiring of neural circuits.
Fig. 5.
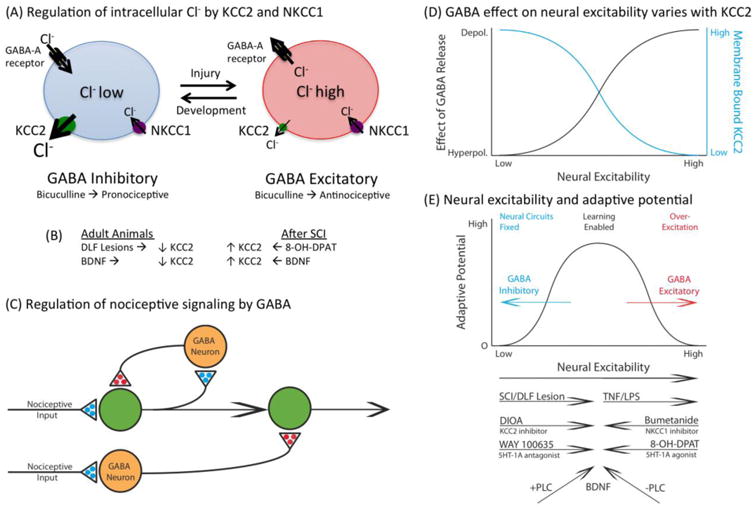
(A) Intracellular Cl- concentrations are controlled by the co-transporters KCC2 and NKCC1, which regulate the outward and inward flow of Cl-, respectively. In adult animals (left), the outward flow of Cl- through the KCC2 channel maintains a low intracellular concentration of Cl-. Under these conditions, engaging the GABA-A receptor allows Cl- to enter the cell, which has a hyperpolarizing effect. Early in development, and after SCI (right), the relative concentration of membrane-bound KCC2 is lower. As a result, the inward flow of Cl- causes a rise in the intracellular Cl- concentration. Now, engaging the GABA-A receptor can allow Cl- to exit the cell, which can have a depolarizing effect. (B) Membrane-bound KCC2 in adult animals is also reduced by DLF lesions and BDNF. After SCI, pretreatment with a 5HT-1A agonist (8-OH-DPAT) or BDNF can induce an increase in KCC2. (C) Nociceptive activity within the spinal cord is regulated by GABAergic neurons that normally inhibit neural excitation. A reduction in membrane-bound KCC2 in the postsynaptic cell can flip how GABA input affects neural excitation, causing it to foster neural activity. (D) The effect of GABA on neural excitability is inversely related to the level of membrane-bound KCC2. As KCC2 declines (blue line), so too does GABA-dependent hyperpolarization (hyperpolerization). At low levels of KCC2, the rise in intracellular Cl- concentrations can cause GABA to have a depolarizing (depolerization) effect (black line). (E). Evidence suggests that there is a non-monotonic relation between neural excitability and adaptive potential within the spinal cord. In adult uninjured animals, neural plasticity is low due to GABA-dependent inhibition. SCI can remove this brake to allow spinal cord learning. In the absence brain-dependent regulatory systems, noxious input can induce a state of over-excitation that saturates plasticity and disrupts adaptive learning. How various treatments are thought to impact neural excitability are indicated immediately below. In adult animals, SCI, DIOA (i.t.) or WAY 100635 (i.t.) are hypothesized to increase neural excitability and enhance learning. TNF (i.t.) or LPS (i.t.) can over-excite the system and thereby impair learning. After SCI, bumetanide (i.t.) and 8-OH-DPAT quell over-excitation, which allows learning. BDNF has a homeostatic effect that fosters plasticity. Evidence suggests that this bidirectional effect depends upon the presence (+) versus absence (-) of PLCγ.
Recent studies suggest that SCI may alter GABA function because it reduces the expression of membrane-bound KCC2 (Drew et al., 2004, Cramer et al., 2008, Boulenguez et al., 2010, Hasbargen et al., 2010). This would reduce the outward flow of Cl- and induce a rise in the intracellular concentration of Cl-, producing a state akin to that observed early in development, when GABA can have a depolarizing effect (Ben-Ari et al., 2012, Kaila et al., 2014a). Electrophysiological studies have shown that just a 10-20% reduction in membrane-bound KCC2 can bring about a 10-mV depolarizing shift in the GABA-A receptor mediated inhibitory postsynaptic potential (EIPSP) (Boulenguez et al., 2010).
This alteration in GABA function has been shown to impact neural excitability after chronic SCI and contribute to spasticity and chronic pain (Cramer et al., 2008, Lu et al., 2008, Boulenguez et al., 2010, Medina et al., 2014). Reduced KCC2 expression has also been implicated in the loss of neural inhibition, and the development of chronic pain, in an animal model of diabetes (Jolivalt et al., 2008). To determine whether a shift in KCC2 contributes to the effects described earlier, we tested if SCI affects membrane-bound KCC2 24 hrs after injury (Huang, 2016). KCC2 was assessed by separating the membrane and cytoplasmic fractions and then assaying levels of the inactive and active (phosphorylated) forms. We found that SCI reduced both forms within the dorsal horn membrane fraction of the lumbosacral spinal cord.
If a change in Cl- transport determine how bicuculline affects nociceptive sensitization, we should be able to alter the effect of bicuculline using drugs that inhibit KCC2 or NKCC1 function. For example, in intact animals, blocking KCC2 with DIOA should emulate the effect of spinal cord injury, and cause a rise in intracellular Cl- (Jolivalt et al., 2008). Under these conditions, engaging the GABA-A receptor should promote the flow of Cl- out of the cell, yielding a depolarizing effect that fosters the development of nociceptive sensitization. In this case, blocking the GABA-A receptor should undermine the development of capsaicin-induced EMR. We found that pretreatment with DIOA has this effect, causing bicuculline to have an antinociceptive, rather than pronociceptive, effect in uninjured rats (Fig. 4C) (Huang et al., 2016). Conversely, in injured animals, blocking NKCC1 with bumetanide should reduce the inward flow of Cl- (Cramer et al., 2008, Hasbargen et al., 2010). This should reduce the intracellular concentration of Cl-, producing a state akin to that observed in adult animals. Under these conditions, bicuculline should promote nociceptive reactivity and enhance capsaicin-induced EMR. To test this, spinally transected rats were given bumetanide or its vehicle (i.t.) prior to bicuculline (Huang et al., 2016). As usual, bicuculline alone had an antinociceptive effect that blocked the development of capsaicin-induced EMR (Fig. 4D). In rats pretreated with bumetanide, bicuculline had a pronociceptive effect analogous to that observed in uninjured animals.
In uninjured animals, GABA has an inhibitory effect that quells neural excitation (Fig. 5C). Our findings suggest that SCI reduces this inhibitory effect by down-regulating membrane-bound KCC2, which enables neural excitability within the spinal cord and NMDAR-mediated plasticity. When challenged with VIS or capsaicin, this fosters the development of nociceptive sensitization.
5.3 SCI transforms how BDNF affects ionic plasticity
We have suggested that BDNF has a protective effect that enables learning within the spinal cord and counters the development of central sensitization (Grau et al., 2012, Huie et al., 2012b). The former is consistent with other work demonstrating that BDNF can promote locomotor recovery after injury (Boyce et al., 2007, Boyce et al., 2012, Boyce and Mendell, 2014). Yet, our finding that BDNF attenuates VIS-induced EMR stands in contrast to a large body of literature demonstrating that BDNF fosters the development of central sensitization (Coull et al., 2005a, Merighi et al., 2008b, Lu et al., 2009, Beggs and Salter, 2013). Given this, we posited that SCI may also transform how BDNF affects nociceptive circuits. To evaluate this possibility, we tested the effect of BDNF on capsaicin-induced EMR (Huang et al., 2017). BDNF attenuated the EMR elicited by capsaicin in spinally transected rats. In contrast, in sham operated rats, BDNF treatment enhanced mechanical reactivity and amplified capsaicin induced EMR. An analogous pattern of results was observed at the cellular level, where we found that BDNF increased capsaicin-induced ERK phosphorylation in sham operated rats, but attenuated ERK phosphorylation after SCI. Interestingly, here too we observed that SCI per se amplified the development of central sensitization.
Other work has related the BDNF-induced EMR observed in uninjured animals to a down-regulation in KCC2, which would have an effect similar to SCI and reduce GABA-dependent inhibition (Coull et al., 2005a, Merighi et al., 2008b, Lu et al., 2009, Beggs and Salter, 2013). We hypothesized that the effect of BDNF may depend upon the background level of neural excitability and injury, a notion that had some empirical support (Shulga et al., 2008, Boulenguez et al., 2010). The implication is that BDNF may have a homeostatic effect that acts to re-establish GABA-dependent inhibition after SCI by increasing membrane bound KCC2. To evaluate these claims, we assessed the effect of BDNF on KCC2 in sham operated and transected rats (Huang et al., 2017). In uninjured rats, i.t. BDNF down-regulated KCC2, whereas it increased expression after injury.
Normally, pretreatment with a GABA agonist, such as muscimol, attenuates capsaicin-induced EMR in uninjured animals (Hwang and Yaksh, 1997, Kaneko and Hammond, 1997). Our results suggest that BDNF reduces membrane-bound KCC2, which should attenuate the inhibitory effect of muscimol. To evaluate this possibility, we examined how BDNF and muscimol impact capsaicin-induced EMR in uninjured and injured rats. In uninjured rats, muscimol alone had an antinociceptive effect. Pretreatment with BDNF enhanced capsaicin-induced EMR and eliminated the antinociceptive effect of muscimol. Exactly the opposite pattern of results was observed after SCI. In injured rats, BDNF attenuated capsaicin-induced EMR and reinstated the antinociceptive effect of muscimol.
In summary, BDNF has opposite effects on KCC2 expression in uninjured and injured animals (Huang et al., 2017). In intact animals, BDNF down-regulates membrane-bound KCC2 and fosters the development of central sensitization. After SCI, BDNF upregulates membrane-bound KCC2, which should augment GABA-dependent neural inhibition and quell central sensitization. How can BDNF affect KCC2 in opposite ways? A potential answer was provided by Rivera and his colleagues (Rivera et al., 2004, Rivera et al., 2005, Ferrini and De Koninck, 2013). BDNF binds to the TrkB receptor, which can engage multiple signal pathways, including SHC and PLCγ. How these signal pathways affect KCC2 appears to depend upon PLCγ: BDNF down-regulates KCC2 when PLCγ is present, but up-regulates KCC2 when PLCγ is absent. This hypothesis is consistent with the finding that SCI down-regulates the expression of PLCγ which should reverse the effect of BDNF and cause it to up-regulate KCC2. Further work is needed to explore the generality of this hypothesis, the cell types involved, and the temporal dynamics of the underlying processes.
5.4 Descending serotonergic (5HT) fibers regulate ionic plasticity
Our studies indicate that SCI transforms how GABA affects spinal function. Why does injury have this effect? A preliminary answer was suggested from work exploring the fiber pathways that regulate neural excitability within the dorsal horn (Sandkuhler, 2009). As noted above, electrical stimulation of the sciatic nerve can induce LTP within the dorsal horn in spinally transected animals. Interestingly, in uninjured animals, this same stimulus does not induce LTP, which implies that descending fibers normally inhibit neural excitability (Gjerstad et al., 2001). Likewise, VIS does not induce a learning impairment in awake uninjured animals (Washburn et al., 2007), which implies that descending pathway dampens nociceptive plasticity. Further work linked these effect to serotonergic (5HT) fibers within the dorsal lateral funiculus (DLF), which inhibit neural excitability within the dorsal horn through the 5HT-1A receptor (Bardin et al., 2000, Crown and Grau, 2005, Gjerstad et al., 2001, Hains et al., 2001a, Hains et al., 2001b, Hains et al., 2001c).
We posited that lesioning the DLF allows afferent input to over-drive nociceptive systems within the dorsal horn because it lowers membrane-bound KCC2, removing a GABA-dependent brake on neuronal plasticity. This is consistent with data from the developmental literature that shows that the switch in GABA function, from depolarizing to hyperpolarizing, coincides with the innervation of descending fibers and that a depolarizing shift is not observed if the spinal cord is transected at an early age, before these fibers reach the caudal spinal cord (Jean-Xavier et al., 2006). Based on these observations, we hypothesized that descending 5HT fibers regulate neural excitability within the spinal cord through a form of GABA-dependent ionic plasticity. To explore this possibility, we tested whether blocking 5HT receptors within the spinal cord in intact animals affects GABA function. Based on past pharmacological work (Crown and Grau, 2005), we targeted the 5HT-1A receptor using the antagonist WAY-100635. Our prediction was that i.t. administration of this 5HT antagonist would have an effect analogous to spinal transection, inducing a shift in GABA function wherein GABA fuels, rather than inhibits, the development of central sensitization. If that is true, WAY-100635 should reverse how bicuculline affects the development of capsaicin-induced sensitization, unveiling an antinociceptive effect that blocks the development of EMR. We found this pattern of results (Huang, submitted). We reinforced our behavioral data with cellular assays, demonstrating that bicuculline attenuates capsaicin-induced pERK expression in rats that received WAY-100635. Conversely, we predicted that administration of a 5HT-1A agonist (8-OH-DPAT) would increase membrane-bound KCC2 in spinally transected rats and eliminate the antinociceptive effect of bicucuclline, and this is what we found (Fig. 4E). We then showed that lesions limited to the DLF were sufficient to down-regulate KCC2 expression and that these lesions reversed how bicuculline affects nociceptive sensitization. In sham controls, bicuculline had an allodynic effect and enhanced the development of capsaicin-induced EMR. In DLF lesioned rats, bicuculline had an antinociceptive effect that attenuated capsaicin-induced EMR. These results imply that lesions limited to the DLF are sufficient to induce a shift in GABA function.
An important feature of DLF lesions is that they have minimal effect on ascending pain pathways within the spinothalamic tract. This allowed us to explore an issue that was otherwise intractable: Does the spinally-mediated change in GABA function affect pain transmission to the brain? We addressed this issue using a place conditioning procedure (King et al., 2009). Over the course of two days, sham operated and DLF lesioned animals were exposed to two distinct contexts. Before each, the rats were treated with either i.t. bicuculline or its vehicle. Half the rats then had capsaicin applied to one hind paw. In uninjured rats, i.t. bicuculline should increase neural excitability in the dorsal horn and enhance pain transmission to the brain, increasing the aversion to the bicuculline-paired context. As predicted, the sham operated rats spent less time in the bicuculline-paired context (Huang, submitted). If lesioning the DLF reverses how bicuculline affects nociceptive signaling within the dorsal horn, bicuculline should have an antinociceptive effect that reduces pain. As a result, DLF-lesioned rats should exhibit a preference for the bicuculline-paired context, and this is what we found.
Our findings are generally consistent with other studies exploring the regulation of neural excitability within the spinal cord and alterations in GABA function (Cramer et al., 2008, Boulenguez et al., 2010, Gwak and Hulsebosch, 2011). We go beyond these studies by demonstrating: 1) that SCI produces an acute shift in GABA function that is evident within the first 24 hrs; 2) that SCI alters how both a GABA agonist and antagonist affect nociceptive processing; 3) that SCI transforms how BDNF affects nociceptive processing and GABA function; 4) that the change in GABA function is related to a loss of 5HT fibers within the DLF that influence GABA function through the 5HT-1A receptor; and 5) that these changes in spinal processing impact a brain-dependent measure of pain. Further work is needed to decipher how these processes interact within the spinal cord and the particular fiber pathways that regulate GABA function.
6. Summary and implications
We have shown that neurons within the spinal cord can learn and that experience can have a lasting effect on adaptive potential. Exposure to uncontrollable/unpredictable stimulation induces a state that impairs learning (negative transfer) (Grau et al., 1998). This involves a general (extrinsic) effect that retards learning when spinal neurons are challenged in new ways (Crown et al., 2002b, Joynes et al., 2003). We coupled the learning impairment to the development of central sensitization, C-fiber input, and inflammation (Ferguson et al., 2006, Baumbauer et al., 2008, Ferguson et al., 2012a, Ferguson et al., 2012b). The alteration has a lasting effect that depends upon the NMDAR and protein synthesis (Patton et al., 2004, Ferguson et al., 2006). We further showed that learning impairment involves non-neuronal cells, TNF, and mGlu (Ferguson et al., 2008, Vichaya et al., 2009, Huie et al., 2012a), which fits well with other work implicating these processes in the maintenance of central sensitization (Watkins et al., 1997, Neugebauer et al., 1999, Schafers et al., 2008).
Exposure to controllable/predictable stimulation has the opposite effect—it enables learning (positive transfer) (Crown et al., 2002a, Grau et al., 2012). Controllable/predictable stimulation also prevents, and reverses, the learning impairment and EMR induced by capsaicin (Hook et al., 2008, Baumbauer and Grau, 2011, Baumbauer et al., 2012). Again, this represents a general (extrinsic) effect that impacts how spinal neurons process stimulation applied to other dermatomes. Here too, training has a lasting effect that depends upon the NMDAR and protein synthesis (Joynes et al., 2004, Baumbauer et al., 2009). We linked the beneficial effect of training to the up-regulation of BDNF (Gomez-Pinilla et al., 2007, Baumbauer and Grau, 2011, Huie et al., 2012b), which is consistent with studies demonstrating that this ligand can promote the recovery of locomotor behavior and breathing after SCI (Baker-Herman et al., 2004, Boyce and Mendell, 2014, Fields and Mitchell, 2015).
These examples of positive and negative transfer meet the criteria outlined above for behavioral metaplasticity (Schmidt et al., 2013, Grau et al., 2014). Each has a lasting effect and and each is observed when behavioral reactivity has returned to baseline and when care has been taken to equate initial flexion force and response duration (Crown et al., 2002b, Grau, 2006, Baumbauer et al., 2009). Further, these examples of behavioral metaplasticity impact learning when the system is challenged in new ways, implying that each involves an extrinsic effect rather than a kind of intrinsic additivity. Importantly, the phenomena are reversible. The enabling/restorative effect of predictable/controllable stimulation is blocked by a BDNF inhibitor (TrkB-IgG) (Gomez-Pinilla et al., 2007, Baumbauer et al., 2009, Huie et al., 2012b). Likewise, the expression of the learning impairment is blocked by BDNF, bicuculline, a mGlu receptor antagonist, and a TNF inhibitor (Ferguson et al., 2003, Ferguson et al., 2008, Huie et al., 2012a, Huie et al., 2012b), which implies that the circuitry needed for learning is still functional.
6.1 SCI, BDNF, and the regulation of GABA function
How does BDNF affect learning potential? The traditional answer focused on how BDNF affects NMDAR-mediated plasticity (Gottmann et al., 2009, Grau et al., 2012). It seems likely that this contributes. But the present studies, and other work, suggest that BDNF can also have an indirect effect on neural excitability/plasticity that is mediated by an alteration in GABA function (Smith, 2014). In uninjured animals, BDNF reduces the hyperpolarizing effect of GABA by down-regulating membrane-bound KCC2, setting the stage for nociceptive sensitization (Coull et al., 2005a, Merighi et al., 2008b, Beggs and Salter, 2013). These studies posed a challenge to our work because they imply that BDNF should potentiate, rather than inhibit, central sensitization. A similar problem arose from our work demonstrating that the GABA-A receptor antagonist bicuculline blocks the induction of the learning deficit (Ferguson et al., 2003). Given that bicuculline promotes nociceptive sensitization in uninjured animals (Roberts et al., 1986), if the deficit reflects a form of over-excitation, this drug treatment should foster the development of the deficit, not block it.
To address these puzzles, we posited that SCI transforms how BDNF and bicuculline affect neural function caudal to injury. As predicted by our earlier work, we showed that BDNF blocks VIS and capsaicin induced EMR after SCI (Huie et al., 2012b, Huang et al., 2017). Yet, in intact animals, BDNF had a pronociceptive effect that fostered capsaicin-induced EMR and pERK expression (Huang et al., 2017). The same pattern of results was found when we assessed the impact of bicuculline (Huang et al., 2016). After spinal transection, bicuculline blocked the development of EMR in response to VIS, inflammation, or capsaicin. It also blocked capsaicin-induced c-fos and pERK expression (Huang et al., 2016). But in intact animals, bicuculline had a pronociceptive effect. These observations suggest that SCI transforms how GABA affects nociceptive circuits. Others have suggested that a loss of GABA-dependent inhibition may contribute to the development of nociceptive sensitization (Gwak and Hulsebosch, 2011). The fact that bicuculline blocks the development of nociceptive sensitization after SCI implies a more central role—that GABA release can fuel the development of central sensitization, providing an essential driving force.
Others have shown that SCI can transform motor processes within the spinal cord ventral horn by down-regulating KCC2, causing a depolarizing shift in GABA function that contributes to the development of prolonged muscle contraction (spasticity) after SCI (Boulenguez et al., 2010). We posited that a similar alteration might occur within the dorsal horn after acute SCI, removing a GABA-dependent brake on nociceptive activity and potentially inducing a depolarizing shift that causes GABA to have an excitatory effect that drives the development of nociceptive sensitization. Supporting this, we showed that SCI reduces membrane-bound KCC2 within 24 hrs of SCI and that blocking KCC2 with DIOA in uninjured rats emulates the effect of SCI (Huang et al., 2016). Furthermore, a treatment (bumetanide). which should lower intracellular Cl- levels, re-established a state akin to the intact system wherein bicuculline had a pronociceptive rather than an antinociceptive effect. We posited that the change in GABA function was linked to the loss of descending serotonergic fibers within the DLF that regulate nociceptive processes within the dorsal horn through the 5HT-1A receptor. Supporting this, we showed that lesions limited to the DLF cause a down-regulation in KCC2 and that administration of a 5HT agonist after SCI restores membrane bound KCC2 and the pronociceptive effect of bicuculline (Huang, submitted). We concluded by showing that DLF lesions reverse how i.t. bicuculline affects motivated behavior in a place conditioning task; in intact animals, i.t bicuculline enhanced pain whereas after SCI it had an antinocieptive effect. These observations imply that alterations at the level of the spinal cord impact pain transmission to the brain.
The idea that SCI causes a down-regulation in KCC2, that fosters the development of central sensitization, fits with other work linking the development of chronic pain after SCI to a reduction in KCC2 (Cramer et al., 2008, Lu et al., 2008). The findings are also congruent with the observation that diabetes can induce a depolarizing shift in GABA function that reverses how bicuculline affects formalin-induced nociceptive sensitization (Jolivalt et al., 2008). Finally, the work is consistent with research showing that inflammation, and the activation of microglia, can enhance nociceptive processing within the spinal cord through a down-regulation in KCC2 (Coull et al., 2005a). The only difficulty is that this last effect has been linked to an up-regulation in BDNF in uninjured animals. Further work resolved this conceptual puzzle by demonstrating that BDNF can affect KCC2 in opposite ways, depending upon the state of the system (Huang et al., 2017). In intact animals, BDNF down-regulates membrane-bound KCC2, which would lessen GABA-dependent inhibition and promote neural excitability/plasticity. After SCI, when KCC2 has already been down-regulated, BDNF has the opposite effect—it increases membrane-bound KCC2, which would reduce the over-excitation (saturation) of nociceptive circuits and thereby promote adaptive plasticity. Likewise it would promote learning when animals are tested with a higher response criterion (Grau et al., 1998). We suggest that other treatments (e.g., bumetanide, 8-OH-DPAT) that attenuate nociceptive sensitization should also enable learning.
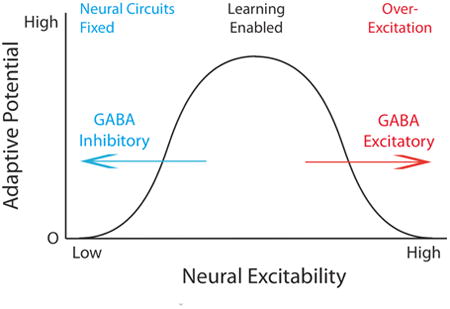
Our results suggest that the relation between spinal cord neural excitability and adaptive potential is non-monotonic in nature (Fig. 5E). In the intact organism, adaptive potential appears to be generally inhibited by GABAergic interneurons. SCI removes this brake, which allows spinal learning. But neural injury also removes a safety-switch that normally inhibits nociceptive activity, which allows noxious stimulation to over-excite spinal circuits and feed the development of central sensitization. BDNF appears to have a homeostatic effect that helps maintain the system within a range that promotes adaptive learning (Turrigiano, 2006, Gomez-Pinilla et al., 2007, Huie et al., 2012b). At a biochemical level, it has been suggested that this reversal in action may depend upon a SCI-induced down-regulation in PLCγ (Rivera et al., 2004, Tashiro et al., 2015). This framework implies that GABA-dependent processes normally block the development of maladaptive plasticity within the spinal cord. The corollary to this is that central sensitization will typically only develop after injury or pathology (e.g., inflammation, diabetes, arthritis, opiate addiction) (Weng et al., 1998, Coull et al., 2005a, Jolivalt et al., 2008, Ferrini et al., 2013).
Our framework posits 3 states (Fig. 5E): 1) a fixed state maintained by GABAergic inhibition; 2) a malleable state that allows for spinal learning; and 3) a state of over-excitation driven, in part, by a depolarizing shift in GABA. An interesting feature of this framework is that physiological processes can hold the system within a particular state over time, providing a kind lock. In adult uninjured animals, GABAergic inhibition is maintained by descending serotonergic fibers. Removing this lock allows spinal learning, which can itself engage a NMDAR/protein synthesis dependent state that counters the development of central sensitization (Grau et al., 2012). Over-excitation engages a third state, which again is preserved over time by means of a NMDAR/protein synthesis dependent process (Ferguson et al., 2012a). While we know that the consequences of over-stimulation fade over time (in 48-96 hrs) (Crown et al., 2002b), we have not assessed the long-term effect of relational learning or BDNF treatment beyond 24 hrs. It is possible that homeostatic processes normally act to maintain this intermediate state over time after SCI, which could promote adaptive learning but also increase susceptibility to over-excitation. The latter effect could set the stage for chronic pain.
6.2 Ionic plasticity regulates adaptive potential
Thirty years ago, we outlined a model that assumed brain-dependent learning and memory influence pain through descending fibers (Grau, 1987), an idea that has found some support (McNally et al., 2011). At that time, it was assumed that lower-level modulatory systems are only engaged in an unconditioned (unlearned) manner (Watkins et al., 1982, Meagher et al., 1990). Subsequent work challenged this proposal, demonstrating that nociceptive systems within the spinal cord are sensitive to Pavlovian relations (Grau et al., 1990, Joynes and Grau, 1996). We also found that spinal cord neurons are sensitive to response-outcome relations and that this learning can have a lasting effect on spinal function (Grau et al., 1998, Grau et al., 2004). These studies complement research within the rehabilitation literature that shows behavioral training can help restore stepping behavior after SCI minus input from the brain (Edgerton et al., 2008), that this system can adapt to an environmental obstacle (Edgerton et al., 1997), and that locomotor function is affected by nociceptive input (Bouffard et al., 2014). Furthermore, locomotor training can reduce pain and spasticity after injury (Tashiro et al., 2015) and this effect has been linked to BDNF (Boyce and Mendell, 2014) and an up-regulation in KCC2 (Cote et al., 2014).
At the same time, it was recognized that evidence for spinal learning came primarily from work using a transection paradigm, which is a necessity for demonstrating that brain mechanisms play no role. While there is evidence that brain-dependent learning can cause a form of rewiring within the spinal cord that impacts reflex function, that is evident after the system is later disconnected from the brain, establishing this effect typically requires extensive training (Wolpaw and Carp, 1990, Wolpaw, 2010). These observations imply that, in uninjured adult animals the spinal cord is a relatively immutable (fixed) system, designed so that sensory/motor systems are relayed in a reliable manner, a state that is maintained by descending fibers and GABAergic inhibition.
We propose that this descending regulatory pathway is not a permanent lock and that it is subject to regulation. Indeed, even surgical anesthesia can remove this brain-dependent lock (Washburn et al., 2007). More subtle modifications may be elicited through unconditioned and conditioned pathways within the brain, which could enable learning and performance within the spinal cord by down-regulating KCC2. To the extent that this descending regulatory process is affected by experience, it too could constitute a form of behavioral metaplasticity.
We have focused on how descending 5HT systems can regulate neural function within the spinal cord to enable plasticity. However, there is little reason to believe that this mode of regulating neural excitability is limited to just the spinal cord. Neural function throughout the brain is inhibited by GABAergic neurons that inhibit neural excitability and limit plasticity (Wigstrom and Gustafsson, 1983, Wang et al., 2006), preserving memories and motor function over time. Just as it makes little sense to overwrite a circuit within the spinal cord that organizes the timing and structure of locomotor function, it would appear maladaptive to overwrite neural circuits within the brain that store motor programs, maps of the environment, and the meaning of words. Like the spinal cord, these neural circuits may shift into an immutable state that helps to preserve the information over time, with over-excitation and neural plasticity held in check through GABA-dependent inhibition (Fig. 5E). And just as neural excitation in the spinal cord exhibits a non-monotonic function, with intermediate levels promoting adaptive plasticity while high levels produce a state of over-excitation that impairs learning, a down-regulation of KCC2 within the brain could foster a state of over-excitation (epilepsy) that impairs function (Kaila et al., 2014b).
Highlights.
Neurons within the spinal cord are sensitive to environmental relations
Relational learning impacts plastic potential (behavioral metaplasticity).
Plastic potential is regulated by BDNF, TNF, and GABA (ionic plasticity)
Spinal cord injury lowers membrane-bound KCC2, which reduces GABAergic inhibition
Released from inhibition, noxious input can sensitize pain circuits
Acknowledgments
Work on this paper was supported by grants from the Craig H. Neilsen Foundation and NIH (NINDS NS091723) to JWG. The authors would like to thank Rachel Baine, Paris Bean, Melissa Henwood, Jacob Davis, Gizelle Fauss, Travis Johnston, Clarence Lim, and Megan Tarbet for their help and input on this paper.
Abbreviations
- AMPA
a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- AMPAR
AMPA receptor
- BDNF
Brain-derived neurotrophic factor
- Bic
Bicuculline
- Bum
Bumetanide
- Cap
Capsaicin
- CPCCOEt
7-(hydroxy-imino)cycloproa[b]chromen-1a-carboxylate ethyl ester
- CPG
Central pattern generator
- Contra
Contralateral
- DHPG
3,5-Dihydroxyphenylglycine
- DIOA
R-(+)-[(2-n-Butyl-6,7-dichloro-2-cyclopentyl-2,3-dihydro-1-oxo-1H- inden-5-yl)oxy]acetic acid
- DLF
Dorsolateral funiculus
- DNIC
Diffuse noxious inhibitory control
- EMR
Enhanced mechanical reactivity
- FT
Fixed time
- GABA
Gamma-aminobutyric acid
- 5HT
Serotonin
- i.t
Intrathecal
- IPSP
Inhibitory postsynaptic potential
- ipsi
ipsilateral
- KCC2
K+-Cl− cotransporter 2
- LPS
Lipopolysaccharide
- LTD
Long-term depression
- LTP
Long-term potentiation
- mGlu
Metabotropic glutamate
- mGluR1
mGlu receptor 1
- mGluR5
mGlu receptor 5
- MPEP
2-Methyl-6-(phenylethylnyl)pyridine
- NKCC1
Na+-K+- Cl− cotransporter 1
- NMDA
N-methyl-D-aspartate
- NMDAR
NMDA receptor
- 8-OH-DPaT
8-Hydroxy-2-dipropylaminotetralin hydrobromide
- pERK
Phosphorylated extracellular signal-regulated kinases
- PLCγ
Phospholipase C gamma
- SCI
Spinal cord injury
- 5HT
Serotonin
- T2
Second thoracic
- Veh
Vehicle
- VIS
Variable intermittent shock
- VT
Variable time
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nature Reviews Neuroscience. 2008;9:387–399. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends in neurosciences. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- Baba H, Ji RR, Kohno T, Moore KA, Ataka T, Wakai A, Okamoto M, Woolf CJ. Removal of GABAergic inhibition facilitates polysynaptic A fiber-mediated excitatory transmission to the superficial spinal dorsal horn. Molecular and cellular neurosciences. 2003;24:818–830. doi: 10.1016/s1044-7431(03)00236-7. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nature Neuroscience. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Bardin L, Schmidt J, Alloui A, Eschalier A. Effect of intrathecal administration of serotonin in chronic pain models in rats. European journal of pharmacology. 2000;409:37–43. doi: 10.1016/s0014-2999(00)00796-2. [DOI] [PubMed] [Google Scholar]
- Baumbauer KM, Grau JW. Timing in the Absence of Supraspinal Input III: Regularly Spaced Cutaneous Stimulation Prevents and Reverses the Spinal Learning Deficit Produced by Peripheral Inflammation. Behavioral Neuroscience. 2011;125:37–45. doi: 10.1037/a0022009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbauer KM, Hoy KC, Huie JR, Hughes AJ, Woller SA, Puga DA, Setlow B, Grau JW. Timing in the absence of supraspinal input I: Variable, but not fixed, spaced stimulation of the sciatic nerve undermines spinally-mediated instrumental learning. Neuroscience. 2008;155:1030–1047. doi: 10.1016/j.neuroscience.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbauer KM, Huie JR, Hughes AJ, Grau JW. Timing in the Absence of Supraspinal Input II: Regularly Spaced Stimulation Induces a Lasting Alteration in Spinal Function That Depends on the NMDA Receptor, BDNF Release, and Protein Synthesis. Journal of Neuroscience. 2009;29:14383–14393. doi: 10.1523/JNEUROSCI.3583-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbauer KM, Lee KH, Puga DA, Woller SA, Hughes AJ, Grau JW. Temporal regularity determines the impact of electrical stimulation on tactile reactivity and response to capsaicin in spinally transected rats. Neuroscience. 2012;227:119–133. doi: 10.1016/j.neuroscience.2012.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs S, Salter MW. The known knowns of microglia-neuronal signalling in neuropathic pain. Neurosci Lett. 2013;557(Pt A):37–42. doi: 10.1016/j.neulet.2013.08.037. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. Excitatory actions of GABA during development: The nature of the nurture. Nature Reviews Neuroscience. 2002;3:728–739. doi: 10.1038/nrn920. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y. The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience. 2014;279:187–219. doi: 10.1016/j.neuroscience.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Kahle KT, Cherubini E. The GABA Excitatory/Inhibitory Shift in Brain Maturation and Neurological Disorders. Neuroscientist. 2012;18:467–486. doi: 10.1177/1073858412438697. [DOI] [PubMed] [Google Scholar]
- Bienenstock EL, Cooper LN, Munro PW. Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1982;2:32–48. doi: 10.1523/JNEUROSCI.02-01-00032.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggs JE, Lu VB, Stebbing MJ, Balasubramanyan S, Smith PA. Is BDNF sufficient for information transfer between microglia and dorsal horn neurons during the onset of central sensitization? Molecular Pain. 2010;6 doi: 10.1186/1744-8069-6-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouffard J, Bouyer LJ, Roy JS, Mercier C. Tonic pain experienced during locomotor training impairs retention despite normal performance during acquisition. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34:9190–9195. doi: 10.1523/JNEUROSCI.5303-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulenguez P, Liabeuf S, Bos R, Bras H, Jean-Xavier C, Brocard C, Stil A, Darbon P, Cattaert D, Delpire E, Marsala M, Vinay L. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nature medicine. 2010;16:302–307. doi: 10.1038/nm.2107. [DOI] [PubMed] [Google Scholar]
- Boyce VS, Mendell LM. Neurotrophins and spinal circuit function. Frontiers in neural circuits. 2014;8:59. doi: 10.3389/fncir.2014.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce VS, Park J, Gage FH, Mendell LM. Differential effects of brain-derived neurotrophic factor and neurotrophin-3 on hindlimb function in paraplegic rats. European Journal of Neuroscience. 2012;35:221–232. doi: 10.1111/j.1460-9568.2011.07950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce VS, Tumolo M, Fischer I, Murray M, Lemay MA. Neurotrophic factors promote and enhance locomotor recovery in untrained spinalized cats. Journal of Neurophysiology. 2007;98:1988–1996. doi: 10.1152/jn.00391.2007. [DOI] [PubMed] [Google Scholar]
- Castellanos DA, Daniels LA, Morales MP, Hama AT, Sagen J. Expansion of formalin-evoked Fos-immunoreactivity in rats with a spinal cord injury. Neurosci Res. 2007;58:386–393. doi: 10.1016/j.neures.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote MP, Azzam GA, Lemay MA, Zhukareva V, Houle JD. Activity-Dependent Increase in Neurotrophic Factors Is Associated with an Enhanced Modulation of Spinal Reflexes after Spinal Cord Injury. Journal of Neurotrauma. 2011;28:299–309. doi: 10.1089/neu.2010.1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote MP, Gandhi S, Zambrotta M, Houle JD. Exercise Modulates Chloride Homeostasis after Spinal Cord Injury. Journal of Neuroscience. 2014;34:8976–8987. doi: 10.1523/JNEUROSCI.0678-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005a;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Coull JAM, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature. 2005b;438:1017–1021. doi: 10.1038/nature04223. [DOI] [PubMed] [Google Scholar]
- Cramer SW, Baggott C, Cain J, Tilghman J, Allcock B, Miranpuri G, Rajpal S, Sun D, Resnick D. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain. 2008;4:36. doi: 10.1186/1744-8069-4-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown ED, Ferguson AR, Joynes RL, Grau JW. Instrumental learning within the spinal cord II. Evidence for central mediation. Physiology & Behavior. 2002a;77:259–267. doi: 10.1016/s0031-9384(02)00859-4. [DOI] [PubMed] [Google Scholar]
- Crown ED, Ferguson AR, Joynes RL, Grau JW. Instrumental learning within the spinal cord: IV Induction and retention of the behavioral deficit observed after noncontingent shock. Behavioral Neuroscience. 2002b;116:1032–1051. doi: 10.1037//0735-7044.116.6.1032. [DOI] [PubMed] [Google Scholar]
- Crown ED, Grau JW. Preserving and restoring behavioral potential within the spinal cord using an instrumental training paradigm. Journal of Neurophysiology. 2001;86:845–855. doi: 10.1152/jn.2001.86.2.845. [DOI] [PubMed] [Google Scholar]
- Crown ED, Grau JW. Evidence that descending serotonergic systems protect spinal cord plasticity against the disruptive effect of uncontrollable stimulation. Exp Neurol. 2005;196:164–176. doi: 10.1016/j.expneurol.2005.07.016. [DOI] [PubMed] [Google Scholar]
- Czeschik JC, Hagenacker T, Schaefers M, Buesselberg D. TNF-alpha differentially modulates ion channels of nociceptive neurons. Neuroscience Letters. 2008;434:293–298. doi: 10.1016/j.neulet.2008.01.070. [DOI] [PubMed] [Google Scholar]
- Dougherty KJ, Hochman S. Spinal cord injury causes plasticity in a subpopulation of lamina I GABAergic interneurons. Journal of Neurophysiology. 2008;100:212–223. doi: 10.1152/jn.01104.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew GM, Siddall PJ, Duggan AW. Mechanical allodynia following contusion injury of the rat spinal cord is associated with loss of GABAergic inhibition in the dorsal horn. Pain. 2004;109:379–388. doi: 10.1016/j.pain.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Edgerton VR, Courtine G, Gerasimenko YP, Lavrov I, Ichiyama RM, Fong AJ, Cai LL, Otoshi CK, Tillakaratne NJ, Burdick JW, Roy RR. Training locomotor networks. Brain Res Rev. 2008;57:241–254. doi: 10.1016/j.brainresrev.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton VR, Roy RR, DeLeon R, Tillakaratne N, Hodgson JA. Does motor learning occur in the spinal cord? Neuroscientist. 1997;3:287–294. [Google Scholar]
- Edgerton VR, Tillakaratne NJK, Bigbee AJ, de Leon RD, Roy RR. Plasticity of the spinal neural circuitry after injury. Annual Review of Neuroscience. 2004;27:145–167. doi: 10.1146/annurev.neuro.27.070203.144308. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Bolding KA, Huie JR, Hook MA, Santillano DR, Miranda RC, Grau JW. Group I Metabotropic Glutamate Receptors Control Metaplasticity of Spinal Cord Learning through a Protein Kinase C-Dependent Mechanism. Journal of Neuroscience. 2008;28:11939–11949. doi: 10.1523/JNEUROSCI.3098-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Crown ED, Grau JW. Nociceptive plasticity inhibits adaptive learning in the spinal cord. Neuroscience. 2006;141:421–431. doi: 10.1016/j.neuroscience.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Huie JR, Crown ED, Baumbauer KM, Hook MA, Garraway SM, Lee KH, Hoy KC, Grau JW. Maladaptive spinal plasticity opposes spinal learning and recovery in spinal cord injury. Frontiers in physiology. 2012a;3 doi: 10.3389/fphys.2012.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Huie JR, Crown ED, Grau JW. Central nociceptive sensitization vs. spinal cord training: opposing forms of plasticity that dictate function after complete spinal cord injury. Frontiers in physiology. 2012b;3 doi: 10.3389/fphys.2012.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AR, Washburn SN, Crown ED, Grau JW. GABA(A) receptor activation is involved in noncontingent shock inhibition of instrumental conditioning in spinal rats. Behavioral Neuroscience. 2003;117:799–812. doi: 10.1037/0735-7044.117.4.799. [DOI] [PubMed] [Google Scholar]
- Ferrini F, De Koninck Y. Microglia control neuronal network excitability via BDNF signalling. Neural plasticity. 2013;2013:429815. doi: 10.1155/2013/429815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, Trang T, Mattioli TA, Laffray S, Del'Guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De Koninck Y. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 2013;16:183–192. doi: 10.1038/nn.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields DP, Mitchell GS. Spinal metaplasticity in respiratory motor control. Frontiers in neural circuits. 2015;9:2. doi: 10.3389/fncir.2015.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YJ, Xu ZZ, Liu YC, Wen YR, Decosterd I, Ji RR. The c-Jun N-terminal kinase 1 (JNK1) in spinal astrocytes is required for the maintenance of bilateral mechanical allodynia under a persistent inflammatory pain condition. Pain. 2010;148:309–319. doi: 10.1016/j.pain.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway SM, Turtle JD, Huie JR, Lee KH, Hook MA, Woller SA, Grau JW. Intermittent Noxious Stimulation Following Spinal Cord Contusion Injury Impairs Locomotor Recovery and Reduces Spinal Brain-Derived Neurotrophic Factor-Tropomyosin- Receptor Kinase Signaling in Adult Rats. Neuroscience. 2011;199:86–102. doi: 10.1016/j.neuroscience.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway SM, Woller SA, Huie JR, Hartman JJ, Hook MA, Miranda RC, Huang YJ, Ferguson AR, Grau JW. Peripheral noxious stimulation reduces withdrawal threshold to mechanical stimuli after spinal cord injury: Role of tumor necrosis factor alpha and apoptosis. Pain. 2014;155:2344–2359. doi: 10.1016/j.pain.2014.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjerstad J, Tjolsen A, Hole K. Induction of long-term potentiation of single wide dynamic range neurones in the dorsal horn is inhibited by descending pathways. Pain. 2001;91:263–268. doi: 10.1016/S0304-3959(00)00448-6. [DOI] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Huie JR, Ying Z, Ferguson AR, Crown ED, Baumbauer KM, Edgerton VR, Grau JW. BDNF and learning: Evidence that instrumental training promotes learning within the spinal cord by up-regulating BDNF expression. Neuroscience. 2007;148:893–906. doi: 10.1016/j.neuroscience.2007.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla F, Ying Z, Roy RR, Molteni R, Edgerton VR. Voluntary exercise induces a BDNF-mediated mechanism that promotes neuroplasticity. Journal of Neurophysiology. 2002;88:2187–2195. doi: 10.1152/jn.00152.2002. [DOI] [PubMed] [Google Scholar]
- Gottmann K, Mittmann T, Lessmann V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Experimental brain research Experimentelle Hirnforschung Experimentation cerebrale. 2009;199:203–234. doi: 10.1007/s00221-009-1994-z. [DOI] [PubMed] [Google Scholar]
- Grau JW. The Central Representation of an Aversive Event Maintains the Opioid and Nonopioid Forms of Analgesia. Behavioral Neuroscience. 1987;101:272–288. doi: 10.1037//0735-7044.101.2.272. [DOI] [PubMed] [Google Scholar]
- Grau JW. Spinal neurons exhibit a surprising capacity to learn and a hidden vulnerability when freed from the brain's control. Current Neurology and Neuroscience Reports. 2006;6:177–180. doi: 10.1007/s11910-006-0001-3. [DOI] [PubMed] [Google Scholar]
- Grau JW. Learning from the spinal cord: How the study of spinal cord plasticity informs our view of learning. Neurobiology of Learning and Memory. 2014;108:155–171. doi: 10.1016/j.nlm.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau JW, Barstow DG, Joynes RL. Instrumental learning within the spinal cord: I. Behavioral properties. Behavioral Neuroscience. 1998;112:1366–1386. doi: 10.1037//0735-7044.112.6.1366. [DOI] [PubMed] [Google Scholar]
- Grau JW, Crown ED, Ferguson AR, Washburn SN, Hook MA, Miranda RC. Instrumental learning within the spinal cord: underlying mechanisms and implications for recovery after injury. Behavioral and cognitive neuroscience reviews. 2006;5:191–239. doi: 10.1177/1534582306289738. [DOI] [PubMed] [Google Scholar]
- Grau JW, Huang YJ, Turtle JD, Strain MM, Miranda RC, Garraway SM, Hook MA. When Pain Hurts: Nociceptive Stimulation Induces a State of Maladaptive Plasticity and Impairs Recovery after Spinal Cord Injury. J Neurotrauma. 2017;34:1873–1890. doi: 10.1089/neu.2016.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau JW, Huang YJ, Turtle JD, Strain MM, Miranda RM, Garraway SM, Hook MA. When Pain Hurts: Nociceptive Stimulation Induces a State of Maladaptive Plasticity and Impairs Recovery after Spinal Cord Injury. J Neurotrauma. 2016 doi: 10.1089/neu.2016.4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau JW, Huie JR, Garraway SM, Hook MA, Crown ED, Baumbauer KM, Lee KH, Hoy KC, Ferguson AR. Impact of behavioral control on the processing of nociceptive stimulation. Frontiers in physiology. 2012;3 doi: 10.3389/fphys.2012.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau JW, Huie JR, Lee KH, Hoy KC, Huang YJ, Turtle JD, Strain MM, Baumbauer KM, Miranda RM, Hook MA, Ferguson AR, Garraway SM. Metaplasticity and behavior: how training and inflammation affect plastic potential within the spinal cord and recovery after injury. Frontiers in neural circuits. 2014;8:100. doi: 10.3389/fncir.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau JW, Salinas JA, Illich PA, Meagher MW. Associative Learning and Memory for an Antinociceptive Response in the Spinalized Rat. Behavioral Neuroscience. 1990;104:489–494. doi: 10.1037//0735-7044.104.3.489. [DOI] [PubMed] [Google Scholar]
- Grau JW, Washburn SN, Hook MA, Ferguson AR, Crown ED, Garcia G, Bolding KA, Miranda RC. Uncontrollable stimulation undermines recovery after spinal cord injury. Journal of Neurotrauma. 2004;21:1795–1817. doi: 10.1089/neu.2004.21.1795. [DOI] [PubMed] [Google Scholar]
- Groves PM, De Marco R, Thompson RF. Habituation and sensitization of spinal interneuron activity in acute spinal cat. Brain Res. 1969;14:521–525. doi: 10.1016/0006-8993(69)90129-2. [DOI] [PubMed] [Google Scholar]
- Gwak YS, Hulsebosch CE. GABA and central neuropathic pain following spinal cord injury. Neuropharmacology. 2011;60:799–808. doi: 10.1016/j.neuropharm.2010.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Fullwood SD, Eaton MJ, Hulsebosch CE. Subdural engraftment of serotonergic neurons following spinal hemisection restores spinal serotonin, downregulates serotonin transporter, and increases BDNF tissue content in rat. Brain Research. 2001a;913:35–46. doi: 10.1016/s0006-8993(01)02749-4. [DOI] [PubMed] [Google Scholar]
- Hains BC, Johnson KM, McAdoo DJ, Eaton MJ, Hulsebosch CE. Engraftment of serotonergic precursors enhances locomotor function and attenuates chronic central pain behavior following spinal hemisection injury in the rat. Experimental Neurology. 2001b;171:361–378. doi: 10.1006/exnr.2001.7751. [DOI] [PubMed] [Google Scholar]
- Hains BC, Yucra JA, Hulsebosch CE. Reduction of pathological and behavioral deficits following spinal cord contusion injury with the selective cyclooxygenase-2 inhibitor NS-398. Journal of Neurotrauma. 2001c;18:409–423. doi: 10.1089/089771501750170994. [DOI] [PubMed] [Google Scholar]
- Hasbargen T, Ahmed MM, Miranpuri G, Li L, Kahle KT, Resnick D, Sun D. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Annals of the New York Academy of Sciences. 2010;1198:168–172. doi: 10.1111/j.1749-6632.2010.05462.x. [DOI] [PubMed] [Google Scholar]
- Hook MA, Huie JR, Grau JW. Peripheral inflammation undermines the plasticity of the isolated spinal cord. Behavioral Neuroscience. 2008;122:233–249. doi: 10.1037/0735-7044.122.1.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Grau JW. Descending serotongeric fibers regulate nociceptive sensitization through KCC2 after spinal injury submitted. [Google Scholar]
- Huang YJ, Lee KH, Murphy L, Garraway SM, Grau JW. Spinal cord injury (SCI) transforms how GABA affects nociceptive plasticity. Experimental Neurology. 2016 doi: 10.1016/j.expneurol.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Lee KH, Grau JW. Complete spinal cord injury (SCI) transforms how brain derived neurotrophic factor (BDNF) affects nociceptive sensitization. Exp Neurol. 2017;288:38–50. doi: 10.1016/j.expneurol.2016.11.001. [DOI] [PubMed] [Google Scholar]
- Huang YJ, Lee KH, Murphy L, Garraway SM, Grau JW. Acute spinal cord injury (SCI) transforms how GABA affects nociceptive sensitization. Exp Neurol. 2016;285:82–95. doi: 10.1016/j.expneurol.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long-term potentiation. Science. 1992;255:730–733. doi: 10.1126/science.1346729. [DOI] [PubMed] [Google Scholar]
- Huie JR, Baumbauer KM, Lee KH, Bresnahan JC, Beattie MS, Ferguson AR, Grau JW. Glial Tumor Necrosis Factor Alpha (TNF alpha) Generates Metaplastic Inhibition of Spinal Learning. Plos One. 2012a;7 doi: 10.1371/journal.pone.0039751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie JR, Garraway SM, Baumbauer KM, Hoy KC, Beas BS, Montgomery KS, Bizon JL, Grau JW. Brain-Derived Neurotrophic Factor Promotes Adaptive Plasticity within the Spinal Cord and Mediates the Beneficial Effects of Controllable Stimulation. Neuroscience. 2012b;200:74–90. doi: 10.1016/j.neuroscience.2011.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie JR, Stuck ED, Lee KH, Irvine KA, Beattie MS, Bresnahan JC, Grau JW, Ferguson AR. AMPA receptor phosphorylation and synaptic co-localization on motor neurons drive maladaptive plasticity below complete spinal cord injury. eNeuro. 2015 doi: 10.1523/ENEURO.0091-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hulme SR, Jones OD, Abraham WC. Emerging roles of metaplasticity in behaviour and disease. Trends in neurosciences. 2013;36:353–362. doi: 10.1016/j.tins.2013.03.007. [DOI] [PubMed] [Google Scholar]
- Hwang JH, Yaksh TL. The effect of spinal GABA receptor agonists on tactile allodynia in a surgically-induced neuropathic pain model in the rat. Pain. 1997;70:15–22. doi: 10.1016/s0304-3959(96)03249-6. [DOI] [PubMed] [Google Scholar]
- Jean-Xavier C, Pflieger JF, Liabeuf S, Vinay L. Inhibitory postsynaptic potentials in lumbar motoneurons remain depolarizing after neonatal spinal cord transection in the rat. J Neurophysiol. 2006;96:2274–2281. doi: 10.1152/jn.00328.2006. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends in neurosciences. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Jolivalt CG, Lee CA, Ramos KM, Calcutt NA. Allodynia and hyperalgesia in diabetic rats are mediated by GABA and depletion of spinal potassium-chloride co-transporters. Pain. 2008;140:48–57. doi: 10.1016/j.pain.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joynes RL, Ferguson AR, Crown ED, Patton BC, Grau JW. Instrumental learning within the spinal cord: V. Evidence the behavioral deficit observed after noncontingent nociceptive stimulation reflects an intraspinal modification. Behavioural Brain Research. 2003;141:159–170. doi: 10.1016/s0166-4328(02)00372-8. [DOI] [PubMed] [Google Scholar]
- Joynes RL, Grau JW. Mechanisms of pavlovian conditioning: Role of protection from habituation in spinal conditioning. Behavioral Neuroscience. 1996;110:1375–1387. doi: 10.1037//0735-7044.110.6.1375. [DOI] [PubMed] [Google Scholar]
- Joynes RL, Grau JW. Instrumental learning within the spinal cord: III. Prior exposure to noncontingent shock induces a behavioral deficit that is blocked by an opioid antagonist. Neurobiology of Learning and Memory. 2004;82:35–51. doi: 10.1016/j.nlm.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Joynes RL, Janjua K, Grau JW. Instrumental learning within the spinal cord: VI The NMDA receptor antagonist, AP5, disrupts the acquisition and maintenance of an acquired flexion response. Behavioural Brain Research. 2004;154:431–438. doi: 10.1016/j.bbr.2004.03.030. [DOI] [PubMed] [Google Scholar]
- Kahle KT, Deeb TZ, Puskarjov M, Silayeva L, Liang B, Kaila K, Moss SJ. Modulation of neuronal activity by phosphorylation of the K-Cl cotransporter KCC2. Trends in neurosciences. 2013;36:726–737. doi: 10.1016/j.tins.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nature Reviews Neuroscience. 2014a;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Current opinion in neurobiology. 2014b;26:34–41. doi: 10.1016/j.conb.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Hammond DL. Role of spinal gamma-aminobutyric acidA receptors in formalin-induced nociception in the rat. The Journal of pharmacology and experimental therapeutics. 1997;282:928–938. [PubMed] [Google Scholar]
- King T, Vera-Portocarrero L, Gutierrez T, Vanderah TW, Dussor G, Lai J, Fields HL, Porreca F. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central Sensitization: A Generator of Pain Hypersensitivity by Central Neural Plasticity. Journal of Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Huang YJ, Grau JW. Learning about time within the spinal cord II: Evidence that temporal regularity is encoded by a spinal oscillator. Frontiers in Behavioral Neuroscience. 2016;10:14. doi: 10.3389/fnbeh.2016.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Turtle JD, Strain MM, Huang YJ, Baumbauer KM, Grau JW. Learning about time within the spinal cord: Evidence that spinal neurons can abstract and store an index of regularity. Frontiers in Behavioral Neuroscience. 2015;9:274. doi: 10.3389/fnbeh.2015.00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu VB, Biggs JE, Stebbing MJ, Balasubramanyan S, Todd KG, Lai AY, Colmers WF, Dawbarn D, Ballanyi K, Smith PA. Brain-derived neurotrophic factor drives the changes in excitatory synaptic transmission in the rat superficial dorsal horn that follow sciatic nerve injury. Journal of Physiology-London. 2009;587:1013–1032. doi: 10.1113/jphysiol.2008.166306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zheng J, Xiong L, Zimmermann M, Yang J. Spinal cord injury-induced attenuation of GABAergic inhibition in spinal dorsal horn circuits is associated with down-regulation of the chloride transporter KCC2 in rat. The Journal of physiology. 2008;586:5701–5715. doi: 10.1113/jphysiol.2008.152348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. Synaptic plasticity in the hippocampus: LTP and LTD. Cell. 1994;78:535–538. doi: 10.1016/0092-8674(94)90517-7. [DOI] [PubMed] [Google Scholar]
- McNally GP, Johansen JP, Blair HT. Placing prediction into the fear circuit. Trends in neurosciences. 2011;34:283–292. doi: 10.1016/j.tins.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meagher MW, Grau JW, King RA. Role of Supraspinal Systems in Environmentally Induced Antinociception - Effect of Spinalization and Decerebration on Brief Shock-Induced and Long Shock-Induced Antinociception. Behavioral Neuroscience. 1990;104:328–338. doi: 10.1037//0735-7044.104.2.328. [DOI] [PubMed] [Google Scholar]
- Medina I, Friedel P, Rivera C, Kahle KT, Kourdougli N, Uvarov P, Pellegrino C. Current view on the functional regulation of the neuronal K+-Cl- cotransporter KCC2. Frontiers in Cellular Neuroscience. 2014;8:18. doi: 10.3389/fncel.2014.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi A, Bardoni R, Salio C, Lossi L, Ferrini F, Prandini M, Zonta M, Gustincich S, Carrnignot G. Presynaptic functional trkB receptors mediate the release of excitatory Neurotransmitters from primary afferent terminals in lamina II (substantia gelatinosa) of postnatal rat spinal cord. Developmental Neurobiology. 2008a;68:457–475. doi: 10.1002/dneu.20605. [DOI] [PubMed] [Google Scholar]
- Merighi A, Salio C, Ghirri A, Lossi L, Ferrini F, Betelli C, Bardoni R. BDNF as a pain modulator. Progress in Neurobiology. 2008b;85:297–317. doi: 10.1016/j.pneurobio.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, Tracey K, Martin D, Maier SF, Watkins LR. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser EI, Moser MB. Is learning blocked by saturation of synaptic weights in the hippocampus? Neuroscience and biobehavioral reviews. 1999;23:661–672. doi: 10.1016/s0149-7634(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Chen PS, Willis WD. Role of metabotropic glutamate receptor subtype mGluR1 in brief nociception and central sensitization of primate STT cells. Journal of Neurophysiology. 1999;82:272–282. doi: 10.1152/jn.1999.82.1.272. [DOI] [PubMed] [Google Scholar]
- Park CK, Lue N, Xu ZZ, Liu T, Serhan CN, Ji RR. Resolving TRPV1-and TNF-alpha- Mediated Spinal Cord Synaptic Plasticity and Inflammatory Pain with Neuroprotectin D1. Journal of Neuroscience. 2011;31:15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton BC, Hook MA, Ferguson AR, Crown ED, Grau JW. The behavioral deficit observed following noncontingent shock in spinalized rats is prevented by the protein synthesis inhibitor cycloheximide. Behavioral Neuroscience. 2004;118:653–658. doi: 10.1037/0735-7044.118.3.653. [DOI] [PubMed] [Google Scholar]
- Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. European Journal of Pain-London. 2000;4:247–257. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Kaila K. Two developmental switches in GABAergic signalling: the K+-Cl- cotransporter KCC2 and carbonic anhydrase CAVII. The Journal of physiology. 2005;562:27–36. doi: 10.1113/jphysiol.2004.077495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipila S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LA, Beyer C, Komisaruk BR. Nociceptive responses to altered GABAergic activity at the spinal cord. Life Sci. 1986;39:1667–1674. doi: 10.1016/0024-3205(86)90164-5. [DOI] [PubMed] [Google Scholar]
- Sandkuhler J. Models and Mechanisms of Hyperalgesia and Allodynia. Physiological reviews. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Sandkuhler J, Chen JG, Cheng G, Randic M. Low-frequency stimulation of afferent Adelta-fibers induces long-term depression at primary afferent synapses with substantia gelatinosa neurons in the rat. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1997;17:6483–6491. doi: 10.1523/JNEUROSCI.17-16-06483.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafers M, Sommer C, Geis C, Hagenacker T, Vandenabeele P, Sorkin LS. Selective stimulation of either tumor necrosis factor receptor differentially induces pain behavior in vivo and ectopic activity in sensory neurons in vitro. Neuroscience. 2008;157:414–423. doi: 10.1016/j.neuroscience.2008.08.067. [DOI] [PubMed] [Google Scholar]
- Schmidt MV, Abraham WC, Maroun M, Stork O, Richter-Levin G. Stress-induced metaplasticity: from synapses to behavior. Neuroscience. 2013;250:112–120. doi: 10.1016/j.neuroscience.2013.06.059. [DOI] [PubMed] [Google Scholar]
- Sehgal M, Song C, Ehlers VL, Moyer JR., Jr Learning to learn - intrinsic plasticity as a metaplasticity mechanism for memory formation. Neurobiol Learn Mem. 2013;105:186–199. doi: 10.1016/j.nlm.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga A, Thomas-Crusells J, Sigl T, Blaesse A, Mestres P, Meyer M, Yan Q, Kaila K, Saarma M, Rivera C, Giehl KM. Posttraumatic GABA(A)-mediated [Ca2+](i) increase is essential for the induction of brain-derived neurotrophic factor-dependent survival of mature central neurons. Journal of Neuroscience. 2008;28:6996–7005. doi: 10.1523/JNEUROSCI.5268-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone DA, Baumann TK, Lamotte RH. Dose-dependent pain and mechanical hyperalgesia in humans after intradermal injection of capsaicin. Pain. 1989;38:99–107. doi: 10.1016/0304-3959(89)90079-1. [DOI] [PubMed] [Google Scholar]
- Sivilotti L, Woolf CJ. The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia in the spinal cord. J Neurophysiol. 1994;72:169–179. doi: 10.1152/jn.1994.72.1.169. [DOI] [PubMed] [Google Scholar]
- Smith PA. BDNF: No Gain Without Pain? Neuroscience. 2014;283:107–123. doi: 10.1016/j.neuroscience.2014.05.044. [DOI] [PubMed] [Google Scholar]
- Sorkin LS, Puig S, Jones DL. Spinal bicuculline produces hypersensitivity of dorsal horn neurons: effects of excitatory amino acid antagonists. Pain. 1998;77:181–190. doi: 10.1016/S0304-3959(98)00094-3. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Tashiro S, Shinozaki M, Mukaino M, Renault-Mihara F, Toyama Y, Liu M, Nakamura M, Okano H. BDNF Induced by Treadmill Training Contributes to the Suppression of Spasticity and Allodynia After Spinal Cord Injury via Upregulation of KCC2. Neurorehabil Neural Repair. 2015;29:677–689. doi: 10.1177/1545968314562110. [DOI] [PubMed] [Google Scholar]
- Thompson RF, Spencer WA. Habituation - A Model Phenomenon for Study of Neuronal Substrates of Behavior. Psychological Review. 1966;73:16–&. doi: 10.1037/h0022681. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG. More than a sidekick: glia and homeostatic synaptic plasticity. Trends Mol Med. 2006;12:458–460. doi: 10.1016/j.molmed.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Vaynman S, Gomez-Pinilla F. License to run: Exercise impacts functional plasticity in the intact and injured central nervous system by using neurotrophins. Neurorehabilitation and Neural Repair. 2005;19:283–295. doi: 10.1177/1545968305280753. [DOI] [PubMed] [Google Scholar]
- Vichaya EG, Baumbauer KM, Carcoba LM, Grau JW, Meagher MW. Spinal glia modulate both adaptive and pathological processes. Brain Behavior and Immunity. 2009;23:969–976. doi: 10.1016/j.bbi.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Gong N, Xu TL. Downregulation of KCC2 following LTP-contributes to EPSP-spike potentiation in rat hippocampus. Biochemical and Biophysical Research Communications. 2006;343:1209–1215. doi: 10.1016/j.bbrc.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Washburn SN, Patton BC, Ferguson AR, Hudson KL, Grau JW. Exposure to intermittent nociceptive stimulation under pentobarbital anesthesia disrupts spinal cord function in rats. Psychopharmacology. 2007;192:243–252. doi: 10.1007/s00213-007-0707-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Molecular and cellular neurosciences. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Cobelli DA, Mayer DJ. Opiate vs non-opiate footshock induced analgesia (FSIA) - Descending and intra-spinal components. Brain Research. 1982;245:97–106. doi: 10.1016/0006-8993(82)90342-0. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Martin D, Ulrich P, Tracey KJ, Maier SF. Evidence for the involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–235. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- Weng HR, Laird JM, Cervero F, Schouenborg J. GABAA receptor blockade inhibits A beta fibre evoked wind-up in the arthritic rat. Neuroreport. 1998;9:1065–1069. doi: 10.1097/00001756-199804200-00019. [DOI] [PubMed] [Google Scholar]
- Wigstrom H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]
- Willis WD. Mechanisms of central sensitization of nociceptive dorsal horn neurons 2001 [Google Scholar]
- Windhorst U. Muscle proprioceptive feedback and spinal networks. Brain Research Bulletin. 2007;73:155–202. doi: 10.1016/j.brainresbull.2007.03.010. [DOI] [PubMed] [Google Scholar]
- Wolpaw JR. What Can the Spinal Cord Teach Us about Learning and Memory? Neuroscientist. 2010;16:532–549. doi: 10.1177/1073858410368314. [DOI] [PubMed] [Google Scholar]
- Wolpaw JR, Carp JS. Memory Traces in Spinal-Cord. Trends in neurosciences. 1990;13:137–142. doi: 10.1016/0166-2236(90)90005-u. [DOI] [PubMed] [Google Scholar]
- Young EE, Baumbauer KM, Elliot A, Joynes RL. Lipopolysaccharide induces a spinal learning deficit that is blocked by IL-1 receptor antagonism. Brain Behavior and Immunity. 2007;21:748–757. doi: 10.1016/j.bbi.2007.02.001. [DOI] [PubMed] [Google Scholar]