

Abstract
Prostate cancer (PCa) responds to therapies that suppress androgen receptor (AR) activity (androgen deprivation therapy, ADT) but invariably progresses to castration-resistant prostate cancer (CRPC). The Tec family nonreceptor tyrosine kinase BMX is activated downstream of phosphatidylinositol-3 kinase and has been implicated in regulation of multiple pathways and in the development of cancers including PCa. However, its precise mechanisms of action, and particularly its endogenous substrates, remain to be established. Here we demonstrate that BMX expression in PCa is suppressed directly by AR via binding to the BMX gene, and that BMX expression is subsequently rapidly increased in response to ADT. BMX contributed to CRPC development in cell line and xenograft models by positively regulating the activities of multiple receptor tyrosine kinases (RTK) through phosphorylation of a phosphotyrosine-tyrosine (pYY) motif in their activation loop, generating pYpY that is required for full kinase activity. To assess BMX activity in vivo, we generated a BMX substrate-specific antibody (anti-pYpY) and found that its reactivity correlated with BMX expression in clinical samples, supporting pYY as an in vivo substrate. Inhibition of BMX with ibrutinib (developed as an inhibitor of the related Tec kinase BTK) or another BMX inhibitor BMX-IN-1 markedly enhanced the response to castration in a PCa xenograft model. These data indicate that increased BMX in response to ADT contributes to enhanced tyrosine kinase signaling and the subsequent emergence of CRPC, and that combination therapies targeting AR and BMX may be effective in a subset of patients.
Keywords: BMX, ETK, prostate cancer, tyrosine kinase, androgen receptor, castration-resistance
Introduction
Tec kinases are a family of non-receptor tyrosine kinases expressed primarily in hematopoietic cells, and are related in structure to SRC kinases in having SH3 followed by SH2 and tyrosine kinase domains, but they lack the C-terminal tyrosine that negatively regulates SRC kinases (1). The Tec kinases are also unique in having a pleckstrin homology (PH) domain, which is located at the N-terminus and mediates membrane targeting in response to phosphatidylinositol-3 kinase (PI3K) activation and subsequent SRC mediated phosphorylation of a tyrosine in the kinase domain that activates the enzyme. BMX (bone marrow tyrosine kinase gene on chromosome X), also termed ETK (epithelial tyrosine kinase), in contrast to other Tec kinases, is broadly expressed by cell types outside the hematopoietic lineage, including in arterial endothelium and epithelial cells (2–5). Similarly to other Tec kinases, BMX can be activated downstream of PI3K by PH domain-mediated membrane targeting and SRC-mediated phosphorylation of a kinase domain tyrosine (Y566) (1,6), and may alternatively be recruited to the membrane by focal adhesion kinase (FAK) (7). BMX-deficient mice have only modest defects related to angiogenesis and inflammation (3,8–11). However, increasing evidence indicates that BMX has roles in modulating multiple cellular processes including proliferation, differentiation, motility, and apoptosis (12–20), and BMX has been implicated in several cancers (18,20–24). BMX has been reported to directly or indirectly regulate the activity of proteins including TNFR2, PAK1, TP53, PIM1, and STAT3 (10,13,15,25–27), and was recently reported to directly phosphorylate BAK (19).
Prostate cancer (PCa) is the most common noncutaneous malignancy in men. The androgen receptor (AR) plays a central role in PCa and most patients with metastatic PCa respond initially to androgen deprivation therapy (ADT). However, they invariably relapse with metastatic disease despite castrate levels of androgen (castration-resistant prostate cancer, CRPC), and treatment of this advanced stage of the disease is a therapeutic challenge. Responses can be obtained by further suppression of androgen synthesis using agents such as abiraterone or by AR antagonists such as enzalutamide, but most of these patients still relapse within 1-2 years, likely via multiple mechanisms (28–31). Further responses may be obtained with taxanes, radium 223, or immunotherapy in subsets of patients, but these also are not generally durable. BMX is expressed in primary PCa and metastatic CRPC (2,4,22), and transgenic overexpression of BMX in mouse prostate epithelium results in lesions resembling prostate intraepithelial neoplasia (PIN) (22). BMX expression may be increased in response to androgen deprivation therapy, and its ectopic overexpression can confer resistance to castration (23). In addition to its increased expression, BMX in PCa cells may be activated downstream of PTEN loss, EGF family receptors, IL-6, and several neuropeptides (6,32,33).
The progression of PCa to higher grades and to metastatic CRPC also is associated with increased MAPK pathway activation (34), which in many cancers is mediated by receptor tyrosine kinases (RTKs), but activating mutations or copy number changes in RTKs are uncommon in primary or advanced PCa (35). We previously identified phosphotyrosine-tyrosine (pYY) as a novel substrate motif for BMX, with BMX generating pYpY (36). Significantly, this motif is found in the activation loop of multiple tyrosine kinases, and we found that BMX could enhance the activation of these kinases by phosphorylating this second tyrosine, which is required for full activity. The physiological importance of this activity is supported by increased expression of insulin receptor in BMX-deficient mice (36), but whether BMX regulates RTK activity in cancer is unknown. We hypothesized that increased BMX in response to ADT may function to augment basal and growth factor stimulated RTK activity and thereby contribute to CRPC. Our objectives in this study were to test this hypothesis and to assess the efficacy of BMX inhibition in combination with ADT.
Materials and Methods
Antibodies and plasmids
Anti-pFAK (pY576/577), anti-pIGF1R (pY1165/1166)/InsR (pY1189/1190), anti-pMET (pY1234/1235), anti-pS6s235/236, anti-pPYK2Y579/580 and anti-pErbB2Y1221/1222 were from Cell Signaling Technology. Anti-Flag M2, anti-FAK, anti-pFAK (pY576), anti-pFAK (pY577), anti-BMX (H-220, sc20711, rabbit polyclonal) and anti-β-actin were from Santa Cruz Biotechnology. Anti-pTyr (4G10) and anti-tubulin were from Millipore. 3xFlag-FAK was produced by inserting FAK (pCMV-SPORT6-FAK, Open Biosystems) into p3XFlag-CMV (Sigma) between HindIII and BamH1 sites, and mutants were then generated using QuikChange Site-Directed Mutagenesis Kit (Stratagene). The BMX substrate antibody was generated in collaboration with Cell Signaling Technology (Beverly, MA). Rabbits were immunized with a BMX motif peptide, XX(D/E/S/T)pYpYXX (where X is any amino acid). The antibodies then were depleted of reactivity to the unphosphorylated and single phosphorylated peptides, and absorbed and eluted from resin conjugated with the dually phosphorylated peptide.
Cell culture and transfection
HEK293 and VCaP cells were cultured in DMEM/10% FBS (Hyclone). LNCaP, DU145, and CWR22 RV1 cells were cultured in RPMI-1640/10% FBS. Cells were obtained from ATCC (Manassas, VA) and used within 6 months of thawing for studies. They were authenticated by STR profiling, and were free of Mycoplasma. For 3D cultures, approximately 4,000 cells were seeded onto chamber slides (LAB-TEK) coated with growth factor-reduced Matrigel (BD Biosciences), and then cultured in basal medium (RPMI1640 or DMEM) replenished with 2% FBS and 2% Matrigel. The medium was replaced every 4 days. Transfections were performed using Lipofectamine 2000 (Invitrogen) according to manufacturer instructions. For cell proliferation assays, cells were grown with or without drug treatment for 72 hours. Viable cells were counted or analyzed using Cell Counting Kit-8 (Dojindo).
Immunoblotting and immunoprecipitation
Cells were lysed with RIPA buffer (Thermo Scientific) containing Pierce protease inhibitor and phosphatase inhibitor cocktails (Thermo Scientific). 1mM of sodium orthovanadate was added when necessary. Lysates were sonicated for 10 s and centrifuged at 13,000 rpm at 4 °C for 15 min. Protein extracts were mixed with 2X Laemmli sample buffer and boiled for 5 minutes and then resolved on 4-15% Mini-Protean TGX precast gels (Bio-Rad) followed by membrane transfer. Membranes were blocked with 5% milk (or 5% BSA for phosphospecific Abs) in TBS/0.1% Tween 20 (TBS/T) at room temperature for 1 hr and incubated with primary antibodies overnight at 4 °C. Membranes were then incubated with secondary antibodies at room temperature for 1 hr and developed by ECL. For immunoprecipitation, equal amounts of protein extracts (1-5 mg) were mixed with 5 μg of pYpY antibody and 20 μl of protein A agarose beads and incubated at 4 °C overnight with continuous agitation. The beads were washed extensively with RIPA buffer followed by TBS buffer, and beads were eluted with 2X Laemmli sample buffer.
Immunohistochemistry (IHC) in xenograft, patient samples and PCa tissue microarrays
Tissues were fixed with 10% formalin and then paraffin-embedded. The sections were analyzed by Hematoxylin and Eosin (H&E) staining and by standard IHC staining using the pYpY antibody (clone BL10920, BL10921; Cell Signaling Technology). IHC for BMX was carried out with a mouse monoclonal IgG1 antibody (BD Science #610792) versus nonspecific IgG1 (DakoCytomation). CRPC patient samples were from a clinical trial of abiraterone combined with dutasteride (28). Written informed consent was obtained from patients for research use of excess tissue, and the studies were conducted in accordance with recognized ethical guidelines as outlined in the U.S. Common Rule. Tissue microarrays (TMAs) were from US Biomax. The immunostained TMAs were then evaluated and scored by a study pathologist (X. Yuan).
RT-PCR and chromatin-immunoprecipitation (ChIP) Assay
Quantitative real-time RT-PCR amplification was performed on RNA extracted from tissue samples or cell lines using RNeasy mini kit (Qiagen). RNA (50ng) was used for each reaction and the result was normalized by co-amplification of 18S RNA. Reactions were performed on an ABI Prism 7700 Sequence Detection System using Taqman one-step RT-PCR reagents. For ChIP assay, cells were formalin fixed, lysed and sonicated. Anti-AR (Santa Cruz), anti-p300 (Santa Cruz), anti-FOXA1 (Abcam), anti-OCT1 (Santa Cruz), anti-LSD1 (Abcam), anti-H3K4me1 (Abcam), antiH3K4me2 (Upstate), anti-H3K4me3 (Abcam), or rabbit IgG (Santa Cruz) were used to precipitate chromatin fragments from cell extracts. Quantitative real time PCR was used to analyze binding to the ABS1 and ABS2. We used real time quantitative PCR (SYBR green) to amplify the DNA fragment in the antibody precipitated DNA and the un-precipitated input DNA to calculate ΔCT values. The RQ values (RQ=2−ΔCT) are presented and reflect the precipitated DNA as a percentage of the input DNA. Results are represented as mean ± STD for replicate samples. Data are representative of at least three experiments. Significant differences are indicated (*) in the experiments.
Mouse xenograft studies
All animal experiments were approved by Beth Israel Deaconess Medical Center (BIDMC) Institutional Animal Care and Use Committee (IACUC) and were performed in accordance with institutional and national guidelines. About 2-5 million PCa cells (CWR-22RV1, DU145 and VCaP) were suspended in serum free medium supplemented with 50% Matrigel (BD Biosciences) and were implanted subcutaneously into the dorsal flank of 4-6 week old male ICR SCID mice (Taconic). Once the tumors reach approximately 250 mm3 in size, mice were injected intraperitoneally with BMX-IN-1, ibrutinib or vehicle at indicated doses once per day. Tumor volumes were measured twice weekly using calipers. At the end of the study, all mice were humanely euthanized, tumors were collected and analyzed. For VCaP xenograft model, mice were castrated once the tumor reached ~250 mm3 and the BMX inhibitor therapy was immediately started.
Analysis of public domain RNA-seq and microarray data
75th-percentile normalized gene-level RSEM values for the prostate TCGA tumor and normal cases (mRNA-seq V2) were downloaded directly from the NCI Genomic Data Commons. CRPC data was downloaded from dbGaP for the Beltran 2016 (37) and Robinson 2015 (38) data sets (phs000909 and phs000915). Unpublished RNA-seq data corresponding to the LuCaP cases described previously (39) were graciously provided by Dr. Eva Corey (U. of Washington). For the Beltran, Robinson, and LuCaP datasets, CRPC cases were identified by excluding cases previously marked as neuroendocrine or otherwise having high levels of SYP or CHGA mRNA expression. LuCaP data was pre-processed by the removal of ambiguous reads originating from mouse tissue as described previously (40). All datasets were then processed through the TCGA mRNA-seq V2 alignment protocol, in which raw data was adaptor trimmed, aligned with MapSplice, sorted by read group and reference name with SamTools, translated and filtered to the transcriptome, quantified with RSEM, and normalized to the 75th percentile of each sample. Outlier analyses were performed using GraphPad Prism version 7. For microarrays, log2 microarray data of 4 LuCaP PDX cases (41) were downloaded from GEO, accession ID GSE85672. Values corresponding to BMX were grouped by condition and compared within each group by one-way ANOVA. Samples within each group were compared against each other using Tukey’s multiple comparison test. Statistical analyses were performed using GraphPad Prism version 7.
Statistical Analysis
Results are expressed as mean ± SE. Statistical significance was determined by a two-sided Student’s t-test, with p<0.05 considered statistically significant.
Results
BMX inhibition suppresses the growth of CRPC cells in vitro and in vivo
Previous studies using BMX inhibitors or RNAi have indicated that blocking BMX can suppress the growth or induce apoptosis of PCa cell lines and xenografts (42–44). We also previously reported on development of a Tec kinase (BMX and BTK) selective irreversible covalent inhibitor (BMX-IN-1, Bi1), and showed that it could suppress the growth and/or induce apoptosis of several PCa cell lines (45). Consistent with this previous study, BMX-IN-1 suppressed the proliferation of CWR22Rv1 cells (AR dependent but androgen independent PCa cell line) with an IC50 of ~5 μM, while the reversible noncovalent analogue of BMX-IN-1 (BMX-IN-1R, Bi1R) was much less active (Figure 1A). Ibrutinib is a covalent inhibitor of BMX and BTK, which can also target EGFR family kinases at ~10-fold higher concentrations, and is an approved BTK inhibitor used for the treatment of B cell malignancies (BTK not being expressed at detectable levels in PCa cell lines, see below). It had similar effects on proliferation (Figure 1B), although the concentrations required were substantially higher than for suppression of BTK in CLL cells (10-100 nM). More dramatically, BMX-IN-1 and ibrutinib blocked CWR22Rv1 cell colony formation in Matrigel-based 3D cultures (Figure 1C), with the compounds reducing both colony number and size at 2 weeks (Figure 1D, E).
Figure 1. BMX inhibition suppresses the growth of CRPC cells in vitro and in vivo.
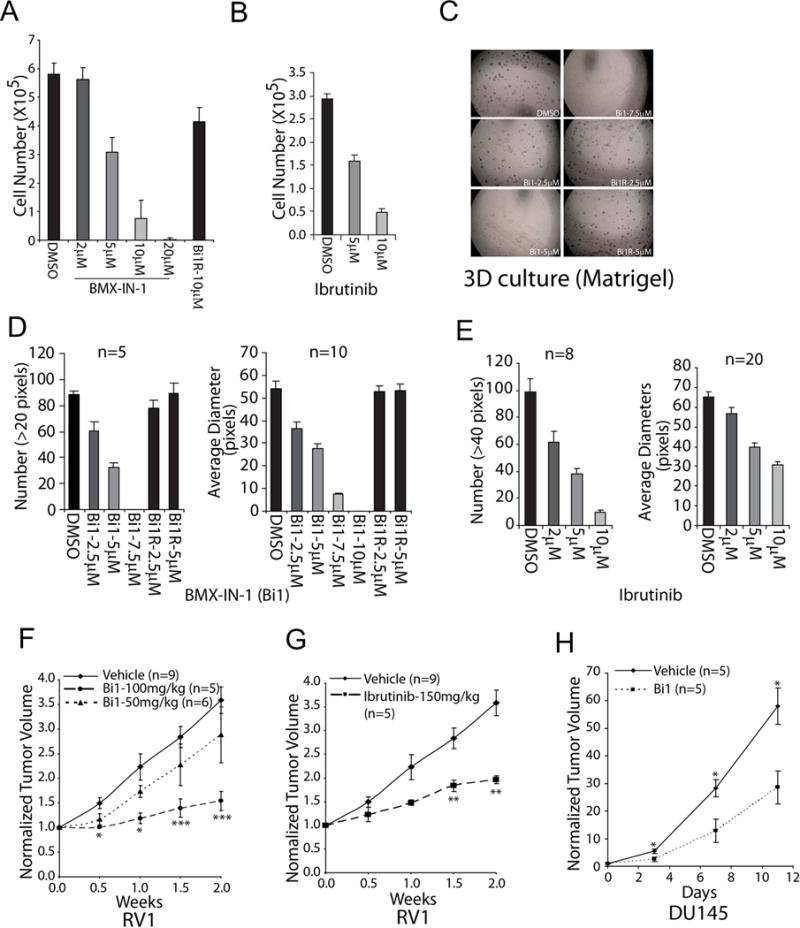
(A) CWR22RV1 cells were treated with BMX-IN-1(Bi1) or BMX-IN-1R (BilR) as a negative control for 72 hr and then measured for viability (mean ± SE, n = 4). (B) CWR22RV1 cells were treated with ibrutinib for 72 hr and then measured for viability (mean ± SE, n = 4). (C) CWR22RV1 cells were cultured in Matrigel-based 3D system with indicated BMX-IN-1 or control treatment for 2-3 weeks and (D) analyzed for colony number (left panel) and average diameters (right panel) (mean ± SE, n=5 for left panel and n=10 for right panel). (E) CWR22RV1 cells were cultured in Matrigel-based 3D system with ibrutinib or control treatment for 2 weeks and colonies were analyzed for numbers and average diameters (mean ± SE, n=8 for left panel and n=20 for right panel). (F) CWR22RV1 xenografts bearing mice were administered 0, 50 or 100 mg/kg of BMX-IN-1(Bi1) and growth was monitored (mean ± SE; n=9 for vehicle, n=5 for 100mg/kg/d, and n=6 for 50mg/kg). (G) CWR22RV1 xenografts bearing mice were administered 0 or 150 mg/kg/d of ibrutinib and tumor growth was monitored (mean ± SE; n=9 for vehicle, n=5 for 150mg/kg/d of ibrutinib). (H) DU145 xenografts bearing mice were administered with 0 or 100mg/kg/d of BMX-IN-1(Bi1) and growth was monitored (mean ± SE, n=5). *p<.05, **p<.01, ***p<.001
To determine whether BMX may similarly contribute to PCa growth in vivo, we generated subcutaneous CWR22Rv1 xenografts in immunodeficient male mice. When xenografts reached ~250 mm3, mice were randomized to daily treatment with BMX-IN-1 (50 or 100 mg/kg), ibrutinib (150 mg/kg), or vehicle by intraperitoneal injection. Both BMX-IN-1 (Figure 1F) and ibrutinib (Figure 1G) significantly suppressed the growth of these CWR22Rv1 xenografts. This response was not clearly related to AR, as BMX-IN-1 also suppressed the growth of xenografts derived from the AR negative DU145 PCa cell line (Figure 1H).
To determine whether these drug-induced effects are consistent with what is observed upon inhibiting BMX expression in vivo, we also generated CWR22Rv1 sublines expressing doxycycline inducible BMX shRNA. In two independent lines, doxycycline induction of the BMX shRNA resulted in reduced growth in both 2D cultures and in Matrigel-based 3D models when compared to uninduced counterparts (Supplemental Figure S1A, B). Of note, basal BMX levels in these lines differed, and the growth rates prior to (as well as after) doxycycline induction of the shRNA in the two lines were correlated with BMX expression, consistent with a growth-stimulatory function. Finally, we injected cells from one of these lines (line 12) subcutaneously into immunodeficient male mice, and randomized the mice to receive doxycycline or vehicle in the drinking water. Consistent with the in vitro results, doxycycline suppressed growth of these CWR22Rv1 xenografts (Supplemental Figure S1C).
BMX inhibition in PCa cells blocks the downstream dual tyrosine phosphorylation of multiple tyrosine kinases in vitro
We previously reported that BMX showed a dramatic preference for substrates with a priming phosphotyrosine at position −1(pYY), to yield pYpY (36). The activation loop of multiple tyrosine kinases contains a tandem YY that must be dually phosphorylated for full activity (46,47), and we further found that BMX could thereby enhance the activity of multiple tyrosine kinases through phosphorylation of the second tyrosine in the activation loop after a priming tyrosine autophosphorylation at the −1 position (36). Based on these findings we proposed that BMX, which is activated downstream of PI3K and SRC, functions to modulate tyrosine kinase activity and subsequently downstream signaling (see schematic, Supplemental Figure S2). Consistent with this function, BMX depletion by siRNA in CWR22Rv1 cells impaired activation of the MAPK and PI3K pathways in response to serum stimulation (Figure 2A). This impairment was observed with a pool of BMX siRNA, and was more dramatic using a single BMX siRNA that more effectively depleted BMX (Supplemental Figure S3 and Figure 2A, respectively).
Figure 2. BMX inhibition in PCa cells blocks the downstream dual tyrosine phosphorylation signal of multiple tyrosine kinases in vitro.
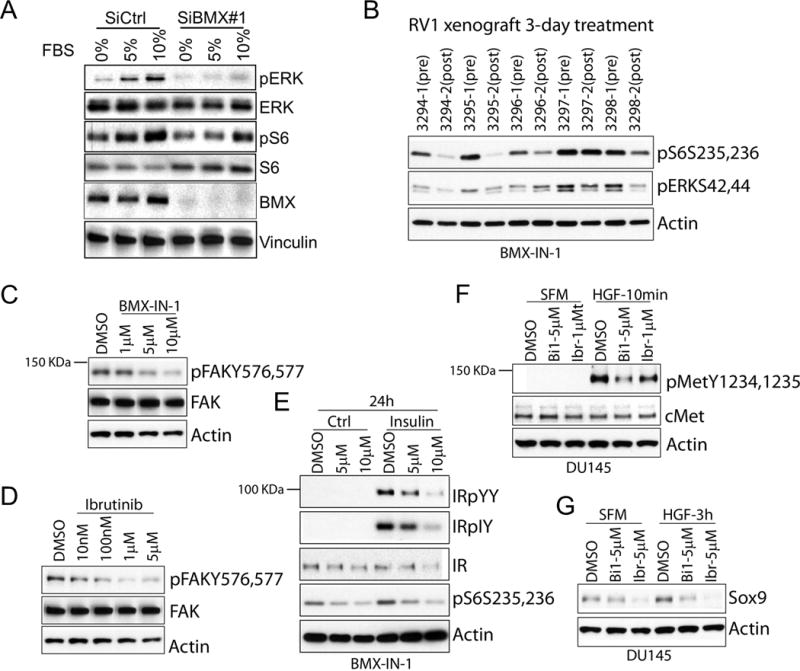
(A) CWR22Rv1 cells were treated with BMX or control siRNA for 48 hours, serum starved overnight, and then stimulated by addition of FBS to 10% for 10 minutes. (B) CWR22 RV1 tumor bearing mice were administered with BMX-IN-1 for 3 days; biopsies prior to and after treatment were immunoblotted for indicated proteins. (C, D) CWR22RV1 cells in complete medium were treated with BMX-IN-1 (C) or ibrutinib (D) for 4 hr. (E) CWR22RV1 cells were serum starved for 24 hrs, then pre-incubated with BMX-IN-1 in serum free medium for 2 hrs, and then stimulated with insulin for 10 min. (F, G) DU145 cells serum starved for 24 hr were pre-incubated with BMX inhibitors in serum free medium for 2 hr, then stimulated with Hepatocyte Growth Factor (HGF), and immunoblotted for pMET after 10 minutes (F) or SOX9 after 3 hr (G).
We next assessed the effects of the BMX inhibition on PCa in vivo. Similarly to the in vitro results, by immunoblotting we found that levels of phospho-S6 (serine 235, 236) and phospho-ERK (threonine 202, tyrosine 204) were reduced after 3-days of therapy with BMX-IN-1 in the CWR22Rv1 xenografts (Figure 2B), supporting a block in both the PI3K and MAPK pathways. Therefore, we next determined the effect of BMX inhibitors on the phosphorylation of individual tyrosine kinases in CWR22Rv1 and DU145 PCa cells. BMX inhibitors diminished pYpY levels on FAK and on insulin-stimulated Insulin Receptor/Insulin Growth Factor (IR/IGFR1), with a resultant decrease in S6 phosphorylation (Figure 2C-E). BMX-IN-1 and ibrutinib also suppressed pYpY levels in MET after HGF stimulation (Fig. 2F), as well as the increase in SOX9 that occurs downstream of MET activation (Figure 2G) (48). Taken together these findings suggested that BMX may contribute to driving PCa growth in vivo through this enhancement of tyrosine kinase activation.
Generation of a BMX substrate antibody
In addition to a pY at the −1 position, we previously found that BMX showed a preference for acidic residues at position −2 (also consistent with the activation loop of multiple tyrosine kinases) (36). Therefore, in collaboration with Cell Signaling Technology, we generated an affinity purified rabbit antibody that recognizes (D/E/S/T)pYpY (herein referred to as pYpY antibody) as a potential reagent to assess for BMX activity (Figure 3A). In order to determine the specificity of this antibody (which was extensively absorbed against the unphosphorylated and monophosphorylated peptides), we first co-transfected FAK, a BMX substrate, with empty vector (EV), BMX wild type (WT) or BMX kinase dead (KD), and then treated with or without ibrutinib. BMX WT increased levels of dual-phosphorylated pY576,p577 FAK, as assessed by a specific phospho-FAK pY576, p577 antibody, and markedly increased reactivity with the pYpY antibody (Figure 3B). The reactivity was not increased by kinase dead BMX, and was markedly suppressed by ibrutinib. We further mutated FAK at tyrosines 576 and/or 577, and found that both single and double tyrosine mutations completely abrogated the pYpY signal (as well as reactivity with the phospho-FAK pY576, p577 antibody) (Figure 3C). Moreover, the 4G10 antibody (recognizing pY) showed modest basal reactivity that was increased by BMX on the control FAK constructs, but not on the Y576A or Y577A constructs.
Figure 3. Generation of a dual phospho-tyrosine (pYpY) antibody that indicates BMX activity.
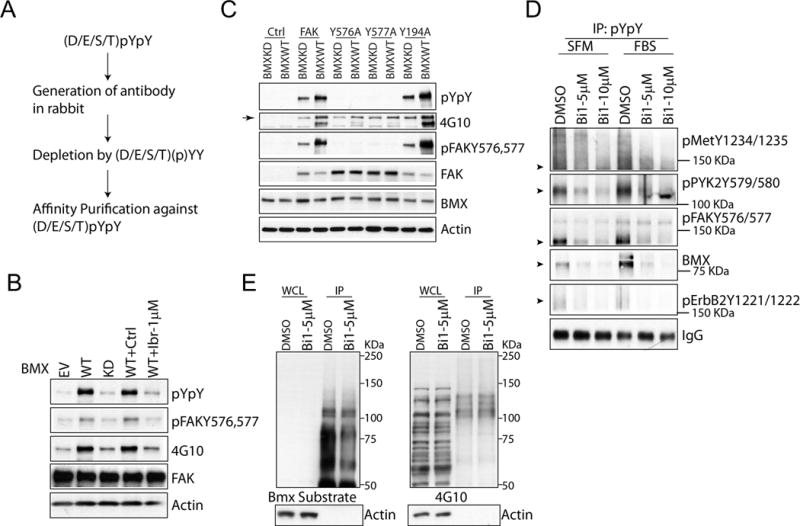
(A) Schematic for (D/E/S/T)pYpY BMX substrate antibody generation. (B) HEK 293 cells double transfected with wild type FAK and wild type or kinase dead BMX (or empty vector, EV) were treated with or without 1 μM ibrutinib for 4 hr. (C) HEK 293 cells were double transfected with wild type or mutant FAK (or empty vector), and with wild type or kinase dead BMX. (D) CWR22RV1 cells serum starved for 24 hr were pre-treated with BMX-IN-1(Bi1) for 4 hr, and then stimulated with or without FBS (to 10%) for 10 min. Proteins were immunoprecipitated with the pYpY antibody and then blotted for dual tyrosine phosphorylation of the indicated tyrosine kinases or for total BMX. Arrowheads show predicted molecule weights. (E) CWR22RV1 cells treated with or without 5 μM of BMX-IN-1were were immunoprecipitated using the pYpY BMX substrate antibody. Whole cell lysate (WCL) or immunoprecipitated proteins were then blotted with the pYpY (left panel) or phospho-tyrosine (4G10; right panel) antibodies.
We next used the pYpY antibody to assess the effects of BMX on a series of tyrosine kinases. Serum starved CWR22Rv1 cells were treated overnight with BMX-IN-1 or vehicle, and then treated with serum for 10 minutes. Proteins were then immunoprecipitated with the pYpY antibody followed by immunoblotting using individual antibodies that recognize the pYpY in specific tyrosine kinases including MET, FAK, PYK, ERBB2 and BMX itself. For each kinase the BMX inhibitor decreased the pYpY signal in the serum starved and serum stimulated cells (Figure 3D). We also immunoblotted the pYpY immunoprecipitated proteins with the pYpY antibody or with 4G10 (recognizing a single phosphotyrosine). BMX inhibition suppressed dual tyrosine phosphorylation but failed to inhibit single tyrosine phosphorylation, further supporting the substrate specificity of BMX (Figure 3E).
Reactivity with BMX substrate antibody correlates with BMX level and activity in vivo
We next performed immunohistochemistry (IHC) for BMX and with the pYpY antibody on castration-resistant VCaP xenografts (which express increased levels of BMX, see below) and patient samples. For the BMX IHC we used a mouse monoclonal that had been validated previously for IHC (24), and further confirmed its specificity by immunoblotting and by IHC on CWR22Rv1 cell blocks from cells treated with BMX siRNA (Supplemental Figure S4A, B). The castration-resistant VCaP xenografts were positive for BMX and with the pYpY antibody, and the latter staining could be competed with excess pYpY peptide (Figure 4A). To further assess BMX function in vivo, we biopsied castration-resistant VCaP xenografts and then treated for 4 days with BMX-IN-1 (100 mg/kg/day). Significantly, the pYpY signal by IHC was substantially reduced in the BMX-IN-1 treated xenograft tumors compared to the pre-treated counterparts from the same mice (Figure 4B). We next stained several CRPC bone marrow biopsies and a primary PCa tissue microarray (TMA) (Figure 4C, D). Importantly, the pYpY signal correlated with BMX expression in the biopsies and the TMA (Pearson coefficient 0.7743 in the TMA). Figure 4E shows the distribution of staining in the TMA. Taken together, these data strongly support the conclusion that BMX functions in vivo to phosphorylate pYY substrates including multiple tyrosine kinases, and may thereby contribute to the increased signaling through downstream pathways.
Figure 4. Reactivity with pYpY antibody correlates with BMX activity in vivo.
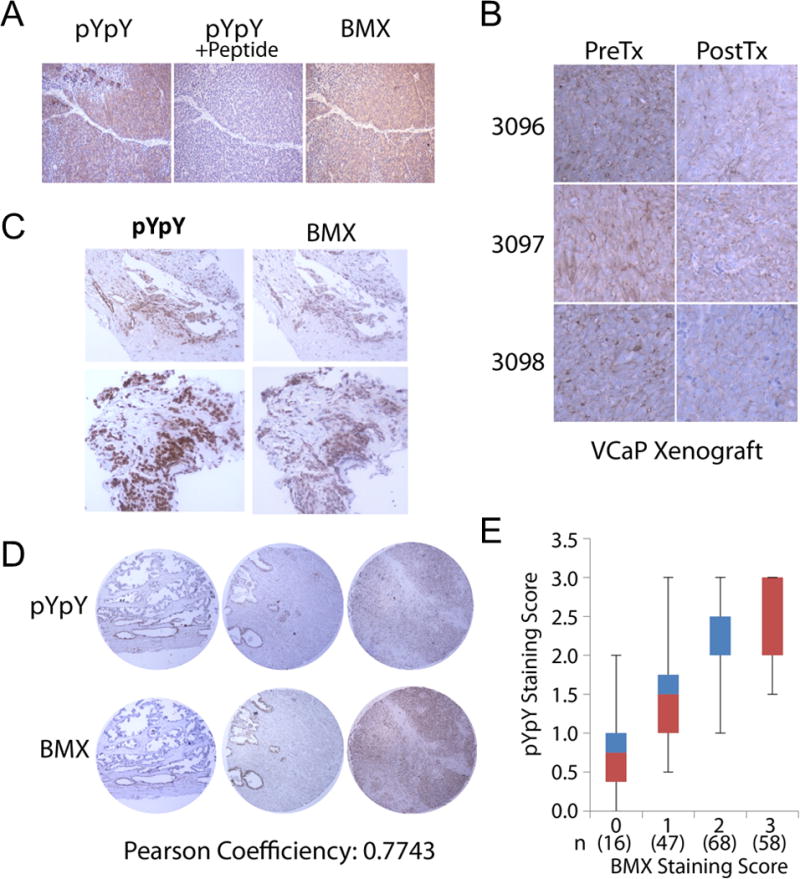
(A) Castration resistant VCaP xenografts were immunostained for total BMX or pYpY, the latter with or without (D/E/S/T)pYpY peptide blocking. (B) Castration resistant VCaP xenograft bearing mice were administered 100 mg/kg/d of BMX-IN-1 for 4 days. Pretreatment biopsies and post treatment tissues were immunostained for pYpY. (C) Bone metastasis biopsies from patients with CRPC were immunostained for pYpY and BMX. (D and E) Tissue microarrays of human PCa were immunostained for pYpY and BMX (D) and scored for correlation between pYpY and BMX (E).
BMX is upregulated in castration-resistant and abiraterone resistant PCa
A previous study found that BMX protein was increased in a series of castration-resistant transurethral resection of prostate (TURP) samples versus untreated samples, and in PCa xenografts and mouse prostate after castration (23). We therefore examined BMX mRNA levels in primary PCa and metastatic CRPC RNA-seq datasets. Significantly, median BMX mRNA levels in the TCGA dataset of primary PCa were low and not increased relative to normal prostate, although there was a substantial outlier population (Figure 5A) (49). BMX mRNA levels were higher in two metastatic CRPC datasets, although they were still lower than for a series of other kinases (Figure 5B). Consistent with these results, although overall levels were low, BMX mRNA was increased in CRPC bone marrow metastases versus primary untreated PCa samples on Affymetrix microarrays we reported previously (Supplemental Figure S5) (50).
Figure 5. BMX is upregulated in castration-resistant and abiraterone resistant PCa.
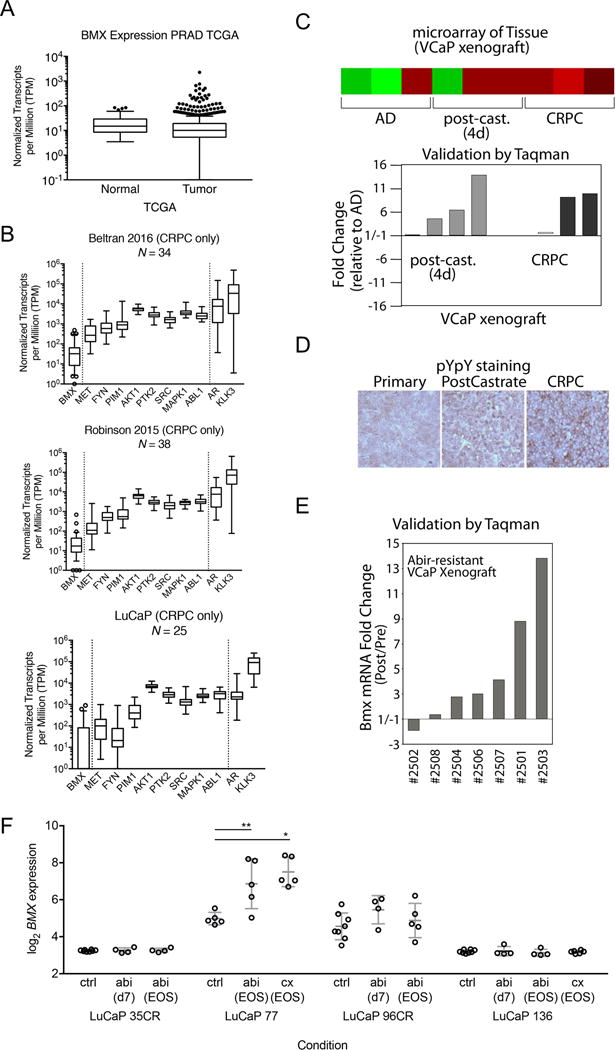
(A) Tukey’s method for a box-and-whiskers plot of normal (N=53) and tumor (N=493) RNA expression data from the prostate cancer TCGA, where the boxes depict the 25th percentile, median, and 75th percentile, and whiskers depict the lowest value and the 75th percentile + 1.5 times the interquartile range. Dots depict outlier values greater than the 75th percentile + 1.5 times the interquartile range. (B) Box-and-whiskers plots for three metastatic CRPC RNA-seq data sets. For BMX expression, whiskers depict the 10th to 90th percentiles, while open circles depict outlier values outside the 10th and 90th percentiles. For all other genes, whiskers depict the minimum and maximum values. (C) VCaP xenografts were biopsied at three stages: androgen-dependent tumor (AD), 4 days after castration (post-cast), and castration-resistant relapsed tumor (CRPC). mRNAs were extracted from the biopsies of tumors and analyzed on Agilent microarrays. BMX expression is shown by heatmap (upper panel) and validated by qRT-PCR analysis (bottom panel). (D) Representative VCaP xenograft biopsied at different stages was immunostained for pYpY. (E) Castration resistant VCaP xenograft bearing mice were treated with abiraterone (i.p. 1 mg/day) for the first week and then 2 mg/day until progression. BMX in pre-abiraterone biopsies versus at progression were analyzed for BMX mRNA expression by qRT-PCR. (F) BMX Affymetrix expression prior to therapy (control), after 7 days of abiraterone (d7), or at relapse on abiraterone or after castration (end of study, EOS) in LuCaP PDX models (41).
To further trace the effects of androgen deprivation on BMX, we established androgen-dependent VCaP xenografts and examined BMX expression prior to castration (androgen dependent, AD), 4 days after castration (post-cast), and in the relapsed CRPC stage. Both Affymetrix microarray data (Figure 5C, upper panel heatmap) and quantitative reverse transcriptase PCR (qRT-PCR) validation data (Figure 5C, bottom panel) showed that BMX mRNA was upregulated immediately after castration and persisted at the CRPC stage. Strikingly, corresponding to the increase of BMX expression with tumor progression to CRPC, staining with the pYpY antibody was also markedly intensified (Figure 5D). It is also noteworthy that the pYpY signal was diffusely expressed in the cytoplasm prior to castration, started to be enriched in the plasma membrane area immediately after castration, and was highly abundant in plasma membrane at the CRPC stage, consistent with membrane localized RTKs downstream of BMX being activated during the progression to CRPC.
To determine whether BMX expression was altered in response to further androgen deprivation with abiraterone, we next administered abiraterone to a cohort of castrated mice bearing castration-resistant VCaP xenografts. This treatment resulted in tumor regression or stabilization for ~4-6 weeks followed by progression, at which time these abiraterone-resistant VCaP xenografts were similarly analyzed for BMX expression in comparison with tumor biopsies taken prior to initiating abiraterone treatment. Results from qRT-PCR showed that the abiraterone treatment further increased BMX expression above the levels in the CRPC xenografts (Figure 5E). Finally, we examined BMX expression in a series of LuCaP patient derived xenografts (PDXs) that were given AR targeted therapy (41). In a castration-sensitive PDX (LuCaP-77), BMX mRNA was significantly increased in tumors that progressed after castration or abiraterone treatment (Figure 5F). In contrast, BMX mRNA was not increased by castration or abiraterone in another castration-sensitive PDX (LuCaP-136). In the castration-resistant LuCaP-96CR (established in castrated mice), treatment with abiraterone increased BMX during an initial response after 7 days, while there was no effect in another castration-resistant PDX (LuCaP-35CR). Overall these findings indicate that increased BMX may contribute to PCa progression after androgen deprivation therapy in at least a subset of tumors.
AR negatively regulates BMX gene transcription
A previous study found that BMX was repressed by androgen in LNCaP cells, and found by chromatin immunoprecipitation (ChIP) that AR was recruited to sites in the BMX gene, suggesting AR may directly repress the BMX gene (23). We examined our previous Affymetrix microarray data generated to identify genes that are stimulated versus repressed by androgen in vitro in VCaP cells and in a subline of VCaP derived from a castration-resistant xenograft (VCS2 cells) (51), and found that BMX also was repressed by DHT in both cell lines (Supplemental Figure S6). To confirm this result, VCaP cells in steroid depleted medium were treated with DHT for 24 hours. This treatment markedly decreased BMX mRNA, and this decrease was blunted by co-treatment with an AR antagonist (bicalutamide) (Figure 6A). Analysis by immunoblotting similarly showed BMX protein was markedly reduced by DHT, and could be further increased by treatment with the AR antagonist enzalutamide (presumably reflecting blockade of residual androgen in the steroid depleted medium) (Figure 6B). Consistent with AR mediated repression of BMX, the expression of BMX protein under basal conditions (complete medium containing androgen) was markedly lower in the AR positive LNCaP and VCaP cell lines versus the AR negative PC3 and DU145 cell lines, although it was also higher in the AR positive CWR22Rv1 cell line (which expresses high levels of an AR splice variant) (Figure 6C). We further confirmed that this band was BMX by immunoblotting after treatment with a series of BMX siRNA (Supplemental Figure S7). BTK was undetectable in any of the PCa lines (Figure 6C).
Figure 6. AR negatively regulates BMX gene transcription.
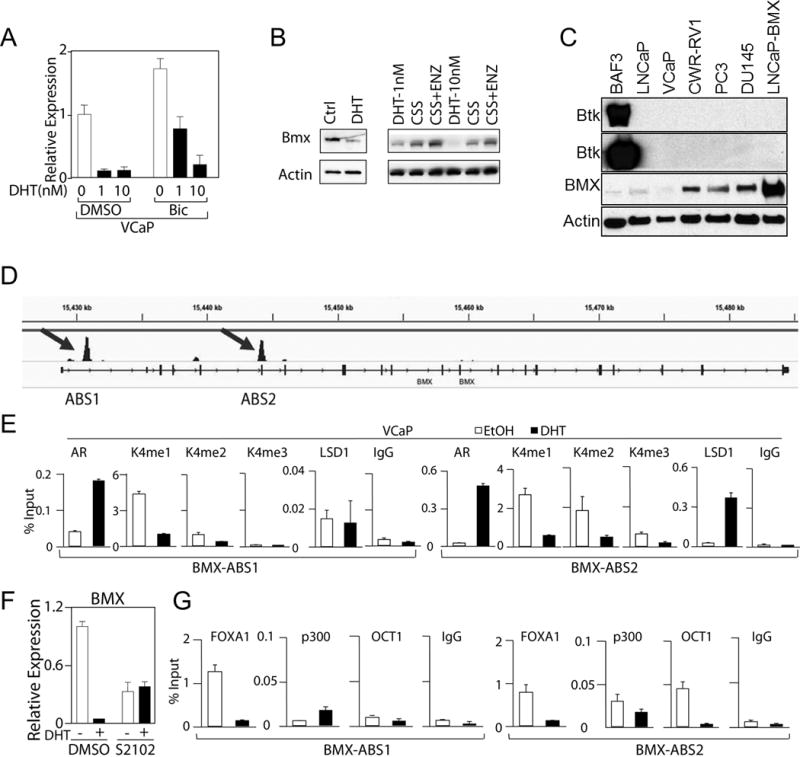
(A) VCaP cells in steroid depleted medium were treated with DHT, without or with 10 μM of Bicalutamide (Bic) for 24 hr and BMX mRNA was measured using qRT-PCR. (B) VCaP cells were cultured in Charcoal-Stripped Serum (CSS) containing medium (androgen depleted) with or without 10 nM DHT for 72 hr (Left panel). VCaP cells in CSS medium for 48 h were treated with vehicle, DHT, or enzalutamide (ENZ, 10μM) for 24 hr. (C) BMX and BTK protein expression in a series of PCa cell lines (LNCaP-BMX cells are stably transfected with BMX). (D) AR binding sites in BMX loci after 4 hr DHT stimulation identified by AR ChIP-seq. (E) VCaP cells in CSS medium were treated with or without 10 nM DHT for 4 hrs followed by CHIP with the indicated antibodies and qPCR for the ABS1 (left) and ABS2 (right) sites (means ± SD of at least 3 biological replicates). (F) VCaP cells were treated with LSD1 inhibitor S2101 (100 μM) for 4 hours with/out 10 nM DHT, and effects on BMX mRNA were analyzed by qRT-PCR (means ± SD, n=3). (G)VCaP cells were treated with or without 10 nM DHT for 4 hrs followed by CHiP-qPCR for FOXA1, p300 or OCT1 binding to ABS1 (left) and ABS2 (right) sites (means ± SD of at least 3 biological repeats).
We next examined our previous AR ChIP-seq data in VCaP cells (52) and identified two AR binding sites linked to the BMX gene, designated as ABS1 and ABS2, which are located in intron 1 and overlapping exon 5, respectively (Figure 6D and Supplemental Figure S8). To confirm these binding sites in VCaP cells, we designed primers spanning each region and performed ChIP coupled with quantitative PCR (ChIP-qPCR) to measure AR binding. DHT induced significant AR binding at both sites, which was more dramatic on ABS2 (Figure 6E). To determine whether AR binding had local effects on chromatin, we next assessed for changes in histone marks associated with active transcription (H3K4 mono-, di- and tri-methylation). Substantial H3K4 mono-methylation (H3K4me1) was detected at both sites, H3K4 di-methylation (H3K4me2) was abundant at the ABS2 site but minimal at ABS1 site, and tri-methylation (H3K4me3, generally associated with gene promoters) was only observed at the ABS2 site (Figure 6E). Significantly, DHT treatment (10 nM for 4 hours) caused a decrease in H3K4 methylation at both sites, consistent with transcriptional repression.
As demethylation of H3K4me1 and H3K4me2 are mediated by lysine specific demethylase 1 (LSD1, KDM1A), we also examined LSD1 binding and found that it was higher at the ABS1 site prior to DHT, but markedly increased at the ABS2 site in response to DHT (Figure 6E). We have similarly reported an association between AR-mediated LSD1 recruitment and H3K4 demethylation at several other AR repressed genes including the AR gene (51). Consistent with a role for LSD1, treatment with an LSD1 inhibitor (S2102) prevented the DHT-induced suppression of BMX (Figure 6F). Finally, to further characterize these two sites we examined binding of FOXA1 (AR pioneer factor), p300 (major AR coactivator), and OCT1 (transcription factor frequently associated with AR at enhancer sites). Substantial basal levels of FOXA1 were associated with both sites, and these levels were markedly decreased at both sites in response to DHT (Figure 6G). In contrast, basal OCT1 and p300 were higher at ABS2, and were decreased in response to DHT at ABS2, but not ABS1. By immunoblotting we found that the DHT treatment did not change total cellular levels of LSD1 (or CoREST), FOXA1, or p300, while AR protein were initially increased at 6 hrs and then markedly decreased at 24 hours (consistent with previous data showing that DHT rapidly stabilizes AR protein, but subsequently causes repression of the AR gene) (Supplemental Figure S9A and S9B, respectively) (51). Overall, these findings support the conclusion that ABS2 contains a BMX enhancer that is negatively regulated by the agonist-liganded AR, and contributes to the increase of BMX after ADT.
BMX is a driver of castration resistance in vitro and in vivo
Our data so far have shown that BMX is upregulated immediately after castration and that this is associated with increased levels pYpY, suggesting a role for BMX at early stages in the development of castration resistance. In order to determine whether BMX contributes to tumor progression after castration, we first treated androgen starved VCaP cells with BMX inhibitors to see whether BMX inhibition could further suppress cell recovery. Both BMX-IN-1 and ibrutinib dose dependently decreased cell recovery (Figure 7A, B), and there was at least an additive effect when ibrutinib was combined with enzalutamide to further suppress AR activity (Fig. 7C). The results from Matrigel-based 3D cultures further supported the combinatorial effect between BMX inhibitors and AR antagonist, although VCaP cells did not generate well-formed colonies in this 3D model (Figure 7D). Most strikingly, simultaneous administration of BMX inhibitors (BMX-IN-1 or ibrutinib) along with castration in VCaP xenografts led to marked tumor regression compared to castration alone (Figure 7E).
Figure 7. BMX is a driver of castration resistance in vitro and in vivo.
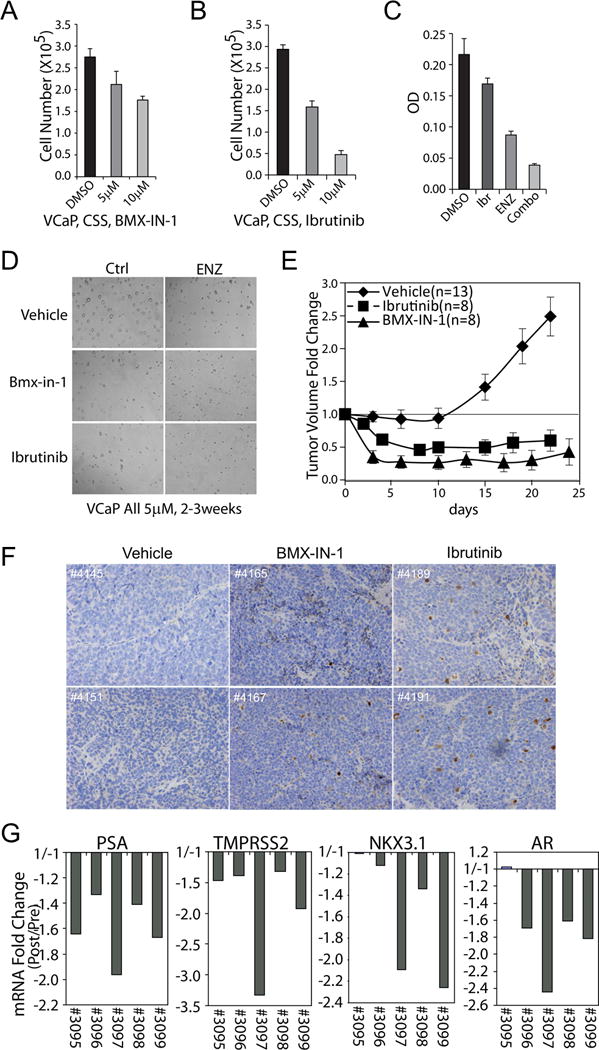
(A, B) VCaP cells in CSS medium were treated with BMX-IN-1 (A) or ibrutinib (B) for 5 days and counted (means ± SE, n=4). (C) VCaP cells in CSS medium were treated with 5 μM enzalutamide (ENZ), 5 μM ibrutinib (Ibr), or the combination (Combo) for 5 days (means ± SE, n=4). (D) Representative images of VCaP cells cultured for 2-3 weeks in Matrigel based 3D system with 5 μM enzalutamide, BMX-IN-1, and/or ibrutinib. (E) VCaP subcutaneous xenografts were established in ICR-SCID mice. The mice were castrated once the tumors reach 500 mm3 and then administered vehicle (n=13), ibrutinib (n=8) or BMX-IN-1 (n=8) (each at 100mg/kg/d) by i.p. injection. The results are means ± SE. (F, G) Castration resistant VCaP xenografts were established and then treated for 4 days with vehicle or 100 mg/kg/d BMX-IN-1 or ibrutinib via i.p. injection. (F) Representative sections were stained for cleaved caspase 3. (G) Biopsies prior to or post 4-day treatment were analyzed for PSA, TMPRSS2, NKX3.1 or AR mRNA expression by qRT-PCR.
We showed previously that progression to castration-resistance in the VCaP xenograft model is associated with restoration of AR activity (53,54). A previous study indicated that BMX may directly or indirectly interact with AR and enhance its stability or activity, possibly through tyrosine phosphorylation, suggesting that BMX may be contributing to AR activity in CRPC (23). To test this hypothesis, we treated a cohort of castration-resistant VCaP xenografts with BMX-IN-1 for 4 days and assessed for gene expression in pretreatment biopsies versus post-treatment tumor. Significantly, IHC for cleaved caspase 3 showed that this 4-day treatment with BMX-IN-1, or with ibrutinib, resulted in a marked increase in apoptosis (Figure 7F). By qRT-PCR we found decreased expression of AR regulated genes (PSA, TMPRSS2, NKX3.1), but also found decreases in AR mRNA, indicating that BMX inhibition may decrease AR activity by transcriptional as well as posttranslational mechanisms (Figure 7G). In any case, taken together these data support the conclusion that BMX plays a role at early stages in the development of CRPC, with increased phosphorylation of regulatory sites in multiple tyrosine kinases being a mechanism that contributes to its survival function.
Discussion
BMX is activated downstream of PTEN loss and PI3K pathway stimulation, and has been implicated in several cancers including PCa, but mechanisms through which it may drive tumor growth remain to be established. We previously used a positional scanning peptide library screening approach to determine that BMX had a marked preference for a priming phosphotyrosine in the −1 position, which indicated that BMX substrates may include multiple tyrosine kinases that are fully activated by pYpY sites in the kinase domain (36). However, the extent to which BMX functions in vivo through this mechanism has not been clear. In this study we initially found that BMX inhibitors decrease pYpY levels in FAK, IR, and MET in PCa cell lines. We then generated and characterized a BMX motif antibody against a (D/E/S/T)pYpY peptide, and showed that treatment of VCaP xenografts with a BMX inhibitor caused a rapid decrease in the level of pYpY. Moreover, immunostaining of clinical samples showed a strong correlation between BMX and pYpY levels, further supporting the conclusion that pYY is a BMX substrate in vivo.
Together these findings indicate that BMX contributes to activation of tyrosine kinase signaling in vivo in PCa, with subsequent increases in MAPK and PI3K pathway activation. Specifically, we propose a positive feedback loop wherein BMX enhances the activation of multiple RTKs by phosphorylation of pYY sites in their activation loop (with phosphorylation of the −1 tyrosine being mediated by autophosphorylation), and that subsequent activation of RAS, PI3K, and SRC further enhances BMX activity. It should be noted that BMX may be activated by alternative mechanisms (7,32,33), and may have additional direct substrates. In particular, BMX has been linked to increased STAT3 phosphorylation in both PCa and glioblastoma (22,24,27,44), which could be direct (although STAT3 does not have the activation loop YY motif) or indirect through RTKs. Other potential BMX substrates that may contribute to our observed responses to BMX inhibition include AR (23) and BAK (19).
Consistent with a previous report (23), we also found that BMX expression was repressed by androgen and increased by androgen deprivation. By analysis of ChIP-seq data we then identified and validated two AR binding sites in the BMX gene, which were distinct from those examined in the previous study. Both sites had features of enhancers that were repressed in response to androgen, with the ABS2 (overlapping exon 5) appearing to be most repressed in response to androgen. These findings indicate that AR is a direct negative regulator of BMX, and that increased expression of BMX may be a very rapid adaptation in response to castration that contributes to tumor cell survival and eventual emergence of CRPC. Indeed, we found that BMX expression was markedly increased at 4 days after castration in a series of VCaP xenografts. Moreover, this expression persisted when xenografts became castration-resistant, and was then further increased in xenografts that developed resistance to abiraterone. Significantly, this increase in BMX was associated with an increase in staining with the BMX substrate antibody.
Based on these findings, we hypothesized that treatment with a BMX inhibitor may enhance responses to ADT. Indeed, BMX inhibition markedly decreased the recovery of androgen deprived VCaP cells in vitro. Moreover, treatment with BMX inhibitors (ibrutinib or BMX-IN-1) dramatically enhanced the response to castration in VCaP xenografts. It should again be noted that these inhibitors also target BTK, but this kinase is B cell specific and not expressed at detectable levels in the PCa cells we have examined. However, we cannot rule out effects on another Tec kinase, or the possibility that other effects of these inhibitors contribute to their efficacy. Nonetheless, these findings indicate that BMX contributes to tyrosine kinase pathway activation in at least a subset of primary PCa, and that its contribution may be greater in CRPC. Moreover, the rapid induction of BMX after ADT suggests that BMX may be particularly critical for tumor cell survival immediately after ADT. Therefore, these studies provide support for clinical trials of BMX inhibitors (such as ibrutinib) in CRPC, possibly with selection for patients based on BMX expression or activity. In addition, these studies suggest that combining BMX inhibition with initial ADT, or with secondary ADT (abiraterone or enzalutamide) may be particularly effective.
Supplementary Material
Acknowledgments
Portions of this research utilized the computational resources of the NIH HPC Biowulf cluster.
Financial Support: This work was supported by grants to S.P. Balk (NIH P50 CA090381, NIH P01 CA163227, R01 CA168393, DoD W81XWH-09-1-0435, and W81XWH-16-1-0431), S. Chen (DoD W81XWH-14-1-0126), H. Ye (Prostate Cancer Foundation Young Investigator Award), X. Yuan (R01 CA168393), and A. G. Sowalsky (supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute).
Footnotes
Conflicts of Interest: S. Chen, N.S. Gray, and S.P. Balk have joint patents on BMX-IN-1 and related BMX inhibitors. S.P. Balk has consulted for Sanofi and Pfizer. H. Ye has consulted for Janssen.
References
- 1.Qiu Y, Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19(49):5651–61. doi: 10.1038/sj.onc.1203958. [DOI] [PubMed] [Google Scholar]
- 2.Chott A, Sun Z, Morganstern D, Pan J, Li T, Susani M, et al. Tyrosine kinases expressed in vivo by human prostate cancer bone marrow metastases and loss of the type 1 insulin-like growth factor receptor. Am J Pathol. 1999;155(4):1271–9. doi: 10.1016/S0002-9440(10)65229-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rajantie I, Ekman N, Iljin K, Arighi E, Gunji Y, Kaukonen J, et al. Bmx tyrosine kinase has a redundant function downstream of angiopoietin and vascular endothelial growth factor receptors in arterial endothelium. Mol Cell Biol. 2001;21(14):4647–55. doi: 10.1128/MCB.21.14.4647-4655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson D, He F, Pretlow T, Kung HJ. A tyrosine kinase profile of prostate carcinoma. Proc Natl Acad Sci U S A. 1996;93(12):5958–62. doi: 10.1073/pnas.93.12.5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamagnone L, Lahtinen I, Mustonen T, Virtaneva K, Francis F, Muscatelli F, et al. BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene. 1994;9(12):3683–8. [PubMed] [Google Scholar]
- 6.Qiu Y, Robinson D, Pretlow TG, Kung HJ. Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95(7):3644–9. doi: 10.1073/pnas.95.7.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen R, Kim O, Li M, Xiong X, Guan JL, Kung HJ, et al. Regulation of the PH-domain-containing tyrosine kinase Etk by focal adhesion kinase through the FERM domain. Nat Cell Biol. 2001;3(5):439–44. doi: 10.1038/35074500. [DOI] [PubMed] [Google Scholar]
- 8.He Y, Luo Y, Tang S, Rajantie I, Salven P, Heil M, et al. Critical function of Bmx/Etk in ischemia-mediated arteriogenesis and angiogenesis. J Clin Invest. 2006;116(9):2344–55. doi: 10.1172/JCI28123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang R, Xu Y, Ekman N, Wu Z, Wu J, Alitalo K, et al. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. 2003;278(51):51267–76. doi: 10.1074/jbc.M310678200. [DOI] [PubMed] [Google Scholar]
- 10.Pan S, An P, Zhang R, He X, Yin G, Min W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: role in endothelial cell migration and angiogenesis. Mol Cell Biol. 2002;22(21):7512–23. doi: 10.1128/MCB.22.21.7512-7523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottar-Guillier M, Dodeller F, Huesken D, Iourgenko V, Mickanin C, Labow M, et al. The tyrosine kinase BMX is an essential mediator of inflammatory arthritis in a kinase-independent manner. J Immunol. 2011;186(10):6014–23. doi: 10.4049/jimmunol.1002813. [DOI] [PubMed] [Google Scholar]
- 12.Tu T, Thotala D, Geng L, Hallahan DE, Willey CD. Bone marrow X kinase-mediated signal transduction in irradiated vascular endothelium. Cancer Res. 2008;68(8):2861–9. doi: 10.1158/0008-5472.CAN-07-5743. [DOI] [PubMed] [Google Scholar]
- 13.Jiang T, Guo Z, Dai B, Kang M, Ann DK, Kung HJ, et al. Bi-directional regulation between tyrosine kinase Etk/BMX and tumor suppressor p53 in response to DNA damage. J Biol Chem. 2004;279(48):50181–9. doi: 10.1074/jbc.M409108200. [DOI] [PubMed] [Google Scholar]
- 14.Kim O, Yang J, Qiu Y. Selective activation of small GTPase RhoA by tyrosine kinase Etk through its pleckstrin homology domain. J Biol Chem. 2002;277(33):30066–71. doi: 10.1074/jbc.M201713200. [DOI] [PubMed] [Google Scholar]
- 15.Bagheri-Yarmand R, Mandal M, Taludker AH, Wang RA, Vadlamudi RK, Kung HJ, et al. Etk/Bmx tyrosine kinase activates Pak1 and regulates tumorigenicity of breast cancer cells. J Biol Chem. 2001;276(31):29403–9. doi: 10.1074/jbc.M103129200. [DOI] [PubMed] [Google Scholar]
- 16.Chau CH, Chen KY, Deng HT, Kim KJ, Hosoya K, Terasaki T, et al. Coordinating Etk/Bmx activation and VEGF upregulation to promote cell survival and proliferation. Oncogene. 2002;21(57):8817–29. doi: 10.1038/sj.onc.1206032. [DOI] [PubMed] [Google Scholar]
- 17.Semaan N, Alsaleh G, Gottenberg JE, Wachsmann D, Sibilia J. Etk/BMX, a Btk family tyrosine kinase, and Mal contribute to the cross-talk between MyD88 and FAK pathways. J Immunol. 2008;180(5):3485–91. doi: 10.4049/jimmunol.180.5.3485. [DOI] [PubMed] [Google Scholar]
- 18.Potter DS, Galvin M, Brown S, Lallo A, Hodgkinson CL, Blackhall F, et al. Inhibition of PI3K/BMX Cell Survival Pathway Sensitizes to BH3 Mimetics in SCLC. Mol Cancer Ther. 2016;15(6):1248–60. doi: 10.1158/1535-7163.MCT-15-0885. [DOI] [PubMed] [Google Scholar]
- 19.Fox JL, Storey A. BMX Negatively Regulates BAK Function, Thereby Increasing Apoptotic Resistance to Chemotherapeutic Drugs. Cancer Res. 2015;75(7):1345–55. doi: 10.1158/0008-5472.CAN-14-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Potter DS, Kelly P, Denneny O, Juvin V, Stephens LR, Dive C, et al. BMX acts downstream of PI3K to promote colorectal cancer cell survival and pathway inhibition sensitizes to the BH3 mimetic ABT-737. Neoplasia. 2014;16(2):147–57. doi: 10.1593/neo.131376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paavonen K, Ekman N, Wirzenius M, Rajantie I, Poutanen M, Alitalo K. Bmx tyrosine kinase transgene induces skin hyperplasia, inflammatory angiogenesis, and accelerated wound healing. Mol Biol Cell. 2004;15(9):4226–33. doi: 10.1091/mbc.E04-03-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dai B, Kim O, Xie Y, Guo Z, Xu K, Wang B, et al. Tyrosine kinase Etk/BMX is up-regulated in human prostate cancer and its overexpression induces prostate intraepithelial neoplasia in mouse. Cancer Res. 2006;66(16):8058–64. doi: 10.1158/0008-5472.CAN-06-1364. [DOI] [PubMed] [Google Scholar]
- 23.Dai B, Chen H, Guo S, Yang X, Linn DE, Sun F, et al. Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 2010;70(13):5587–96. doi: 10.1158/0008-5472.CAN-09-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guryanova OA, Wu Q, Cheng L, Lathia JD, Huang Z, Yang J, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19(4):498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xie Y, Xu K, Dai B, Guo Z, Jiang T, Chen H, et al. The 44 kDa Pim-1 kinase directly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells from apoptosis induced by chemotherapeutic drugs. Oncogene. 2006;25(1):70–8. doi: 10.1038/sj.onc.1209058. [DOI] [PubMed] [Google Scholar]
- 26.Saharinen P, Ekman N, Sarvas K, Parker P, Alitalo K, Silvennoinen O. The Bmx tyrosine kinase induces activation of the Stat signaling pathway, which is specifically inhibited by protein kinase Cdelta. Blood. 1997;90(11):4341–53. [PubMed] [Google Scholar]
- 27.Tsai YT, Su YH, Fang SS, Huang TN, Qiu Y, Jou YS, et al. Etk, a Btk family tyrosine kinase, mediates cellular transformation by linking Src to STAT3 activation. Mol Cell Biol. 2000;20(6):2043–54. doi: 10.1128/mcb.20.6.2043-2054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen EJ, Sowalsky AG, Gao S, Cai C, Voznesensky O, Schaefer R, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015;21(6):1273–80. doi: 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ware KE, Garcia-Blanco MA, Armstrong AJ, Dehm SM. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr Relat Cancer. 2014;21(4):T87–T103. doi: 10.1530/ERC-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yuan X, Cai C, Chen S, Chen S, Yu Z, Balk SP. Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis. Oncogene. 2014;33(22):2815–25. doi: 10.1038/onc.2013.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15(12):701–11. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee LF, Guan J, Qiu Y, Kung HJ. Neuropeptide-induced androgen independence in prostate cancer cells: roles of nonreceptor tyrosine kinases Etk/Bmx, Src, and focal adhesion kinase. Mol Cell Biol. 2001;21(24):8385–97. doi: 10.1128/MCB.21.24.8385-8397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang X, Borgesi RA, McKnight NC, Kaur R, Carpenter CL, Balk SP. Activation of nonreceptor tyrosine kinase Bmx/Etk mediated by phosphoinositide 3-kinase, epidermal growth factor receptor, and ErbB3 in prostate cancer cells. J Biol Chem. 2007;282(45):32689–98. doi: 10.1074/jbc.M703412200. [DOI] [PubMed] [Google Scholar]
- 34.Weber MJ, Gioeli D. Ras signaling in prostate cancer progression. J Cell Biochem. 2004;91(1):13–25. doi: 10.1002/jcb.10683. [DOI] [PubMed] [Google Scholar]
- 35.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161(5):1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S, Jiang X, Gewinner CA, Asara JM, Simon NI, Cai C, et al. Tyrosine kinase BMX phosphorylates phosphotyrosine-primed motif mediating the activation of multiple receptor tyrosine kinases. Sci Signal. 2013;6(277):ra40. doi: 10.1126/scisignal.2003936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22(3):298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson D, Van Allen Eliezer M, Wu Y-M, Schultz N, Lonigro Robert J, Mosquera J-M, et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell. 2015;161(5):1215–28. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang X, Coleman IM, Brown LG, True LD, Kollath L, Lucas JM, et al. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2015;21(20):4698–708. doi: 10.1158/1078-0432.CCR-15-0157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahdesmaki MJ, Gray SR, Johnson JH, Lai Z. Disambiguate: An open-source application for disambiguating two species in next generation sequencing data from grafted samples. F1000Res. 2016;5:2741. doi: 10.12688/f1000research.10082.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lam HM, McMullin R, Nguyen HM, Coleman I, Gormley M, Gulati R, et al. Characterization of an Abiraterone Ultraresponsive Phenotype in Castration-Resistant Prostate Cancer Patient-Derived Xenografts. Clin Cancer Res. 2017;23(9):2301–12. doi: 10.1158/1078-0432.CCR-16-2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dai B, Chen H, Guo S, Yang X, Linn DE, Sun F, et al. Compensatory upregulation of tyrosine kinase Etk/BMX in response to androgen deprivation promotes castration-resistant growth of prostate cancer cells. Cancer Res. 2010;70(13):5587–96. doi: 10.1158/0008-5472.CAN-09-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo W, Liu R, Bhardwaj G, Ma AH, Changou C, Yang JC, et al. CTA095, a Novel Etk and Src Dual Inhibitor, Induces Apoptosis in Prostate Cancer Cells and Overcomes Resistance to Src Inhibitors. PLoS One. 2013;8(8):e70910. doi: 10.1371/journal.pone.0070910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo W, Liu R, Bhardwaj G, Yang JC, Changou C, Ma AH, et al. Targeting Btk/Etk of prostate cancer cells by a novel dual inhibitor. Cell Death Dis. 2014;5:e1409. doi: 10.1038/cddis.2014.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu F, Zhang X, Weisberg E, Chen S, Hur W, Wu H, et al. Discovery of a selective irreversible BMX inhibitor for prostate cancer. ACS Chem Biol. 2013;8(7):1423–8. doi: 10.1021/cb4000629. [DOI] [PubMed] [Google Scholar]
- 46.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15(2):954–63. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol. 2001;8(12):1058–63. doi: 10.1038/nsb721. [DOI] [PubMed] [Google Scholar]
- 48.van Leenders GJ, Sookhlall R, Teubel WJ, de Ridder CM, Reneman S, Sacchetti A, et al. Activation of c-MET induces a stem-like phenotype in human prostate cancer. PLoS One. 2011;6(11):e26753. doi: 10.1371/journal.pone.0026753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 51.Cai C, He HH, Chen S, Coleman I, Wang H, Fang Z, et al. Androgen receptor gene expression in prostate cancer is directly suppressed by the androgen receptor through recruitment of lysine-specific demethylase 1. Cancer Cell. 2011;20(4):457–71. doi: 10.1016/j.ccr.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, et al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell reports. 2014;9(5):1618–27. doi: 10.1016/j.celrep.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cai C, Wang H, Xu Y, Chen S, Balk SP. Reactivation of androgen receptor-regulated TMPRSS2:ERG gene expression in castration-resistant prostate cancer. Cancer Res. 2009;69(15):6027–32. doi: 10.1158/0008-5472.CAN-09-0395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao S, Ye H, Gerrin S, Wang H, Sharma A, Chen S, et al. ErbB2 Signaling Increases Androgen Receptor Expression in Abiraterone-Resistant Prostate Cancer. Clin Cancer Res. 2016;22(14):3672–82. doi: 10.1158/1078-0432.CCR-15-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.