

Abstract
Prion diseases are fatal neurodegenerative diseases caused by misfolding of the prion protein (PrP). These conditions affect humans and animals, including endemic forms in sheep and deer. Bovine, rodents, and many zoo mamma ls also developed prion diseases during the “mad-cow” epidemic in the 1980’s. Interestingly, rabbits, horses, and dogs show unusual resistance to prion diseases, suggesting that specific sequence changes in the corresponding endogenous PrP prevents the accumulation of pathogenic conformations. In vitro misfolding assays and structural studies have identified S174, S167, and D159 as the key residues mediating the stability of rabbit, horse, and dog PrP, respectively. Here, we expressed the WT forms of rabbit, horse, and dog PrP in transgenic Drosophila and found that none of them is toxic. Replacing these key residues with the corresponding amino acids in hamster PrP showed that mutant horse (S167D) and dog (D159N) PrP are highly toxic, whereas mutant rabbit (S174N) PrP is not. These results confirm the impact of S167 and D159 in local and long-range structural features in the globular domain of PrP that increase its stability, while suggesting the role of additional residues in the stability of rabbit PrP. Identifying these protective amino acids and the structural features that stabilize PrP can contribute to advance the field towards the development of therapies that halt or reverse the devastating effects of prion diseases.
Keywords: Prion protein, neurotoxicity, susceptibility, Drosophila, transgenics, amino acid substitution
Introduction
Prion diseases are a diverse group of neurodegenerative conditions affecting humans and other mammals (Colby and Prusiner, 2011; Ironside et al., 2017; Prusiner, 1998). Sporadic Creutzfeldt-Jacob disease is the most common prion disease in humans and is characterized mainly by aggressive dementia (Knight, 2017). Other sporadic, inherited, and transmitted prion diseases can present with motor deficits or sleep perturbations (Knight, 2017). Prion diseases are the only human neurodegenerative disorders with true correlates in mammals (Zlotnik and Rennie, 1965), making rodents ideal models to understand the mechanisms of disease transmission and pathogenesis. Scrapie, bovine spongiform encephalopathy (BSE), and chronic wasting disease (CWD) are prion diseases described in sheep and goats, bovine, and cervids, respectively (Mathiason, 2017). One common link to all these conditions is the characteristic spongiform brain pathology. The second main feature is the brain deposition of misfolded, insoluble conformations of the prion protein (PrP) resistant to proteases termed scrapie PrP (PrPSc) (Prusiner, 1998). Prion transmission requires the catalytic conversion of cellular PrP (PrPc) by PrPSc, but this interaction may be hindered by sequence differences, an observation that underlies the “species barrier” phenomenon (Telling et al., 1996; Telling et al., 1995). It is well known that the C-terminal globular domain, which contains three α-helices and two small β-pleated domains, plays a key role in PrP pathogenesis (Donne et al., 1997; Inouye and Kirschner, 1998; James et al., 1997). An increase in β-sheet content is proposed to alter PrP solubility, aggregation, and neurotoxicity, three new features of PrP during pathogenesis (Aguzzi et al., 2008; Inouye and Kirschner, 1998; Prusiner, 1998). Despite major efforts to understand the mechanisms regulating PrP conversion, transmission, and pathogenesis, there are currently no treatments to stop or reverse these diseases.
Over 50 pathogenic mutations and several artificial mutations have provided important but limited clues about how the amino acid sequence modulates PrP conformational dynamics and pathogenesis (Lloyd et al., 2011). In addition, nature provides an understudied source of sequence variants: the PrP zoo. Most amino acid changes accumulate during evolution by neutral drift, although some variants may be subject to selection if they alter critical PrP functions. The challenge to identify key residues mediating PrP conversion from these natural variants is that several changes accumulate even in closely related species. For instance, mice and hamsters are rodents sensitive to prion diseases that accumulate 13 amino acid differences in the 202 amino acids of mature PrP (amino acids 29–231), four of them in the globular domain. Although many of these substitutions are conservative (similar properties), their contribution to PrP conformation cannot be dismissed. In fact, many pathogenic mutations responsible for dominantly inherited prion diseases in humans are conservative too, particularly in hydrophobic residues (e.g., V180I, V203I, V210I) (Lloyd et al., 2011). PrP from a few animals known to be resistant to prion diseases - rabbits, horses, and dogs - are an underutilized resource that can teach us about the intrinsic determinants of PrP conformational changes and toxicity. Although PrP from these three animals also show several amino acid changes compared to hamster PrP, focusing on unique changes in the globular domain can identify the critical residues that confer structural stability to PrP and prevent pathogenesis.
Scrapie is an endemic disease described in sheep and goat more than 250 years ago. Although scrapie is not transmissible to humans, it can be transmitted experimentally to rodents (mouse, rat, hamster, bank vole) (Chandler, 1971; Chandler and Fisher, 1963; Zlotnik and Rennie, 1963; Zlotnik and Rennie, 1965), but not to rabbits (Gibbs and Gajdusek, 1973) even after passage through mice (Barlow and Rennie, 1976). In contrast to sheep and goats, there is no endemic prion disease in bovine. Unfortunately, the human-mediated transmission of scrapie to bovine resulted in the mad-cow epidemics in the mid-80’s (Wells et al., 1987). From that initial event, bovine prions spread to humans as a new disease called variant Creutzfeldt-Jakob diseas (Will et al., 1996). Bovine prions also spread to several domestic and zoo animals, including felines, ferrets, and others (Kirkwood and Cunningham, 1994), demonstrating the susceptibility of many mammals with no known endemic prion disease. Horses and dogs were also exposed to prions during the mad-cow epidemics through bovine-contaminated feed. Despite this exposure, not a single case of prion disease has been described among horses and dogs, so far (Kirkwood and Cunningham, 1994). These are domestic animals with good veterinarian care and prion diseases are easily diagnosed in any animal due to the typical spongiform degeneration of the brain followed by confirmatory molecular tests. Two hypotheses have been proposed to explain the resistance of a few animals to prion diseases: intrinsic factors (differences in PrP sequence) or extrinsic factors (cellular factors). In this regard, rabbit cells can convert transgenic PrP from susceptible animals (Courageot et al., 2008) while fragments of rabbit PrP (RaPrP) are sufficient to prevent conversion of chimeric rabbit-mouse PrP (Gibbs and Gajdusek, 1973). These results argue for a normal cellular environment in rabbit cells, suggesting that the inefficient conversion of RaPrP is due to protective substitutions in its endogenous sequence. Thus, it is likely that prion-resistant mammals carry “protective” amino acid substitutions that stabilize PrPc conformations and hinder conversion into pathogenic conformations. Identifying the key amino acid changes responsible for stabilizing the tertiary structure of rabbit, horse, and dog PrP can reveal key insight to understand PrP misfolding and pathogenesis.
In addition to these limited experimental manipulations and natural history of prion diseases, NMR structure is available for the globular domain of PrP from rabbits, horses, and dogs (Lysek et al., 2005; Perez et al., 2010; Wen et al., 2010b), while high-resolution X-ray crystallography is only available for (RaPrP) (Khan et al., 2010). In RaPrP, serine (Ser, S) at 174 (human numbering, see Fig. 1A) (Vorberg et al., 2003) is proposed to form a helix-capping motif that stabilizes the β2-α2 loop and distal helix 3 (Khan et al., 2010). Introducing an asparagine (Asn, N) found in most animals at 174 (S174N) increases RaPrP conformational instability in vitro (Khan et al., 2010), supporting the protective role of S174. We recently confirmed the conformational stability and lack of toxicity of RaPrP expressed in Drosophila, which is in sharp contrast to the high toxicity of hamster PrP (Femandez-Funez et al., 2010; Fernandez-Funez et al., 2011). Recent studies challenged the traditional idea that rabbit is resistant to prions, although only 20% of animals inoculated with rabbit-adapted prions developed disease after long incubations on second passage (Chianini et al., 2012). Furthermore, structural studies also identified candidate protective residues in horse and dog PrP, including S167 in horse PrP (Equus, EqPrP) and aspartic acid (Asp, D) at 159 in dog PrP (Canis, CaPrP) (Lysek et al., 2005; Perez et al., 2010). In vitro conversion studies have shown that both rabbit and dog PrP can be converted into infectious isoforms that induce disease in mice expressing bovine and human PrP (Vidal et al., 2013). But these laboratory conditions do not indicate that rabbits and dogs can be infected with prions under natural conditions. We recently demonstrated that the N159D substitution increases the conformational stability of mouse PrP and reduces its toxicity in Drosophila, supporting the protective role of D159 (Sanchez-Garcia et al., 2016). Overall, Drosophila has shown to be a model highly sensitive to the expression of PrP from different species (hamster, mouse, ovine, rabbit, human), making this a powerful model to study PrP folding and toxicity (Fernandez-Funez et al., 2017; Fernandez-Funez et al., 2011; Thackray et al., 2012).
Figure 1. Sequence alignment and 3D structure of the globular domain of PrP from susceptible and resistant mammals.

A, Sequence alignment (ClustalW) for the PrP globular domain from human (Homo), Syrian hamster, mouse, rabbit, horse, and dog. The three main substitutions in rabbit, horse, and dog PrP are indicated with blue circles. The box indicates the clustering of the three substitutions. The color-coded amino acids indicate properties relevant for protein structure (size and charge). B and C, 90˚ views of the globular domain of hamster (Ha) PrP indicating the position of the three critical residues at 159, 167, and 174. D-F, MNR structure of the globular domain of rabbit (Ra, D), horse (Eq, E), and dog (Ca, F) PrP. The relevant amino acid changes in each backbone are indicated. Note the elongation of helix 2 in RaPrP and helix 1 in CaPrP (D and F, arrows), and the decrease in β-sheet content in horse and dog PrP (E and F, arrows). NMR structures were downloaded from RCSB-PDB and visualized in PyMOL.
Here, we show that transgenic flies expressing WT PrP from rabbit, horse, and dog demonstrate no toxicity. We also examined the in vivo consequences of mutating the suspected protective residues in their respective backbones to the corresponding residues in hamster PrP (HaPrP): RaPrP-S174N, EqPrP-S167D, and CaPrP-D159N. Despite previous in vitro evidence (Khan et al., 2010), we found that RaPrP-S174N was not toxic in transgenic flies. In contrast, replacing single amino acids in horse and dog PrP resulted in dramatic locomotor dysfunction and shortened lifespan. Interestingly, EqPrP-S167D and CaPrP-D159N induced different phenotypes in degenerating brain neurons, suggesting the accumulation of different pathogenic conformers that perturb distinct cellular pathways. These studies demonstrate the role of the primary sequence of rabbit, horse, and dog PrP in conferring structural stability, which explains the resistance of these animals to prion diseases under natural conditions. Also, S167 and D159 seem to introduce local and global changes in the PrP globular domain that result in decreased β-sheet content and increased conformational stability. Overall, this knowledge can be leveraged to search for genetic or pharmacologic agents that can stabilize these domains and prevent PrP conversion, hence protecting against prion diseases.
Results
Identification of protective residues in the globular domain of PrP
To identify key residues mediating resistance to PrP misfolding and disease, we aligned the sequence of the globular domains of PrP from susceptible and resistant mammals, including human (Hu), Syrian hamster (Ha), mouse (Mo), rabbit (Ra), horse (Eq), and dog (Ca) PrP. These sequences are highly conserved, with most changes due to conservative substitutions in hydrophobic residues (Fig. 1A). Since we use human PrP (HuPrP) as the reference for the alignment, all PrP sequences are numbered according to HuPrP throughout this paper to avoid confusion. The alignment shows that most of the variability accumulates around the loops (except the α2-α3 loop) and the distal end of helix 3. Interestingly, the β2-α2 loop or “rigid loop” is physically close to in the distal α3 in the 3D structure of the folded protein (Fig. 1B). This 3D domain is proposed to be key for regulating PrP stability and the interaction site with a yet unknown chaperone termed protein-X (Kaneko et al., 1997). Since there are no common substitutions in the three prion-resistant animals, we focused on the residues identified in the literature as key regulators of the stability of rabbit (S174), horse (S167), and dog (D159) PrP (Khan et al., 2010; Lysek et al., 2005; Perez et al., 2010; Vorberg et al., 2003).
For experimental purposes, we used HaPrP structure as control because we have shown that it is more neurotoxic and conformationally unstable than mouse PrP (MoPrP) when expressed in Drosophila (Fernandez-Funez et al., 2010). Still, is is important to note that hamster, mouse, and human PrP share the same residues at positions 159, 167 and 174. The 3D structure of the globular domain of HaPrP has been resolved by crystallography and NMR, with good agreement between the two techniques (Donne et al., 1997; Inouye and Kirschner, 1998; James et al., 1997). The well-known globular domain of HaPrP consists of three α helices and one short β-pleated sheet composed of two four-amino acid β-strands (Fig. 1B). Figure 1 shows the corresponding position of the three protective amino acids in HaPrP (N174, D167, and N159) in two angles (Fig. 1B and C).
NMR structures are also available for the globular domains of rabbit, horse, and dog PrP (Lysek et al., 2005; Perez et al., 2010; Wen et al., 2010a). All thre share the basic architecture of HaPrP with subtle differences, including a longer helix 2 in RaPrP (Fig. 1D, blue arrow), shorter β-sheets in horse and dog PrP (Fig. 1E and F, magenta arrows), and a longer helix 1 in CaPrP (Fig. 1F, blue arrow). More details about the structural features of rabbit, horse, and dog PrP are described below.
S174 stabilizes helix 2 in rabbit PrP
Previous research identified S174 in helix 2 of RaPrP as a critical regulator of PrP stability in crystallographic and NMR studies (Khan et al., 2010; Wen et al., 2010b). Most animals carry Asn at 174, which has weak interactions with N171 located in the loop just outside of helix 2 (Fig. 2A). In contrast, the smaller size and charge of S174 in RaPrP aligns closer to N171, enabling the formation of two hydrogen bonds, one with the side chain of S174 and one with its backbone. This strong interaction creates a helix-capping domain that stabilizes helix 2 and elongates helix 2 to include N171 (Fig. 2C) (Khan et al., 2010; Wen et al., 2010a). Additionally, S174 seems to also alter the position of the β2-α2 loop. In HaPrP, the β2-α2 loop is relatively distant from helix 3 as indicated by the position of D167 (Fig. 2B). In contrast, in RaPrP the β2-α2 loop is closer to helix 3, as indicated by the downward orientation of D167 (Fig. 2D). Additionally, valine (Val, V) at 166 is deeply buried in RaPrP, potentially contributing to the stability of the hydrophobic core (Fig. 2D). In vitro experiments with recombinant RaPrP carrying the S174N substitution showed decreased resistance to denaturing agents and increased propensity to populate the β-state, supporting the role of S174 in the structural stability of RaPrP (Khan et al., 2010). To visualize the structural changes caused by S174N, the Chakrabartty group generated the NMR structure (Khan et al., 2010). Although the NMR structure shows that misalignment of N174 and N171 preventing the formation of the helix capping domain, S174N still preserves the longer helix 2 and the tight contact between the β2-α2 loop and helix 3 (Fig. 2E and F). The 310 mini-helix within the rigid loop now lies parallel to helix 3, indicating a close interaction between these two domains. Since there is currently no evidence for the protective role of S174 in vivo, we introduced the RaPrP-S174N substitution in flies to determine the ability of a single amino acid substitution to change the biological properties of RaPrP.
Figure 2. S174 stabilizes helix 2 in rabbit PrP.

A and B, Detail of the NMR structure of HaPrP showing the position of N171 and N174 at the start of helix 2. N171 is right outside of helix 2. The β2-α2 loop is has an open conformation as indicated by the position of V166 and D167. C and D, Detail of the NMR structure of RaPrP-S174 showing the helix-capping domain formed by the aligned SI74 and N171. N171 is now included in the extended helix 2. The β2-α2 loop is now closer to helix 3 than in HaPrP as indicated by the position of V166 and D167. E and F, Detail of the NMR structure of RaPrP-S174N lacking the helix-capping domain. N171 is still included in the expanded helix 2 and the β2-α2 loop maintains its proximity to helix 3.
S167 stabilizes the β2-α2 loop in horse PrP
EqPrP carries two consecutive changes within the β2-α2 loop, S167 and glutamate (Glu, E) at 168. This sequence reverses the order of acidic - polar residues found in rodents, rabbits, and dogs (DQ167,168) (Fig. 1A). Interestingly, HuPrP contains two acidic residues at this position (DE167,168), suggesting that the relevant protective substitution for EqPrP is S167 since E168 is present in the highly susceptible HuPrP (Perez et al., 2010). The NMR of EqPrP shows less β-sheet content (Fig. 3D-F) together with a more compact and organized β2-α2 loop than in HaPrP (Fig. 3A-C). In EqPrP, S167 is closer to helix 3 and the orientation of S167 favors a deeper burying of V166, creating a more compact hydrophobic region (Fig. 3D-F). These structural features are similar to those described for RaPrP, although in this case the substitution is actually in the loop. Interestingly, the side chains of several conserved and horse-specific amino acids in helix 3 are oriented outwardly (Y218-Y222-Q226 on one side and E221-P225 on the other side), creating the space to cradle the loop closer to helix 3 (Fig. 3D and E). In HaPrP, Y225 is oriented towards the loop, occupying the space left by the more distant loop or, alternatively, pushing the loop away from helix 3 (Fig. 3A and B). Although EqPrP carries several substitutions in helix 3 that interact closely with the loop, S167 has been proposed to be the key substitution responsible for the stability of EqPrP (Perez et al., 2010). So far, the role of S167 in the stability of EqPrP has not been tested in vivo. To determine the biological relevance of S167, we replaced this domain with the HaPrP sequence (SE167,168DQ) and introduced the mutant EqPrP in transgenic flies.
Figure 3. S167 stabilizes the loop in horse PrP.

A-C, Detail of the β2-α2 loop in HaPrP showing the position of D167. The loop is far from helix 3 as indicated by D167 (A, arrow). A simplified image of the β2-α2 loop (C) shows the open conformation of V166 and D167 (arrows). D-E, Detail of the loop in EqPrP showing the position of S167 (D, arrow). The β2-α2 loop is closer to helix 3, which flanks the loop with several charged residues (Y-Y-Q) oriented outward and a Phe underneath. The simplified model (F) shows the downward position of S167 and the buried V166 (arrows). Notice also the shorter β-pleated sheet in EqPrP (D-F).
D159 alters surface charge in dog PrP
The CaPrP sequence contains a prominent acidic substitution at 159 (D/E159) only present in the canid family (Fig. 1A), two mustelids, and one bat (Lysek et al., 2005; Stewart et al., 2012). We showed recently that introducing N159D in the MoPrP backbone decreased both the accumulation of pathogenic conformations and neurotoxicity, indicating the protective activity of N159 (Sanchez-Garcia et al., 2016). As with rabbit and horse PrP, the globular domain of CaPrP maintains the same general 3D structure as that of HaPrP, although the NMR structure indicates a longer helix 1 and the complete loss of β-sheet content (Fig. 4A and B). This model is consistent with the high resistance of CaPrP to denaturing agents and the lowest propensity to populate the β-state (Khan et al., 2010; Lysek et al., 2005). Locally, the side chain of D159 is oriented outwards, creating a new negatively charged domain on the protein surface (Lysek et al., 2005). It is currently unclear how the charge / orientation of the side chain at 159 favors the expansion of helix 1 and destabilizes the short β-sheet, suggesting dramatic local and long-range effects (Fig. 4A and B). CaPrP carries a nearby substitution at Y155 that is conserved in mouse, rabbit, horse, and dog PrP, but not in hamster and human PrP (N155). Y155 is unlikely to contribute to the expansion of helix 1 since helix 1 is shorter in mouse and horse PrP; thus, Y155 is not sufficient for the elongation of helix 1 in CaPrP. To determine the effect of Y155 and D159 on PrP structure and toxicity, we introduced only CaPrP-D159N (similar to MoPrP) and the double substitution CaPrP-YD155,159NN (similar to HaPrP) in transgenic flies.
Figure 4. D159 stabilizes the loop in dog PrP.
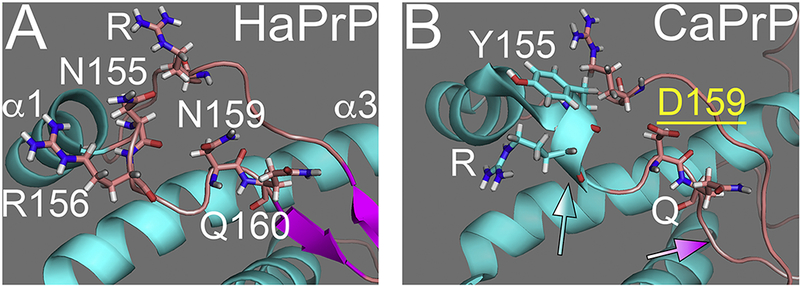
A, Detail of the αl-β2 loop in HaPrP showing the position of N155 (outside of helix 1) and N159. B, Detail of the loop in CaPrP NMR structure showing the position of Y155 (part of extended helix 1, blue arrow) and D159. NMR data suggest the elimination of the β-sheet despite perfect sequence conservation (magenta arrow).
Rabbit, horse, and dog PrP undergo normal biogenesis in flies
We generated transgenic flies carrying the WT alleles for rabbit, horse, and dog PrP as well as the specific substitutions replacing the putative protective residues: S174N, SE167,168DQ, and D159N, along with YD155,159NN in their respective backbones. We next selected lines robustly expressing PrP and matched the levels of WT and mutant alleles for each species to directly compare the consequences of the mutations in PrP biogenesis, subcellular distribution, locomotor behavior, and neurotoxicity. We first examined the biogenesis and posttranslational modifications of WT and mutant alleles by electrophoretic mobility to visualize potential effects of the substitutions. PrP has two facultative N-glycosylation sites, rendering up to three isoforms: un-, mono-, and diglycosylated. The relative abundance of each glycoform depends on the accessibility (i.e., local conformation) of the glycosylation sites during biogenesis and maturation in the ER and Golgi. To examine PrP biogenesis, we prepared homogenates from flies expressing each of the constructs, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and incubated the membranes with the 6H4 antibody that recognizes rabbit, horse, and dog PrP. WT rabbit, horse, and dog PrP each accumulate in three isoforms, but the relative abundance of glycoforms are different in each case (Fig. 5A-C), suggesting subtle structural differences. This is a sharp contrast to the lack of unglycosylated isoform in HaPrP expressed in Drosophila (Fernandez-Funez et al., 2009; Fernandez-Funez et al., 2010). Rabbit and horse PrP show similar glycosylation profiles, with high levels of mono- and diglycosylated PrP and lower levels of the unglycosylated isoform. In contrast, dog PrP accumulates higher levels of the unglycosylated isoform and lower levels of the diglycosylated isoform. RaPrP-S174N, EqPrP-SE167,168DQ, CaPrP-D159N, and CaPrP-YD155,159NN exhibit the same glycopatterns as their respective WT sequences, indicating that the substitutions have no overt effect on PrP biogenesis (Fig. 5-C).
Figure 5. PrP biogenesis and glycoforms.
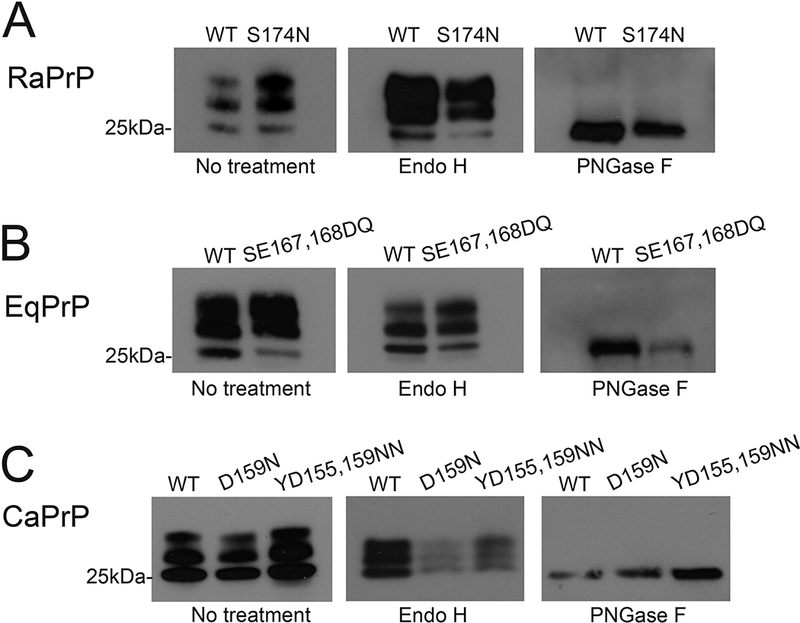
A, RaPrP-WT and -S174N accumulate in three glycoforms, with lower levels for the unglycosylated isoform Treatments with Endo H and PNGase indicate that both WT and mutant RaPrP are completely processed and carry mature glycosylations. B, EqPrP-WT and -SE167DQ accumulate in three glycoforms, with lower levels for the unglycosylated isoform Treatments with Endo H and PNGase indicate that both EqPrP carry mature glycosylations. C, CaPrP-WT, and -D159N, and YD155,159NN accumulate in three glycoforms, with lower levels for the diglycosylated isoform Treatments with Endo H and PNGase indicate that all CaPrP mature glycosylations.
Next, to verify that all these WT and mutant PrP are processed correctly during maturation in the ER and Golgi, we performed deglycosylation assays. Treatment with Endo H eliminates only immature glycosylation: significant digestion of glycosylated PrP by Endo H indicates immature glycoforms stuck in the ER/Golgi system due to aberrant folding or misfolding. We found that Endo H has no effect on the electrophoretic mobility of any of the WT or mutant PrP tested, indicating that all PrP are completely processed in the ER/Golgi system (Fig. 5A-C). Next, digestion with PNGase F eliminates all glycosylations (mature and immature) and results in a single 27kDa band corresponding to unglycosylated PrP (Fig. 5A). In our experiments, PNGase F digestion produces a single band consistent with the expected size of unglycosylated PrP for each WT and mutant PrP tested, supporting the normal biogenesis of all the PrP constructs (Fig. 5A-C). Overall, WT and mutant rabbit, horse, and dog PrP are processed normally in Drosophila with slight differences in the glycosylation pattern, indicating subtle conformational differences. However, the mutants do not introduce significant alteration in PrP biogenesis and structure.
Rabbit, horse, and dog PrP exhibit distinct subcellular distribution
To further understand the differences between the WT alleles of rabbit, horse, and dog PrP and the substitutions of putative protective residues, we analyzed the subcellular distribution of each construct in Drosophila brain neurons. We have shown before that both hamster and mouse PrP accumulate in the membrane, but small amounts of PrP can be detected in the Golgi, likely due to early misfolding (Fernandez-Funez et al., 2010). We also described previously the unusual diffuse distribution of RaPrP-WT throughout the ER and membrane (Fig. 6A) (Fernandez-Funez et al., 2010; Fernandez-Funez et al., 2011). To examine the cellular distribution of the new PrP constructs, we co-expressed them with CD8-GFP, a fusion that labels plasma membrane as well as ER during its production, in brain interneurons under the control of the OK107-Gal4 driver. Then, we collected the images by optical sectioning of whole mount larval brains (see details in Methods). RaPrP-S174N shows the same subcellular distribution as RaPrP-WT (Fig. 6B), suggesting that the substitution does not induce dramatic changes on the cellular behavior of RaPrP. EqPrP-WT accumulates in a few intracellular puncta consistent with partial retention in the Golgi (Fig. 6C), similar to the distribution previously described for hamster and mouse PrP in flies (Fernandez-Funez et al., 2010). EqPrP-SE167,168DQ displays a similar distribution in large puncta (Fig. 6D), indicating that the mutations introduce no significant changes. CaPrP-WT exhibits a distinct distribution in small puncta in Drosophila brain interneurons (Fig. 6E). Both CaPrP mutants accumulate in both small and large puncta, suggesting that D159N may affect CaPrP folding and / or stability (Fig. 6F and G). Panel H shows a control neuron not expressing PrP (Fig. 6H). Overall, these results indicate that WT rabbit, horse, and dog PrP display distinct subcellular distribution, and that the D159N substitution alters the cellular distribution of CaPrP.
Figure 6. Rabbit, horse and dog PrP exhibit distinct subcellular distribution.
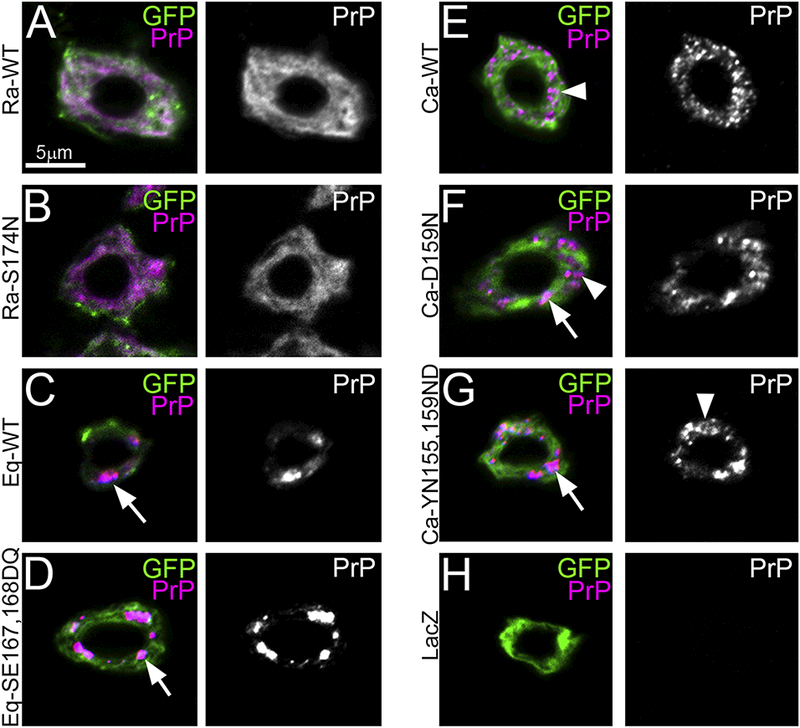
A-H, Cellular distribution of PrP (magenta) and CD8-GFP (green) in brain neurons under the control of OK107-Gal4. A and B, RaPrP-WT and -S174N show diffuse distribution throughout the ER and the plasma membrane. C and D, EqPrP-WT and -SE167,168DQ accumulate in large puncta in the Golgi apparatus. E, CaPrP-WT accumulates in small puncta throughout the ER and Golgi. F and Q, CaPrP-D159N and -YD 155,159NN accumulate in small and large puncta in the secretory pathway. H, Control neuron expressing LacZ and negatively stained for PrP. All panels were imaged at the same magnification (630x), as indicated by scale bar in A.
Mutant horse and dog PrP induce locomotor dysfunction
To determine the functional consequences of expressing WT and mutant PrP targeting the putative protective residues, we expressed all the constructs in motor neurons and analyzed the locomotor activity of the flies over time. We showed previously that expression of RaPrP-WT had no effect on the locomotor output of flies (Fernandez-Funez et al., 2010). Our current results show normal age-dependent locomotor dysfunction in flies expressing RaPrP-WT, which exhibit 50% climbing index by day 27 and 0% climbing by day 37, similar to control flies expressing LacZ (Fig. 7A, p = 0.017). Expression of RaPrP-S174N shows similar locomotor activity as the flies expressing the LacZ reporter (p = 0115) or the WT allele (p = 0.026), with 50% climbing at day 26 and 0% climbing by day 41 (Fig. 7A). Thus, S174N does not increase neurotoxicity of RaPrP-WT.
Figure 7. Mutant horse and dog PrP induce locomotor dysfunction and shorter survival.
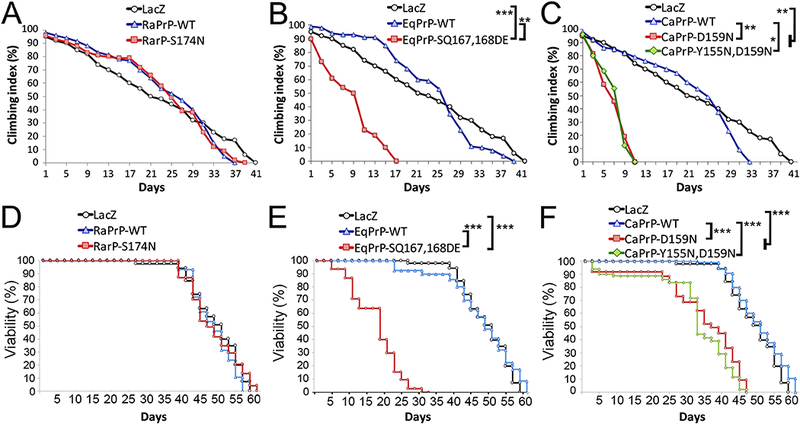
A-C, Locomotor performance. A, Flies expressing RaPrP-S174N pan-neurally show the same climbing performance (50% climbing at 26 days) than flies expressing RaPrP-WT (50% climbing at 27 days) and the reporter LacZ (50% climbing at 28 days). B, Flies expressing EqPrP-WT (50% climbing at 28 days) exhibit a similar locomotor profile than control flies expressing LacZ. However, flies expressing EqPrP-SQ167,168DE show a prominent decrease in climbing performance (50% climbing at 9 days). C, Flies expressing CaPrP-WT (50% climbing at 28 days) exhibit a similar locomotor profile than control flies expressing LacZ. In contrast, flies expressing CaPrP-D159N or -YD155,159NN show a prominent decrease in climbing performance (50% climbing at 7.5 days). The LacZ controls are the same in A-C because all climbing experiments were performed at the same time. D-F, Longevity. D, Flies expressing RaPrP-S174N pan-neurally show similar survival curves than flies expressing RaPrP-WT and the reporter LacZ. E, Flies expressing EqPrP-WT exhibit a similar survival profile than control flies expressing LacZ. However, flies expressing EqPrP-SQ167,168DE show a prominent reduction in viability. F, Flies expressing CaPrP-WT exhibit a similar survival profile than control flies expressing LacZ. In contrast, flies expressing CaPrP-D159N or -YD155,159NN show a prominent decrease in longevity. Notes: The LacZ controls are the same in A-C and D-F because all climbing and longevity experiments were performed at the same time. After Bonferroni post hoc correction, adjusted α = 0.00625. Not significant differences (p > 0.00625) are not indicated for clarity of the panels; *p < 0.00625; ** p < 0.001; *** p < 0.0001.
Next, expression of the EqPrP constructs shows that the WT allele induces no locomotor dysfunction compared with LacZ (p = 0.018), with flies reaching 50% climbing index by day 28 and 0% climbing by day 39 (Fig. 7B). In contrast, expression of EqPrP-SE167,168DQ results in a dramatic reduction in locomotor activity compared to LacZ (p = 4×10−5) and to the WT allele (p = 0.0002), with flies reaching 50% climbing by day 9 and 0% climbing by day 17 (Fig. 7B). Expression of CaPrP-WT shows a similar locomotor profile as the other WT alleles and LacZ (p = 0.97), with flies reaching 50% climbing index by day 28 and 0% climbing by day 33 (Fig. 7C). Expression of either CaPrP-D159N or - YD155,159NN induce robust locomotor dysfunction compared to LacZ (D159N p = 0.00015; YD155,159NN, p = 0.00024) and to the WT allele (D159N p = 0.0009; YD155,159NN, p = 0.0012), reaching 50% climbing index by day 7.5 and 0% climbing by day 11 (Fig. 7C). Since the single and double CaPrP mutants exert similar effects on locomotor dysfunction, D159N seems to be solely responsible for suppressing CaPrP neurotoxicity. Overall, these results support the idea that rabbit, horse, and dog PrP-WT are intrinsically non-toxic, indicating that specific amino acid substitutions in their sequences promote structural stability. Our results support the protective role of S167 and D159 in horse and dog PrP, respectively. Surprisingly, these experiments do not support the protective role of S174 in RaPrP despite the obvious changes in structural stability demonstrated in vitro (Khan et al., 2010).
Mutant horse and dog PrP shorten lifespan
We next analyzed the longevity of flies expressing the PrP constructs ubiquitously at low levels under the control of da-Gal4. Combined analysis of the eight genotypes shows strong statistical differences (log-rank p < 0.001; Wilcoxson p < 0.001). The longevity of flies expressing RaPrP-WT is comparable to that of flies expressing the LacZ reporter (log-rank p = 0.066; Wilcoxson p = 0.32), with 50% survival at day 50 (Fig. 7D). Expression of RaPrP-S174N has no effect on the longevity of flies compared to LacZ (log-rank p = 0.82; Wilcoxson p = 0.5) or RaPrP-WT (log-rank p = 0.127; Wilcoxson p = 0.95), with flies reaching 50% viability by 50 days consistent with the normal locomotor function (Fig. 7D). The 50% viability of flies expressing EqPrP-WT is 48 days, similar to that of control flies expressing LacZ (log-rank p < 0.307; Wilcoxson p < 0.88) (Fig. 7E). However, expression of EqPrP-SE167,168DQ dramatically shortens the longevity of the flies compared to LacZ and EqPrP-WT (log-rank p < 0.001; Wilcoxson p < 0.001), reaching 50% viability by day 18. Expression of CaPrP-WT has no effect on the longevity of the flies compared to Laz (log-rank p < 0.022; Wilcoxson p < 0.144), with flies reaching 50% survival by day 50 (Fig. 7F). But flies expressing CaPrP-D159N or -YD155,159NN exhibit reduced longevity compared to LacZ and CaPrP-WT (log-rank p < 0.001; Wilcoxson p < 0.001), with 50% survival at 32 and 36 days, respectively. In summary, the WT alleles for rabbit, horse, and dog PrP have no effect on longevity, but the S167D and D159N residues dramatically shorten viability in horse and dog PrP, respectively. Consistent with the lack of effect in the locomotor task, the S174N substitution in RaPrP has no effect on viability.
Mutant dog and horse PrP induce degeneration of axonal lobes in mushroom body neurons
We next directed the expression of the PrP constructs to specific brain neurons to examine in more detail the consequences of expressing WT and mutant alleles for rabbit, horse, and dog PrP. For these experiments, we studied the mushroom bodies, the high order brain centers implicated in olfactory learning and memory (Davis, 2005; Davis, 2011). The mushroom body neurons are composed of large bilateral clusters (~2,500 Kenyon cells) in the posterior brain that project their dendrites in compact structures in the underlying neuropil (calyces). Their axons project towards the anterior brain, where they split into dorsal (α) and medial (β and γ) lobes (Fig. 8A) (Davis, 2005; Tanaka et al., 2008). We first examined the axonal projections, which we have shown previously undergo dramatic progressive degeneration following expression of HaPrP, but not RaPrP (Fernandez-Funez et al., 2010). Here, we use expression of LacZ as control and monitor the architecture of the axonal lobes by co-expressing CD8-GFP under the control of OK107-Gal4
Figure 8. Mutant horse and dog PrP induce degeneration of mushroom body axonal terminals.

A, 3D visualization of a complete Drosophila mushroom body. The cell bodies (Kenyon cells, Kc, dark blue) are located in the posterior head. The dendrites project in the neuropil underneath the Kc (calyx, ca, cyan). The axons project to the anterior brain and branch into dorsal (α, orange) and medial (β, yellow and γ, orange) terminals. B-L, Maximum intensity projections of the axonal terminals of the mushroom bodies in flies expressing CD8-GFP (OK107-Gal4/UAS-CD8-GFP) with or without PrP. B, Control flies expressing LacZ aged for 40 days exhibit normal architecture of the mushroom body axonal projections. C, Flies expressing RaPrP-WT aged for 40 days show normal axonal terminals. D and E, Flies expressing RaPrP-S174N show normal axonal terminals at days 1 and 40. F, Flies expressing EqPrP-WT aged for 40 days show normal axonal terminals. G and H, Flies expressing EqPrP-SE167,168DQ show normal axonal terminals at day 1 (G), but flies aged for 40 days show shorter dorsal and medial projections (H, arrows). I, Flies expressing CaPrP-WT aged for 40 days show normal axonal terminals. J and K, Flies expressing CaPrP-D159N show normal axonal terminals at day 1 (J), but flies aged for 40 days show thinner dorsal and medial projections (K, arrows) and membrane blebbing (arrowhead). L, Flies expressing CaPrP-YD155,159NN aged for 40 days show thinner projections (arrows) and membrane blebbing (arrowhead). M, Quantification of the surface of the mushroom body axonal projections at days 1 (black bars) and 40 (grey bars). N ≥ 10. Nates: Samples were tested for within genotype differences (time-dependent) (black brackets) and inter-genotype differences (same time) (red brackets). Not significant differences (p > 0.00625) are not indicated for clarity of the panels; *** p < 0.0001.
Flies expressing LacZ show robust axonal α, β, and γ lobes that expand slightly in 40 day-old flies (Fig. 8B and M). Notice the round cap at the end of the dorsal lobes and the broad medial lobes (Fig. 8B). Flies expressing RaPrP-WT and -S174N exhibit slightly smaller axonal lobes than those of flies expressing LacZ, but they preserve the terminal structures (α lobe cap) and expand by day 40 (WT p = 0.075; S174N p = 0.015), supporting the lack of toxicity of both proteins (Fig. 8C-E and M). The size of the lobes is comparable between WT and S174N at days 1 (p = 0.56) and 40 (p = 0.24) (Fig. 8M), which supports the lack of toxicity of this substitution.
Flies expressing EqPrP-WT display normal architecture of mushroom body axonal lobes at days 1 and 40 with a significant increase in surface at day 40 (p = 2.3 10−5) (Fig. 8F and M). Although flies expressing EqPrP-SE167,168DQ have normal axonal lobes at day 1, by day 40 the dorsal and medial lobes show significant loss of volume as evidenced by the overall thinning of axonal lobes and the loss of the α lobe cap (p = 6.4 10−8) (Fig. 8G, H and M). Flies expressing WT and SE167,168DQ show similar mushroom body lobe surface at day 1 (p = 0.16), but they are significantly smaller in the flies expressing SE167,168DQ at day 40 (p = 2.4 10−15) (Fig. 8M), supporting the toxicity of this substitution.
Finally, flies expressing CaPrP-WT exhibit large axonal lobes at days 1 and 40, with a slight (not significant) increase in surface in the older flies (p = 0.24) (Fig. 8I and M). Flies expressing CaPrP-D159N and -YD155,159NN show large axonal lobes at day 1, but display progressive loss of volume over 40 days, resulting in thin lobes with abundant blebbing, a sign of nerve degeneration (D159N p = 1.02 10−7; YD155,159NN p = 5.1 10−10) (Fig. 8J-M). Although WT and D159N show comparable mushroom body lobes at day 1 (p = 0.52), flies expressing YD155,159NN start with smaller lobes (p = 5.1 10−9 (Fig. 8M).
When flies expressing CaPrP constructs age, the surface of mushroom body lobes is significantly smaller at day 40 in flies expressing D159N (p = 2.4 10−7) or YD155,159NN (p = 5.1 10−9) (Fig. 8M). Overall, these results support our observations that WT rabbit, horse, and dog PrP, and RaPrP-S174N are not toxic, whereas EqPrP-S167D and CaPrP-D159N are highly toxic in Drosophila brain neurons.
Mutant dog and horse PrP induce distinct degenerative phenotypes in cell bodies
The cell bodies of the mushroom body neurons form tight clusters in the posterior brain with a characteristic triangular shape (Fig. 8A and9A). These clusters expand slightly during aging mostly due to cell reorganization since the adult brain has no neuronal cell division. Expression of LacZ results in large, triangular clusters that expand slightly over 40 days (p = 0.0035) (Fig. 9B and M). Expression of RaPrP-WT produce smaller clusters that fail to expand in older flies (p = 0.42) (Fig. 9C and M), suggesting some alterations in the behavior of these neurons. Expression of RaPrP-S174N has essentially the same failure to expand over time as the WT allele (p = 0.58) (Fig. 9D, E and M), indicating that S174N has no effect on RaPrP. We also find no differences between the WT allele and S174N at days 1 (p = 0.56) and 40 (p = 0.27) (Fig. M), supporting similarities between the two constructs.
Figure 9. Mutant horse and dog PrP induce degeneration of mushroom body cell bodies.

A-L, Single optical planes of Kenyon cell clusters (Kc) and calyces (ca) in flies expressing CD8-GFP (OK107-Gal4/UAS-CD8-GF8) with or without PrP. A and B, Control flies expressing LacZ aged for 40 days show dense and tight Kc clusters at day 1 (A) and expansion of Kc clusters and calyces after aging for 40 days (B). C, Flies expressing RaPrP-WT aged for 40 days show small Kc clusters. D and E, Flies expressing RaPrP-S174N show small Kc clusters at days 1 and 40. F, Flies expressing EqPrP-WT aged for 40 days show normal Kc clusters. G and H, Flies expressing EqPrP-SE167,168DQ show normal Kc clusters at day 1 (G), but flies aged for 40 days show significantly expanded Kc clusters (H, dotted outline) and weak calyces (H, arrow). Insets in panels F and H show details of the Kc, which are significantly larger in day 40 flies expressing EqPrP-SE167,168DQ. I, Flies expressing CaPrP-WT aged for 40 days show normal Kc clusters. J and K, Flies expressing CaPrP-D159N show normal Kc clusters at day 1 (J), but flies aged for 40 days exhibit smaller Kc clusters with disorganized calyces (K, arrow). L, Flies expressing CaPrP-YD155,159NN aged for 40 days show smaller Kc clusters. M, Quantification of the surface of the Kc clusters at days 1 (black bars) and 40 (grey bars). N ≥ 10. Nates: Samples were tested for within genotype differences (time-dependent) (black brackets) and inter-genotype differences (same time) (grey brackets). Not significant differences (p > 0.00625) are not indicated for clarity of the panels; * 0.001 >p< 0.00625; *** p < 0.0001.
Expression of EqPrP-WT has no effect on the size of the Kenyon cell clusters, which expand in older flies (p = 0.001) (Fig. 9F and M). Young flies expressing EqPrP-SE167,168DQ exhibit Kenyon cell clusters of the same size as flies expressing the WT allele or LacZ (Fig. 9G and M). However, older flies expressing EqPrP-SE167,168DQ show a dramatic expansion of the Kenyon cell clusters compared to day 1 (p = 4.5 10−8) and to older flies expressing EqPrP-WT (p = 7.1 10−9) (Fig. 9H and M). This expansion is not due to the increase in the number of cells or to relocation of cells (flattening), but to the swelling of the cell bodies (Fig. 9, F and H, insets), a cellular pathology similar to that described in flies expressing the Aβ42 peptide (Ling et al., 2009). Additionally, the loss of integrity of the calyx supports the neurotoxicity of EqPrp-SE167,168DQ (Fig. 9H, arrow). Finally, expression of CaPrP-WT results in Kenyon cell clusters that fail to expand in older flies (p = 0.48) (Fig. (9I and M). But expression of CaPrP-D159N or CaPrP-YD155,159NN result in significantly smaller Kenyon cell clusters due to massive cell loss compared to day 1 (D159N p = 1.9 10−6; YD155,159NN p = 4.2 10−8) and to older flies expressing CaPrP-WT (D159N p = 1.6 10−5; YD155,159NN p = 6.6 10−7) (Fig. 9K-M). Thus, horse and dog PrP mutants induce distinct Kenyon cell pathologies, suggesting the perturbation of different endogenous pathways.
Discussion
Computational and biophysical techniques had previously identified unique protective residues (S174, S167, and D159) in PrP from three prion-resistant animals: rabbit, horse, and dog (Khan et al., 2010; Lysek et al., 2005; Perez et al., 2010). We demonstrate here that WT PrP from rabbit, horse, and dog are not toxic in transgenic flies, as indicated by locomotor, longevity, and mushroom body architecture assays. Interestingly, each PrP exhibits unique glycosylation patterns and subcellular distribution, indicating subtle differences in biogenesis and / or conformation. Overall, WT rabbit, horse, and dog PrP demonstrate intrinsic conformational stability in Drosophila, supporting the presence of protective residues in their sequence. These results argue against the hypothesis that extrinsic factors (the cellular environment, expression of cofactors that prevent PrP conversion) contribute to or dictate the high conformational stability of PrP in these animals. These results are consistent with our previous experiments in Drosophila expressing RaPrP-WT (Fernandez-Funez et al., 2010; Fernandez-Funez et al., 2011), mice expressing chimeric mouse / rabbit PrP (Vorberg et al., 2003), and recombinant rabbit, horse and dog PrP (Khan et al., 2010). Thus, these three PrP contain critical amino acid substitutions that make them intrinsically more stable structurally, less prone to misfold, and less neurotoxic, explaining the resistance of host animals to prion diseases.
Our work provides the first evidence to support the protective role of D159 and S167 in vivo. Previous work showed that S174N decreased the conformational stability of recombinant RaPrP, although RaPrP-S174N still populated the β-state at pH 4.0 and 4 M urea, conditions similar to those required to misfold RaPrP-WT (Khan et al., 2010). Surprisingly, our Drosophila studies indicate that RaPrP-S174N is not neurotoxic. Interestingly, this result is consistent with the lack of structural changes in the 3D NMR model for RaPrP-S174N (Fig. 2E). The model still shows a longer helix 2 and a tight interaction between the β2-α2 loop and helix 3, two key structural features of RaPrP-WT, despite the lack of the helixcapping domain. Our results suggest that S174 is not the key residue mediating neuroprotection in RaPrP. Other than disrupting the helix-capping domain, the S174N substitution does not appear to introduce additional long-range structural changes in RaPrP. RaPrP contains other unique substitutions in the globular domain that could contribute to its structural stability. It is important to note that rabbits were recently infected with prions following a long process of strain adaptation in vitro (Chianini et al., 2012). Despite showing the possibility of infecting rabbits with a new prion, the efficiency of intracerebral transmission was very low (20%) after second passage, further arguing for the special properties of RaPrP that prevents endemic or naturally transmitted disease.
In vitro studies have demonstrated the structural stability of EqPrP (Khan et al., 2010) and NMR studies had identified the contribution of S167 to its unique structural features (Perez et al., 2010). Here, we show for the first time that EqPrP-S167D is highly toxic in Drosophila. In fact, this mutant is very aggressive, inducing dramatic reductions in locomotor activity and survival. Additionally, EqPrP-S167D induces a unique pattern of neuronal degeneration that we and others have observed in flies expressing the Aβ42 peptide. This swelling of cell bodies together with degeneration of axonal and dendritic terminals was interpreted as a cellular phenotype caused by blocked autophagy and the accumulation of autophagic vesicles in the cell body (Ling et al., 2009). Overall, these neurotoxic phenotypes support the relevance of S167 in the conformational stability of EqPrP. Since the side chain of Asp is larger and more charged than that of Ser, D167 forces the loop to separate from helix 3 in HaPrP. In contrast, S167 forms a tighter 3D domain in EqPrP that buries V166 deeper into the hydrophobic core, which strengthens its structural stability. S167 does not seem to disrupt the conserved structure of the rigid β2-α2 loop, which has been proposed to play a key role in PrP conformational change as suggested by numerous studies on sequence variants found in sheep and cervid PrP (Bett et al., 2012; Kurt et al., 2009; Sigurdson et al., 2010). In addition to these local changes in the loop, the NMR structure of EqPrP indicates a lower β-sheet content, suggesting that the changes in the loop have long-range effects that perturb the alignment of the short β-sheet domain. This loss of β-pleated content further supports and explains the conformational stability of EqPrP.
Similar to EqPrP, in vitro misfolding studies and NMR structure identified a unique substitution - D159 - as the determining change conferring high structural stability to CaPrP (Lysek et al., 2005; Nystrom and Hammarstrom, 2015). Moreover, MDCK kidney dog cells and coyote brain extracts are refractory to prion infection, further supporting the resistance of canids to prion diseases (Kurt et al., 2009; Polymenidou et al., 2008). These observations invite to speculate about the possibility that canids have adapted PrP-intrinsic and -extrinsic mechanisms to prevent the transmission of prions from sheep and goats, their natural prey. Our recent efforts have been directed to understanding the contribution of PrP-intrinsic vs PrP-extrinsic mechanisms that confer this resistance to CaPrP. We recently introduced the N159D substitution in the MoPrP backbone and found that it increases structural stability and reduces neurotoxicity, thus supporting the protective activity of D159 (Sanchez-Garcia et al., 2016). Here, we completed the converse substitution in CaPrP and demonstrate that CaPrP-D159N induces dramatic toxicity in several assays in flies, with phenotypes similar to those described in flies expressing HaPrP. The original NMR study of CaPrP proposed that the outward orientation of D159 mainly impacted the surface charge, potentially disrupting the interactions of CaPrP with co-factors (Lysek et al., 2005). Additionally, the NMR structure of CaPrP indicates the complete loss of β-sheet content despite perfect sequence conservation of this domain (Fig. 1A and4B) (Lysek et al., 2005). It is, thus, likely that the local perturbations introduced by D159 spread regionally to alter the stability of the short β-sheet. The impact of D159 extends to helix 1 as well, which appears to expand for almost a complete extra turn (Y155 and R156) (Fig. 4B). Although Y155 is not present in human and hamster PrP, it is found in mouse, rabbit, horse, and dog PrP, and neither rabbit nor horse PrP exhibit the extended helix 1. Thus, Y155 does not appear to be directly responsible for the elongation of helix 1. However, it is also possible that the combination of Y155 and D159 provides the ideal conditions for the extension of helix 2 only found in CaPrP. Still, our work demonstrates that D159N is sufficient to induce toxicity in CaPrP and N159D is sufficient to decrease MoPrP toxicity, indicating that D159 plays a major role in CaPrP conformational stability. In any event, D159 has a significant impact on the α1-β2 loop consistent with higher structural stability. These long-distance structural changes indicate that D159 introduces global perturbations in several key domains that, together, stabilize CaPrP. Unfortunately, we cannot propose at this time a mechanism that explains these long-range structural changes mediated by D159, indicating the need for further studies.
Overall, here we identify two critical residues promoting the stability of PrP from two animals classically known to be resistant to prion diseases: D159 in CaPrP and S167 in EqPrP. These residues induce local changes that spread to nearby regions that reduce the β-sheet content and promote the structural stability of horse and dog PrP. Remarkably, D159 and S167 induce long distance effects that promote the stability of the 3D domain created by the rigid loop and helix 3. Thus, these two residues different mechanisms selected over evolution to promote the conformational stability of PrPc. Additional functional demonstration of the protective activity of D159, S167, and other proposed protected polymorphisms can be done in future studies taking advantage of the highly toxic human PrP that we recently introduced in Drosophila (Fernandez-Funez et al., 2017). Identifying these protective amino acids can increase the safety of herds for human consumption by creating transgenic animals that carry one or multiple protective residues. Moreover, understanding the structural features that stabilize PrP can lead to promising advances in structure-based drug design (e.g., molecular docking) and the identification of compounds that halt or reverse prion diseases.
Materials and Methods
Sequence alignment
The alignment of the globular domain of human, hamster, mouse, rabbit, horse, and dog prion protein sequences was done using ClustalW2 (www.ebi.ac.uk/Tools/clustalw2). We used HuPrP as reference and amino acid numbering for all species refers to the corresponding amino acid in HuPrP (see Fig. 1A). Amino acid sequences for the six species were obtained from NCBI with the following accession numbers: AAH22532 (human), B34759 (Syrian hamster), AAA39996 (mouse), AAD01554 (rabbit), ACG59277 (horse), and ACO71291 (dog). The color-coded amino acids indicate properties relevant for protein structure (size and charge). To generate 3D views of Syrian hamster, rabbit, horse, and dog PrP, we opened in PyMOL (pymol.org) the published NMR structures for HaPrP (1B10), RaPrP-WT (2FJ3), RaPrP-S174N (2JOH), EqPrP (2KU4), and CaPrP (1XYK) deposited in the RSCB Protein Data Bank (rcsb.org/pdb). We displayed the proteins in Cartoon formats showing only relevant amino acids to optimize their visualization.
Generation of transgenic flies and genetics
Flies expressing RaPrP-WT were described previously (Fernandez-Funez et al., 2010). The constructs carrying RaPrP-S174N, EqPrP-WT, EqPrP-SE167,168DQ, CaPrP-WT, CaPrP-D159N, and CaPrP-YD155,159DN cDNAs were synthesized at GenScript and cloned between EcoRI and NotI sites onto the pUAST Drosophila expression vector (Brand and Perrimon, 1993). The pUAST-based constructs were injected into yw embryos at Rainbow Transgenics following standard procedures (Rubin and Spradling, 1982) to generate multiple independent transgenic lines for each plasmid. The driver strains OK107-Gal4 (mushroom bodies), BG380-Gal4 (motor neurons), and da-Gal4 (ubiquitous), and the reporters UAS-LacZ and UAS-CD8-GFP were obtained from the Bloomington Drosophila Stock Center (http://fly.bio.indiana.edu). Fly stocks were maintained on standard Drosophila medium at 25°C. For experiments, homozygous females for the Gal4 strains were crossed with UAS males to generate progeny expressing PrP in the desired tissue. Crosses were placed at 25°C for two days, transferred to 28°C until the progeny completed development, and adults were aged at 28°C, unless otherwise indicated. All assays were performed using females.
Drosophila homogenates and western blot
One fly per genotype and time point was used for analysis. Single flies were homogenized in 30 μl of RIPA buffer containing Complete protease inhibitors (Roche) using a motorized pestle and centrifuged for 1 min at 1,000 rpm. 25 μl of supernatant was mixed with loading buffer and resolved by SDS-PAGE in 4–12 % Bis-Tris gels (Invitrogen) under reducing conditions and electro-blotted into nitrocellulose membranes. Membranes were blocked in TBS-T containing 5% non-fat milk and probed against the primary antibodies: anti-PrP 6D11 (1: 10,000, Covance), anti-PrP 6H4 (1: 10,000, Prionics), anti-αTubulin (1: 250,000, Sigma). The secondary antibody was anti-Mouse-HRP (1: 2000) (Sigma). Immunoreactive bands were visualized by enhanced chemiluminescence (ECL, Amersham). The protein biochemistry protocols are described in more detail in (Sanchez-Garcia et al., 2013).
PrP processing (glycosylation)
For de-glycosylation assays, 9 μl of cleared homogenates were incubated in the presence/absence of PNGase F or Endo H (NEB) according to the manufacturer’s instructions and analyzed by immunoblotting as indicated above.
Immunofluorescence and microscopy
For subcellular localization, we co-expressed the PrP constructs and CD8-GFP as a membrane marker in intemeurons of the larval ventral ganglion under the control of OK107-Gal4 (UAS-CD8-GFP; OK107-Gal4/UAS-PrP). Whole-mount immunohistochemistry of fixed larval brains was conducted by fixing in 4% formaldehyde, washing with PBT, and blocking with 3% bovine serum albumin before incubating with the primary antibody as described previously (Fernandez-Funez et al., 2010). We incubated first with the 6H4 anti-PrP antibody (1: 1,000) followed by the secondary antibody anti-mouse-Cy3 (Molecular Probes) at 1:600. We mounted the stained larval brain in Vectashield antifade (Vector) mounting medium for microscopic observation and documentation. Images of single neurons were taken at and 630x (see details below).
We collected fluorescent images by optical sectioning using AxioVision (Zeiss) software in an Axio-Observer Z1 microscope (Zeiss) equipped with ApoTome (structured light microscopy) with 40x NA: 1.3 (air) and 63x NA: 1.4 oil objectives. Images were combined using Adobe Photoshop; processing included trimming of non-informative edges and brightness / contrast adjustment to whole images.
Mushroom body degeneration
We crossed OK107-Gal4; CD8-GFP flies with LacZ alone (negative control) or with rabbit, horse, and dog PrP constructs (UAS-CD8-GFP; OK107-Gal4/UAS-PrP) at 27˚C. Adult flies were collected at day 1 post eclosion followed by aging for 40 days. We then, imaged cell bodies (Kenyon cells) and axonal lobes labeled with CD8-GFP at days 1 and 40 by dissecting, fixing, and mounting the brains as described above. To quantify the surface of mushroom body axonal lobes, we imaged them as z-stacks with the 40x objective, flattened the Z-stacks (maximum intensity projections), and manually selected the maximum area from at least 10 images. To quantify the surface of the Kenyon cell clusters, we collected Z-stacks with the 40x objective for 10 or more mushroom bodies. Then, the surface of the Kenyon cell clusters in a single optical section was traced manually. The average surface for each condition and time point were entered in Excel to create the graphs (Fernandez-Funez et al., 2010). Finally, data series were analyzed for statistical significance in GraphPad Prism using a two-way ANOVA with Bonferroni post hoc pair-wise analysis of significance. Since we used 8 groups (samples), the adjusted level of significance (α) was 0.00625. P-values below 0.00625 were considered significant.
Locomotor and longevity assays
Flies carrying LacZ (control) or PrP transgenes were crossed with BG380-Gal4 at 25°C and the progeny was tested for their ability to move vertically in an empty vial (climbing assay) at 25°C (Le Bourg and Lints, 1992). Briefly, 25 newborn adult females were placed in empty vials in duplicate and forced to the bottom by firmly tapping against the surface. After 10 sec, the number of flies that climb above 5 cm was recorded. This was repeated 10 times to obtain the average climbing index each day. At the end of the assay, the climbing index (flies above line/total flies x 100) was plotted as a function of age in Excel. Finally, data series for locomotor activity were analyzed for statistical significance in Excel using a 2-way ANOVA with Replication. Since ANOVA showed strong significance for genotype and age, we calculated pair-wise t-test significance with Bonferroni multiple comparisons post-hoc test to minimize type I error. To avoid inflating differences between individual climbing sets, we first averaged the replicates and then averaged the climbing index for two consecutive days. Then, we performed pairwise t-test comparing each genotype to the control flies (LacZ) and each PrP mutant to its corresponding WT allele. Since the total samples was 8, the adjusted level of significance (α) was 0.00625. P-values below 0.00625 were considered significant.
For the longevity assay, flies carrying PrP transgenes or LacZ were crossed with the da-Gal4 driver. Fifty females of each genotype were collected and kept in groups of ten at 28˚ C. Flies were counted every day and transferred to a new tube until all the flies died. Then, longevity was calculated as % of flies alive at each time point and plotted in JMP Pro as a Kaplan-Maier survival plot. Data series for longevity were analyzed for statistical significance in JMP Pro by log-rank and Wilcoxon tests. Both tests indicated strong differences among the eight genotypes analyzed globally (p < 0.001). Post-hoc Bonferroni adjustments of pair-wise analyses were also conducted to examine differences between each WT allele and their corresponding mutations. P-values were considered significant if < 0.00625 as indicated above.
Highlights.
Rabbits, horses and dogs are rare mammals resistantto prion diseases
S174, S167, and D159 are unique residues in the globular domain of the prion protein (PrP) in rabbits, horses and dogs (respectively) compared with the corresponding conserved sequences in humans, rodents and other animals. S174, S167, and D159 introduce significant changes to the 3D conformation of the globular domain that support their protective activity
Expressing WT PrP from rabbits, horses and dogs in Drosophila brain neurons has no effect: these WT PrP alleles show an intrinsic resistance to accumulating in toxic conformations
Introducing the S174N, S167N, and D159 substitutions in the sequence of rabbit, horse and dog PrP, respectively, should make these mutant proteins toxic when expressed in a living organism
Expression of horse PrP-S167N and dog PrP-D159 in Drosophila causes robust neurodegenerative changes, including lower locomotor activity and longevity, and progressive degeneration of brain neurons. In contrast, rabbit PrP-S174N shows the same properties as the wild-type allele.
In conclusion, S167 and D159 are key protective residues in horse and dog PrP that provide conformational stability and prevent the conversion into pathogenic conformations
Acknowledgements
We thank the Bloomington Drosophila Stock Center (NIH P40OD018537) for transgenic flies, the RSCB Protein Data Bank, ClustalW2, and PyMOL for free data and software, and Richard Melvin (Univ. Minnesota Medical School, Duluth) for his assistance with statistical analysis. This work was supported by the NIH grants DP2 OD002721–01 and 7R21NS096627–02 to PF-F. JS-G was supported by a postdoctoral fellowship from the Department of Education of the Basque Country (Spain).
Abbreviations
- α3
helix 3
- Ca
Canis, dog PrP
- EqPrP
Equus, horse PrP
- PrP
Prion protein
- RaPrP
rabbit PrP
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- WT
wild type
Footnotes
Competing Interests
The authors declare that no competing interests exist.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguzzi A, et al. , 2008. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol. 3, 11–40. [DOI] [PubMed] [Google Scholar]
- Barlow RM, Rennie JC, 1976. The fate of ME7 scrapie infection in rats, guinea-pigs, and rabbits. Res. Vet. Sci 21, 110–111. [PubMed] [Google Scholar]
- Bett C, et al. , 2012. Structure of the beta2-alpha2 loop and interspecies prion transmission. FASEB J. 26, 2868–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N, 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 118, 401–15. [DOI] [PubMed] [Google Scholar]
- Chandler RL, 1971. Experimental transmission of scrapie to voles and Chinese hamsters. Lancet. 1, 232–3. [DOI] [PubMed] [Google Scholar]
- Chandler RL, Fisher J, 1963. Experimental Transmission of Scrapie to Rats. Lancet. 2, 1165. [PubMed] [Google Scholar]
- Chianini F, et al. , 2012. Rabbits are not resistant to prion infection. Proc Natl Acad Sci U S A. 109, 5080–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB, 2011. Prions. Cold Spring Harb Perspect Biol. 3, a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courageot MP, et al. , 2008. A cell line infectible by prion strains from different species. J Gen Virol. 89, 341–7. [DOI] [PubMed] [Google Scholar]
- Davis RL, 2005. Olfactory memory formation in Drosophila: from molecular to systems neuroscience. Annu Rev Neurosci. 28, 275–302. [DOI] [PubMed] [Google Scholar]
- Davis RL, 2011. Traces of Drosophila memory. Neuron. 70, 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donne DG, et al. , 1997. Structure of the recombinant full-length hamster prion protein PrP(29–231): the N terminus is highly flexible. Proc Natl Acad Sci U S A. 94, 13452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, et al. , 2009. In vivo generation of neurotoxic prion protein: role for hsp70 in accumulation of misfolded isoforms. PLoS Genet. 5, e1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, et al. , 2017. Drosophila models of prionopathies: insight into prion protein function, transmission, and neurotoxicity. Curr Opin Genet Dev. 44, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, et al. , 2010. Sequence-dependent prion protein misfolding and neurotoxicity. J Biol Chem. 285, 36897–36908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, et al. , 2011. Pulling rabbits to reveal the secrets of the prion protein. Commun Integr Biol. 4, 262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs CJ Jr., Gajdusek DC, 1973. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science. 182, 67–68. [DOI] [PubMed] [Google Scholar]
- Inouye H, Kirschner DA, 1998. Polypeptide chain folding in the hydrophobic core of hamster scrapie prion: analysis by X-ray diffraction. J Struct Biol. 122, 247–55. [DOI] [PubMed] [Google Scholar]
- Ironside JW, et al. , 2017. Prion diseases. Handb Clin Neurol. 145, 393–403. [DOI] [PubMed] [Google Scholar]
- James TL, et al. , 1997. Solution structure of a 142-residue recombinant prion protein corresponding to the infectious fragment of the scrapie isoform. Proc Natl Acad Sci U S A. 94, 10086–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K, et al. , 1997. Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A. 94, 10069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MQ, et al. , 2010. Prion disease susceptibility is affected by beta-structure folding propensity and local side-chain interactions in PrP. Proc Natl Acad Sci U S A. 107, 19808–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood JK, Cunningham AA, 1994. Epidemiological observations on spongiform encephalopathies in captive wild animals in the British Isles. Vet Rec. 135, 296–303. [DOI] [PubMed] [Google Scholar]
- Knight R, 2017. Infectious and Sporadic Prion Diseases. Prog Mol Biol Transl Sci. 150, 293–318. [DOI] [PubMed] [Google Scholar]
- Kurt TD, et al. , 2009. Trans-species amplification of PrP(CWD) and correlation with rigid loop 170N. Virology. 387, 235–43. [DOI] [PubMed] [Google Scholar]
- Le Bourg E, Lints FA, 1992. Hypergravity and aging in Drosophila melanogaster. 4. Climbing activity. Gerontology. 38, 59–64. [DOI] [PubMed] [Google Scholar]
- Ling D, et al. , 2009. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS ONE. 4, e4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd S, et al. , 2011. Genetics of prion disease. Top Curr Chem. 305, 1–22. [DOI] [PubMed] [Google Scholar]
- Lysek DA, et al. , 2005. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc Natl Acad Sci U S A. 102, 640–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiason CK, 2017. Scrapie, CWD, and Transmissible Mink Encephalopathy. Prog Mol Biol Transl Sci. 150, 267–292. [DOI] [PubMed] [Google Scholar]
- Nystrom S, Hammarstrom P, 2015. Generic amyloidogenicity of mammalian prion proteins from species susceptible and resistant to prions. Sci Rep. 5, 10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez DR, et al. , 2010. Horse prion protein NMR structure and comparisons with related variants of the mouse prion protein. J Mol Biol. 400, 121–8. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, et al. , 2008. Canine MDCK cell lines are refractory to infection with human and mouse prions. Vaccine. 26, 2601–14. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, 1998. Prions. Proc Natl Acad Sci U S A. 95, 13363–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC, 1982. Genetic transformation of Drosophila with transposable element vectors. Science. 218, 348–53. [DOI] [PubMed] [Google Scholar]
- Sanchez-Garcia J, et al. , Protein Misfolding in Drosophila Models of Prionopathies. Protein purification and analysis I: methods and applications. iConcept Press, 2013. [Google Scholar]
- Sanchez-Garcia J, et al. , 2016. A single amino acid (Asp159) from the dog prion protein suppresses the toxicity of the mouse prion protein in Drosophila. Neurobiol Dis. 95, 204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson CJ, et al. , 2010. A molecular switch controls interspecies prion disease transmission in mice. J Clin Invest. 120, 2590–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart P, et al. , 2012. Genetic predictions of prion disease susceptibility in carnivore species based on variability of the prion gene coding region. PLoS One. 7, e50623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka NK, et al. , 2008. Neuronal assemblies of the Drosophila mushroom body. J Comp Neurol. 508, 711–55. [DOI] [PubMed] [Google Scholar]
- Telling GC, et al. , 1996. Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev. 10, 1736–50. [DOI] [PubMed] [Google Scholar]
- Telling GC, et al. , 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 83, 79–90. [DOI] [PubMed] [Google Scholar]
- Thackray AM, et al. , 2012. Ovine PrP transgenic Drosophila show reduced locomotor activity and decreased survival. Biochem J. 444, 487–95. [DOI] [PubMed] [Google Scholar]
- Vidal E, et al. , 2013. Bovine spongiform encephalopathy induces misfolding of alleged prion-resistant species cellular prion protein without altering its pathobiological features. J Neurosci. 33, 7778–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorberg I, et al. , 2003. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J Virol. 77, 2003–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells GA, et al. , 1987. A novel progressive spongiform encephalopathy in cattle. Vet Rec. 121, 419–20. [DOI] [PubMed] [Google Scholar]
- Wen Y, et al. , 2010a. Solution structure and dynamics of the I214V mutant of the rabbit prion protein. PLoS One. 5, e13273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, et al. , 2010b. Unique structural characteristics of the rabbit prion protein. J Biol Chem. 285, 31682–31693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will RG, et al. , 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 347, 921–5. [DOI] [PubMed] [Google Scholar]
- Zlotnik I, Rennie JC, 1963. Further observations on the experimental transmission of scrapie from sheep and goats to laboratory mice. J Comp Pathol. 73, 150–62. [DOI] [PubMed] [Google Scholar]
- Zlotnik I, Rennie JC, 1965. Experimental Transmission of Mouse Passaged Scrapie to Goats, Sheep, Rats and Hamsters. J Comp Pathol. 75, 147–57. [DOI] [PubMed] [Google Scholar]