

Abstract
BACKGROUND:
In genome-wide screening studies for de novo mutations underlying autism and intellectual disability, mutations in the ADNP gene are consistently reported among the most frequent. ADNP mutations have been identified in children with autism spectrum disorder comorbid with intellectual disability, distinctive facial features, and deficits in multiple organ systems. However, a comprehensive clinical description of the Helsmoortel-Van der Aa syndrome is lacking.
METHODS:
We identified a worldwide cohort of 78 individuals with likely disruptive mutations in ADNP from January 2014 to October 2016 through systematic literature search, by contacting collaborators, and through direct interaction with parents. Clinicians filled in a structured questionnaire on genetic and clinical findings to enable correlations between genotype and phenotype. Clinical photographs and specialist reports were gathered. Parents were interviewed to complement the written questionnaires.
RESULTS:
We report on the detailed clinical characterization of a large cohort of individuals with an ADNP mutation and demonstrate a distinctive combination of clinical features, including mild to severe intellectual disability, autism, severe speech and motor delay, and common facial characteristics. Brain abnormalities, behavioral problems, sleep disturbance, epilepsy, hypotonia, visual problems, congenital heart defects, gastrointestinal problems, short stature, and hormonal deficiencies are common comorbidities. Strikingly, individuals with the recurrent p.Tyr719* mutation were more severely affected.
CONCLUSIONS:
This overview defines the full clinical spectrum of individuals with ADNP mutations, a specific autism subtype. We show that individuals with mutations in ADNP have many overlapping clinical features that are distinctive from those of other autism and/or intellectual disability syndromes. In addition, our data show preliminary evidence of a correlation between genotype and phenotype.
Keywords: ADNP, Autism, Genetics, Helsmoortel-Van der Aa syndrome, Intellectual disability, Neurodevelopmental disorder
Autism spectrum disorder (ASD) is a condition defined by deficits in social interaction, communication, and selected behaviors (1). Each aspect of the disorder may vary in presentation, range, and severity, cumulating in a broad clinical spectrum. The frequency of the disorder is under continuous debate, but ASD may affect up to 1.5% of the population (2). Although a genetic contribution to its etiology has been firmly demonstrated (3), it took the introduction of trio-based whole-exome sequencing to truly accelerate substantially the identification of ASD genes. In these studies, individuals are screened along with their parents, enabling the unbiased detection of de novo mutations in large ASD cohorts (4–6). These initiatives are complemented by targeted resequencing of larger cohorts (7). Studies in ASD cohorts comorbid with intellectual disability (ID) collectively demonstrate an unprecedented genetic heterogeneity of ASD, with no single gene responsible for more than a fraction of the total population. Several of the identified genes appear to cluster in a subset of cellular networks, including networks enriched for chromatin remodeling and synaptic functioning (5,8). Overlap between ASD genes and genes causative for other neurodevelopmental disorders, including ID and seizures, is common (9,10).
Despite the high heterogeneity and observed molecular overlap, there is preliminary evidence for the existence of clinical ASD subtypes. For instance, mutations in the chromatin remodeler CHD8 cause an ASD/ID subtype with specific physical characteristics, such as macrocephaly and significant gastrointestinal problems (11,12). In contrast, individuals with a mutation in DYRK1A, a gene duplicated in Down syndrome, have ASD/ID, microcephaly, intrauterine growth retardation, febrile seizures in infancy, impaired speech, stereotypic behavior, hypertonia, and a distinctive facial gestalt (13). Yet the clinical delineation of ASD/ID syndromes has lagged behind their respective molecular definition. Because possible future treatment may be based on targeting the underlying molecular defect rather than on the basis of the clinical presentation, it is of primary importance to define autism subtypes correctly at the molecular level (14).
ADNP was one of the most frequently mutated genes across multiple recent whole-exome sequencing and targeted molecular inversion probe sequencing studies in ASD/ID cohorts (6,7). The ADNP gene plays a role in embryonic development, especially during the time of neuronal tube closure, and is involved in chromatin remodeling (15–18). Based on the first 10 individuals identified with ADNP-related ASD/ID, ADNP mutations were estimated to explain one or two per 1000 ASD/ ID cases. Some shared clinical features were suggested (19). Since that time, a number of case reports have expanded the phenotype of the Helsmoortel-Van der Aa syndrome (Online Mendelian Inheritance in Man [OMIM] identification 615873) (20–23). Here, we describe the clinical details of a cohort of 78 individuals from 16 countries with a likely disruptive mutation in ADNP. We define a novel subtype of ASD/ID, and at the same time we present evidence for a significant genotype-phenotype correlation.
METHODS AND MATERIALS
Participants
The study was performed at the University of Antwerp, Belgium. Individuals were identified through exome sequencing in our own center or gathered from genetic centers worldwide that offer exome-wide or targeted genetic screening in a clinical or a research setting. Additional individuals were collected on the website http://humandiseasegenes.nl/adnp/. A minority of the individuals were previously described in case reports (19–23). All individuals were enrolled between January 1,2014, and October 1, 2016. Inclusion required a clinical geneticist-confirmed diagnosis of a nonsense or frameshift mutation in the ADNP gene and presence of clinical information in at least three of the following domains: demographics, development, craniofacial features, and behavior. Essentially all mutations were identified by next-generation sequencing of individuals with autism and/or developmental delay, often in combination with additional syndromic features. In part, the ADNP mutations were identified in individuals in preassembled ASD/ID cohorts that were subjected to trio-based whole-exome sequencing or targeted molecular inversion probe sequencing as described in Helsmoortel et al. (19). The remainder of our cohort was assembled from the individuals in whom an ADNP mutation was diagnosed after genetic testing using either neurodevelopmental gene panels or trio-based whole-exome sequencing. After the identification of a causative ADNP mutation in an individual, the patient’s clinical geneticist asked for consent to be included in this study. In each case, the mutation that was identified using next-generation sequencing was independently verified using Sanger sequencing either in our own or in the referring laboratory. Individuals carrying a missense mutation in ADNP were excluded from this study. All gene annotations have been made according to reference sequence NM_015339.2 (human genome version 19). Approval for this study was obtained from the Ethics Committee of the Antwerp University Hospital. Pictures were published only if the parents provided written informed consent on behalf of their child.
Procedures
Collaborating physicians were asked to fill out an extensive questionnaire with clinical and molecular information about the individuals they had identified and assessed. We specifically asked for the results of the test the individuals had been given, including but not limited to IQ test and Autism Diagnostic Observation Schedule. Medical specialist reports and magnetic resonance imaging data were collected and systematically reevaluated to refine the interpretation of the findings. To compare the data that were collected in various parts of the world, which did not in all cases use the same tests and terminology, we curated all incoming data and recontacted the collaborating clinicians to harmonize the medical information. The ADNPkids Facebook community (24) helped us contact clinicians and parents so we could complete and verify the details of the clinical information.
Statistical Analysis
Associations between reported clinical features were systematically tested in a pairwise analysis using one-way analysis of variance (ANOVA), Pearson correlation, and Fisher’s exact tests, depending on the nature of the variables. A listing of all 170 variables included in our analysis is provided in Supplemental Table S1. For one-way ANOVA, features for which only a single level was available were excluded. If ANOVA resulted in significant results (p < .05), post hoc Tukey honest significant difference testing was applied to identify significant differences in mean. For Fisher’s exact tests, a minimum of two levels per tested category and >10 records per tested condition were required. If either category contained three or more levels, p values were calculated via a Monte Carlo simulation using 10,000 replicates. The association between demographic features, including gender, age, and clinical features, was analyzed similarly. Additionally, we evaluated the presence of genotype-phenotype correlations. First, the three most frequent mutations were analyzed separately: p.Tyr719* (17 individuals), p.Leu831Ilefs*82/ p.Asn832Lysfs*81 (14 individuals), and p.Arg730* (5 individuals). Subsequently, mutations were grouped according to gene location: in the N terminus (25 individuals), at the center of the gene (49 individuals), and in the C terminus (4 individuals). Finally, we analyzed mutations per domain. For each analysis, prevalence or extent of all individual clinical features was compared between the selected subcohort and the remaining individuals. Multiple testing correction was performed via the false discovery rate method [Qvalue add-on package in R, version 2.6.0 (25)]. All calculations were performed in the software package in R version 3.3.1 (26). Significant correlations are indicated at the appropriate Results section.
RESULTS
We included 78 individuals with a disruptive mutation in ADNP, including 44 male subjects and 34 female subjects (Figures 1 and 2). The mean age of our cohort is 8 years 2 months, with a range of 1 to 40 years. Individuals were from 44 clinics in 16 countries. Parental consanguinity was not reported, and no sibling was diagnosed with a mutation in ADNP. Five individuals have nonidentical healthy twin siblings. We found 46 unique mutations on the DNA level, of which 25 were nonsense and 21 frameshift (Supplemental Table S2). All but three mutations were located in the fifth and last exon of the ADNP gene and were predicted to escape nonsense-mediated decay. On the protein level, three mutations, including the p.Tyr719* mutation, were present in $5 individuals. Sixty-eight mutations in our cohort were confirmed de novo, eight mutations were of unknown inheritance, and two C-terminal mutations were inherited.
Figure 1.
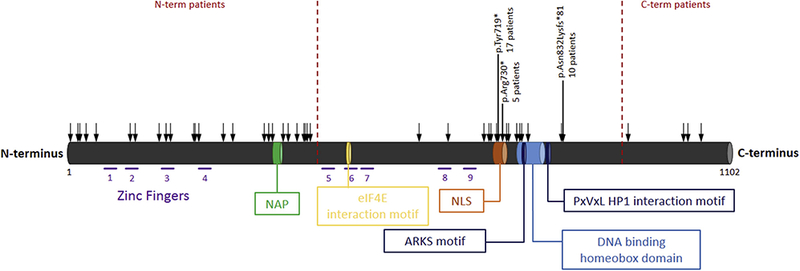
Schematic illustration of ADNP and its functional domains. ADNP consists of five exons and 14 domains, including nine zinc fingers, NAP (a short octapeptide sequence, single letter code, NAPVSIPQ), an eIF4E interaction motif, a nuclear localization signal (NLS), an alanine-arginine-lysine-serine (ARKS) motif, a DNA-binding homeobox domain, and a PxVxL motif (15,17,30). Zinc fingers: AA 74–97, 107–129, 165–188, 221–244, 447–469, 489–510, 512–535, 622–647, 662–686; NAP amino acid (AA) 354–361; eIF4E NAP AA 490–499; NLS AA 716–733; ARKS motif AA 765–768; DNA-binding homeobox domain AA 754–814; PxVxL heterochromatin protein 1 (HP1) interaction motif AA 819–823. Black arrows indicate the location of the mutations in the reported individuals, highlighting the three most frequent mutations.
Figure 2.
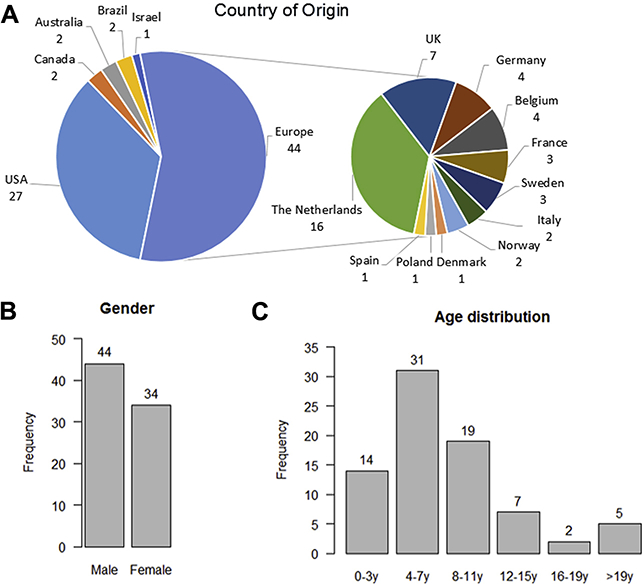
Demographic data of the reported individuals: (A) country of origin, (B) gender, and (C) age distribution.
Pre- and Perinatal Observations and Congenital Abnormalities
Most children were born at term (mean gestational age 38.7 weeks, range 30–42 weeks). Mean maternal and paternal ages at birth were 30 and 32 years, respectively. Intrauterine growth retardation was not reported. Overall, birth weight, height, and head circumference were within normal ranges (Supplemental Table S3, Supplemental Figure S1A-C).
Six individuals (12.5%) were born with renal anomalies (narrow ureters, bilateral vesicoureteral reflux that was surgically repaired) (Table 1). Reported hand and feet abnormalities were nonspecific; they included fetal finger pads, clinodactyly, small fifth fingers, brachydactyly, single palmar crease, sandal gap, pes planus, long or broad halluces, and syndactyly of the second and third toes. Twenty-five percent had nail abnormalities such as thin or small nails, or hypoplastic nails of the fifth digit. Some had widely spaced nipples, pectus excavatum, pectus carinatum, or combined excavatum/carinatum deformity. One child had a submucous cleft palate. Two of the children were born with metopic craniosynostosis, and 1 of them needed surgery. Six children had plagiocephaly, of whom 3 wore a cranial-molding helmet.
Table 1.
Clinical Features of the Reported Individuals With Mutation in the ADNP Gene
| Clinical Features | Sample n/ Total N |
|
|---|---|---|
| General Information | ||
| Age at examination, range (mean) | 1–40 years (8 years 2 mo) |
78/78 |
| Gender, female:male, n | 34:44 | 78/78 |
| Gestational age, weeks | 38.7 | 70/70 |
| Age of father at time of birth, years | 32.1 | 65/65 |
| Age of mother at time of birth, years | 29.8 | 67/67 |
| Mutation Information, % | ||
| De novo ADNP mutation | 97.1 | 68/70 |
| Nonsense mutation | 56.4 | 44/78 |
| Frameshift mutation | 43.6 | 34/78 |
| Growth | ||
| Short stature, < −2 SD, % | 23.2 | 16/69 |
| Neurodevelopmental Features | ||
| Developmental delay/ID, % | 100.0 | 73/73 |
| Mild ID | 12.3 | 9/73 |
| Moderate ID | 35.6 | 26/73 |
| Severe ID | 52.1 | 38/73 |
| Motor delay, % | 95.9 | 71/74 |
| Age at sitting independently, years, Mean |
1.1 | 58/58 |
| Walking independently, % | 86.8 | 66/76 |
| Age at walking independently, years, Mean |
2.5 | 64/64 |
| Speech delay, % | 98.6 | 70/71 |
| Age at first words, years, mean | 2.5 | 49/49 |
| No speech, % | 19.4 | 14/72 |
| Autism spectrum disorder including autistic features, % |
92.8 | 64/69 |
| Attention-deficit/hyperactivity disorder, % |
43.9 | 25/57 |
| Loss of skills, % | 20.3 | 12/59 |
| Bladder training delay, % | 81.1 | 43/53 |
| Feeding and Gastrointestinal Problems, % | 83.3 | 60/72 |
| Gastroesophageal reflux (disease) | 58.5 | 38/65 |
| Constipation | 49.3 | 34/69 |
| Oral movement problems | 45.6 | 26/57 |
| Lack of satiation | 41.5 | 22/53 |
| Problems swallowing liquids | 32.2 | 19/59 |
| Frequent vomiting | 29.5 | 18/61 |
| Aspiration difficulties | 21.4 | 12/56 |
| Gastrostomy tube | 12.7 | 8/63 |
| Obesity | 7.5 | 5/67 |
| Neurological Problems and Behavior, % | ||
| Hypotonia | 78.3 | 54/69 |
| Hypertonia | 3.8 | 3/78 |
| Seizures | 16.2 | 12/74 |
| Cerebral imaging-structural brain Abnormalities |
55.9 | 33/59 |
| Wide ventricles | 29.4 | 15/51 |
| Corpus callosum Underdevelopment |
18.4 | 9/49 |
| Cerebral atrophy | 17.8 | 8/45 |
| Delayed myelination | 8.9 | 4/45 |
| White matter lesions | 7.5 | 4/53 |
| Cortical dysplasia | 3.8 | 2/52 |
| MRI brain abnormalities, unspecified |
36.2 | 17/46 |
| Behavioral problems | 77.6 | 38/49 |
| Temper tantrums/aggression | 83.3 | 20/24 |
| Obsessive-compulsive behavior | 64.0 | 16/25 |
| Mood disorder | 56.3 | 9/16 |
| Self-injurious behavior | 20.0 | 2/10 |
| Insensitivity to pain | 63.6 | 35/55 |
| Sensory processing disorder | 66.7 | 28/42 |
| Sleep problems | 65.2 | 45/69 |
| Visual System, % | 73.6 | 53/72 |
| Strabismus | 49.2 | 31/63 |
| Cerebral visual impairment | 41.2 | 14/34 |
| Hypermetropia | 40.3 | 25/62 |
| Ptosis | 24.2 | 15/62 |
| Nystagmus | 11.7 | 9/77 |
| Myopia | 7.9 | 5/63 |
| Colobomata | 5.6 | 4/72 |
| Ear-Nose-Throat System, % | 32.1 | 25/78 |
| Narrow hearing canal | 87.5 | 7/8 |
| Frequent otitis media | 85.7 | 12/14 |
| Hearing tubes | 73.3 | 11/15 |
| Hearing loss | 11.7 | 7/60 |
| Obstructive sleep apnea syndrome | 6.6 | 5/76 |
| Cardiovascular System, % | 37.7 | 26/69 |
| Atrial septal defect | 15.9 | 11/69 |
| Patent ductus arteriosus | 8.7 | 6/69 |
| Mitral valve prolapse | 5.8 | 4/69 |
| Patent foramen ovale | 5.8 | 4/69 |
| Ventricular septal defect | 4.3 | 3/69 |
| Tetralogy of Fallot | 1.4 | 1/69 |
| Cardiac defect, unspecified | 8.7 | 6/69 |
| Urogenital System, % | 28.0 | 21/75 |
| Cryptorchidism | 34.3 | 12/35 |
| Renal anomalies | 12.5 | 6/48 |
| Small genitalia | 5.4 | 4/74 |
| Endocrine System, % | 24.5 | 12/49 |
| Early puberty | 30.0 | 3/10 |
| Thyroid hormone problems | 15.2 | 7/46 |
| Growth hormone deficiency | 10.9 | 5/46 |
| Musculoskeletal System, % | 54.9 | 39/71 |
| Joint hypermobility | 37.7 | 23/61 |
| Scoliosis | 17.2 | 11/64 |
| Hip problems (hip dysplasia, Perthes’ disease, dislocated hips) |
7.5 | 4/53 |
| Thorax abnormalities | 22.2 | 12/54 |
| Pectus excavatum | 14.8 | 8/54 |
| Pectus carinatum | 5.6 | 3/54 |
| Narrow thorax | 1.9 | 1/54 |
| Abnormal skull shape | 13.9 | 10/72 |
| Plagiocephaly | 8.3 | 6/72 |
| Trigonocephaly | 2.8 | 2/72 |
| Brachycephaly | 4.2 | 3/72 |
| Hand and Foot Abnormalities, % | 62.3 | 43/69 |
| Finger abnormalities (prominent distal phalanges, prominent interphalangeal joints, polydactyly, interdigital webbing, 2–3 toe syndactyly, fifth finger clinodactyly, small fifth finger or absent distal phalanx of fifth finger, tapering fingers, brachydactyly, broad fingers, fetal fingertip pads) |
46.3 | 31/67 |
| Single palmar crease | 10.8 | 7/65 |
| Nail anomalies | 25.0 | 14/56 |
| Sandal gap | 19.6 | 11/56 |
| Toe abnormalities (broad halluces, 2–3 toe syndactyly, brachydactyly) |
10.8 | 7/65 |
| Other, % | ||
| Early teeth | 71.1 | 32/45 |
| Frequent infections | 50.7 | 35/69 |
| Widely spaced nipples | 20.4 | 11/54 |
| Umbilical/inguinal hernia | 8.5 | 5/59 |
ID, intellectual disability; MRI, magnetic resonance imaging.
Failure to thrive in early childhood was noted in a number of individuals. Some of them appeared to have severe cardiac problems, requiring open heart surgery. Thirty-eight percent had one or more congenital cardiac defects. These were diverse: atrial septal defect, patent ductus arteriosus, patent foramen ovale, mitral valve prolapse, ventricular septal defect, and other cardiovascular malformations such as a right aortic arch, dysplastic aortic valve, tetralogy of Fallot, ductus arteriosus aneurysm, quadricuspidal aortic valve, aortic ectasia, and a mild pulmonary valve stenosis were found (Figure 3A).
Figure 3.
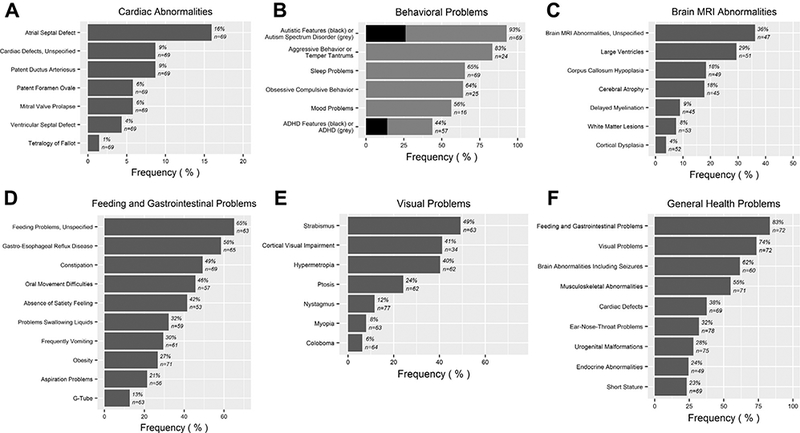
Clinical features reported in individuals with ADNP mutation: (A) cardiac abnormalities, (B) behavioral problems, (C) brain magnetic resonance imaging (MRI) abnormalities, (D) feeding and gastrointestinal problems, (E) visual problems, and (F) general health problems. ADHD, attention-deficit/ hyperactivity disorder.
Facial Appearance
Individuals shared similar facial features, including a prominent forehead with a high anterior hairline, a wide and depressed nasal bridge, and a short nose with full, upturned nasal tip (Figure 4, Supplemental Table S4). One third of the individuals had downslanted palpebral fissures and prominent eyelashes. Ear malformations were observed in nearly half of individuals. Abnormalities included small or dysplastic, low-set, and posteriorly rotated ears. The philtrum was long in 39.3% of study cohort. Seventy percent of individuals had a thin upper lip, often combined with an everted lower lip and a pointed chin that appeared more pronounced at younger age (Figure 5). One third have widely spaced teeth.
Figure 4.

Facial features of individuals with mutations in ADNP. Frontal and lateral views. Note the prominent forehead with high anterior hairline, the wide and depressed nasal bridge, and short nose with full, upturned nasal tip. Informed consent has been obtained for publication of all images present in this paper. (Individual numbers from Supplemental Table S2 corresponding to the pictures: A = 49, B = 34, C = 44, D = 21, E = 17, F = 63, G = 28, H = 29, I = 45, J = 11, K = 38, L = 15, M = 48, N = 50, O = 60, P = 36, Q = 58, R = 33, S = 51, T = 39, U = 42, V = 31, W = 41, X = 10, Y = 70, Z = 27).
Figure 5.
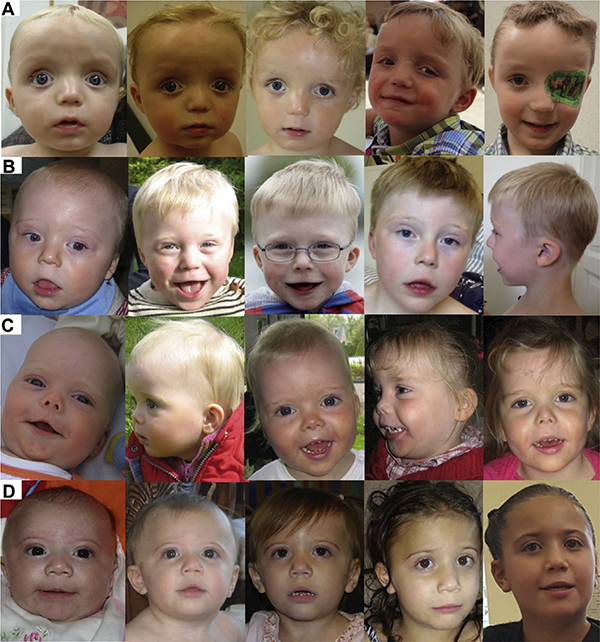
Individuals at different ages showing evolution with age. (A) Individual 1 at 10 months, 15 months, 29 months, 4 years, and 6 years of age; (B) individual 32 at 13 months, 26 months, 3 years 10 months, 5 years 11 months, 5 years 11 months of age; (C) individual 40 at 4 months, 13 months, 13 months, 3 years 6 months, 3 years 6 months of age; (D) individual 65 at 3 months, 10 months, 24 months, 4 years 9 months, 8 years 9 months of age. Informed consent has been obtained for publication of all images present in this paper.
Growth and Endocrine System
Twenty-three percent of the individuals had short stature (height < −2 SD, range 2–23 years old) (Supplemental Table S3, Supplemental Figure S1E). Nine individuals had hormonal deficiencies (Table 1). Of these, 2 had isolated growth hormone deficiency, 4 had hypothyroidism, and 3 a combination of both hormonal deficiencies. One 29-year-old woman had a narrow thorax with breast hypoplasia. Signs of early puberty were present in 3 of 10 individuals older than 6 years for whom information was available; 1 boy and 1 girl had pubic hair growth at the age of 7 and 8 years, respectively, and 1 girl had menarche at 8 years of age.
Development and Neurology
Fifty-two percent of the individuals in this cohort presented with severe ID at the age of assessment, 36% had a moderate disability, and 12% had a mild disability. Developmental delay was present in all individuals, with motor delay being one of the key features. The average age to sit up independently was 12.8 months [cohort range 6–60 months, normal range 4–9 months (27)] (Supplemental Figure S2A). Delayed age of walking independently [after 18 months of age (27)] (Supplemental Figure S2B) was observed in 86.8% of the children, with an average age of 2 years 5.5 months (cohort range 15–72 months).
Interestingly, individuals with a p.Tyr719* mutation started walking at a mean of 3.5 years, significantly later than the 2 years 2 months of the remainder of the cohort (p < .0001, oneway ANOVA). Seventy-eight percent of the children had hypotonia, while hypertonia was present in 3 children. Standing unassisted for long periods of time or walking long distances is difficult for many of the children. The walking pattern can be abnormal (e.g., broad-based or tiptoe gait, foot slap). Six children learned to walk with support between 5.5 and 8 years of age, after many years of physiotherapy. A minority were not able to walk at the time of last evaluation.
Another key feature was speech delay, which presented in 98.6% of individuals. The mean age of first words was 30 months (cohort range 7–72 months, as opposed to a normal range of 12–18 months) (Supplemental Figure S2C). Nineteen percent had no language development at all. Apparent loss of acquired abilities was reported in 12 children for skills such as speaking, counting, riding a bicycle, or being toilet trained. Eighty-one percent of the children had a considerable delay in bladder training and many were still not toilet trained when approaching puberty.
Sixteen percent had seizures, including absence seizures, focal seizures with reduced awareness, epilepsy with continuous spike and waves during slow-wave sleep, or unclassified seizures. At least 5 children were reported to have breathholding spells. Some of them were hospitalized for multiple cyanotic episodes causing an acute life-threatening event.
Autistic Features, Behavior, and Sleep
Ninety-three percent of the individuals presented with autistic features (Figure 3B). Sixty-seven percent of them were reported to have a clinical diagnosis of ASD. They had a strong sensory interest, illustrated by putting fingers or objects in their mouth or being attracted to lights or water. Repetitive use of objects, hand and finger mannerisms, and stereotyped movements such as rocking back and forth or hand flapping were common. Some presented with echolalia. Sixty-seven percent had also been diagnosed with sensory processing disorder. A high pain threshold was reported in 63.6% of individuals. Interestingly, all individuals with a p.Tyr719* mutation are included in this group (p = 0.0003, Fisher’s exact test).
Although parents report that 88% of the children were overall happy and friendly, behavioral problems were reported in 77.6% of them. Several presented with obsessive-compulsive behavior, mood disorder, a high anxiety level, temper tantrums, self-injurious behavior, and (verbally) aggressive behavior. Forty-four percent of the individuals were hyperactive or easily distracted. About one third of them had a diagnosis of attention-deficit/hyperactivity disorder. Several individuals were taking behavior-regulating medication such as methylphenidate or atypical antipsychotics such as risperidone or olanzapine to help control behavioral disturbances, particularly aggression.
Sleep problems were present in 65.2%. Some individuals were extremely anxious, with struggles falling asleep and frequent nighttime awakenings. Some were treated with melatonin. Many individuals had a low daytime activity level or excessive daytime sleepiness; a minority had sleep apnea.
Cerebral Imaging
In this cohort, magnetic resonance imaging of the brain was performed in 75.6% of the individuals. Fifty-six percent of them appeared to have cerebral abnormalities, including atypical white matter lesions, delayed myelination, cortical dysplasia or atrophy, perinatal hypoxic ischemic encephalopathy, hydrocephalus, and hippocampal hypoplasticity (Figure 3C).
Magnetic resonance images of 5 individuals were studied in detail. The following abnormalities were seen in multiple individuals: underdevelopment of the frontal lobes with simplified gyral pattern of the cortex and occasional hypoplasia of the bulbus olfactorius and chiasma opticum; a thin and/or short, underdeveloped corpus callosum and inferior vermis hypoplasia; abnormal, often asymmetric opercularization of the Sylvian fissure with sometimes abnormal overlying cortex; dilatation of the lateral ventricles, mostly in the frontal areas; and dilated perivascular spaces of Virchow-Robin in the cerebral white matter (Figure 6).
Figure 6.
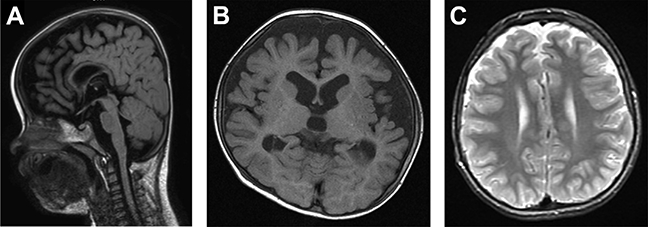
Brain magnetic resonance imaging of a child with a mutation in the ADNP gene. (A) Brain magnetic resonance image of individual 49 performed at 13 months of age, showing generalized and frontal cortical atrophy and a gracile corpus callosum (sagittal, T2 weighted fluid-attenuated inversion recovery). (B) Brain magnetic resonance image of individual 49 performed at 19 months of age, showing frontotemporal atrophy (axial, T1-weighted). (C) Brain magnetic resonance image of individual 45 performed at 12 years of age, showing mild frontal atrophy (axial, T2-weighted).
Gastrointestinal Problems
Eighty-three percent of the individuals had feeding or gastrointestinal problems, mainly gastroesophageal reflux, frequent vomiting, and constipation (Figure 3D). A few had excessive appetite. At the age of assessment, 20.9% of the individuals were overweight and 7.5% were obese, according to standard World Health Organization classification (28). Two individuals had Crohn’s disease, 1 of them with a positive familial history. Oral movement problems, with implications for feeding and speech, were common (45.6%) and were significantly more common in individuals with mutations in the nuclear localization signal and C terminal of this domain (p = .0004, Fisher’s exact test). Problems with drinking liquids or aspiration difficulties were frequent. Eight individuals were fed by gastrostomy tube in early childhood. The individuals suffering from gastrointestinal problems presented more often with sleep disturbances (p = .0005, Fisher’s exact test).
Visual Problems
In 73.6% of the individuals, visual problems, especially hypermetropia (40.3%) and strabismus (49.2%), but also myopia and astigmatism, were present (Figure 3E). Many of these individuals were prescribed eyeglasses. Forty-one percent of the individuals had a diagnosis of cerebral visual impairment. Ophthalmologic defects were diverse: ectropion, coloboma, congenital cataracts, nystagmus. Some had an everted or notched eyelid, or mild ptosis, the latter observed particularly in individuals with mutations in the nuclear localization signal and C terminal of this domain (p = .0004, Fisher’s exact test).
Additional Problems
Musculoskeletal problems were common (Figure 3F). In addition to joint hypermobility, mild scoliosis was present in some individuals. Four had hip problems. Thirty-four percent of the male individuals had unilateral or bilateral cryptorchidism; 2 had bilateral inguinal hernias. Fifty-one percent of the individuals had recurrent infections. Many of the children experienced chronic otitis media requiring ventilation tubes. Some individuals (11.7%) were diagnosed with mild hearing loss in childhood. Two children had hearing aids for sensorineural hearing loss. Ear-nose-throat problems, including narrow ear canals, laryngomalacia, and sleep apnea, were present in 32.1% of the individuals.
DISCUSSION
Individuals with mutations in ADNP present with mild to severe ID, autistic features, and a delay in language and motor development (Table 1). In addition, the syndrome may be accompanied by a wide range of medical conditions, including very frequent (>75%) gastrointestinal and feeding problems, hypotonia, and behavioral disturbances. Frequent comorbidities (50%−75%) include visual problems, brain malformations, sleep disturbances, hand/foot and musculoskeletal abnormalities, and frequent infections. Common (25%−50%) associated features include congenital heart disease, otorhinolaryngologic problems, and urogenital defects. Up to 25% of individuals have hormonal deficiencies, short stature, or seizures. The clinical symptoms of Helsmoortel-Van der Aa syndrome show partial overlap with other genetic syndromes that include developmental delay and ASD, as evidenced by genetic testing of our cohort for disorders such as Angelman, Prader-Willi, Kleefstra, Smith-Magenis, or Rett syndromes prior to the diagnosis of an ADNP mutation. As we did not have access to the full clinical data of all individuals in the screening cohorts from which our cohort was assembled, we cannot determine to what extent a possible ascertainment bias has influenced the clinical presentation of the syndrome.
A striking element is the presence of mutational hot spots. The p.Tyr719*, p.Leu831Ilefs*82/p.Asn832Lysfs*81, and p.Arg730* mutations each occurred independently in $5 individuals. Interestingly, we found evidence for a genotype phenotype correlation. We noticed, for instance, that individuals with a p.Tyr719* mutation walked later and had a higher pain threshold than the individuals with other mutations. Individuals with mutations in the C terminal of the nuclear localization signal domain more often had ptosis or oral movement problems than individuals with mutation elsewhere in the gene. Our findings encourage further investigations on larger study cohorts to unveil possible additional genotype-phenotype correlations. We did not find any evidence for gender-, IQ-level- or age-specific correlations.
Social media is increasingly used by parents to connect with one another and with scientists. This has been the case for this syndrome (24). These interactions helped us to collect genetic and clinical information and the parents’ experiences, providing us with important new insights into symptoms, daily struggles, and challenges. While consensus has to grow to determine what level of evidence is required to include parental observations of this type in a scientific publication, some of these hypotheses have been successfully tested in follow-up studies. As an example, the recently reported early teething in individuals with an ADNP mutation started as a parental observation (29).
Conclusions
Through a careful and structured comparison of the clinical symptoms of 78 individuals with a mutation in the ADNP gene, we delineated the clinical presentation of this specific subtype of autism. Our synthesis is indispensable in the decisionmaking process for caretakers and relatives. Moreover, it will significantly improve the interpretation of the clinical relevance of novel rare variants in the gene. The main limitation of our study is the relatively young age of our study cohort. Longterm follow-up studies are necessary to define the developmental path of individuals with a mutation in ADNP. While to date most cases have been found on a genotype-first basis, a specific combination of features such as ID, ASD, speech and motor delay, and additional problems may emerge to screen for ADNP mutations in cohorts including older individuals. Finally, this clinical delineation can be used to monitor effects of potential future treatment, when available.
Supplementary Material
ACKNOWLEDGMENTS AND DISCLOSURES
This work was supported by grants from the European Research Area Networks Network of European Funding for Neuroscience Research through the Research Foundation-Flanders and the Chief Scientist Office-Ministry of Health (to RFK, GV, IG). This research was supported, in part, by grants from the Simons Foundation Autism Research Initiative (Grant No. SFARI 303241 to EEE) and National Institutes of Health (Grant No. R01MH101221 to EEE). This work was also supported by the Italian Ministry of Health and ‘5 per mille’ funding (to CR).
For many individuals, sequencing was provided by research initiatives like the Care4Rare Research Consortium in Canada or the Deciphering Developmental Disorders (DDD) study in the UK. The DDD Study presents independent research commissioned by the Health Innovation Challenge Fund (Grant No. HICF-1009–003), a parallel funding partnership between the Wellcome Trust and the Department of Health, and the Wellcome Trust Sanger Institute (Grant No. WT098051). The views expressed in this publication are those of the author(s) and not necessarily those of the Wellcome Trust or the Department of Health. The study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South Research Ethics Committee, and GEN/284/12 granted by the Republic of Ireland Research Ethics Committee). The research team acknowledges the support of the National Institute for Health Research, through the Comprehensive Clinical Research Network.
We are sincerely thankful to all the individuals and their parents and to the ADNPkids Facebook community. We acknowledge Maarten Lequin, Berten Ceulemans, Sarah Weckhuysen, An Boudewyns, Elena Levtchenko, Miek Claes, Patricia Van de Walle, and Ann Hallemans for clinical advice.
IG is Chief Scientific Officer of Coronis Neurosciences, Israel, a pharmaceutical company developing drugs for Helsmoortel-Van der Aa syndrome. EEE is an investigator of the Howard Hughes Medical Institute. He is on the Scientific Advisory Board of DNAnexus, Inc. GVis a postdoctoral fellowofthe Research Foundation-Flanders. The remaining authors report no biomedical financial interests or potential conflicts of interest.
Appendix
ADNP Consortium Members:
Madhura Bakshi, Department of Genetic Medicine, Westmead Hospital, Sydney, Australia; Meredith Wilson, Department of Clinical Genetics, Children’s Hospital at Westmead, Westmead, Australia; Yemina Berman, Department of Clinical Genetics, Royal North Shore Hospital, Sydney, New South Wales, Australia; Rebecca Dickson, Department of Clinical Genetics, Royal North Shore Hospital, Sydney, New South Wales, Australia; Erik Fransen, Center for Medical Genetics, Antwerp University Hospital and University of Antwerp, Antwerp, Belgium; StatUa Center for Statistics, University of Antwerp, Antwerp, Belgium; Céline Helsmoortel, Department of Medical Genetics, University of Antwerp, Edegem, Belgium; Jenneke Van den Ende, Department of Medical Genetics, University and University Hospital of Antwerp, Edegem, Belgium; Nathalie Van der Aa, Department of Medical Genetics, University of Antwerp, Edegem, Belgium; Marina J. van de Wijdeven, Department of Medical Genetics, University of Antwerp, Edegem, Belgium; Jessica Rosenblum, Department of Medical Genetics, University of Antwerp, Edegem, Belgium; Fabiola Monteiro, Mendelics Genomic Analysis, São Paulo, Department of Medical Genetics, Campinas State University, Campinas, São Paulo, Brazil; Fernando Kok, Mendelics Genomic Analysis, São Paulo, Brazil, Department of Neurology, University of São Paulo, São Paulo, Brazil; Nada Quercia, Division of Clinical and Metabolic Genetics, Hospital for Sick Children, Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada; Sarah Bowdin, Division of Clinical and Metabolic Genetics, Hospital for Sick Children, Toronto, Ontario, Canada; David Dyment, Department of Genetics, Children’s Hospital of Eastern Ontario, Ottawa, Canada; David Chitayat, The Prenatal Diagnosis and Medical Genetics Program, Department of Obstetrics and Gynecology, Mount Sinai Hospital and Division of Clinical and Metabolic Genetics, Department of Pediatrics, Hospital for Sick Children, University of Toronto, Toronto, Ontario, Canada; Ebba Alkhunaizi, The Prenatal Diagnosis and Medical Genetics Program, Department of Obstetrics and Gynecology, Mount Sinai Hospital and Division of Clinical and Metabolic Genetics, Department of Pediatrics, Hospital for Sick Children, University of Toronto, Toronto, Ontario, Canada; Susanne E. Boonen, Clinical Genetic Unit, Department of Paediatrics, Zealand University Hospital, Roskilde Department of Clinical Genetics, Aarhus University Hospital, Aarhus, Denmark; Boris Keren, Departement de Genetique, Hopital de la Pitie-Salpetriere, Assistance Publique-Hopitaux de Paris, Paris Sorbonne Universites, Universite Pierre et Marie Curie Paris 06, Unite de Mixte de Recherche S 1127, Institut du Cerveau et de la Moelle Epiniere, Paris, France; Aurelia Jacquette, Assistance Publique-Hopitaux de Paris, Departement de Genetique, Centre de Reference Deficiences Intellectuelles de Causes Rares, Groupe Hospitalier Pitie-Salpetriere, Paris, France; Laurence Faivre, Centre de Genetique, Centre de Reference Maladies Rares “Anomalies du Developpement et Syndromes Malformatifs”, Hopital d’En-fants, Dijon, France; Stephane Bezieau, Service de Genetique Medicale, Centre Hospitalier Universitaire de Nantes, Nantes, France; Bertrand Isidor, Service de Genetique Medicale, Centre Hospitalier Universitaire de Nantes, Nantes, France; Angelika Rieß, Institute of Medical Genetics and Applied Genomics, University of Tubingen, Tubingen, Germany; Ute Moog, Institute of Human Genetics, Heidelberg University, Heidelberg, Germany; Sally Ann Lynch, Dublin City University, Our Lady’s Children’s Hospital, Crumlin, Dublin, Ireland; Terri McVeigh, Dublin City University, Our Lady’s Children’s Hospital, Crumlin, Dublin, Ireland; Orly Elpeleg, Monique and Jacques Roboh Department of Genetics, Hadassah Hebrew University Medical Center, Jerusalem, Israel; Marie Falkenberg Smeland, Department of Medical Genetics, Division of Child and Adolescent Health, University Hospital of North Norway, Tromsø, Norway; Madeleine Fannemel, Department of Medical Genetics, University of Oslo and Oslo University Hospital, Blindern, Oslo, Norway; Arie van Haeringen, Department of Clinical Genetics, Leiden University Medical Centre, Leiden, The Netherlands; Saskia M. Maas, Department of Clinical Genetics, Academic Medical Center, Amsterdam, The Netherlands; H.E. Veenstra-Knol, University of Groningen, University Medical Center Groningen, Department of Genetics, Groningen, The Netherlands; Meyke Schouten, Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands; Marjolein H. Wil-lemsen, Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands; Carlo L. Marcelis, Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands; Charlotte Ockeloen, Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands; Ineke van der Burgt, Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands; Ilse Feenstra, Department of Human Genetics, Radboud University Medical Center, Nijmegen, The Netherlands; Jasper van der Smagt, Department of Clinical Genetics, University Medical Centre, Utrecht and Donders Institute for Brain, Cognition and Behaviour, Radboud University Medical Center, Nijmegen, The Netherlands; Aleksandra Jezela-Stanek, Department of Medical Genetics, Children’s Memorial Health Institute, Warsaw, Poland; Malgorzata Krajewska-Walasek, Department of Medical Genetics, Children’s Memorial Health Institute, Warsaw, Poland; Domingo Gonzalez-Lamuno, Department of Pediatrics. University of Cantabria and University Hospital Marques de Valdecilla, Santander, Spain; Britt-Marie Anderlid, Department of Clinical Genetics, Karolinska University Hospital and Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden; Helena Malmgren, Department of Clinical Genetics, Karolinska University Hospital and Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden; Magnus Nordenskjold, Department of Clinical Genetics, Karolinska University Hospital and Department of Molecular Medicine and Surgery, Karolinska Institutet, Stockholm, Sweden; Emma Clement, Great Ormond Street Hospital for Children NHS Foundation Trust, United Kingdom; Jane Hurst, Great Ormond Street Hospital for Children NHS Foundation Trust, United Kingdom; Kay Metcalfe, Manchester Centre For Genomic Medicine, Central Manchester University Hospitals NHS Foundation Trust, Manchester Academic Health Science Centre, Manchester, United Kingdom; Sahar Mansour, Thames Regional Genetics Service, St. George’s, University of London, Tooting, London, United Kingdom; Katherine Lachlan, Wessex Clinical Genetics Service, University of Southampton Foundation NHS Trust, Southampton, United Kingdom; Jill Clayton-Smith, Manchester Centre for Genomic Medicine, St Mary’s Hospital, Central Manchester University Hospitals National Health Services Foundation Trust Manchester Academic Health Sciences Centre, Division of Evolution and Genomic Sciences, School of Biological Sciences University of Manchester, Manchester, United Kingdom; Laura G. Hendon, University of Mississippi Medical Center of Jackson, Jackson, Mississippi; Omar A. Abdulrahman, University of Mississippi Medical Center of Jackson, Jackson, Mississippi; Eric Morrow, Department of Molecular Biology, Cell Biology and Biochemistry, Providence, Rhode Island. Developmental Disorders Genetics Research Program, Emma Pendleton Bradley Hospital and Department of Psychiatry and Human Behavior, Alpert Medical School of Brown University, East Providence, Rhode Island. Rhode Island Consortium of Autism Research and Treatment (RI-CART), Providence, Rhode Island; Clare McMillan, Private practice, Rhode Island. Clinically affiliated with Hasbro Children’s Hospital in Providence, Rhode Island; Jennifer Gerdts, Department of Psychiatry and Behavioral Sciences, University of Washington Autism Center, Seattle, Washington; Joseph Peeden, Diagnostic Clinic, East Tennessee Children’s Hospital and University of Tennessee, Knoxville, Tennessee; Samantha A. Schrier Vergano, Division of Medical Genetics and Metabolism, Children’s Hospital of The King’s Daughters, Norfolk, Virginia; Caitlin Valentino, Division of Medical Genetics and Metabolism, Children’s Hospital of The King’s Daughters, Norfolk, Virginia; Wendy K. Chung, Departments of Pediatrics and Medicine, Columbia University, New York, New York; Jillian R. Ozmore, Division of Clinical Genetics, Dartmouth-Hitchcock Medical Center, Lebanon, New Hampshire; Sandra Bedrosian-Sermone, ADNP Kids Research Foundation, Brush Prairie, Washington; Anna Dennis, Graduate Program in Genetic Counseling, University of Colorado Denver, Aurora, Colorado; Kayla Treat, Department of Medical and Molecular Genetics, Indiana University Hospital, Indianapolis, Indiana; Susan Starling Hughes, Children’s Mercy Hospitals and Clinics, Genetics, Kansas City, Missouri; Nicole Safina, Children’s Mercy Hospitals and Clinics, Genetics, Kansas City, Missouri; Jean-Baptiste Le Pichon, Children’s Mercy Hospitals and Clinics, Kansas City, Missouri; Marianne McGuire, Children’s Hospital of Pittsburgh of University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania; Elena Infante, Children’s Hospital of Pittsburgh of University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania; Suneeta Madan-Khetarpal, Children’s Hospital of Pittsburgh of University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania; Sonal Desai, Department of Neurogenetics, Kennedy Krieger Institute, Baltimore, Maryland; Paul Benke, Department of Medical Genetics, Joe DiMaggio Children’s Hospital, Hollywood, Florida; Alyson Krokosky, Pediatric Specialty Clinic, Walter Reed National Military Medical Center, Bethesda, Maryland; Ingrid Cristian, Nemours Children’s Hospital, Orlando, Florida; Laura Baker, Division of Medical Genetics, Nemours/Alfred I. duPont Hospital for Children, Wilmington, Delaware; Karen Gripp, Division of Medical Genetics, Nemours/ Alfred I. duPont Hospital for Children, Wilmington, Delaware; Holly A. Stessman, Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington; Jacob Eichenberger, Children’s Hospital of Georgia at Augusta University, Augusta, Georgia; Parul Jayakar, Division of Genetics and Metabolism, Nicklaus Children’s Hospital, Miami, Florida; Amy Pizzino, Children’s National Health System, Washington, District of Columbia; Melanie Ann Manning, Division of Medical Genetics, Stanford Children’s Health, Stanford, California; Leah Slattery, Division of Medical Genetics, Stanford Children’s Health, Stanford, California.
Footnotes
ARTICLE INFORMATION
Supplementary material cited in this article is available online at http://doi.org/10.1016/j.biopsych.2018.02.1173.
REFERENCES
- 1.American Psychiatric Association (2013): Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Arlington, VA: American Psychiatric Association Publishing. [Google Scholar]
- 2.Lyall K, Croen L, Daniels J, Fallin MD, Ladd-Acosta C, Lee BK, et al. (2016): The changing epidemiology of autism spectrum disorders. Annu Rev Public Health 38:81–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaugler T, Klei L, Sanders SJ, Bodea CA, Goldberg AP, Lee AB, et al. (2014): Most genetic risk for autism resides with common variation. Nat Genet 46:881–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iossifov I, O’Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, et al. (2014): The contribution of de novo coding mutations to autism spectrum disorder. Nature 515:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Ercument Cicek A, et al. (2014): Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Study DDD (2017): Prevalence and architecture of de novo mutations in developmental disorders. Nature 542:433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stessman HA, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. (2017): Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet 49:515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krumm N, O’Roak BJ, Shendure J, Eichler EE (2014): A de novo convergence of autism genetics and molecular neuroscience. Trends Neurosci 37:95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchell KJ (2011): The genetics of neurodevelopmental disease. Curr Opin Neurobiol 21:197–203. [DOI] [PubMed] [Google Scholar]
- 10.Johnson MR, Shkura K, Langley SR, Delahaye-Duriez A, Srivastava P, Hill WD, et al. (2016): Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nat Neurosci 19:223–232. [DOI] [PubMed] [Google Scholar]
- 11.Barnard RA, Pomaville MB, O’Roak BJ (2015): Mutations and modeling of the chromatin remodeler CHD8 define an emerging autism etiology. Front Neurosci 9:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, et al. (2014): Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Bon BW, Coe BP, Bernier R, Green C, Gerdts J, Witherspoon K, et al. (2016): Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol Psychiatry 21:126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stessman HA, Turner TN, Eichler EE (2016): Molecular subtyping and improved treatment of neurodevelopmental disease. Genome Med 8:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zamostiano R, Pinhasov A, Gelber E, Steingart RA, Seroussi E, Giladi E, et al. (2001): Cloning and characterization of the human activity-dependent neuroprotective protein. J Biol Chem 276: 708–714. [DOI] [PubMed] [Google Scholar]
- 16.Mosch K, Franz H, Soeroes S, Singh PB, Fischle W (2011): HP1 recruits activity-dependent neuroprotective protein to H3K9me3 marked pericentromeric heterochromatin for silencing of major satellite repeats. PLoS One 6:e15894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mandel S, Gozes I (2007): Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J Biol Chem 282:34448–34456. [DOI] [PubMed] [Google Scholar]
- 18.Pinhasov A, Mandel S, Torchinsky A, Giladi E, Pittel Z, Goldsweig AM, et al. (2003): Activity-dependent neuroprotective protein: A novel gene essential for brain formation. Brain Res Dev Brain Res 144:83–90. [DOI] [PubMed] [Google Scholar]
- 19.Helsmoortel C, Vulto-van Silfhout AT, Coe BP, Vandeweyer G, Rooms L, van den Ende J, et al. (2014): A SWI/SNF-related autism syndrome caused by de novo mutations in ADNP. Nat Genet 46:380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pescosolido MF, Schwede M, Johnson Harrison A, Schmidt M, Gamsiz ED, Chen WS, et al. (2014): Expansion of the clinical phenotype associated with mutations in activity-dependent neuroprotective protein. J Med Genet 51:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vandeweyer G, Helsmoortel C, Van Dijck A, Vulto-van Silfhout AT, Coe BP, Bernier R, et al. (2014): The transcriptional regulator ADNP links the BAF (SWI/SNF) complexes with autism. Am J Med Genet C Semin Med Genet 166:315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krajewska-Walasek M, Jurkiewicz D, Piekutowska-Abramczuk D, Kucharczyk M, Chrzanowska KH, Jezela-Stanek A, et al. (2016): Additional data on the clinical phenotype of Helsmoortel-Van der Aa syndrome associated with a novel truncating mutation in ADNP gene. Am J Med Genet A 170:1647–1650. [DOI] [PubMed] [Google Scholar]
- 23.Gozes I, Helsmoortel C, Vandeweyer G, Van der Aa N, Kooy F, Sermone SB (2015): The compassionate side of neuroscience: Tony Sermone’s undiagnosed genetic journey—ADNP mutation. J Mol Neurosci 56:751–757. [DOI] [PubMed] [Google Scholar]
- 24.ADNP Syndrome Parents Group. Available at: https://www.facebook.com/groups/ADNPkids/. Accessed July 1, 2016.
- 25.Dabney A, Storey JD, Warnes GR (2016): qvalue: Q-value estimation for false discovery rate control [software package]. R package version 1.38.0. Seattle, WA: University of Washington. [Google Scholar]
- 26.R Development Core Team (2016): R: A language and environment for statistical computing [software] Vienna, Austria: R Foundation for Statistical Computing. [Google Scholar]
- 27.World Health Organization Multicentre Growth Reference Study Group (2006): WHO Motor Development Study: Windows of achievement for six gross motor development milestones. Acta Paediatr Suppl 450:86–95. [DOI] [PubMed] [Google Scholar]
- 28.World Health Organization (2017): The WHO child growth standards. Available at: http://www.who.int/childgrowth/en. Accessed January 31, 2017.
- 29.Gozes I, Van Dijck A, Hacohen-Kleiman G, Grigg I, Karmon G, Giladi E, et al. (2017): Premature primary tooth eruption in cognitive/motor-delayed ADNP-mutated children. Transl Psychiatry 7:e1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malishkevich A, Leyk J, Goldbaum O, Richter-Landsberg C, Gozes I (2015): ADNP/ADNP2 expression in oligodendrocytes: Implication for myelin-related neurodevelopment. J Mol Neurosci 57:304–313. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.