

Abstract
MEK inhibition is of interest in cancer drug development, but clinical activity in metastatic colorectal cancer (mCRC) has been limited. Preclinical studies demonstrated Wnt pathway overexpression in KRAS mutant cell lines resistant to the MEK inhibitor, selumetinib. The combination of selumetinib and cyclosporin A (CsA), a non-canonical Wnt pathway modulator, demonstrated antitumor activity in mCRC patient-derived xenografts (PDX). To translate these results, we conducted a NCI CTEP-approved multicenter Phase I/IB trial (NCT02188264) of the combination of selumetinib and CsA.
Patients with advanced solid malignancies were treated with the combination of oral selumetinib and CsA in the dose escalation phase followed by an expansion cohort of irinotecan and oxaliplatin-refractory mCRC. The expansion cohort utilized a single-agent selumetinib “run-in” to evaluate FZD2 biomarker upregulation and RAS-WT and RAS-MT stratification to identify any potential predictors of efficacy.
Twenty and 19 patients were enrolled in dose escalation and expansion phases, respectively. The most common adverse events and grade 3/4 toxicities were rash, hypertension, and edema. Three dose-limiting toxicities - Grade 3 hypertension, rash and increased creatinine were reported. The maximum tolerated dose was selumetinib 75 mg BID and CsA 2 mg/kg BID on a 28-day cycle. RAS stratification did not identify any differences in response between RAS-WT and RAS-MT cancers. Two partial responses (PR), 18 stable disease (SD), and 10 progressive disease (PD) responses were observed.
Combination selumetinib and CsA is well-tolerated with evidence of activity in mCRC. Future strategies for concept development include identifying better predictors of efficacy and improved Wnt pathway modulation.
Keywords: Phase I, Metastatic colorectal cancer, Selumetinib, Cyclosporin A, MEK inhibitor
INTRODUCTION
Colorectal cancer is the third leading cause of malignancy and the fourth common cause of cancer death worldwide (1). In the United States, colorectal cancer is the fourth most common cancer, and this year, an estimated 135,430 new cases of colorectal cancer will be diagnosed (2). Approximately 20% of patients have metastatic or stage IV disease and only 13.9% of patients are alive at 5 years (2). Current treatment options include initial treatment with a 5-Flurouracil (5-FU) and leucovorin backbone accompanied by oxaliplatin or irinotecan. Bevacizumab, a vascular endothelial growth factor (VEGF) inhibitor is administered along with 5-FU based therapy and is commonly continued beyond progression. Rat Sarcoma (RAS) gene wild-type patients with metastatic colorectal cancer (mCRC) have been shown to benefit from monoclonal antibodies directed against epidermal growth factor receptors (EGFR). Other agents used in later lines of therapy include regorafenib, a multi-kinase inhibitor and TAS-102, a combination of a thymidine-based nucleic acid analogue and a potent thymidine phosphorylase inhibitor.
Despite these therapeutic advances, mCRC is often incurable with a sobering median survival of 28–30 months (3). There is an unmet need for research and development of new and more effective therapies. A better understanding of the resistance mechanisms to targeted therapy has led to rational combination strategies (4). One of the distinctive fundamental capabilities of cancer is the ability to sustain proliferative signaling. The MAPK (Mitogen Activating Protein Kinase) pathway (RAS/RAF/MEK/ERK) is one such proliferation pathway that is frequently dysregulated in cancer through gain of function mutations in the RAS (Rat Sarcoma gene) and RAF (Rapidly Accelerated Fibrosarcoma) proteins. RAS mutations are found roughly 55% of colorectal cancers and its downstream effector pathways include the mitogen activating protein kinase/extracellular signal-regulated kinases (MAPK/ERK), the phosphotidyl inositol 3-kinase (PI3 kinase) and the Ral-GDS pathways. MEK is a critical MAPK enzyme in the downstream pathway from RAS and RAF that phosphorylates and activates Extracellular Signal-Regulated Kinases (ERK/p-ERK), its only known substrate, which in turn translocates to the nucleus where it activates many transcription factors resulting in growth and proliferation (4–6). Unfortunately, activation of this downstream signaling pathway is associated with lack of beneficial responses to EGFR antibody blockade in patients with mutations in these proteins (7,8).
Therefore, MEK inhibition has been an attractive therapeutic target for cancer treatment and has been tested in clinical trials since 2000. The safety, tolerability and efficacy of MEK inhibition has been established from numerous studies investigating selumetinib as well as other MEK inhibitors such as trametinib and cobimetinib (9–13). Single-agent activity has been somewhat modest except for trametinib which demonstrated improved median progression-free survival (4.8 vs 1.5 mos, p < 0.001) and 6-month survival rates (81% vs 67%) in patients with advanced BRAF V600E or V600K mutated melanoma (10,14). This lack of convincing clinical activity of single agent MEK inhibition could be due to simultaneous dysregulation of multiple signaling pathways and/or compensatory pathways that overcome the effect of MEK inhibitors (5,6,15–17). The combination of MEK inhibitors with other targeted agents or chemotherapy may overcome resistance and thus improve efficacy.
Selumetinib (AZD6244; ARRY-142886) is an orally-active small molecule MEK inhibitor that has been studied in many clinical trial settings. In the initial phase I study, selumetinib was found to be well tolerated with a Recommended Phase II Dose (RP2D) of 100 mg BID (16). Bennouna et al conducted a phase II randomized open label study that compared selumetinib at 100 mg BID to 1250 mg/2 twice daily of oral capecitabine in patients with refractory mCRC. Disease progression was described in 80% of patients in both treatment groups with a very modest progression-free survival in both (17). MEK inhibitors have been combined with other therapies to enhance clinical efficacy. Hochster et al combined selumetinib and irinotecan in KRAS mutated CRC patients showing improved clinical activity with the combination, but the study was terminated prior to full accrual (18). The combination of selumetinib and cetuximab has also been shown to be safe and well tolerated in another phase I study but minimal anti-tumor activity was noted in KRAS mutant refractory mCRC (19).
Previous studies have identified the Wingless integrated (Wnt) signaling pathway as a resistance mechanism to MEK inhibition (6). The Wnt pathway is an evolutionarily conserved signal transduction pathway that regulates several cellular processes including stem cell renewal through canonical and non-canonical pathways (6,20,21) (Figure 1). The canonical pathway signals via the frizzled (FZD) family of G-protein coupled receptors. In the absence of Wnt ligand binding to FZD, the β-catenin destruction complex is degraded through proteolytic destruction. Binding of Wnt to the FZD-LRP5/6 co-receptor complex disrupts the APC/Axin/GSK3 complex that is required for the destruction of β-catenin. Stabilized β-catenin translocates to the nucleus where it mediates transcription of target genes (Figure 1A). Aberrant Wnt pathway activation through the loss of function mutation of Adenomatous Polyposis Coli (APC) is an early event in the development of colorectal cancer (20). The two well-known non-canonical or β-catenin independent pathways are the Wnt/Ca2+ pathway (Figure 1B) and the planar cell polarity pathway (Figure 1C). The Wnt/Ca2+ pathway also acts through Wnt-FZD activation of Dsh. Dsh through PLC, activates IP3 which leads to release of intracellular Ca2+, and the Ca2+ in turn activates CamK11 and a serine/threonine phosphatase, calcineurin. Calcineurin induced de-phosphorylation of NFAT results in the translocation of NFAT to the nucleus where it regulates transcription of genes (Figure 1B). Preclinical studies of selumetinib-resistant KRAS mutant CRC cell lines show overexpression of several members of the Wnt pathway including Frizzled (FZD). Both gene set enrichment analysis and a synthetic lethal screen demonstrated that many of the genes involved in the canonical and non-canonical Wnt pathways were upregulated in selumetinib-resistant CRC cell lines (22).
Figure 1. The wingless/integrated (Wnt) signaling pathway.
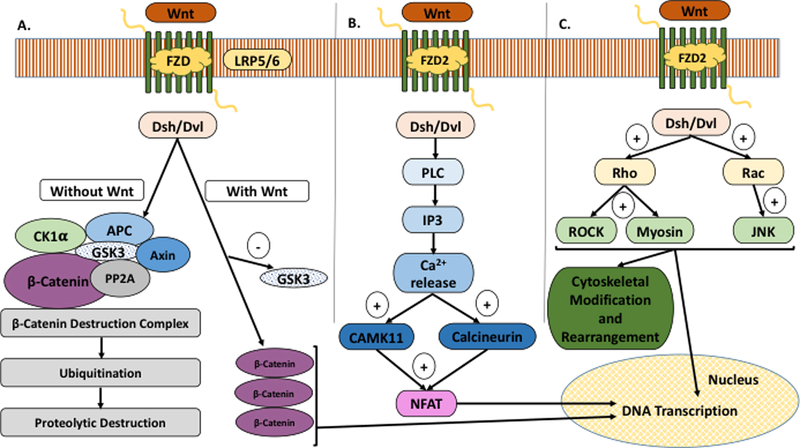
A) The Canonical Pathway signals via the frizzled (FZD) family of G-protein coupled receptors; B) Wnt/Ca2+ Pathway (Non-Canonical); C) Planar Cell Polarity (Non-Canonical).
Recent studies have shown that cyclosporin A (CsA), a calcineurin inhibitor traditionally used for its immunosuppressive effects, inhibits the activity of the non-canonical Wnt/Ca++/NFAT signaling pathway (23–25). DeGregori et al identified the Wnt/Ca2+ pathway genes as being synthetically lethal in combination with imatinib in RNAi based screens and NFAT inhibition by CsA resulted in the sensitization of leukemia cells to Bcr-Abl inhibition (23). Synergistic anti-tumor effects with the combination of CsA and selumetinib were observed in KRAS mutant CRC xenografts that were known to be resistant to selumetinib monotherapy. These xenografts were noted to have increased expression of FZD2 by qRT-PCR when treated with selumetinib monotherapy. Given the predilection of CsA to inhibit P-glycoprotein drug efflux pumps, the above mentioned preclinical study also measured selumetinib concentrations in plasma, tumor and liver when treated with selumetinib alone and in combination with CsA and reported no significant difference in selumetinib or its metabolite (6).
The theoretical rationale and the data suggest that the primary and secondary resistance to selumetinib driven MEK inhibition may be overcome through concurrent non-canonical Wnt inhibition with CsA. Based upon these intriguing preclinical data, we pursued the next step of translation in a multicenter Phase Ib study of selumetinib and CsA with an expansion cohort in patients with mCRC.
PATIENTS AND METHODS
This study was conducted according to the ethical guidelines laid out by the Belmont Report and was approved by the institutional review board. Written informed consent was obtained from patients during the course of the study.
Patients
Patients with a histological or cytopathological diagnosis of an advanced solid cancer that is refractory to standard therapy or for which there is no standard therapy were included in the dose escalation cohort of the study. Once the MTD was identified, patients who had progressed on oxaliplatin- and irinotecan-based therapies with a histological or cytopathological diagnosis of advanced/metastatic unresectable colorectal cancer with known RAS mutational status, no known BRAF mutation and measurable disease were eligible for the expansion cohort. Patients were required to be ≥ 18 years, ECOG performance status 0–1 and have an estimated life expectancy > 3 months. Adequate marrow function, renal function, hepatic function, and serum albumin ≥ 2.5 g/dl was required. Study specific exclusion criteria included chemotherapy or radiotherapy within 4 weeks, unstable brain metastases, less than 1 month from definitive therapy of brain metastases, uncontrolled inter-current illness, known ophthalmological conditions especially current or past history of serous retinopathy or retinal vein occlusion, major surgical procedure within less than 3 weeks or minor surgical procedure within 1 week of first study drug administration, inability to swallow capsules, known history of HIV, Hepatitis B and/or Hepatitis C, pregnancy and electrolyte abnormalities that refractory to therapy.
Study Design, Drug and Treatment
This was a multicenter phase I/IB study with escalation and expansion cohorts. The trial incorporated a standard 3+3 design with a cohort expansion to 6 patients if a dose limiting toxicity (DLT) was reported. All patients were treated with selumetinib and cyclosporin A (CsA). In the dose escalation phase, the starting dose level of selumetinib and CsA was 50 mg BID and 2 mg/kg BID respectively. The CsA trough levels were measured 6–8 days after initiating treatment and the steady state trough level goals were 125 to 250 ng/ml. The MTD was defined as the highest dose at which no more than one patient out of six experienced a DLT. Once the RP2D/maximum tolerated dose was identified, the dose expansion cohort of 20 patients with metastatic irinotecan and oxaliplatin refractory CRC was initiated. KRAS wild-type (WT) and mutant (MT) CRC objective responses were assessed to determine whether there were differential responses between the subsets. All patients in the expansion cohort were required to have a baseline tumor biopsy prior to starting treatment. The first ten patients within the expansion cohort had a 7-day run-in of selumetinib alone at the RP2D followed by a repeat tumor biopsy during the cycle #1 of treatment to assess if there was a correlation between FZD upregulation and tumor response. Five of these ten patients were KRAS MT and the other five were KRAS WT to determine if RAS status was predictive of response. The remaining ten patients in the dose expansion cohort received selumetinib and CsA concurrently from the start of enrollment and had a tumor biopsy at the time of restaging prior to cycle #3.
Toxicities were defined as per the National Cancer Institute’s Common Terminology Criteria for Adverse Events v4.0. Patients were monitored for DLTs through the course of therapy but only DLTs occurring in the first 28 days were used to determine dose escalation and ultimately, MTD. The following AEs if attributed to either selumetinib or CsA were considered a DLT: Grade 4 neutropenia for ≥ 7 days, grade 3 or 4 neutropenia with a single temperature reading ≥ 38.3 C or sustained temperature reading > 38 C for > 1 h, grade 3 thrombocytopenia associated with clinically significant bleeding that required transfusion therapy, Grade 4 thrombocytopenia, ≥ grade 3 nausea or vomiting that persisted beyond 72 h despite use of optimal anti-emetics, ≥ grade 3 diarrhea that persisted beyond 72 h despite use of optimal anti-diarrheal agents, ≥ grade 3 hyponatremia, hypokalemia, hypomagnesemia, hypophosphatemia that persisted for or beyond 7 days despite maximal medical management, any intolerable adverse event regardless of grade that did not resolve within 7 days despite maximal medical management or other non-hematologic toxicities grade 3 or higher except for AEs related to underlying disease, alopecia, fatigue, lymphopenia without significant infection and isolated asymptomatic grade 3 electrolyte abnormalities.
Dose Modifications
Selumetinib
Treatment with selumetinib was withheld if patient experienced a DLT or any intolerable adverse event regardless of grade that was considered related to selumetinib despite optimal supportive care. Treatment was not restarted until the toxicity improved to grade 1 or baseline except in the case of rash where treatment was restarted with a grade 2 rash. Treatment was resumed at the original dose or at a permanently reduced dose at the discretion of the investigator. Drug was withheld and then restarted at a permanently reduced dose in patients who experienced recurrence of a specific toxicity. If previous dose reductions had already taken place due to recurring toxicity or patient was already receiving the lowest possible dose of selumetinib (50 mg once daily), then selumetinib was discontinued.
Cyclosporin A
CsA dosing was changed according to trough levels although the range was not absolute and trough levels outside this range were also accepted at the discretion of the treating physician. If the patient had a known toxicity to CsA such as renal toxicity or hypertension, then the dose adjustment was determined by the toxicity and not the CsA level. Dose adjustment recommendations were: if the CsA trough level was below 125 ng/ml then the dose of CsA was increased by 0.5–1 mg/Kg not to exceed 50 mg per dose adjustment; If the CsA trough levels were above 250 ng/ml then dose was decreased by 0.5–1 mg/kg not to exceed 50 mg per dose adjustment; if the CsA trough level was above 350 mg/ml then CsA was held and levels were monitored until the level was below 125 ng/ml and then CsA was restarted at 67% of previous dose with repeated drug levels at 48–72 h. If patients contracted or were exposed to infectious diseases like herpes viruses or Pneumocystis jiroveci pneumonia (PJP) then CsA was discontinued due to concerns about CsA related immunosuppression.
Pharmacologic Assessments
Pharmacokinetic Assessments
Since the pharmacokinetics of selumetinib and CsA have already been studied in humans, the PK analysis in our study specifically evaluated the PK effects of selumetinib, its active metabolite N-desmethyl AZD6244 and CsA on each other. All patients in cycle 1 of the dose escalation phase received selumetinib alone on Day −7 and CsA on Day −3 with plasma sampling at 0.5, 1, 2, 4, 8 and 24 hours after each of those treatments. PK sampling was also performed at the same time intervals after both drugs were given to patients on day 1 of the first cycle. Steady state PK measurements were performed on weeks 2 and 4 of cycle 1. All plasma samples were analyzed with liquid chromatography tandem mass spectrometry assays (26,27).
Pharmacodynamic Analyses
Biomarker analysis for p-ERK and FZD1/2 was performed in the expansion cohort of 20 patients to determine if there were any associations between p-ERK and FZD1/2 expression and anti-tumor activity of the selumetinib-CsA combination. All patients had tumor biopsies at baseline. The first 10 patients had a second biopsy after the selumetinib run-in while the other ten patients had a second biopsy at the time of restaging. Two core biopsies of tumor were analyzed by immunohistochemistry (IHC) for p-ERK and FZD1/2. P-ERK IHC utilized a primary p44/p42 ERK1/2 rabbit monoclonal antibody while FZD1/2 IHC was achieved with Santa Cruz Goat Polyclonal IgG FZD1/2 primary antibody. A light to dark brown staining of the membrane and/or cytoplasm along with a pale to dark blue coloration of the nuclei with hematoxylin counterstaining was considered a positive IHC reaction. Predominantly stained compartments were identified in pre- and post- treatment tissue specimens and Histology-scores (H-scores) were calculated using the proportion and intensity of stained tumor cells. The H-scores pre- and post- treatment were then compared. The H-score range was from 0–300 and a value over 50 was considered to be positive while anything below that was considered a negative test.
Statistical Analyses
All patients who had received at least one dose of the study medication were included in the safety analyses. The primary objective of the study was to find the MTD and the MTD was defined as the highest dose at which 0 or 1 out of 6 patients or 2 out of 12 patients has a DLT. Descriptive statistics were used to analyze patient characteristics, safety, pharmacodynamics and efficacy. Adverse events were tabulated by type and grade. Antitumor activity was assessed based on objective tumor response per Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 criteria and Progression Free Survival (PFS). Response rates were tabulated with 95% exact binomial confidence intervals. PFS was evaluated using the product limit method of Kaplan and Meier. Pharmacokinetic analyses included examination of the area under the curve (AUC) and Cmax of the combination therapy of the study drugs. Pharmacodynamic studies included pre- and post- treatment evaluation of MEK activity and non-canonical Wnt signaling by using IHC to measure p-ERK and FZD1/2 respectively. Post-treatment specimens were expected to demonstrate ≥ 30% inhibition in p-ERK to be considered significant while a 1.5-fold or greater increase in FZD1/2 was considered a cut-off for the selumetinib only arm.
RESULTS
Patient Characteristics
Twenty patients were enrolled in the dose escalation cohort while nineteen patients were enrolled in the dose expansion cohort (Table 1). The majority of patients were 18 to 64 years of age with a smaller proportion of patients being over the age of 65. Colorectal cancer was the most common tumor type and comprised 31 (79.5%) patients included in the study. Other tumor types that were included in the dose escalation cohort were renal cell (1), prostate (1), hepatocellular (1), cervical (1), endometrial (1), ovarian (2) and pancreatic cancer (2). The rates of RAS mutation on the study were similar to what is found in the general population. Enrollment occurred over a period of 24 months and patients were on the study for a median duration of 3.15 months.
Table 1.
: Patient Baseline Characteristics
| Characteristics | Escalation Cohort (n = 20) | Expansion Cohort (n = 19) |
|---|---|---|
| No. of patients (%) | No. of patients (%) | |
| Age (years) | ||
| 18–64 | 13 (68.4%) | 12 (57.1%) |
| 65+ | 6 (31.6%) | 9 (42.8%) |
| Sex | ||
| Male | 9 (47.4%) | 10 (47.6%) |
| Female | 10 (52.6%) | 11 (52.4%) |
| Tumor Primary Site | ||
| Colorectal | 13 | |
| Renal | 1 | |
| Prostate | 1 | |
| Pancreas | 1 | |
| ECOG Performance Status | ||
| 0 | 7 (36.8%) | 10 (47.6%) |
| 1 | 12 (63.2%) | 11 (52.4%) |
Drug Exposure
All patients received at least one dose of study medication. 6 patients received the 50 mg BID dosing while the rest of the patients received the 75 mg BID dosing of selumetinib.
CsA was maintained within trough levels of 125–250 ng/ml in all patients during the course of the study. The dosing of CsA was maintained at 2 mg/kg throughout the study.
Safety and Tolerability
Dose-limiting toxicities were grade 3 hypertension, rash and elevated creatinine. The grade 3 hypertension was noted at dose levels of 75 mg PO BID of selumetinib and 2 mg/kg PO BID of CsA. The grade 3 rash and the grade 3 elevated creatinine occurred at the same above-mentioned dose levels as well. Hence, MTD was determined to be 75 mg BID of selumetinib and 2 mg/kg of CsA, and this is also determined to be the RP2D.
Treatment related adverse events are described in Table 2. Adverse events such as acneiform rash, maculopapular rash, diarrhea and edema can be attributed to selumetinib as these AE have been reported by other MEK inhibitor trials while hypertension and elevated creatinine are well known side effects of CsA. Nine grade 3 and one grade 4 toxicity were reported on the study. The grade 3 adverse events comprised of hypertension, rash, decreased lymphocyte count, anemia, hyponatremia, fatigue, anorexia, anal mucositis, myositis, peripheral motor and sensory neuropathy, peripheral edema, acute kidney injury, lung infection, dyspnea, and acute coronary syndrome. Of those hypertension, anemia, peripheral edema and rash were definitely attributed to study drugs. The grade 4 adverse events included hyponatremia and increased ALT and AST. Hyponatremia was possibly related to the study drug but the elevated AST and ALT were thought to be unrelated. Two patients died while still on study, one due to intracranial hemorrhage and the other due to tumor progression but none of the deaths were study drug-related.
Table 2:
Treatment Related Adverse Events Occurring in more than 10% of Patients
| Adverse Event | No. of patients (%) |
|---|---|
| Any Grade ≥ 3 | 10 (26%) |
| Blood and Lymphatic System disorders | |
| Anemia | 11 (28%) |
| Neutropenia | 5 (13%) |
| Thrombocytopenia | 7 (18%) |
| Gastrointestinal Disorders | |
| Abdominal Pain | 8 (21%) |
| Nausea | 26 (67%) |
| Vomiting | 15 (38%) |
| Diarrhea | 18 (46%) |
| Constipation | 8 (21%) |
| Anorexia | 9 (23%) |
| Mucositis | 7 (18%) |
| Dry mouth | 5 (13%) |
| Hepatic Disorders | |
| AST increased | 14 (36%) |
| ALT increased | 5 (13%) |
| ALP increased | 8 (21%) |
| Total Bilirubin increased | 5 (13%) |
| Generalized Disorders of Well-Being | |
| Fatigue | 19 (49%) |
| Hypoalbuminemia | 8 (21%) |
| Weight gain | 5 (13%) |
| Skin Disorders | |
| Rash acneiform | 17 (44%) |
| Rash maculopapular | 16 (41%) |
| Renal Disorders | |
| Elevated Creatinine | 13 (33%) |
| Peripheral Edema | 17 (44%) |
| Facial Edema | 7 (18%) |
| Hypertension | 19 (49%) |
| Respiratory disorders | |
| Dyspnea | 10 (26%) |
| Neurological disorders | |
| Dizziness | 7 (18%) |
| Headache | 6 (15%) |
| Peripheral sensory neuropathy | 5 (13%) |
| Electrolyte Abnormalities | |
| Hyponatremia | 5 (13%) |
| Hypomagnesemia | 11 (28%) |
Pharmacokinetics
Since the pharmacokinetics of selumetinib and CsA have already been studied in humans, the PK analysis in our study specifically evaluated the PK effects of selumetinib and CsA on each other. PK for selumetinib was evaluated on cycle 1 day-7 when it was administered alone and on cycle 1 day 1 when it was administered along with CsA (Figure 2A and2B). When administered alone, the mean t1/2 of selumetinib at the MTD was 6.4 h while mean Cmax was observed to be 1550 ng/ml. For CsA, mean Cmax on day −3 was 858 ng/ml while it was noted to be 919 ng/ml on C1D1 when it was administered with selumetinib (Figure 2C and2D). Mean t1/2, Cmax and area under the plasma concentration-time curve increased with increase in dosing of selumetinib (Figure 3A). Cmax for the MTD of selumetinib when given with CsA was 1250 ng/ml. The mean Cmax of N-desmethyl-AZD6244, an active metabolite of selumetinib was 70 ng/ml when selumetinib was given alone and 58 ng/ml on C1D1 when selumetinib was given along with CsA. Hence, it is appropriate to conclude that there is no substantial difference in the pharmacokinetics of selumetinib when given alone versus in conjunction with CsA (Figure 3B and3C).
Figure 2.
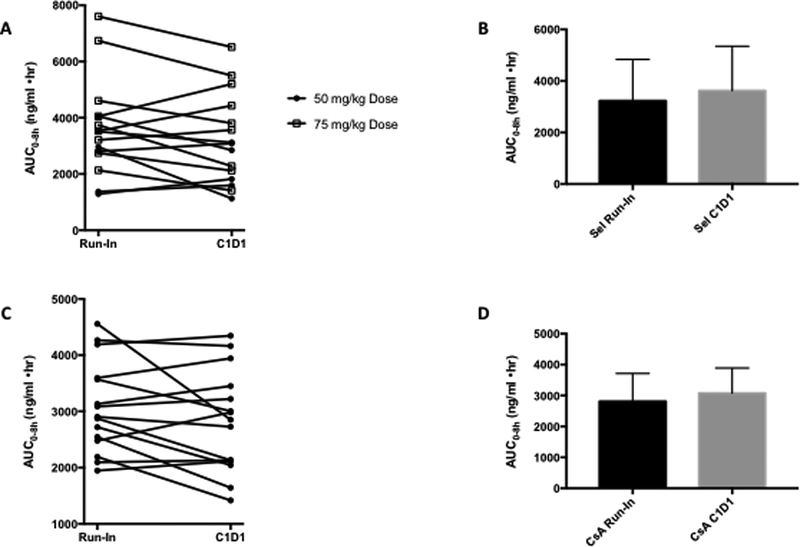
A) and B) AUC for selumetinib run-in and C1D1 when administered with CsA; C) and D) AUC of CsA alone and C1D1 when administered with selumetinib.
Figure 3.
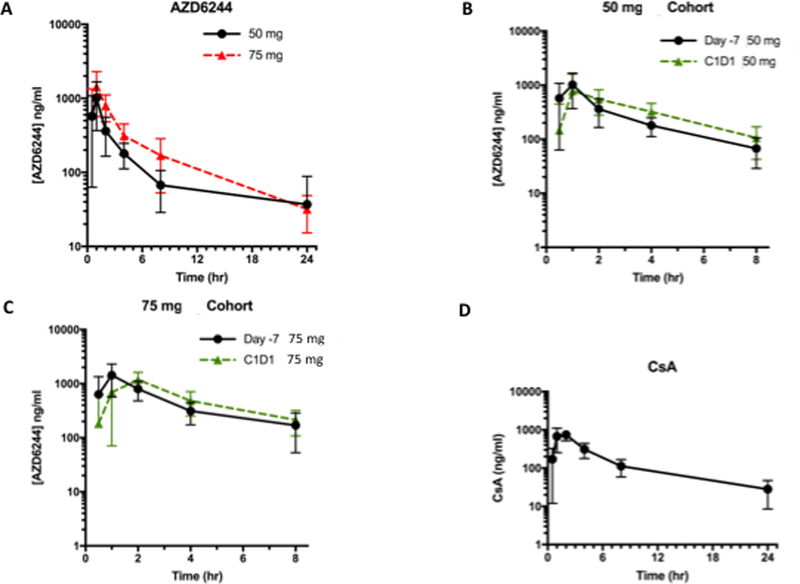
A) AUC over a 24h period of selumetinib alone at the 50 mg and 75 mg dose; B) AUC over an 8 h period of selumetinib 50 mg dosing when administered alone at day −7 and on C1D1 with CsA; C) AUC over an 8 h period of selumetinib 75 mg dosing when administered alone at day −7 and on C1D1 with CsA; D) AUC over a 24h period of CsA at 2 mg/kg dosing when administered alone.
Mean half-life of CsA was determined to 8.24 h on day −3 when it was administered alone (Figure 3D). Selumetinib does not appear to affect the pharmacokinetics of CsA when they are administered together (Figure 2 and 3).
(Steady state PK measurements were done on weeks 2 and 4 of cycle 1. All plasma samples were analyzed with liquid chromatography tandem mass spectrometry assays.)
Pharmacodynamics
p-ERK and FZD1/2, the two biomarkers of interest, were evaluated pre- and post- treatment because p-ERK downregulation and FZD overexpression was expected with MEK inhibition based on prior preclinical studies. FZD overexpression post-treatment with selumetinib alone would confirm the hypothesis that Wnt pathway upregulation is a means of resistance to MEK inhibition. Decreased FZD expression with CsA would be an indication of cellular anti-tumor activity.
Twenty-one patients received pre-treatment biopsies per protocol. Of these, 4 pre-treatment biopsies were not evaluable because partner block was not submitted, or no tumor cells were detected on evaluation. Pre-treatment, p-ERK was positive in all 17 patients but FZD1/2 testing was positive in only 11 patients. Fifteen of the 21 pre-treatment biopsy patients had second biopsies either in the run-in arm or in the combined therapy arm. When patients from the run-in and the concurrent treatments were combined together for post-treatment biomarker assessment, 14 out of 15 patients were positive for p-ERK and 11 out of 15 patients were positive for FZD1/2 testing. One patient who was positive for p-ERK at baseline converted to being negative on the biopsy post treatment with selumetinib alone on the run-in arm. Of all the patients who were positive for p-ERK, 46.6% demonstrated ≥30% inhibition of p-ERK post-treatment. None of the patients who were positive for FZD1/2 at baseline converted to being negative for FZD1/2 post-treatment. One patient who was previously negative for FZD1/2 at baseline became positive post-treatment. Post treatment with selumetinib alone, 28.6% of patients had a 1.5-fold or greater increase in FZD1/2 expression.
Overall, p-ERK downregulation was noted but statistically significant inhibition was seen in approximately half of the evaluable patients. FZD1/2 overexpression with selumetinib treatment was noted (Figure 4A and4B) but correlation between FZD overexpression and overall response rate was not evaluable from our study because of limited sequentially associated biopsy samples.
Figure 4.
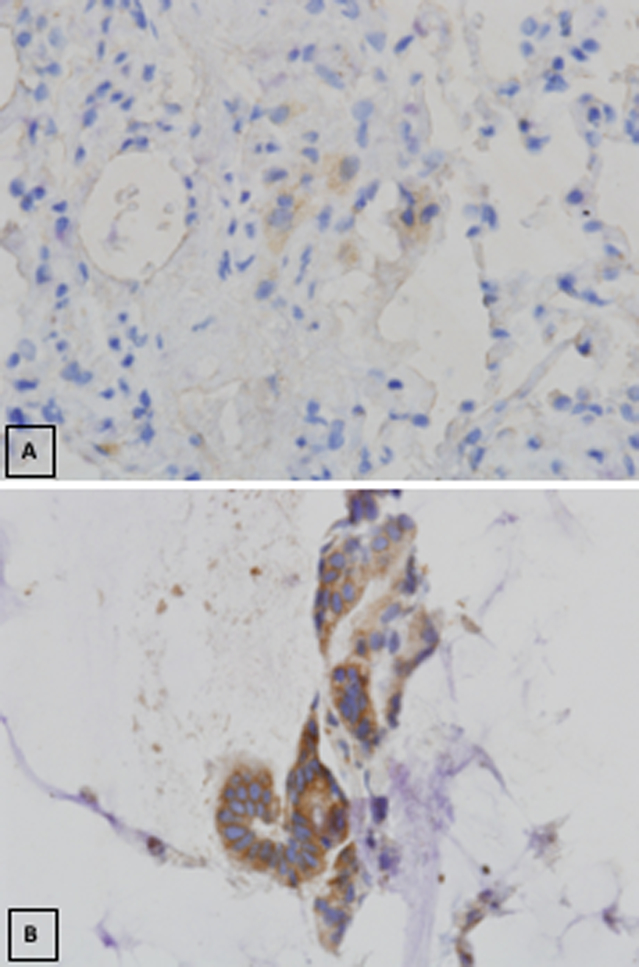
A) FZD 1/2 expression at baseline prior to selumetinib therapy; B) Elevated FZD 1/2 expression post treatment with selumetinib.
Antitumor Effect
Objective response rates per RECIST v1.1 were measurable in 30 out of 39 patients enrolled in both dose escalation and expansion cohorts. Twenty-six of the 30 patients with measurable disease had metastatic colorectal cancer. Among the 30 patients, there were no complete responses (CR), two colorectal cancer patients had partial responses (PR) and 18 other patients had stable disease (SD) (Figure 5). The combined clinical benefit rate (CR + PR + SD) for both cohorts was 67%. The dose expansion cohort alone comprised of 16 metastatic CRC patients with measurable disease. Nine of the 16 patients had stable disease with no partial or complete responses. The clinical benefit rate for the dose expansion cohort alone was 56%.
Figure 5. Objective response rate per RECIST criteria v1.1 by patient.
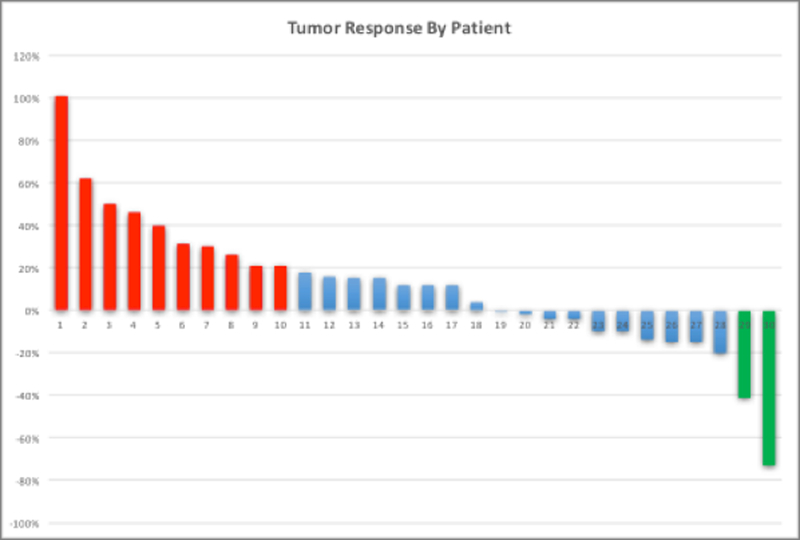
Green bars indicate partial response, Blue bars indicate stable disease, and red bars indicate progressive disease.
KRAS mutations were observed in 58% of study patients with CRC which is in keeping with what is seen in the general population. One CRC patient who had a KRAS-MT had a partial response. Stable disease was noted in 15 patients out of which 10 were KRAS-MT and progressive disease was observed in a total of 8 patients of which 3 patients were KRAS-MT. No statistically significant variations were noted in anti-tumor effect between KRAS-MT and KRAS-WT populations.
Median PFS, a secondary endpoint was calculated to be 3.15 months (95% CI 2.48 – 3.82) in all patients combined from the dose escalation and dose expansion cohorts. However, the value of PFS in an early phase clinic trial is limited.
DISCUSSION
In recent years, new treatment options have been developed for metastatic colorectal cancer but the gains in overall survival have been limited with median survival being 28 to 30 months. At this time, more effective and personalized therapies are needed to improve survival and limit toxicity. In line with these objectives, preclinical studies were done to identify means of resistance to various therapies including MEK inhibition which showed canonical and non-canonical Wnt pathway overexpression in MEK inhibitor resistant KRAS-MT CRC cell lines and this preclinical work formed the basis of our trial. The primary objective of this study was to identify a MTD and determine DLTs. The study design of the expansion cohort was adapted to not only evaluate safety, PKs, and response but also clinically validate Wnt pathway overexpression seen in the preclinical data, enunciate molecular evidence of Wnt suppression with CsA and differentiate efficacy based on the presence or absence of RAS mutations.
This study recommends a phase II dose of 75 mg BID and 2 mg/kg BID for selumetinib and CsA respectively. The dose limiting toxicities were hypertension, elevated creatinine and rash. Hypertension and elevated creatinine are well known toxicities of CsA while rash is a common toxicity associated with MEK inhibitors including selumetinib. Studies have shown that cyclosporin causes a decrease in the glomerular filtration rate through vasoconstriction of glomerular arterioles which results in increased creatinine and hypertension. Transplant patients on chronic cyclosporin treatment are typically managed with calcium channel blockers that can prevent the renal vasoconstriction (28,29). Patients in the study with hypertension were managed with anti-hypertensives with good effect, and there was no known long-term renal dysfunction secondary to cyclosporin on the study.
The dose limiting toxicity of selumetinib in the original single-agent phase I trial was grade 3 acneiform rash and pleural effusion, and the most common adverse events were fatigue, acneiform dermatitis, nausea, diarrhea and peripheral edema (30). All these adverse events were commonly noted in this study as well. Visual changes, a well-known class effect of MEK inhibitors, was noted in only 6% of patients on our study compared to 12% in the phase I study of selumetinib but this is probably because selumetinib dosing did not exceed 75 mg BID(30). The maximum tolerated dose identified in this combination trial for selumetinib was 75 mg BID which is in keeping with findings of the phase I trial of selumetinib monotherapy.
The phase I trial of selumetinib alone has previously shown the mean half-life (t½) of selumetinib administered in the capsule form to be 5 to 8 h. Our study showed a similar t1/2 life for the single agent as well as the combination (30). CsA, which is primarily metabolized through the liver, also had a similar half-life when administered alone and in combination with selumetinib (31). Overall, the PK profile supports the recommended phase II dosing scheme.
Preclinical studies have demonstrated Wnt pathway overexpression with MEK inhibition. Pre-treatment p-ERK positivity is indicative of an active MAPK pathway in the tumors and therefore an active substrate for selumetinib. Phase I studies of selumetinib have previously shown significant p-ERK inhibition but in our study significant p-ERK inhibition post-treatment could be demonstrated in only half the patients suggesting that the MAPK pathway may have still been activated despite treatment with selumetinib. Differences in duration of MEK inhibition, types of sample obtained, variation in methodology of testing for p-ERK and phosphorylation of ERK by kinases other than MEK1/2 could be some of the reasons why all patients did not show significant inhibition of p-ERK compared to prior studies investigating selumetinib (30). The extent of ERK inhibition in our study is also limited by selumetinib which is a second-generation MEKi. There are other MEK inhibitors in development that have shown greater potency in inhibiting MEK1/2 (9,32). We observed FZD expression in 65% of patients at baseline which suggests an activated Wnt pathway. About 30% of tissue specimens obtained from patients post treatment with selumetinib monotherapy showed significant upregulation of FZD2 which is in keeping with the hypothesis of our study that Wnt pathway overexpression is a means of resistance to MEK inhibition therapy in CRC. All tumor specimens did not display FZD2 overexpression with MEK inhibition which confirms our current understanding that multiple pathways are involved in treatment resistance. Our PD analyses could not clearly establish suppression of the Wnt pathway with CsA treatment at the molecular level because of inadequate sample size.
The two partial responses and the eighteen patients with stable disease noted on this study are consistent with the promising activity seen in the preclinical studies. The clinical benefit rate for the combination of selumetinib and CsA in this study is modest but encouraging given that the response rate is higher than would be expected for either single-agent alone (33). However, the results do not mirror the robust responses seen in our preclinical models, thus highlighting the difficulty of translating promising preclinical data into clinical trials. Therefore, it is critically important to investigate resistance mechanisms in patient samples obtained during the course of the study, and these studies are ongoing at this time.
In summary, this phase I study establishes that the combination of selumetinib and CsA has a manageable safety profile at the RP2D for future studies and provides preliminary evidence of antineoplastic activity. Future directions include not only studying the selumetinib/CsA combination in a phase II setting but also combining MEK inhibition with canonical Wnt inhibitors. Many inhibitors of the canonical Wnt signaling pathway are currently being investigated in the preclinical and early clinical studies. Antibodies to Wnt ligands, overexpression of naturally occurring Wnt ligand antagonists, FZD antibodies, promotion of beta-catenin degradation are just some of the many ways in which the Wnt pathway is being targeted (21).
STATEMENT OF TRANSLATIONAL RELEVANCE.
This NCI CTEP-approved Phase I/IB trial (NCT02188264) is part of a larger translational research effort and the primary preclinical data, which was published in Clinical Cancer Research in 2013 by Spreafico et al that demonstrated Wnt pathway overexpression in KRAS mutant cell lines resistant to the MEK inhibitor selumetinib. The combination of selumetinib and cyclosporin A (CsA), a non-canonical Wnt pathway modulator, demonstrated antitumor activity in patient-derived xenograft (PDX) models of mCRC. Findings translate preclinical studies combining selumetinib and cyclosporin into a phase I first-in-human clinical trial of such a combination in patients with advanced solid malignancies.
Acknowledgments
Financial Support: (Source and number of grants for each author): NIH K23 CA190849–01A1 to, C.H. Lieu, NIH 1UM1CA186688 to C.H. Lieu, W.A. Messersmith, and S.G. Eckhardt, Conquer Cancer Foundation Career Development Award to C.H. Lieu, NCI P30 CA047904 for Early Phase Clinical Research Support to J. J. Lee, NCI UM1 CA099168 grant support to J. J. Lee
Footnotes
The following authors declare potential conflicts of interest: Hanna K. Sanoff: Bayer AG and Merck & Co, Inc., Mark N. Stein: Merck Sharpe & Dohme Corp.
REFERENCES
- 1.Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017;66:683–91 [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute - Surveillance E, and End Results Program Cancer Stat Facts: Colon and Rectum Cancer. [Google Scholar]
- 3.Venook AP, Niedzwiecki D, Lenz HJ, Innocenti F, Fruth B, Meyerhardt JA, et al. Effect of First-Line Chemotherapy Combined With Cetuximab or Bevacizumab on Overall Survival in Patients With KRAS Wild-Type Advanced or Metastatic Colorectal Cancer: A Randomized Clinical Trial. JAMA 2017;317:2392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74 [DOI] [PubMed] [Google Scholar]
- 5.Martinelli E, Morgillo F, Troiani T, Ciardiello F. Cancer resistance to therapies against the EGFR-RAS-RAF pathway: The role of MEK. Cancer Treat Rev 2017;53:61–9 [DOI] [PubMed] [Google Scholar]
- 6.Spreafico A, Tentler JJ, Pitts TM, Tan AC, Gregory MA, Arcaroli JJ, et al. Rational combination of a MEK inhibitor, selumetinib, and the Wnt/calcium pathway modulator, cyclosporin A, in preclinical models of colorectal cancer. Clin Cancer Res 2013;19:4149–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008;26:1626–34 [DOI] [PubMed] [Google Scholar]
- 8.Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–65 [DOI] [PubMed] [Google Scholar]
- 9.Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol 2012;13:782–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med 2012;367:107–14 [DOI] [PubMed] [Google Scholar]
- 11.Ascierto PA, McArthur GA, Dreno B, Atkinson V, Liszkay G, Di Giacomo AM, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)-mutant melanoma (coBRIM): updated efficacy results from a randomised, double-blind, phase 3 trial. Lancet Oncol 2016;17:1248–60 [DOI] [PubMed] [Google Scholar]
- 12.Larkin J, Ascierto PA, Dreno B, Atkinson V, Liszkay G, Maio M, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med 2014;371:1867–76 [DOI] [PubMed] [Google Scholar]
- 13.Ribas A, Gonzalez R, Pavlick A, Hamid O, Gajewski TF, Daud A, et al. Combination of vemurafenib and cobimetinib in patients with advanced BRAF(V600)-mutated melanoma: a phase 1b study. Lancet Oncol 2014;15:954–65 [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol 2014;11:385–400 [DOI] [PubMed] [Google Scholar]
- 15.Balmanno K, Chell SD, Gillings AS, Hayat S, Cook SJ. Intrinsic resistance to the MEK1/2 inhibitor AZD6244 (ARRY-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int J Cancer 2009;125:2332–41 [DOI] [PubMed] [Google Scholar]
- 16.Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol 2008;26:2139–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bennouna J, Lang I, Valladares-Ayerbes M, Boer K, Adenis A, Escudero P, et al. A Phase II, open-label, randomised study to assess the efficacy and safety of the MEK1/2 inhibitor AZD6244 (ARRY-142886) versus capecitabine monotherapy in patients with colorectal cancer who have failed one or two prior chemotherapeutic regimens. Invest New Drugs 2011;29:1021–8 [DOI] [PubMed] [Google Scholar]
- 18.Hochster HS, Uboha N, Messersmith W, Gold PJ, BH ON, Cohen D, et al. Phase II study of selumetinib (AZD6244, ARRY-142886) plus irinotecan as second-line therapy in patients with K-RAS mutated colorectal cancer. Cancer Chemother Pharmacol 2015;75:17–23 [DOI] [PubMed] [Google Scholar]
- 19.Deming DA, Cavalcante LL, Lubner SJ, Mulkerin DL, LoConte NK, Eickhoff JC, et al. A phase I study of selumetinib (AZD6244/ARRY-142866), a MEK1/2 inhibitor, in combination with cetuximab in refractory solid tumors and KRAS mutant colorectal cancer. Invest New Drugs 2016;34:168–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Novellasdemunt L, Antas P, Li VS. Targeting Wnt signaling in colorectal cancer. A Review in the Theme: Cell Signaling: Proteins, Pathways and Mechanisms. Am J Physiol Cell Physiol 2015;309:C511–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang K, Wang X, Zhang H, Wang Z, Nan G, Li Y, et al. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest 2016;96:116–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tentler JJ, Nallapareddy S, Tan AC, Spreafico A, Pitts TM, Morelli MP, et al. Identification of predictive markers of response to the MEK1/2 inhibitor selumetinib (AZD6244) in K-ras-mutated colorectal cancer. Mol Cancer Ther 2010;9:3351–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gregory MA, Phang TL, Neviani P, Alvarez-Calderon F, Eide CA, O’Hare T, et al. Wnt/Ca2+/NFAT signaling maintains survival of Ph+ leukemia cells upon inhibition of Bcr-Abl. Cancer Cell 2010;18:74–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim J, Kim DW, Chang W, Choe J, Kim J, Park CS, et al. Wnt5a is secreted by follicular dendritic cells to protect germinal center B cells via Wnt/Ca2+/NFAT/NF-kappaB-B cell lymphoma 6 signaling. J Immunol 2012;188:182–9 [DOI] [PubMed] [Google Scholar]
- 25.Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology 2000;47:119–25 [DOI] [PubMed] [Google Scholar]
- 26.Bachtel JC, Pendergraft JS, Rosychuk RA, Gustafson DL, Hansen RJ, Lunghofer PJ. Comparison of the stability and pharmacokinetics in dogs of modified ciclosporin capsules stored at −20 degrees C and room temperature. Vet Dermatol 2015;26:228–e50 [DOI] [PubMed] [Google Scholar]
- 27.Denton CL, Gustafson DL. Pharmacokinetics and pharmacodynamics of AZD6244 (ARRY-142886) in tumor-bearing nude mice. Cancer Chemother Pharmacol 2011;67:349–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamas S Cellular mechanisms of vascular injury mediated by calcineurin inhibitors. Kidney Int 2005;68:898–907 [DOI] [PubMed] [Google Scholar]
- 29.Ruggenenti P, Perico N, Mosconi L, Gaspari F, Benigni A, Amuchastegui CS, et al. Calcium channel blockers protect transplant patients from cyclosporine-induced daily renal hypoperfusion. Kidney Int 1993;43:706–11 [DOI] [PubMed] [Google Scholar]
- 30.Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res 2010;16:1613–23 [DOI] [PubMed] [Google Scholar]
- 31.Novartis. NEORAL: Prescribing Information. 2009
- 32.Patel SP, Kim KB. Selumetinib (AZD6244; ARRY-142886) in the treatment of metastatic melanoma. Expert Opin Investig Drugs 2012;21:531–9 [DOI] [PubMed] [Google Scholar]
- 33.Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004;22:4456–62 [DOI] [PubMed] [Google Scholar]