

Abstract
Recovery from injury to the central nervous system (CNS) is limited in the mammalian adult. Nonetheless, some degree of spontaneous recovery occurs after partial CNS injury. Compensatory axonal growth from uninjured neurons, termed sprouting, contributes to this naturally occurring recovery process and can be modulated by molecular intervention. Extensive studies have depicted a long-held hypothesis that oligodendrocyte-derived Nogo restricts axonal sprouting and functional recovery after CNS injury. However, cell type-specific function of Nogo in compensatory sprouting, spinal axon repair or functional recovery after CNS injury has not been reported. Here we present data showing that inducible, cell type-specific deletion of Nogo from oligodendrocytes led to a ~50% increase in the compensatory sprouting of corticospinal tract (CST) axons in the cervical spinal cord after unilateral pyramidotomy in mice. In contrast to a previously proposed growth-promoting role of neuronal Nogo in the optic nerve, deleting neuronal Nogo did not significantly affect CST axon sprouting in the spinal cord. Sprouting axons were associated with the expression of synaptic marker VGLUT1 in both the oligodendrocytic Nogo deletion and control mice. However, we did not detect any functional improvement in fine motor control associated with the increased sprouting in oligodendrocytic Nogo deletion mice. These data show for the first time with genetic evidence that Nogo specifically expressed by oligodendrocytes restricts compensatory sprouting after CNS injury, supporting a longstanding but heretofore untested hypothesis. While implicating a focus on sprouting as a repair mechanism in the translational potential of targeting the myelin inhibitory pathway, our study illustrates the challenge to harness enhanced structural plasticity for functional improvement.
Keywords: axon growth, sprouting, CNS injury, myelin inhibitor, neural repair, corticospinal tract (CST)
INTRODUCTION
Injury to the CNS often results in permanent functional impairment largely due to the limited ability of axons to grow after injury. There are two forms of injury-induced axonal growth: regeneration, axonal growth from injured neurons; and sprouting, compensatory axonal growth from uninjured neurons (Fig. 1) (Lee and Zheng, 2008; Tuszynski and Steward, 2012). In comparison to the very limited axon regeneration in the CNS, sprouting occurs spontaneously to some extent and is thought to contribute to partial recovery after brain or spinal cord injury (Bradbury and McMahon, 2006; Fouad et al., 2001; Rosenzweig et al., 2010; Ueno et al., 2012; Weidner et al., 2001). Harnessing and enhancing this innate form of structural plasticity in the CNS represents an important strategy in promoting repair and recovery after injury.
Figure 1. Compensatory axon sprouting is a CNS repair mechanism distinct from axon regeneration.
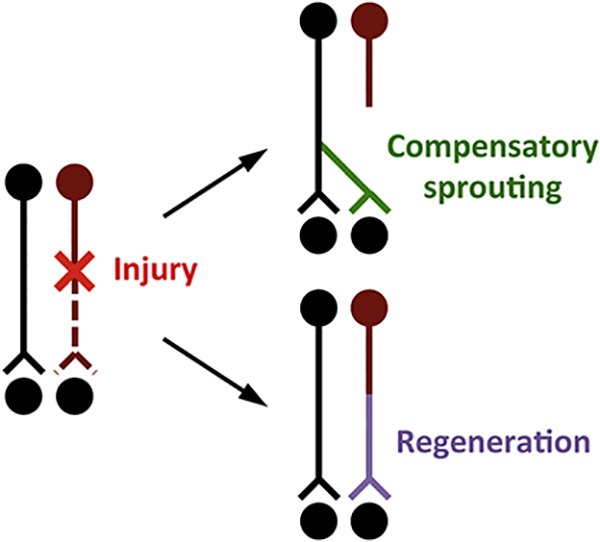
There are two forms of injury-induced axonal growth: regeneration, axonal growth from injured neurons; and sprouting, compensatory axonal growth from uninjured neurons. After a partial CNS injury that severs axons from a subset of neurons (as exemplified by the neuron in dark red), uninjured neurons (black) may exhibit spontaneous compensatory axon sprouting into denervated regions (green), a process distinct from axon regeneration from injured neurons (purple).
CNS myelin is considered inhibitory to axon growth due to the presence of a variety of factors expressed by oligodendrocytes (Geoffroy and Zheng, 2014; Schwab and Strittmatter, 2014). Nogo is one such protein. This membrane-bound protein binds to receptors expressed on the surface of neurons including NgR1 and PirB (Atwal et al., 2008; Fournier et al., 2001; Yiu and He, 2006). The Nogo gene encodes three major isoforms, Nogo-A,B,C, through alternative splicing and alternative promoter usage (Chen et al., 2000; GrandPre et al., 2000). Two domains of Nogo-A, Nogo-66 and NogoΔ20, are inhibitory to neurite outgrowth in vitro and destabilize the cytoskeleton via Rho-A signaling (Fournier et al., 2001; Joset et al., 2010; Niederöst et al., 2002; Oertle et al., 2003). All three Nogo isoforms contain Nogo-66, the ligand for NgR1 and PirB, while only Nogo-A contains NogoΔ20.
Function-blocking antibodies and peptides against Nogo-A increase axon growth and functional recovery in rodent and non-human primate models of spinal cord injury (Gonzenbach and Schwab, 2008; Zörner and Schwab, 2010). While pharmacological studies implicated enhanced axon regeneration as the underlying mechanism, genetic deletion of Nogo or its receptors NgR1 and PirB failed to demonstrate substantial and consistent regeneration of the corticospinal tract (CST), a commonly used model system in studying axonal repair after spinal cord injury (Ji et al., 2008; Kim et al., 2004; Kim et al., 2003; Lee et al., 2009; Nakamura et al., 2011; Simonen et al., 2003; Song et al., 2004; Steward et al., 2007; Zheng et al., 2005; Zheng et al., 2003). Nonetheless, Nogo deletion consistently resulted in increased compensatory axon sprouting after partial injury (Cafferty and Strittmatter, 2006; Lee et al., 2010).
In addition to oligodendrocytes, Nogo is also expressed by neurons. Neuronal expression of Nogo is most widespread during development, but persists in highly plastic regions of the adult CNS such as the hippocampus and cortex (Huber et al., 2002). Unexpectedly, silencing or deleting Nogo-A in retinal ganglion cells reduces axon regeneration and the expression of growth-related molecules after optic nerve crush, while Nogo-A overexpression promotes regeneration, implicating a growth-promoting role for neuronal Nogo-A (Pernet et al., 2012; Vajda et al., 2015). This raises the intriguing possibility that neuronal Nogo and oligodendrocytic Nogo have opposing roles, which has not been tested in the context of compensatory axonal sprouting in the CNS or any form of axonal repair in the spinal cord.
Genetic studies on Nogo in axonal repair in the spinal cord have exclusively relied on germline Nogo deletion, leaving open the possibility that developmental compensation has obscured the effect of Nogo deletion on anatomical or functional recovery from injury. Using inducible cell type-specific deletion of Nogo, here we formally test the hypothesis that Nogo expressed by oligodendrocytes limits CST axon sprouting and functional recovery from injury, whereas neuronal Nogo promotes CST axon sprouting. We found that oligodendrocyte-specific deletion of Nogo promoted CST sprouting but not functional recovery, while neuronal Nogo deletion had no significant effect on CST sprouting. These results provide the first evidence that oligodendrocytic but not neuronal Nogo inhibits compensatory axonal sprouting after CNS injury, and illustrate that other strategies are required to harness the functional benefits of this enhanced structural plasticity.
MATERIALS & METHODS
Generation of Nogo conditional mice
A conditional allele of Nogo was generated in which the first two exons common to all three major Nogo isoforms (Nogo-A,B,C) were flanked by loxP sites (“floxed”). Cre-mediated excision of these two exons would lead to a frame shift for all downstream exons and was predicted to result in a null for Nogo-C and at least a severe hypomorph for Nogo-A and Nogo-B. Importantly, this strategy would eliminate the expression of the growth inhibitory Nogo-66 amino acid loop shared by all three Nogo isoforms (GrandPre et al., 2000), and targeting a similar region of Nogo was previously shown to result in a null or severe hypomorph for all three major Nogo isoforms (Zheng et al., 2003). A targeting vector containing the floxed exons preceded by a floxed Neomycin selection cassette (thus with a total of three loxP sites) was targeted to the endogenous Nogo locus by homologous recombination in AB2.2 mouse embryonic stem cells to generate the targeted allele, which was then transmitted through the germline. Targeted events were initially identified by Southern blot analysis. The expected loxP sites in the targeted allele were verified by polymerase chain reaction (PCR) and sequencing analysis. The targeted mutant was bred to a universal deleter strain Actin-Cre (Lewandoski et al., 1997) to excise the Neomycin cassette via Cre-mediated partial excision, generating mice harboring the conditional allele of Nogo (Fig. 2A) as determined by PCR genotyping. Nogo conditional mutant mice were backcrossed to C57BL/6 at least 10 generations. To generate inducible oligodendrocyticspecific Nogo gene knockout mice, mice homozygous for the Nogo conditional allele (Nogof/f) were bred to PLPCreERT mice (Doerflinger et al., 2003) and backcrossed to generate Nogof/f and Nogof/f;PLPCreERT breeders that were used to generate littermate Nogof/f control and Nogof/f;PLPCreERT mice for sprouting studies. To generate conditional Nogo mice with gene knockout from CST neurons, conditional Nogo mice were bred to a tdTomato reporter line (Madisen et al., 2010) to generate littermate Nogo+/f control and Nogof/f mice all heterozygous for the inducible tdTomato reporter gene and were injected with AAV-Cre into the sensorimotor cortex to delete Nogo from CST neurons (as detailed below).
Figure 2. Generation and characterization of Nogof/f;PLPCreERT mice.
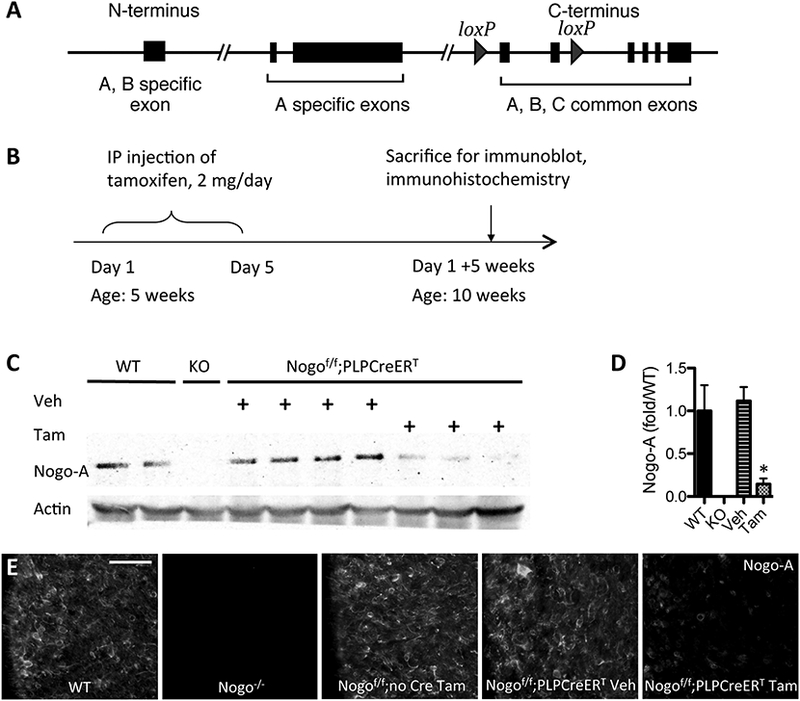
(A) Conditional Nogo allele in which two exons common to all Nogo isoforms is flanked by loxP sites. (B) Timeline of procedures for expression characterization. (C) Immunoblot against Nogo-A in spinal cord of WT, germline KO, control mice, and inducible KO mice. (D) Quantification of immunoblot in (C); *p<0.05 compared to WT and to Veh via ANOVA. Error bar, SEM. (E) Immunostain against Nogo-A in spinal cord white matter of WT, germline KO, control mice, and inducible KO mice. Scale bar = 50 μm.
Tamoxifen administration
Animals were administered 2 mg of tamoxifen (VWR) dissolved in sunflower oil or sunflower oil vehicle via intraperitoneal (IP) injection daily (2 mg/mouse/day) for 5 days beginning at 5 weeks of age for all studies. For sprouting and behavior studies, both Nogof/f;PLPCreERT mice and Nogof/f (Cre-less) controls were administered tamoxifen.
Western blot
Spinal cord protein was extracted by homogenization in 20 mM Tris-HCl (pH 7.5), 50 mM NaCl, 0.5% TritonX-100, 0.25% DOC, 1% SDS, 5 mM NaF, 1 mM EDTA (pH 8.0), 1 mM PMSF, and Complete Mini (Roche). After sitting on ice for 30 min, tissue homogenate was centrifuged at 11,000g for 30 min at 4°C. The protein concentration of the supernatant was determined using the Bradford assay (Bio-Rad) and 30 μg of protein in NuPAGE LDS sample buffer (Invitrogen) was separated with SDS-PAGE, and transferred to nitrocellulose membrane (Amersham). Membranes were washed in TBS-T 4×5 minutes, blocked with 5% milk for 1 hour at room temperature, and incubated overnight at 4°C with primary antibody. The next day, membranes were washed in TBS-T 4×5 minutes and incubated in appropriate HRP-conjugated secondary antibody (1:50,000, Peirce) while shaking for 1 hour at room temperature. After washing in TBST 4×5 minutes, membranes were incubated in Supersignal West Dura (Peirce) chemiluminescence solution for 5 min and then exposed to film (Genesee Scientific). For loading control detection, membrane portion containing actin was cut and detected as described above. Primary antibodies used were rabbit anti-Nogo-A at 1:500 (Zheng et al., 2003) and mouse anti-actin at 1:1000 (Millipore).
Tissue processing
Tissue processing was performed as described previously with minor modifications (Lee et al., 2009; Lee et al., 2010). Mice were administered a lethal dose of Fatal plus and perfused transcardially with 4% paraformaldehyde. Brain and spinal cord were dissected out, and the tissues were post-fixed overnight at 4°C in the same fixative solution. Tissues were incubated in 30% sucrose for cryo-protection. Brain, medulla, and C1-C7 cervical spinal cord were embedded in OCT compound and frozen on dry ice. Tissues were sectioned with a cryostat at a thickness of 20 μm. For sprouting studies, transverse sections of the medullas were processed to obtain estimates of the total number of CST axons labeled to control for labeling efficiency (see below). For tissues containing biotinylated dextran amine (BDA) labeled axons, cervical spinal cord and medulla sections were incubated in Vectastain ABC solution (Vector Laboratories) overnight at 4°C, washed in PBS, and mounted on gelatin-coated slides. BDA was detected with TSA Plus Fluorescein System (10 min, room temperature, 1:200, PerkinElmer). For tissues containing tdTomato-labeled axons, cervical spinal cord and medulla sections were processed without immunostaining for tdTomato. For immunohistochemistry, the following antibodies were used: purified polyclonal rabbit anti-Nogo-A (1:500) as described previously (Zheng et al., 2003), mouse anti-APC clone CC-1 (1:200, Millipore), rabbit anti-VGLUT1 (1:500, Abcam), biotinylated mouse anti-NeuN (1:100, Millipore), and rat anti-GFAP (1:200, Invitrogen). For sprouting studies, selected transverse sections of cervical spinal cord (C7) were immunostained for PKCγ (1:100, Santa Cruz Biotechnology) to examine the completeness of the lesion for each animal, as described previously (Lee et al., 2010). Mice with incomplete lesion were excluded from the study. A total of 1 animal was excluded based on this criteria of the genotype Nogof/f;PLPCreERT.
Immunofluorescence quantifications
To quantify the intensity of Nogo-A immunofluorescence in white and gray matter of cervical spinal cord, the mean intensity of the entire white and gray matter regions of transverse spinal cord sections were measured in AxioVision and averaged across five sections per animal. Mean intensity of the white and gray matter of germline Nogo knockout tissue was subtracted as the background for white and gray matter measurements. To quantify the number of APC+ cells that express Nogo-A, the total number of APC+ cells and the number of APC+Nogo-A+ cells were counted in a 335 μm x 448 μm sample region of cervical gray matter and of cervical white matter by an observer blinded to genotype for n=3 Nogof/f;no Cre and n=3 Nogof/f;PLPCreERT mice. To quantify the percent of BDA+ axons expressing VGLUT1, the total number of BDA+ axons and the number of BDA+VGLUT1+ axons were counted in a 168 μm x 224 μm sample region of cervical spinal cord gray matter with percent averaged between 3 sections per animal by an observer blinded to genotype for n=3 Nogof/f;no Cre and n=3 Nogof/f;PLPCreERT mice. To calculate the percent of tdTomato+ cells expressing NeuN, APC and GFAP and the percent of each cell type expressing tdTomato, the following were counted in a 335 μm x 448 μm sample cortical region near the injection site (where overall tdTomato expression rate was roughly 75% of DAPI+ cells): the total number of cells expressing tdTomato, NeuN, APC, and GFAP, and the total number of cells expressing both tdTomato and each cell type marker for n=3 Nogof/f;no Cre and n=3 Nogof/f;PLPCreERT mice. Percent calculations from 3 regions were averaged for each animal.
Surgical procedures
Pyramidotomy and biotinylated dextran amine (BDA) tracer injection were performed as described previously with minor modifications (Geoffroy et al., 2015; Lee et al., 2010; Liu et al., 2010; Starkey et al., 2005). Surgeons performing the surgeries were blinded to genotype. All animals were anesthetized with 2.5% Avertin (Sigma) and incision wounds were closed using Vetbond (3M). Pyramidotomy was performed at 2.5 months of age for anatomy studies (neuronal Nogo-A deletion, oligodendrocytic Nogo-A deletion without injury) and at approximately 4 months of age for behavior studies after completion of behavioral pre-training and baseline performance assessment. An incision was made overlying the trachea and the pyramidal tracts were accessed at the base of the skull (Starkey et al., 2005). A Feather micro scalpel was used to lesion the entire left pyramidal tract just caudal to the foramen magnum. Sham surgeries consisted of accessing the pyramidal tract in the same manner without lesioning the pyramidal tract. Cortical injection of BDA (10%, Invitrogen) was performed 2 weeks prior to sacrifice for all studies using the same injection volumes and coordinates as for AAVinjection (see Viral production and injection). Animals were sacrificed at approximately 4 months of age for anatomy studies and at approximately 8 months of age for behavior studies.
Viral production and injection
AAV-Cre was produced at the Salk Institute Viral Vector Core as described previously (Geoffroy et al., 2015; Liu et al., 2010). Viral concentration titer was determined to be 0.5 × 1012 TU/ml via qPCR. Virus was delivered via a modified 10 μL Hamilton syringe attached to a fine glass pipette mounted on a stereotaxic device for cortical injection. 5 week old animals were anesthetized with 2.5% Avertin (Sigma) and a total of 1.2 μL of AAV-Cre was injected into the sensorimotor cortex at 3 sites (0.4 μL per site). The right sensorimotor cortex targeting the left forelimb was injected 0.7 mm below the cortical surface at the following coordinates relative to bregma: 0.5 mm anterior, 1.2 mm lateral; 0.3 mm posterior, 1.2 mm lateral; and 0.1 mm anterior, 2.2 mm lateral.
Quantification of sprouting index and percent midline crossing axons
To determine the sprouting index and percent midline crossing axons, transverse spinal cord sections were taken from C5-C7 cervical levels and imaged for BDA and tdTomato (see Tissue Processing). Lines were drawn parallel to the midline dorsoventral axis at 50 and 100 microns from the midline, then every 100 microns laterally in the denervated gray matter. The number of axons crossing each line was manually counted in five randomly chosen sections per animal by a blinded observer. Counts were averaged for each animal and normalized against the total labeled CST axon count in medulla (Lee et al., 2010) to obtain the sprouting index and percent midline crossing axons, which was plotted as a function of distance from the midline. Numbers of mice for each experiment are as follows: oligodendrocyte-specific Nogo knockout mice with pyramidotomy (Figure 3): n=8 Nogof/f;no Cre mice and n=12 Nogof/f;PLPCreERT mice (with 1 additional Nogof/f;PLPCreERT mouse excluded from analysis due to incomplete pyramidotomy as described above under “Tissue processing”); oligodendrocyte-specific Nogo knockout mice without injury (Figure 5): n=3 Nogof/f;no Cre mice and n=3 Nogof/f;PLPCreERT mice; AAV-Cre mediated, cortical neuronal deletion of Nogo with pyramidotomy (Figure 9): n=5 Nogo+/f control mice and n=7 Nogof/f mice.
Figure 3. Inducible deletion of Nogo from oligodendrocytes promotes compensatory sprouting of corticospinal tract axons.
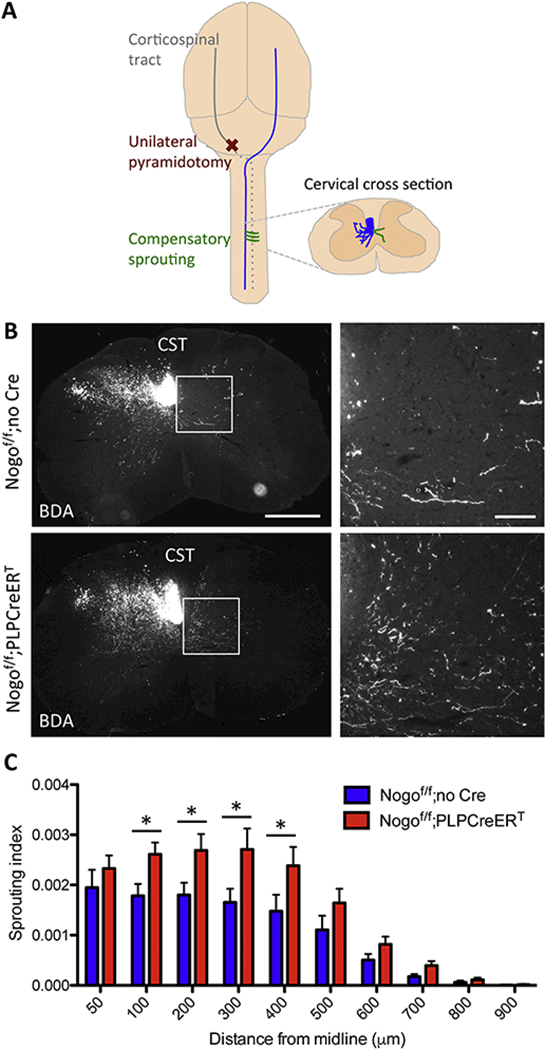
(A) Drawing of the unilateral pyramidotomy model with a dorsal view of the brain. Note that, at the level of the pyramid, unilateral pyramidotomy is performed at the most ventral part of the brain. (B) BDA-labeled CST axons in cervical sections from Nogof/f;no Cre and Nogof/f;PLPCreERT mice after unilateral pyramidotomy. White box depicts enlarged region shown in right panels. Scale bars = 500 μm (low mag, left), 100 μm (high mag, right). (C) Quantification of the number of BDA-labeled axons at specific distances from the midline in the denervated side of the spinal cord normalized to the total labeled CST axon count at the medulla in Nogof/f;no Cre (n=8) and Nogof/f;PLPCreERT (n=12) mice. *p <05, two-way RM ANOVA followed by Fisher’s least significant difference test. Error bar, SEM.
Figure 5. Inducible deletion of Nogo from oligodendrocytes does not affect axon counts in the absence of injury.
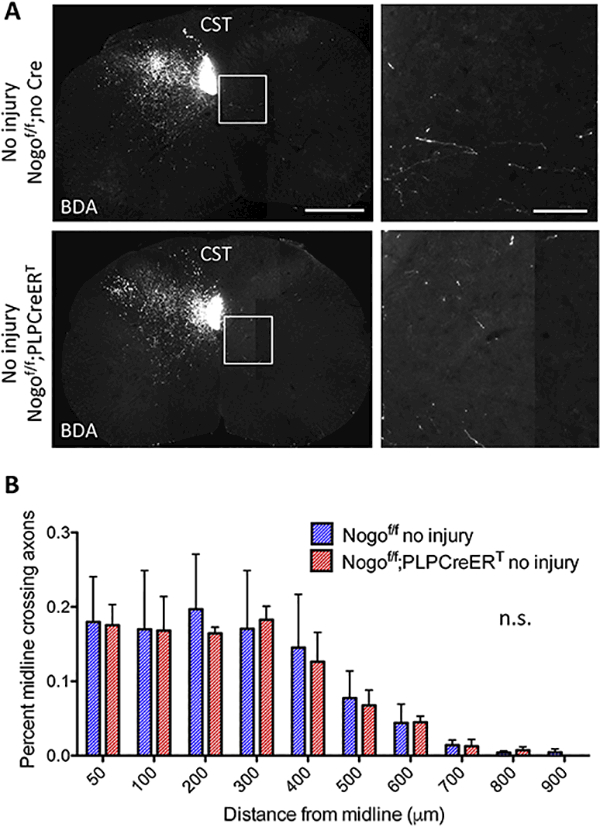
(A) BDA-labeled CST axons in cervical sections from Nogof/f;no Cre and Nogof/f;PLPCreERTmice without injury. White boxes depict enlarged regions shown in the right panels. Scale bars = 500 μm (low mag, left), 100 μm (high mag, right). (B) Quantification of the number of BDA-labeled axons at specific distances from the midline contralateral to the main labeled CST normalized to the total labeled CST axon count at the medulla in Nogof/f;no Cre (n=3) and Nogof/f;PLPCreERT(n=3) mice. n.s, not significant by two-way RM ANOVA. Error bar, SEM.
Figure 9. Deletion of Nogo from cortical neurons via AAV2-Cre injection does not significantly affect CST sprouting.
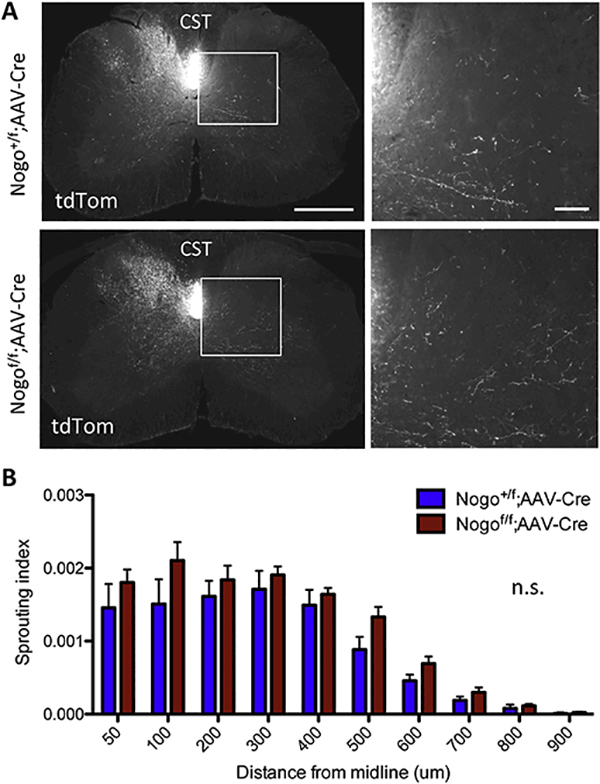
(A) tdTomato-labeled CST axons in cervical sections from Nogo+/f;tdTomato control and Nogof/f;tdTomato mice after cortical injection of AAV2-Cre and unilateral pyramidotomy. White boxes depict enlarged regions shown on the right. Scale bars = 500 μm (low mag, left), 100 μm (high mag, right). (B) Quantification of the number of tdTomatopositive axons at specific distances from the midline in the denervated side of the spinal cord normalized to the total labeled CST axon count in the medulla in Nogo+/f control (n=5) and Nogof/f (n=7) mice. n.s, not significant by two-way RM ANOVA.
Behavioral assessment
All behavioral tests were performed and quantified by observers blinded to genotype. For both staircase reaching and pasta handling tests, mice were deprived of food at approximately 5pm and tested at approximately 9am the following morning resulting in approximately 16 hours food deprivation. Animals were weighed weekly to monitor for excessive weight loss (greater than 10%). For staircase reaching, mice were pre-trained and tested as described previously with slight modifications (Baird et al., 2001; Kloth et al., 2006; Starkey et al., 2005). For habituation prior to pre-training, animals were exposed to dyed sucrose pellets (Fisher Scientific Dustless Precision Pellets 14 mg, dyed with AmeriColor Student Soft Gel Paste Food Color) in their home cages and habituated to the testing environment, experimenter, and staircase chambers (Campden Instruments) for approximately 10 minutes per day for 3 days. Staircase chambers were modified to prevent build up of displaced pellets on lower steps by removing a plastic barrier at the base of the staircase and by adding a plastic barrier between the staircase chamber and the main chamber. During pre-training, animals were placed in sucrose-pellet baited chambers for 10 minutes once daily for 4–5 days per week until performance plateau, for a total of approximately 25 sessions. For all pre-training and testing sessions, staircases were baited with 4 pellets on each step other than the top two steps to prevent usage of the tongue to retrieve pellets. Initial pre-training sessions were baited with additional pellets on the top steps and the central platform to encourage exploration and pellet retrieval. Throughout pre-training and testing, performance was monitored to ensure pellets were retrieved with forepaws and not tongues. After pre-training, animals were tested once every 7 days on day 3, 10, 17, etc. after pyramidotomy (n=12 Nogof/f;PLPCreERT, Nogof/f mice and n=16 Nogof/f;PLPCreERT mice). Test sessions were 10 minutes, after which the number and location of remaining pellets was recorded to calculate pellets eaten (successful retrievals) and pellets eaten relative to pellets displaced (success rate). For pasta handling, mice were tested as described previously (Tennant et al., 2010). Briefly, mice were video-recorded while eating a 2.5 cm piece of dry capellini pasta and the number of adjustments made with each paw was quantified by watching frame-by-frame video playback (n=7 Nogof/f;no Cre mice and n=5 Nogof/f;PLPCreERT mice). Adjustments consisted of paw release and re-contact with the pasta or any movement of the paw or digits while in contact with the pasta as described previously (Tennant et al., 2010). Mice were habituated to the testing setup for 2 sessions prior to testing. Pasta handling testing occurred 10 weeks after pyramidotomy after the conclusion of staircase testing.
Experimental design and statistical analysis
Western blot data were analyzed via one-way ANOVA. Sprouting index, percent midline crossing axons, staircase reaching, pasta handling and Nogo-A immunofluorescence data were analyzed via two way repeated measures ANOVA with Bonferroni or Fisher post-tests as indicated. VGLUT1 expression data was analyzed via unpaired t-test. For all statistical analyses, GraphPad Prism 7 was used with p < 0.05 set as the threshold for statistically significance, denoted by asterisks in graphs. All data is displayed as means with error bars denoting SEM. Specific n values for each study are listed in figure legends. For analysis of sprouting index and percent midline crossing axons, five cervical sections and two medulla sections were quantified for each animal (see Quantification of sprouting index and percent midline crossing axons for specifi cdetails). A mix of male and female mice in an approximately 1:1 ratio was used for all experiments.
RESULTS
Establishing inducible, oligodendrocyte-specific Nogo gene knockout mice
To investigate the effects of deleting Nogo from specific cell types after development is complete, we designed and generated a conditional allele of Nogo using the Cre-loxP sitespecific recombination system (Fig. 2A). The Nogo gene encodes three major isoforms, Nogo-A, B and C, through alternative splicing and alternative promoter usage (Chen et al., 2000; GrandPre et al., 2000). We designed a targeting strategy (see Materials and Methods for details) similar to what we previously used to disrupt all three isoforms simultaneously (Zheng et al., 2003).
Mice harboring this allele were crossed to the PLPCreERT mouse line in which an inducible Cre transgene is expressed under the myelin proteolipid protein (PLP) promoter, which is highly expressed in mature oligodendrocytes (Doerflinger et al., 2003). To assess the extent of Nogo deletion using this approach, Nogof/f;PLPCreERT mice and Nogof/f (Cre-less) controls were administered 2 mg/mouse/day of tamoxifen or sunflower oil vehicle via IP injection for 5 days beginning at 5 weeks of age. Five weeks later, mice were sacrificed for immunoblot and immunohistochemical analyses (Fig. 2B). Nogo-A protein levels in whole spinal cord of vehicle-treated Nogof/f;PLPCreERT mice remained similar to wild-type (WT) levels, while tamoxifen administration led to an ~87% reduction in Nogo-A protein levels relative to vehicletreated Nogof/f;PLPCreERT mice [F(2,6) = 9.951, p = 0.0124, ANOVA, WT vs. TAM and Veh vs. TAM p < 0.05] (Fig. 2C,D). The incomplete loss of total Nogo-A signal is likely due to the presence of Nogo-A in other cell types including neurons and also possibly incomplete removal of Nogo-A from oligodendrocytes or myelin (see below). Likewise, immunostaining against Nogo-A in cervical spinal cord revealed reduction of Nogo-A levels in Nogof/f;PLPCreERT mice treated with tamoxifen but not in vehicle-treated Nogof/f;PLPCreERT mice or tamoxifen-treated Nogof/f (Cre-less) control mice (Fig. 2E).
Inducible Nogo deletion from oligodendrocytes promotes corticospinal tract axon sprouting
To provide a rigorous genetic test on the long-held hypothesis that oligodendrocytic Nogo inhibits axonal sprouting, we assessed the effect of deleting all three Nogo isoforms from PLPexpressing cells in young adult mice on corticospinal tract (CST) axon sprouting after unilateral pyramidotomy. In the unilateral pyramidotomy injury model, one side of the CST is lesioned at the level of the medullary pyramids, and compensatory sprouting of CST axons into the contralateral, denervated side of the cervical spinal cord is assessed (Fig. 3A). After the same induction method described previously in which mice were administered tamoxifen beginning at 5 weeks of age (Fig. 2B), unilateral pyramidotomy was performed at approximately 4 months of age and animals were sacrificed two weeks after cortical injection of axon tracer biotinylated dextran amine (BDA) at approximately 8 months of age. This protracted timeline was necessary to allow time for behavioral pre-training and assessment. Axon tracer was visualized in the cervical spinal cord of Nogof/f control and Nogof/f;PLPCreERT mice by ABC and TSA amplification of BDA (Fig. 3B).
Axon sprouting indices were quantified by counting the numbers of axons crossing predetermined distances from the midline in the contralateral cervical cord, normalized against the total CST axon count in the medulla as previously described (Fig. 3C) (Geoffroy et al., 2015; Lee et al., 2010). Nogof/f;PLPCreERT mice exhibited an increase in the number of axons crossing defined distances from the midline relative to tamoxifen-treated Nogof/f littermate control mice not expressing Cre [F(9,162) = 2.11, p = 0.0313, two-way repeated measures (RM) ANOVA] (Fig. 3C). This effect was largest at 300 μm from the midline, where normalized axon counts were increased by 64% [F(9,162) = 2.11, p = 0.0313 for two-way RM ANOVA; Fisher’s least significant difference post-test p = 0.0251 at 100 μm, 0.0163 at 200 μm, 0.0046 at 300 μm, and 0.0144 at 400 μm].
To confirm efficient deletion of Nogo-A from white matter, from gray matter, and from oligodendrocytes in these animals, cervical sections were doubly immunostained for Nogo-A and APC, a mature oligodendrocyte marker (Fig. 4A). Total Nogo-A expression was significantly reduced in both white and gray matter [F(1,4)=49.69, p = 0.0021 for two-way RM ANOVA; Bonferroni’s post-test, p < 0.0001 for white matter and p = 0.0123 for gray matter] (Fig. 4B). Since neuronal Nogo-A was expected to remain in Nogof/f;PLPCreERT mice, the reduction of Nogo-A immunoreactivity was more noticeable in the white matter than in the gray matter (Fig. 4A,B). Furthermore, Nogo-A was below detectable levels in ~91% of gray matter APC+ cells in tamoxifen-treated Nogof/f;PLPCreERT mice, confirming efficient Nogo deletion in oligodendrocytes even in the gray matter [F(1,4)=700.8, p < 0.0001 for two-way RM ANOVA; Bonferroni’s post-test, p < 0.0001 for both white and gray matter] (Fig. 4C).
Figure 4. Validation of Nogo deletion from experimental animals.
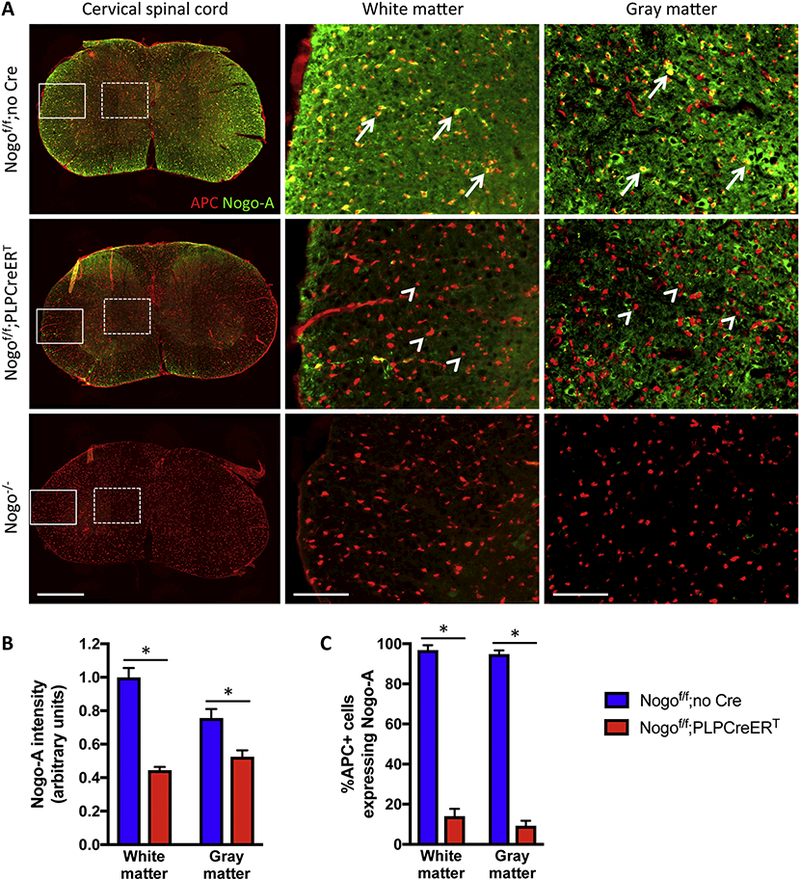
(A) Double immunostain of APC and Nogo-A in experimental animals from Figure 3 and germline Nogo KO (Nogo−/−) tissue. White boxes depict regions shown in the middle panels; white dashed boxes depict regions show in the right panels. Arrows indicate APC+Nogo-A+ cells; arrowheads indicate APC+ cells that are negative for Nogo-A. Note that, in the gray matter, neuronal Nogo-A is expected to remain in the oligodendrocyte-specific Nogo knockout (tamoxifen treated Nogof/f;PLPCreERT) mice. Scale bars = 500 μm (low mag, left panels), 100 μm (high mag, middle and right panels). (B,C) Quantification of background-subtracted Nogo-A immunofluorescence (B) and percent of APC+ cells expressing Nogo-A (C) in Nogof/f;no Cre (n=3) and Nogof/f;PLPCreERT(n=3) mice. *p < 0.05, two-way RM ANOVA with Bonferroni post-tests. Error bar, SEM.
Importantly, sham lesioned Nogof/f;PLPCreERT and tamoxifen-treated Nogof/f littermate controls in which the medullary pyramids were surgically accessed but not lesioned exhibited no difference in midline crossing axons [F(9,36) = 0.11, p = 0.9994, two-way RM ANOVA] (Fig. 5A,B). Together, these data indicate that inducible, oligodendrocyte-specific Nogo deletion enhances CST sprouting across the midline in the cervical spinal cord after unilateral pyramidotomy.
To assess the potential for sprouted axons to form synapses, we examined the expression of the pre-synaptic marker VGLUT1 in conjunction with BDA tracing of CST axons in Nogof/f control and Nogof/f;PLPCreERT mice (Fig. 6). In both control Nogof/f and Nogof/f;PLPCreERT mice, VGLUT1 staining is evident along BDA-labeled axons, consistent with the formation of synaptic structures by sprouting axons (Fig. 6A,B,C). A substantial proportion of BDA-labeled axons expressed VGLUT1 in both genotypes, with Nogof/f;PLPCreERT mice exhibiting a trend for a higher percentage of co-labeling than control Nogof/f mice that did not reach statistical significance [~79% and ~67% respectively, t(4) = 2.553, p = 0.0631, unpaired t-test] (Fig. 6D).
Figure 6. Sprouting axons form synapses.
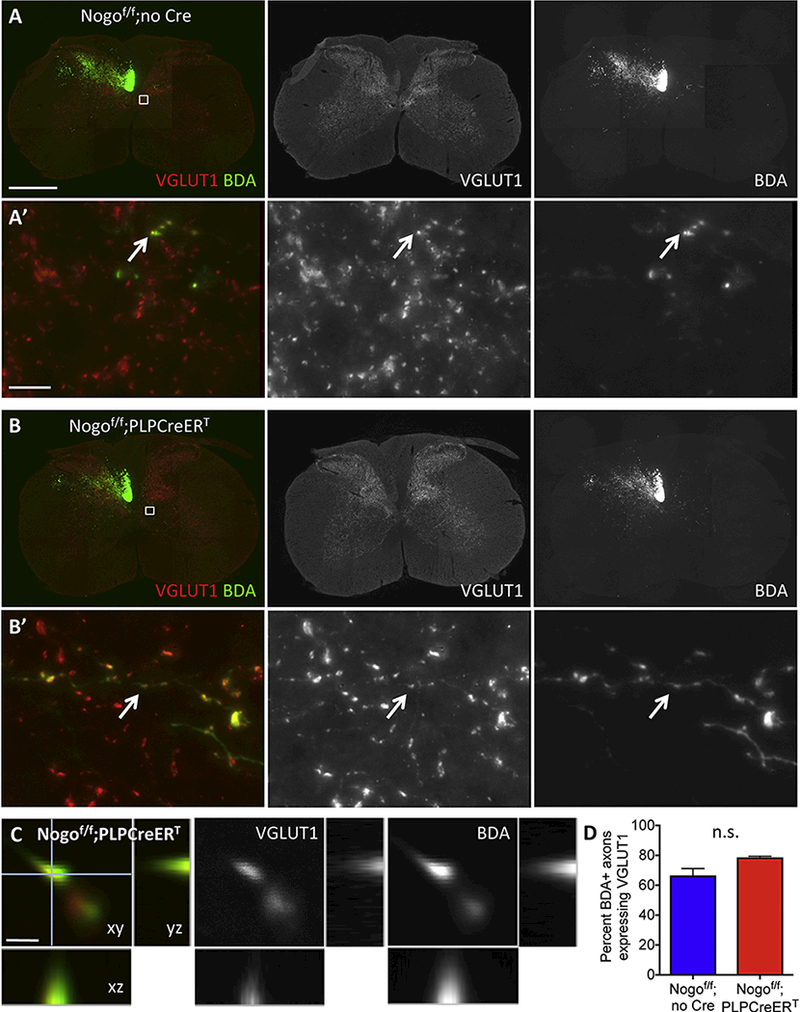
(A-B’) VGLUT1 and BDA double labeling in cervical sections from Nogof/f;no Cre (A and A’) and Nogof/f;PLPCreERT mice (B and B’) after unilateral pyramidotomy. (C) Confocal image showing colocalization of VGLUT1 and BDA. (D) Quantification of the percent of BDA+ axons expressing VGLUT1 in Nogof/f;no Cre (n=3) and Nogof/f;PLPCreERT(n=3) mice. Small white boxes in (A) and (B) depict enlarged regions shown in (A’) and (B’) respectively. Arrows in (A’) and (B’) exemplify VGLUT1 and BDA co-labeled axons. Scale bars = 500 μm (A’), 10 μm (A’), 1 μm (C). n.s, not significant by unpaired t-test.
Inducible Nogo deletion from oligodendrocytes does not promote functional recovery after injury
To determine whether the increase in CST sprouting was associated with improved functional recovery from injury, we tested Nogof/f control and Nogof/f;PLPCreERT mice in the staircase reaching test and the pasta handling test, two assays of fine motor control (Fig. 7A,D) (Baird et al., 2001; Tennant et al., 2010). Both groups of mice showed similar pre-injury and initial postinjury performance in staircase reaching (Fig. 7B,C). Furthermore, while this assay was able to detect significant functional impairment in both groups, Nogof/f control mice showed significant recovery by 6 weeks post-injury whereas Nogof/f;PLPCreERT mice did not (Fig. 7B,C). [Successful retrievals: two-way repeated measures ANOVA with Bonferroni post-tests on week 6 versus −1 and 0, F(2,52) = 38.12, p < 0.0001, week 6 vs. −1 p < 0.01 for both groups, week 6 versus 0 p < 0.05 for Nogof/f but not Nogof/f;PLPCreERT. Success rate: two-way repeated measures ANOVA with Bonferroni post-tests on week 6 versus −1 and 0, F(2,52) = 30.93, p < 0.0001, week 6 vs. −1 p < 0.05 for both groups, week 6 versus 0 p < 0.05 for Nogof/f but not Nogof/f;PLPCreERT.] Similarly, the affected paw showed impairment in pasta handling 10 weeks after injury as evidenced by increased adjustments relative to the intact paw with no significant difference between groups [F(1,7) = 12.57, p = 0.0094, two-way repeated measures ANOVA with Bonferroni post-tests] (Fig. 7E).
Figure 7. Inducible deletion of Nogo from oligodendrocytes does not promote functional recovery from injury.
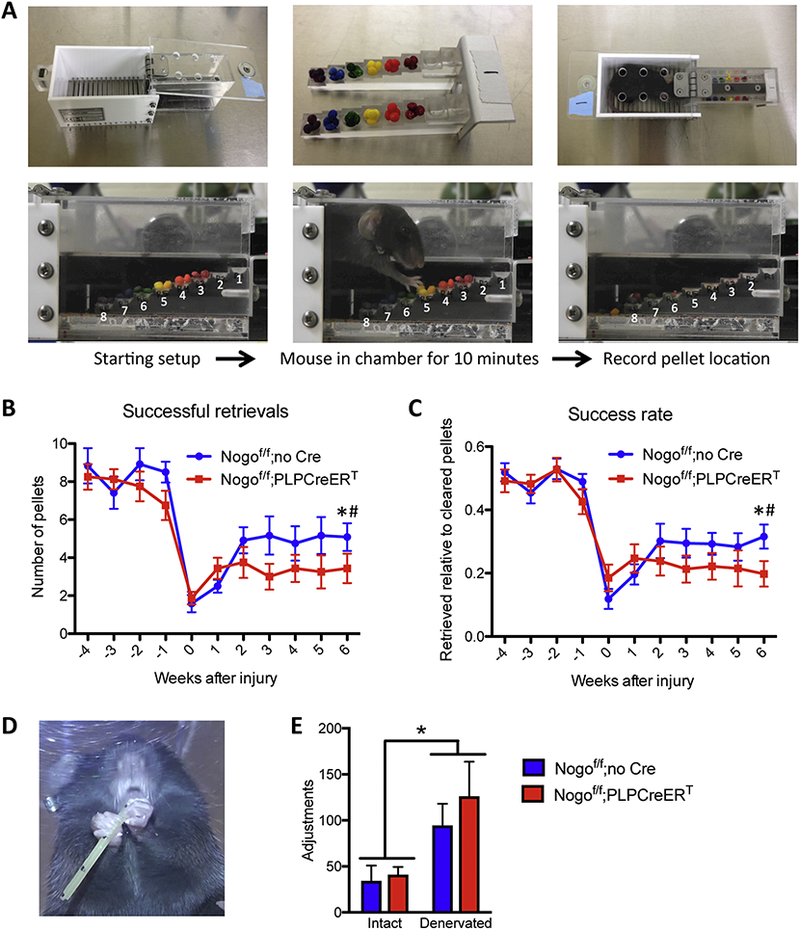
(A) Staircase reaching test in which the mouse is allowed to retrieve color-coded sucrose pellets from steps of varying heights over the course of 10 minutes. (B) Number of pellets retrieved using denervated paw during 10-minute sessions each week after injury. Week 6 performance is significantly reduced relative to baseline week −1 for both genotypes, whereas week 6 performance is significantly improved from week 0 for control Nogof/f;no Cre mice but not for Nogof/f;PLPCreERT mice. (C) Success rate for denervated paw during 10-minute sessions each week after injury. Week 6 performance is significantly reduced relative to baseline week −1 for both genotypes, whereas week 6 performance is significantly improved from week 0 for Nogof/f;no Cre mice but not Nogof/f;PLPCreERT mice. (D) Mouse performing pasta-handling test where the impaired paw exhibits more adjustments than the intact paw. (E) Number of adjustments made with intact vs. denervated paw during pastahandling test 10 weeks after injury. For (B) and (C) two-way RM ANOVA with Bonferroni posttests on week 6 versus −1 and 0, * p < 0.05 week 6 vs. 0 and 1 for Nogof/f;no Cre, # p < 0.05 week 6 vs. −1 for Nogof/f;PLPCreERT, Nogof/f n=12, Nogof/f;PLPCreERT n=16. For (E) two-way RM ANOVA with Bonferroni post-tests p = 0.0094 for paw, Nogof/f;no Cre n=7, Nogof/f; PLPCreERT n=5.
Neuronal deletion of Nogo does not significantly affect sprouting
Previous studies indicate that in the optic nerve system, neuronal Nogo-A and oligodendrocytic Nogo-A have opposite roles in regulating axon growth following injury, which was proposed as a contributing factor to the less-than-expected effect of germline Nogo deletion on axonal repair after CNS injury (Pernet et al., 2012; Vajda et al., 2015). We therefore assessed whether deletion of Nogo from corticospinal neurons themselves affects sprouting. Nogo conditional mice were bred to a tdTomato reporter line to generate littermate Nogo+/f control and Nogof/f mice both of which were also heterozygous for the inducible tdTomato reporter gene (Madisen et al., 2010). Upon AAV2-Cre injection in the motor cortex, cortical cells expressed tdTomato and concomitantly exhibited a substantial reduction of Nogo-A immunoreactivity in neurons identified through NeuN staining (~94% reduction in the percentage of tdTomato+NeuN+ cells that express Nogo-A) (Fig. 8A,B). Furthermore, based on tdTomato reporter expression near the injection sites, ~95% of AA2-Cre transduced cells were NeuN+, ~0.3% were APC+, and ~0.8% were GFAP+; meanwhile, transduction efficiencies for cells expressing each cell type marker were found to be ~74% for NeuN, ~1.2% for APC, and ~8% for GFAP (Fig. 8C,D). TdTomato expression in cervical sections enabled tracing of CST axons only from neurons that had been infected with AAV2-Cre (Fig. 9A). There was no statistically significant difference in the degree of CST sprouting between control and neuronal Nogo-A conditional gene deletion mice (Fig. 9B) [F(1,10) = 2.28, p = 0.1618, two-way repeated measures ANOVA], indicating that neuronal Nogo is unlikely to be a significant factor, at least by itself, in regulating CST axon sprouting after injury.
Figure 8. AAV2-Cre cortical injection induces tdTomato expression and deletes neuronal Nogo-A in Nogof/f;tdtom mice.
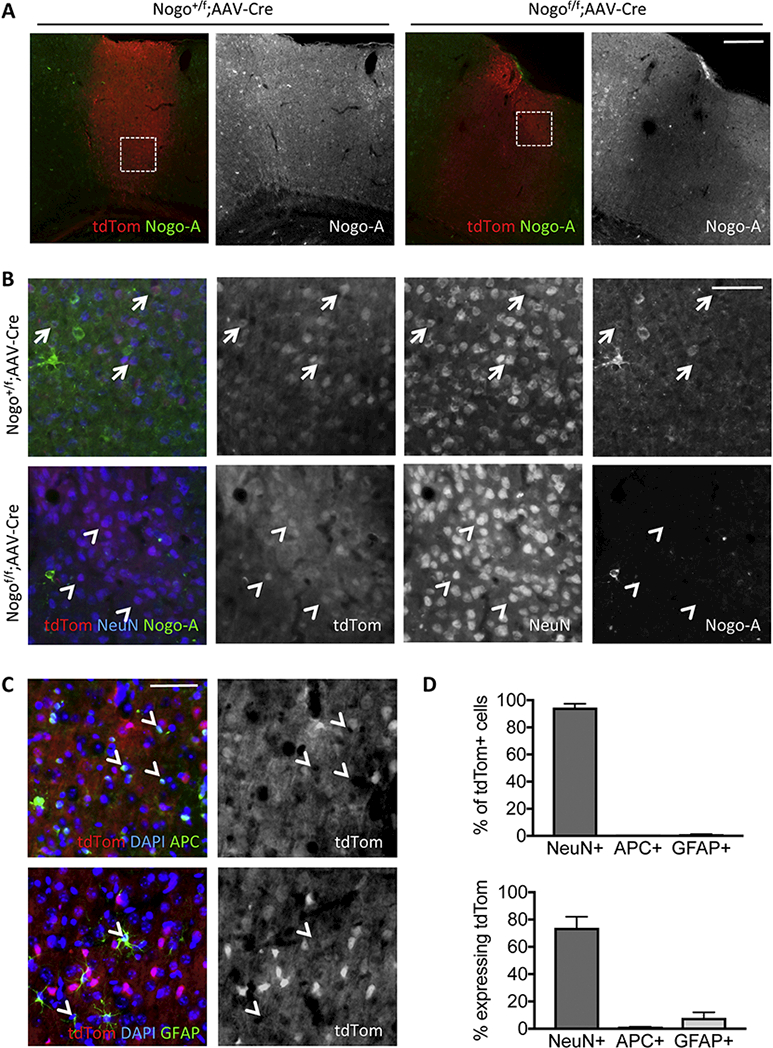
(A) Coronal brain slices from Nogo+/f;tdTomato (“Nogo+/f”) and Nogof/f;tdTomato (“Nogof/f”) mice near cortical AAV2-Cre injection sites immunostained for Nogo-A showing induction of tdTomato reporter and deletion of Nogo-A. White boxes depict enlarged regions shown in (B). (B) High magnification images showing Nogo-A signal in tdTomato, NeuN positive cells in control Nogo+/f mice and loss of Nogo-A signal in Nogof/f mice following AAV-Cre mediated recombination. Arrows indicate tdTomato+NeuN+Nogo-A+ cells; arrowheads indicate tdTomato+NeuN+ cells that are negative for Nogo-A. (C) High magnification images showing relative lack of transduction of APC+ and GFAP+ cells by AAV2-Cre as evidenced by tdTomato reporter (white arrowheads). (D) Quantification of the percent of tdTomato+ cells expressing each of the cell type markers NeuN, APC, GFAP (top panel) and the percent of each cell type expressing tdTomato near cortical injection sites (bottom panel). Scale bars = 200 μm (A), 50 μm (B, C).
DISCUSSION
In the present study we investigated the role of Nogo expressed by oligodendrocytes and by neurons in the compensatory sprouting of CST axons and functional recovery from CNS injury using inducible and cell type-specific gene deletion in mice. In doing so we tested a longstanding hypothesis that Nogo expressed by oligodendrocytes limits anatomical and functional recovery of the spinal cord after injury. We found that Nogo expressed by oligodendrocytes limits CST sprouting, while neuronal Nogo does not significantly modulate CST sprouting. Our results provide the first evidence with rigorous genetic analyses that oligodendrocytic Nogo restricts compensatory axon sprouting in the mammalian CNS after injury.
Oligodendrocytic Nogo inhibits compensatory sprouting of CST axons while neuronal Nogo has no effect
In this study, we focused exclusively on the compensatory sprouting of CST axons after a partial CNS injury. We found that deleting Nogo from oligodendrocytes using inducible Nogof/f;PLPCreERT mice resulted in a ~50% increase in the number of sprouting CST axons after pyramidotomy. This level of sprouting enhancement is comparable to — and at the upper limit of— that from previous studies from our laboratory indicating an up to ~50% increase in CST sprouting following the same pyramidotomy injury in germline Nogo knockout mice (Geoffroy et al., 2015; Lee et al., 2010). Together, these results indicate that oligodendrocytic Nogo is primarily responsible for its growth-inhibitory effect on CST sprouting. As a reference point, neuronal Pten deletion consistently leads to a ~2 fold or more increase in a similar CST sprouting paradigm (Geoffroy et al., 2015; Liu et al., 2010). Indeed, Pten deletion led to a higher level of CST sprouting enhancement in a side-by-side comparison with germline Nogo deletion (Geoffroy et al., 2015). Thus, while we did not conduct a side-by-side comparison in the current study, it is apparent that oligodendrocytic Nogo deletion produces a more modest effect on CST sprouting than neuronal Pten deletion.
In previous studies, deleting or silencing Nogo-A in retinal ganglion cells reduced axon regeneration after optic nerve crush (Pernet et al., 2012; Vajda et al., 2015). In contrast, in the present study we did not detect a growth-reducing effect of neuronal Nogo deletion in CST sprouting (if anything, neuronal Nogo deletion may have slightly increased CST sprouting although the difference did not reach statistical significance). This was unlikely due to a floor effect of CST sprouting after pyramidotomy injury since we previously were able to detect a consistent level of reduction in CST sprouting in MAG knockout mice with the same injury model (Lee et al., 2010). The apparent discrepancy in the effect of neuronal Nogo deletion between our study and Vajda et al. (2015) could reflect the distinct roles of Nogo in different neuron subtypes (e.g., corticospinal neurons versus retinal ganglion cells) or in different forms of axon growth after injury (sprouting versus regeneration). On the one hand, optic nerve injury leads to substantial retinal ganglion cell death (Li et al., 1999) whereas experimental spinal cord injury does not cause significant corticospinal neuron death (Nielson et al., 2010; Nielson et al., 2011). On the other hand, our current study focused exclusively on the compensatory axon sprouting from uninjured neurons using an injury model (unilateral pyramidotomy) that is specifically designed to evaluate CST sprouting; in contrast, Vajda et al. (2015) focused on axon regeneration (albeit short distance regeneration) from injured retinal ganglion neurons based on the commonly accepted definitions of regeneration and sprouting (Fig. 1) (Lee and Zheng, 2008; Tuszynski and Steward, 2012), even though they referred to this form of regeneration as “regenerative sprouting” (Vajda et al., 2015). Notably, the gene targeting strategy employed by Vajda et al. (2015) to conditionally delete Nogo-A undesirably led to substantial Nogo-B upregulation, confounding the interpretation of their data regardless of the focus on regeneration or sprouting (Vajda et al., 2015). In contrast, our gene targeting strategy deletes critical exons for all 3 Nogo isoforms (Nogo-A,B,C), leaving no ambiguity in our conclusions on the cell typespecific function of Nogo.
Lack of functional benefit of deleting Nogo from oligodendrocytes
We were not able to detect a functional benefit resulting from deletion of oligodendrocytic Nogo in two independent measures of forelimb function. Similarly, germline deletion of Nogo did not improve various measures of recovery from pyramidotomy including forepaw preference during rearing, ladder rung missteps, or tape removal assays, but has been shown to increase affected paw usage in food pellet retrieval (Cafferty and Strittmatter, 2006; Geoffroy et al., 2015; Lee et al., 2010). Nonetheless, a large body of literature supports that pharmacologically or functionally blocking Nogo-A in rats and non-human primates promotes functional recovery from CNS injury including pyramidotomy, an effect that appears to be distinct from that of oligodendrocytic deletion of Nogo we evaluated here (Schwab and Strittmatter, 2014; Thallmair et al., 1998; Z’Graggen et al., 1998). In fact, in the present study control mice showed some degree of spontaneous recovery from injury, whereas oligodendrocytic Nogo knockout mice did not. Nonetheless, sprouted axons on the denervated side in both groups expressed the pre-synaptic marker VGLUT1, suggesting an ability to form functional synapses, consistent with previous reports of VGLUT1 expression along pyramidotomy-induced sprouting axons (Maier et al., 2008; Starkey et al., 2012).
Thus, enhancing axon sprouting alone may not be sufficient to promote functional recovery from injury. Indeed, in certain cases increased sprouting even has been correlated with worse functional recovery from injury (Wang et al., 2015). Rehabilitative training could be an important factor in harnessing the enhanced sprouting for functional improvement (García-Alías et al., 2009). In the case of Nogo-A manipulations, the timing of rehabilitative training might be an additional important consideration, as differently timed rehabilitative training may either enhance or worsen functional recovery after CNS injury (Chen et al., 2017; Wahl et al., 2014). Additionally, the identity and functionality of the cells contacted by sprouting CST axons requires further investigation. It remains possible that these newly formed synapses are non-functional or are made with cells and networks that do not enhance motor function. Our results indicate that removal of oligodendrocytic Nogo is sufficient to promote anatomical plasticity after CNS injury, but alone may not be sufficient to promote functional recovery. Whether rehabilitative training will unmask a functional benefit of deleting oligodendrocytic Nogo remains unknown. Future investigation is certainly required to understand the relationship between enhanced sprouting and functional improvement.
Conclusions
Here we showed that inducible, cell type-specific Nogo deletion in oligodendrocytes enhances compensatory sprouting of CST axons after a partial CNS injury. This study represents the first rigorous genetic test demonstrating that oligodendrocytic Nogo inhibits compensatory sprouting and that oligodendrocytic Nogo inhibits axonal repair in the spinal cord. The results have important implications for ongoing clinical trials testing anti-Nogo antibodies, emphasizing the potential for enhanced sprouting while highlighting the challenge in harnessing this structural plasticity for functional recovery.
Highlights.
Inducible cell type-specific gene deletion validates growth inhibitory role of Nogo
Oligodendrocytic Nogo inhibits corticospinal axon sprouting after CNS injury
Neuronal Nogo does not have a growth-promoting role in corticospinal axon sprouting
Functional recovery remains a challenge even with enhanced structural plasticity
Acknowledgments
We thank Vung Khai for assisting in tissue processing. We thank Brian Popko for providing the PLPCreERT mice. The Nogo conditional allele was initially generated by B.Z in Marc Tessier-Lavigne’s lab and then characterized in B.Z’s lab. This work was supported by grants from the National Institutes of Health (NS054734, NS093055), Wings for Life (WFL-US-27/17) and Craig H. Neilsen Foundation (384971) to B.Z. J.M.M. was supported by the Neuroplasticity of Aging Training program at UC San Diego (T32 AG000216). Microscopy imaging supported by NINDS P30 NS047101 (UC San Diego Neuroscience Microscopy Imaging Core). The authors declare no competing financial interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M, 2008. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science (New York, N.Y.) 322, 967–970. [DOI] [PubMed] [Google Scholar]
- Baird AL, Meldrum A, Dunnett SB, 2001. The staircase test of skilled reaching in mice. Brain Res. Bull 54, 243–250. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, McMahon SB, 2006. Spinal cord repair strategies: why do they work? Nature Reviews Neuroscience 7, 644–653. [DOI] [PubMed] [Google Scholar]
- Cafferty WBJ, Strittmatter SM, 2006. The Nogo-Nogo receptor pathway limits a spectrum of adult CNS axonal growth. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 12242–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Marsh BC, Cowan M, Al’Joboori YD, Gigout S, Smith CC, Messenger N, Gamper N, Schwab ME, Ichiyama RM, 2017. Sequential therapy of anti-Nogo-A antibody treatment and treadmill training leads to cumulative improvements after spinal cord injury in rats. Exp. Neurol 292, 135–144. [DOI] [PubMed] [Google Scholar]
- Chen MS, Huber a.B., van der Haar ME, Frank M, Schnell L, Spillmann a.a., Christ F, Schwab ME, 2000. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403, 434–439. [DOI] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B, 2003. Inducible site-specific recombination in myelinating cells. Genesis (New York, N.Y. : 2000) 35, 63–72. [DOI] [PubMed] [Google Scholar]
- Fouad K, Pedersen V, Schwab ME, Brösamle C, 2001. Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Current biology : CB 11, 1766–1770. [DOI] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM, 2001. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409, 341–346. [DOI] [PubMed] [Google Scholar]
- García-Alías G, Barkhuysen S, Buckle M, Fawcett JW, 2009. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nat. Neurosci 12, 1145–1151. [DOI] [PubMed] [Google Scholar]
- Geoffroy CG, Lorenzana a.O., Kwan JP, Lin K, Ghassemi O, Ma a., Xu N, Creger D, Liu K, He Z, Zheng B, 2015. Effects of PTEN and Nogo Codeletion on Corticospinal Axon Sprouting and Regeneration in Mice. J. Neurosci 35, 6413–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoffroy CG, Zheng B, 2014. Myelin-associated inhibitors in axonal growth after CNS injury. Curr. Opin. Neurobiol 27C, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzenbach RR, Schwab ME, 2008. Disinhibition of neurite growth to repair the injured adult CNS: focusing on Nogo. Cellular and molecular life sciences : CMLS 65, 161–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GrandPre T, Nakamura F, Vartanian T, Strittmatter SM, 2000. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403, 439–444. [DOI] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brosamle C, Oertle T, Schwab ME, 2002. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 3553–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji B, Case LC, Liu K, Shao Z, Lee X, Yang Z, Wang J, Tian T, Shulga-Morskaya S, Scott M, He Z, Relton JK, Mi S, 2008. Assessment of functional recovery and axonal sprouting in oligodendrocyte-myelin glycoprotein (OMgp) null mice after spinal cord injury. Mol. Cell. Neurosci 39, 258–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joset A, Dodd DA, Halegoua S, Schwab ME, 2010. Pincher-generated Nogo-A endosomes mediate growth cone collapse and retrograde signaling. The Journal of cell biology 188, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-E, Liu BP, Park JH, Strittmatter SM, 2004. Nogo-66 receptor prevents raphespinal and rubrospinal axon regeneration and limits functional recovery from spinal cord injury. Neuron 44, 439–451. [DOI] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPré T, Qiu D, Strittmatter SM, 2003. Axon regeneration in young adult mice lacking Nogo-A/B. Neuron 38, 187–199. [DOI] [PubMed] [Google Scholar]
- Kloth V, Klein A, Loettrich D, Nikkhah G, Kloth Verena KALDNG, Kloth V, Klein A, Loettrich D, Nikkhah G, Kloth Verena KALDNG, 2006. Colour-coded pellets increase the sensitivity of the staircase test to differentiate skilled forelimb performances of control and 6-hydroxydopamine lesioned rats. Brain Res. Bull 70, 68–80. [DOI] [PubMed] [Google Scholar]
- Lee JK, Chan AF, Luu SM, Zhu Y, Ho C, Tessier-Lavigne M, Zheng B, 2009. Reassessment of corticospinal tract regeneration in Nogo-deficient mice. J. Neurosci 29, 8649–8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Geoffroy CG, Chan AF, Tolentino KE, Crawford MJ, Leal M.a., Kang B, Zheng B, 2010. Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron 66, 663–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JK, Zheng B, 2008. Axon regeneration after spinal cord injury: insight from genetically modified mouse models. Restor. Neurol. Neurosci 26, 175–182. [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M, Meyers EN, Martin GR, 1997. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb. Symp. Quant. Biol 62, 159–168. [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Nickells RW, 1999. Experimental induction of retinal ganglion cell death in adult mice. Invest. Ophthalmol. Vis. Sci 40, 1004–1008. [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z, 2010. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat. Neurosci 13, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, Lein ES, Zeng H, 2010. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier IC, Baumann K, Thallmair M, Weinmann O, Scholl J, Schwab ME, 2008. Constraint-induced movement therapy in the adult rat after unilateral corticospinal tract injury. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 9386–9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Fujita Y, Ueno M, Takai T, Yamashita T, 2011. Paired immunoglobulin-like receptor B knockout does not enhance axonal regeneration or locomotor recovery after spinal cord injury. J. Biol. Chem 286, 1876–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederost B, Oertle T, Fritsche J, McKinney RA, Bandtlow CE, 2002. Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. The Journal of neuroscience : the official journal of the Society for Neuroscience 22, 10368–10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielson JL, Sears-Kraxberger I, Strong MK, Wong JK, Willenberg R, Steward O, 2010. Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. J. Neurosci 30, 11516–11528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielson JL, Strong MK, Steward O, 2011. A reassessment of whether cortical motor neurons die following spinal cord injury. J. Comp. Neurol 519, 2852–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertle T, van der Haar ME, Bandtlow CE, Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L, Brosamle C, Kaupmann K, Vallon R, Schwab ME, 2003. Nogo-A inhibits neurite outgrowth and cell spreading with three discrete regions. The Journal of neuroscience : the official journal of the Society for Neuroscience 23, 5393–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernet V, Joly S, Dalkara D, Schwarz O, Christ F, Schaffer D, Flannery JG, Schwab ME, 2012. Neuronal Nogo-A upregulation does not contribute to ER stress-associated apoptosis but participates in the regenerative response in the axotomized adult retina. Cell Death Differ. 19, 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig ES, Courtine G, Jindrich DL, Brock JH, Ferguson AR, Strand SC, Nout YS, Roy RR, Miller DM, Beattie MS, Havton LA, Bresnahan JC, Edgerton VR, Tuszynski MH, 2010. Extensive spontaneous plasticity of corticospinal projections after primate spinal cord injury. Nat. Neurosci 13, 1505–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab ME, Strittmatter SM, 2014. Nogo limits neural plasticity and recovery from injury. Curr. Opin. Neurobiol. 27C, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der Putten H, Schwab ME, 2003. Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron 38, 201–211. [DOI] [PubMed] [Google Scholar]
- Song XY, Zhong JH, Wang X, Zhou XF, 2004. Suppression of p75NTR does not promote regeneration of injured spinal cord in mice. J. Neurosci. 24, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkey ML, Barritt AW, Yip PK, Davies M, Hamers FPT, McMahon SB, Bradbury EJ, 2005. Assessing behavioural function following a pyramidotomy lesion of the corticospinal tract in adult mice. Exp. Neurol. 195, 524–539. [DOI] [PubMed] [Google Scholar]
- Starkey ML, Bartus K, Barritt AW, Bradbury EJ, 2012. Chondroitinase ABC promotes compensatory sprouting of the intact corticospinal tract and recovery of forelimb function following unilateral pyramidotomy in adult mice. The European journal of neuroscience 36, 3665–3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Zheng B, Banos K, Yee KM, 2007. Response to: Kim et al. , “axon regeneration in young adult mice lacking Nogo-A/B.” Neuron 38, 187–199. Neuron 54, 191–195. [DOI] [PubMed] [Google Scholar]
- Tennant KA, Asay AL, Allred RP, Ozburn AR, Kleim JA, Jones TA, 2010. The vermicelli and capellini handling tests: simple quantitative measures of dexterous forepaw function in rats and mice. Journal of visualized experiments : JoVE, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thallmair M, Metz GA, Z’Graggen WJ, Raineteau O, Kartje GL, Schwab ME, 1998. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat. Neurosci. 1, 124–131. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Steward O, 2012. Concepts and methods for the study of axonal regeneration in the CNS. Neuron 74, 777–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Hayano Y, Nakagawa H, Yamashita T, 2012. Intraspinal rewiring of the corticospinal tract requires target-derived brain-derived neurotrophic factor and compensates lost function after brain injury. Brain : a journal of neurology 135, 1253–1267. [DOI] [PubMed] [Google Scholar]
- Vajda F, Jordi N, Dalkara D, Joly S, Christ F, Tews B, Schwab ME, Pernet V, 2015. Cell type-specific Nogo-A gene ablation promotes axonal regeneration in the injured adult optic nerve. Cell Death Differ. 22, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl AS, Omlor W, Rubio JC, Chen JL, Zheng H, Schroter A, Gullo M, Weinmann O, Kobayashi K, Helmchen F, Ommer B, Schwab ME, 2014. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science 344, 1250–1255. [DOI] [PubMed] [Google Scholar]
- Wang Z, Reynolds A, Kirry A, Nienhaus C, Blackmore MG, 2015. Overexpression of Sox11 promotes corticospinal tract regeneration after spinal injury while interfering with functional recovery. J. Neurosci 35, 3139–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH, 2001. Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc. Natl. Acad. Sci. U. S. A 98, 3513–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z, 2006. Glial inhibition of CNS axon regeneration. Nature reviews. Neuroscience 7, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Z’Graggen WJ, Metz GAS, Kartje GL, Thallmair M, Schwab ME, 1998. Functional Recovery and Enhanced Corticofugal Plasticity after Unilateral Pyramidal Tract Lesion and Blockade of Myelin-Associated Neurite Growth Inhibitors in Adult Rats. The Journal of neuroscience : the official journal of the Society for Neuroscience 18, 4744–4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Atwal J, Ho C, Case L, He X.-l., Garcia KC, Steward O, Tessier-Lavigne M, 2005. Genetic deletion of the Nogo receptor does not reduce neurite inhibition in vitro or promote corticospinal tract regeneration in vivo. Proc. Natl. Acad. Sci. U. S. A 102, 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Ho C, Li S, Keirstead H, Steward O, Tessier-Lavigne M, 2003. Lack of enhanced spinal regeneration in Nogo-deficient mice. Neuron 38, 213–224. [DOI] [PubMed] [Google Scholar]
- Zörner B, Schwab ME, 2010. Anti-Nogo on the go: from animal models to a clinical trial. Ann. N.Y. Acad. Sci 1198 Suppl, E22–34. [DOI] [PubMed] [Google Scholar]