

Abstract
The human pathogen, Vibrio cholerae, belongs to the 10% of bacteria in which the genome is divided. Each of its two chromosomes, like bacterial chromosomes in general, replicates from a unique origin at fixed times in the cell cycle. Chr1 initiates first, and upon duplication of a site in Chr1, crtS, Chr2 replication initiates. Recent in vivo experiments demonstrate that crtS binds the Chr2-specific initiator RctB and promotes its initiator activity by remodeling it. Compared to the well-defined RctB binding sites in the Chr2 origin, crtS is an order of magnitude longer, suggesting that other factors can bind to it. We developed an in vivo screen to identify additional crtS-binding proteins and identified the global transcription factor, Lrp, as one such protein. Studies in vivo and in vitro indicate that Lrp binds to crtS and facilitates RctB binding to crtS. Chr2 replication is severely defective in the absence of Lrp, indicative of a critical role of the transcription factor in licensing Chr2 replication. Since Lrp responds to stresses such as nutrient limitation, its interaction with RctB presumably sensitizes Chr2 replication to the physiological state of the cell.
Keywords: V. cholerae Chr2 replication, replication licensing, crtS, RctB, Lrp, coordination of replication
Introduction
In bacteria, chromosomes initiate replication at fixed times in the cell cycle that vary depending upon the bacteria and their physiological state. Nearly 10% of bacteria from diverse genera possess divided genomes comprising more than one chromosome (Egan et al., 2005). In such bacteria, timely duplication of all chromosomes prior to cell division is crucial for genome maintenance. Vibrio cholerae has emerged as the model organism for studying replication control in multi-chromosome bacteria. It possesses two chromosomes, Chr1, 3 Mb, and Chr2, 1 Mb. Chr1 initiates replication first, and only upon the passage of a Chr1 replication fork across a site, crtS, does Chr2 initiate replication (Val et al., 2016). The crtS site (Chr2 replication triggering site) is thought to function by interacting with and remodeling the Chr2-specific initiator, RctB (Baek and Chattoraj, 2014). It appears that when duplication of a crtS site is prevented within a cell cycle, the site still shows modest activity in licensing Chr2 replication but it is insufficient to do so in a timely fashion (Ramachandran et al., 2018). Duplication of the site as a consequence of a single round of replication increases this activity sufficiently to permit initiation of Chr2 replication in each cell cycle.
The crtS site is essential for Chr2 replication in V. cholerae (Val et al., 2016). Increasing the copy number of crtS increases Chr2 replication in V. cholerae, indicating that the activity of the site is limiting for Chr2 replication. The crtS site also functions in Escherichia coli; the presence of crtS in a plasmid increases copy number of plasmids containing the Chr2 origin of replication (pori2) and a source of RctB (Baek and Chattoraj, 2014).
The structure and function of crtS are fairly well conserved in the Vibrionaceae family (Kemter et al., 2018). The size of crtS (∼153-bp) is rather large for a protein binding site and is much larger than the RctB binding sites in the Chr2 replication origin (12-mers and 39-mers). The region in crtS protected by RctB covers only 18 bp (Baek and Chattoraj, 2014). There is plenty of room for other factors to interact with the site. One such factor is RNA polymerase as crtS possesses a sigma-70 promoter, called PcrtS here, which remains repressed by unknown factors. The repressed promoter allowed us to screen for host genes responsible for that repression and to determine their influence on crtS function.
Here, we show that in addition to RctB, crtS binds Lrp, a global transcription factor that responds to nutritional status (Calvo and Matthews, 1994; Cho et al., 2008). The protein is largely responsible for keeping PcrtS repressed and mediating RctB binding to crtS. In the absence of Lrp, Chr2 replication is severely defective. The regulation of Chr2 replication by a global regulator of nutritional status may provide a link between chromosomal replication and the physiological state of the cell.
Materials and Methods
Bacterial Strains and Growth Conditions
The bacterial strains and plasmids used in this study are listed in Supplementary Tables S1,S2, respectively. All E. coli strains are K12 derivatives and all V. cholerae strains are El Tor N16961 derivatives, and were maintained in lysogeny broth (LB) at 37°C and 30°C, respectively, unless otherwise specified. When required, media was supplemented with antibiotics at the following concentrations for E. coli: 100 μg/ml ampicillin, 25 μg/ml chloramphenicol, 25 μg/ml kanamycin, 40 μg/ml spectinomycin, and 25 μg/ml zeocin. V. cholerae strains were maintained with the same antibiotic concentrations as above except for chloramphenicol, which was used at 5 μg/ml.
Microscopy
Single colonies grown overnight in LB with the appropriate antibiotics were used to inoculate 1X M63 medium supplemented with 1 mM CaCl2, 1 mM MgSO4, 0.001% vitamin B1, 0.2% fructose, 0.1% casamino acids, and 100 μM IPTG (to induce GFP-P1ParB). Cultures were grown at 30°C to an OD600 of 0.3, added to the center of a glass P35 dish (MatTek corporation, Ashland, MA), and overlaid with 1% agarose prepared with the same medium. Dishes were imaged and analyzed as previously described (Ramachandran et al., 2018).
Natural Transformation of V. cholerae to Replace crtS With Δ3′crtS, Δ5′crtS, or Δ5′Δ3′crtS
Natural transformations of CVC3058 (HapR+ derivative of El Tor N16961 with P1parS cloned at +40 kb on Chr2 for visualizing ori2 as GFP-P1ParB foci) and CVC3061 (CVC3058 with extra crtS site cloned 10 kb upstream of native site) were performed as described (Ramachandran et al., 2018). The native copy of crtS was replaced with truncated versions using linear DNA amplified from pPC143, pPC144, and pPC145 containing Δ3′crtS, Δ5′crtS, and Δ5′Δ3′crtS, respectively, flanked by 1 kb of homologous DNA present in plasmid pBJH245. pPC143 was assembled from Δ3′crtS DNA amplified from pBJH188 with primers PNC47 and PNC56 and from pBJH245 amplified with primers PNC46 and PNC54. Primers used here are described in Supplementary Table S3. pPC144 was assembled from Δ5′crtS DNA amplified from pBJH188 with primers PNC51 and PNC55 and from pBJH245 amplified with primers PNC50 and PNC53. pPC145 was assembled from Δ5′Δ3′crtS DNA amplified from pBJH188 with primers PNC51 and PNC56 and from pBJH245 amplified with primers PNC50 and PNC54. Plasmids were assembled using the HiFi DNA assembly kit (NEB).
β-Galactosidase Assay
Plasmid Construction: Truncated crtS species were transcriptionally fused to lacZ in pMLB1109 in order to measure promoter activity. The crtS fragments, Δ3′crtS, Δ5′crtS, and Δ5′Δ3′crtS were amplified from pBJH188 using primers PNC15 and PNC17, PNC16 and PNC18, and PNC16 and PNC18, respectively. The fragments were then ligated into pMLB1109 digested with EcoRI and SmaI to produce plasmids pPC066, pPC067, and pPC068, respectively. E. coli Δlrp strains was complemented with Lrp using plasmid pPC401. Plasmid pPC401 contained Ptrclrp amplified from pJWD-2 using primers PNC139 and PNC140 in a pACYC177 backbone that was amplified using primers PNC141 and PNC142.
Assay Protocol: β-galactosidase assays were performed in 96-well flat bottom plates (Costar 3596) and adapted from Schaefer et al. (2016). Colonies grown overnight on LB plates with appropriate antibiotics were used to inoculate LB. Log-phase culture, 80 μl each, was loaded in duplicates in a 96-well plate to which 120 μl of a custom mix (ONPG + Popculture reagent), prepared as described in (Schaefer et al., 2016), was added. Plates were incubated at 30°C in a Epoch2 plate reader (Biotek, United States) set to double orbital shaking and A420 measurements were taken in one- to 5-min intervals. Equivalent Miller Units (MU)were calculated using a Python program that parses OD600 and A420 values from the plate readers and plots A420 and ΔA420 values as a function of time, to identify maxima. Plotted β-galactosidase activity in MU represent means from three biological replicates and error bars depict SEM.
Screen of Transposon-Insertion Mutants
The EZ-Tn5 transposome kit (Lucigen, WI) was used to generate random Tn insertions in E. coli DH10-β harboring pBJH235. The transformation mixtures were spread first on LB plates with appropriate antibiotics and incubated overnight at 37°C. The following day colonies were patched on MacConkey agar plates (MacConkey Agar Base [Difco, MD] supplemented with 1% (w/v) lactose and 3 mM 2-phenylethyl β-D-thiogalactoside (PETG, [Biosynth, IL], pH adjusted to 7.1). [PETG, a competitive inhibitor of β-galactosidase, was titrated to 3 mM, the concentration at which colonies with 75 MU appear white and those with 180 MU appear red (Supplementary Figure S1)]. Plates were incubated for 16 h at 37°C and colonies were monitored for development of red color. Candidate colonies were grown in LB overnight for genomic DNA isolation. Genomic DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen, CA, United States), digested with EcoRV-HF (NEB, MA, United States), and ligated overnight at 16°C with T4 DNA ligase (NEB). The ligation product was used to transform DH5α(λpir) cells to recover transposon containing circularized genomic DNA, which were replication competent by virtue of the presence of R6Kγori within the transposon. Plasmid DNA was extracted from individual colonies using the QIAprep Spin Miniprep Kit (Qiagen) and sequenced using the primers supplied in the EZ-Tn5TM transposome kit to identify the locations of Tn insertion.
Deletion of lrp in E. coli
Δlrp-787::kan was transduced from E. coli JW0872-2 into E. coli DH10-β and BR8706 (constitutive araE) using P1vir (Miller, 1992). The kan cassette was excised by expressing Flp recombinase from pCP20 and subsequently curing the plasmid by overnight growth at 42°C (Datsenko and Wanner, 2000).
Deletion of lrp in V. cholerae
Deletion of lrp from CVC3058 (derivative of El Tor N16961 with P1parS cloned at +40 kb on Chr2 for visualizing ori2 as GFP-P1ParB foci) was performed in the presence of a plasmid carrying E. coli lrp (pJWD-2), by natural transformation with linear DNA amplified from pPC352 that contained a zeocin cassette flanked by 1 kb upstream and downstream homology sequences. pPC352 was assembled using four DNA fragments: 1 kb upstream homology (amplified from genomic DNA using primers PNC123 and PNC124), 1 kb downstream homology (amplified from genomic DNA using primers PNC121 and PNC122), zeocin cassette (amplified from pEM7-Zeo using primers PNC127 and PNC128) and the backbone (amplified from pEM7-Zeo using primers PNC125 and PNC126). Linear DNA used for natural transformation was amplified from pPC352 using primers PNC131 and PNC132. Deletion of lrp was confirmed by PCR. The plasmid pJWD-2 was cured by growing overnight in the absence of antibiotic and screening colonies that had lost antibiotic resistance, to generate strain CVC3286. The deletion was verified by whole genome sequencing.
Purification of Lrp and MBP-RctB
Lrp was purified from plasmid pJWD-2 (Ernsting et al., 1993). 5 ml of overnight culture of E. coli BL21 containing pJWD-2 was used to inoculate 1 liter of LB supplemented with ampicillin and grown at 37°C. Protein expression was induced at an OD600 nm of 0.8 by adding IPTG to the final concentration of 0.5 mM, and growth was allowed to continue for 2.5 h. The pellet was resuspended in PC Buffer [50 mM phosphate buffer (pH 7.4), 100 mM NaCl, 0.1 mM EDTA, 10 mM β-mercaptoethanol and 10% glycerol, (de los Rios and Perona, 2007)] supplemented with 1× protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO) and lysed by French press. The lysate was clarified by centrifugation for 1 h at 18,000 ×g before loading onto a Hitrap SP HP column (GE Healthcare Life sciences, Chicago, IL, United States), pre-equilibrated with PC Buffer. Lrp was eluted using a gradient of PC Buffer + 1 M NaCl. The fractions containing Lrp were purified further by cation exchange on a Mono S column (GE Healthcare Life sciences) equilibrated with Cat2 buffer (50 mM Hepes (pH 8.0), 1 mM EDTA, 0.2% Tween20, 5% Glycerol and 100 mM NaCl). Lrp was eluted using a gradient of Cat2 buffer + 1 M NaCl. MBP-RctB was purified as described previously (Jha et al., 2017).
Electrophoretic Mobility Shift Assay (EMSA)
Interaction of purified Lrp with crtS was captured in vitro using EMSA. The 153 bp crtS was flanked by ∼100 bp of lambda DNA and amplified from pBJH170 using FAM-labeled primers RR202 and RR214. Non-specific DNA was amplified from pTVC243 using the same primers as above and contained only the 100-bp flanks. Truncated crtS constructs Δ3′crtS, Δ5′crtS, and Δ5′Δ3′crtS were amplified from pPC189, pPC225 and pPC009, respectively, using primers PNC77 and PNC78. Increasing amounts of Lrp protein were added to 20 μl reactions that contained 5 nM each of fluorescent probe and vector DNA, 20 mM Hepes (pH 7.4), 1 mM EDTA, 0.2% Tween20, 5% glycerol, 200 ng poly dI-dC, 1 mM dithiothreitol, 70 mM potassium glutamate and 4 mM magnesium acetate. Leucine was added at 10 mM, when desired. The reaction was incubated at room temperature for 10 min before loading on a 5% native polyacrylamide gel and electrophoresed at 12 V/cm in 0.5 × TBE. The gel was scanned using Typhoon FLA 9500 (GE Healthcare Life Sciences, MA, United States). The image was analyzed, and band intensities quantified using Fiji software (Schindelin et al., 2012). The percent DNA bound was plotted against concentration of protein and KD values were obtained by performing non-linear regression analysis assuming one site specific binding using GraphPad Prism version 7.0 a (La Jolla, CA, United States). Following EMSA of crtS with Lrp and RctB, the super-shifted band was excised from the native polyacrylamide gel and presence of both proteins confirmed by mass spectrometry performed at the Collaborative Protein Technology Resource (CCR, NIH) as previously described (Jha et al., 2017).
Measurement of Plasmid Copy Number
Copy number experiments were performed using either WT E. coli (BR8706, constitutive araE) or Δlrp E. coli (derivatives of BR8706: CVC3260, Δlrp-787::FRT-kan-FRT and CVC3274 Δlrp-787). BR8706 and CVC3260 were transformed with pTVC11 (prctB) and either pTVC243 (vector), pBJH170 (pcrtS), or pBJH239 (pcrtS-10 m). CVC3274 was transformed with pTVC11, pPC401 (plrp), and either pTVC243, pBJH170, or pBJH239. To maintain high levels of RctB, competent cells were grown in 0.2% arabinose before and after transformation with pTVC22 (pori2). Cultures were inoculated at an OD600 nm of 0.005 and grown at 37°C with shaking to an OD600 nm of 0.2. Eight OD units were pelleted and used for plasmid isolation. Relative plasmid copy number was measured essentially as described (Das and Chattoraj, 2004) but normalized to pTVC11.
Whole Genome Sequencing
Genomic DNA was extracted from 1 ml of cells grown overnight at 37°C in LB using DNeasy Tissue Kit (Qiagen, Hilden, Germany). DNA was sequenced on the Illumina MiSeq platform at the NCI CCR genomics sequencing core. 1–6 million reads were obtained for each sample, which were trimmed and mapped to a CVC3058 reference genome using the CLC Genomics Workbench (Qiagen). The reference genome for CVC3058 was constructed by de novo assembly.
Results
5′ Terminal Sequences of crtS Are Important for Licensing Chr2 Replication in V. cholerae
In E. coli, the presence of a plasmid containing 153 bp of V. cholerae Chr1 [coordinates 817947 to 818099 bp of Heidelberg et al. (Heidelberg et al., 2000)] (Figure 1A) increases the copy number of ori2-containing plasmids (pori2) about threefold in the presence of RctB (Baek and Chattoraj, 2014). The 153 bp sequence was called crtS (Val et al., 2016). The central 54 - 123 bp, called Δ5′Δ3′crtS here, also increases the pori2 copy number about twofold in E. coli (Baek and Chattoraj, 2014). To test if Δ5′Δ3′crtS was sufficient to support replication of Chr2 in V. cholerae as well, the crtS sequence in Chr1 was replaced with Δ5′Δ3′crtS using natural transformation. Chr2 replication was followed by visualizing GFP-P1ParB bound to the P1parS site inserted 40 kb away from ori2, as previously described (Ramachandran et al., 2018). We found that Δ5′Δ3′crtS replacement resulted in a loss of ori2 foci in 70% of cells (Figure 1B, top panel). Truncation of the 5′ and 3′ sequences separately revealed that this functional deficiency is due to the 5′ truncation, as truncation of the 3′ did not significantly alter the foci distribution. This suggests that the 1–123 bp of crtS locus spanning chromosomal coordinates 817947 to 818069 is sufficient for licensing Chr2 replication, despite the low conservation of the 5′ bases (Figure 1A). Furthermore, the results were indistinguishable in the Δ5′ constructs, whether or not the 3′ region was present (Figure 1B, top row).
FIGURE 1.

The 5′ but not the 3′ end of crtS is critical for its function. (A) The 153 bp crtS sequence from V. cholerae overlaid with a WebLogo (Crooks et al., 2004), generated from different Vibrio species showing varied conservation along the length of the sequence, as in Kemter et al. (2018). Gray highlighted sequence denotes the minimal 70-bp that retain copy-number enhancement function of crtS in E. coli. The flanking 5′ and 3′ sequences studied here are underlined. Yellow highlighted sequences denote the predicted –35 and –10 elements of the sigma-70 promoter, PcrtS. (B) The effect of crtS and its truncated derivatives on Chr2 replication is shown by histograms of ori2 foci numbers per cell in V. cholerae strains where crtS was replaced at its native locus with the truncated derivatives (top row), and where the same mutant strains had, at 10 kb upstream, a second full length crtS copy (bottom row). On the left of the histogram is shown the approximate location of crtS copies in Chr1, where the native locus is indicated by an empty star and the locus with the added crtS copy by a filled star. The position of the ori1 is denoted by a tick-mark. The strains used were: intact crtS (CVC3058, top; CVC3061, bottom), Δ5′Δ3′crtS (CVC3228, top; CVC3247, bottom), Δ5′crtS (CVC3227, top; CVC3246, bottom), Δ3′crtS (CVC3226, top; CVC3245, bottom). Note that deletion of the upstream 53 bases (Δ5′) severely compromises replication-triggering function of crtS as evidenced by the appearance of cells with zero ori2 foci. Data represent mean ± SEM (standard error of mean) of at least 1000 cells imaged from three biological replicates.
Deletion of crtS leads to suppressor mutations in rctB or fusion of Chr1 and Chr2 (Val et al., 2016). To avoid the selection of suppressors while replacing the native crtS locus with truncated species, we repeated the replacements in strains that also possessed a second functional copy of crtS 10 kb upstream of the native locus (Ramachandran et al., 2018). The presence of two full length copies of crtS causes over-replication of Chr2 (Val et al., 2016) (Figure 1B, bottom row). This over replication was not seen when the native crtS locus was replaced with Δ5′crtS. Replacement with Δ3′crtS did not alter crtS function, as the distribution of ori2 foci was similar to that of cells with two intact crtS copies. In sum, although the exact bounds of crtS remain to be defined, it appears that the 5′ sequence of crtS is essential for licensing replication from ori2.
The Promoter Within crtS Is Repressed
In spite of the importance of the 5′ terminal sequences of crtS, they are not well conserved among the various Vibrio species (Kemter et al., 2018). Apart from AT-richness, the region does not have any known sequence features. crtS, however, possesses −35 and −10 promoter elements in the more conserved central region (Figure 1A). The promoter within crtS, called PcrtS here, was previously shown to be expressed only from Δ5′Δ3′crtS but not from full length crtS (Baek and Chattoraj, 2014). From these results, it appears that the promoter repression and replication enhancement functions of crtS are correlated, and that the promoter repression may be necessary for crtS function. To quantify the promoter repression, we fused a promoterless lacZ gene to the crtS constructs used in Figure 1B. In E. coli, PcrtS activity was as low as in the promoterless vector, but the activity increased fourfold in Δ5′crtS (Figure 2, left panel). These results indicate that an E. coli factor interacts with the 5′ terminal sequences of crtS and represses PcrtS. A test of whether RctB, the only protein previously found to bind crtS (Baek and Chattoraj, 2014), could also repress the promoter showed that it did, but only partially (black vs. white bars, Figure 2). The expression of PcrtS is thus controlled by at least two repressors. The deletion of the 3′ 30 bp had only a marginal effect on promoter activity.
FIGURE 2.

The promoter within crtS (PcrtS) is repressed by an unknown factor common to E. coli and V. cholerae. β-galactosidase activity in E. coli DH10-β (left) and monochromosome V. cholerae MCH1 (right), containing promoterless lacZ in a pBR-based plasmid (none, pMLB1109), or lacZ transcriptionally fused to either crtS (pBJH235), Δ3′crtS (pPC066), Δ5′crtS (pPC067) and Δ5′Δ3′crtS (pPC068). Additionally, the strains had a second plasmid, prctB (pRR24, black bars) supplying RctB or the corresponding empty vector (pPC020, white bars). The x-axis in the two graphs are scaled differently. Both in E. coli and MCH1, the promoter activity dramatically increases upon deletion of the 5′ crtS sequences. Since in both the strains the promoter repression is seen in the absence of RctB, the only factor known to bind crtS, an unknown factor common to two bacteria must be involved in repression of PcrtS. Supplying RctB recovers the repression partially, which indicates that the promoter is normally repressed by RctB as well as the unknown factor. Error bars denote standard deviation of mean from three biological replicates.
In V. cholerae, truncation of the 5′ sequences results in only a slight increase in promoter activity (Supplementary Figure S2). To test whether the lack of increase could be due to the binding of crtS by RctB, the experiments were repeated in a strain of V. cholerae, MCH1, that lacks RctB and where Chr2 is maintained by fusion to Chr1 (Val et al., 2012). In MCH1, PcrtS was expressed threefold higher in Δ5′crtS and Δ5′Δ3′crtS than crtS, mirroring the E. coli results (Figure 2, right panel). Addition of RctB caused partial repression of promoter activity in Δ5′crtS and Δ5′Δ3′crtS, as in E. coli. Together, these results strongly suggest that a factor other than RctB, common to both E. coli and V. cholerae, binds crtS and is responsible for the additional repression of the promoter within crtS.
PcrtS Is Repressed by the Global Regulator Lrp in E. coli and V. cholerae
The putative E. coli factor responsible for repressing PcrtS was identified by performing a transposon (Tn) insertional mutagenesis screen in strains that contained a plasmid with transcriptional-fusion of crtS to lacZ. Colonies with higher lacZ activity were identified by plating on MacConkey agar supplemented with 3 mM PETG, an inhibitor of β-galactosidase, that allowed clearer distinction between red and white colonies (Golding et al., 1991) (Supplementary Figure S1). In most of these colonies, the Tn was found to have inserted into the plasmid expressing lacZ. In one colony, the Tn was found to have inserted within the 5′ untranslated region of the lrp gene. To determine whether Lrp is responsible for the observed repression of PcrtS, the promoter activity was measured in a E. coli Δlrp strain [from Keio collection, (Baba et al., 2006)] and, although crtS was full length, the activity was as high as from Δ5′crtS (Figure 3A). Complementing the Δlrp strain with Lrp using plasmid pJWD-2 (Ernsting et al., 1993) resulted in repression of PcrtS, when present in intact crtS but not when present in Δ5′crtS. These results are fully consistent with Lrp being the factor that, directly or indirectly, keeps PcrtS repressed.
FIGURE 3.

PcrtS is repressed by the global regulator Lrp. (A) Promoter activity was measured after fusion to lacZ in WT (DH10-β), in Δlrp (CVC3259) which is otherwise isogenic, and the same Δlrp strain complemented with plrp (CVC3259/pPC401). The crtS fragments were the same as in Figure 2. The deletion of lrp results in promoter de-repression, which is complemented in the presence of plrp when crtS was full length and not when it was Δ5′. Error bars denote standard error of mean from two biological replicates. (B) Electrophoretic mobility shift assay (EMSA) of fluorescently labeled non-specific DNA (bottom arrow) and crtS DNA (top arrow) in the presence of increasing concentrations Lrp protein. Asterisks indicate positions of Lrp bound species. The fraction of the probe bound (quantified from the loss of intensity of the unbound probe) was plotted as a function of Lrp concentration to generate the binding isotherm that yielded an apparent dissociation constant (KD) of 5.5 ± 1.2 nM.
An in vitro experiment was performed to test whether Lrp itself binds to crtS. E. coli Lrp protein was purified from plasmid pJWD-2 to about 95% purity. The E. coli protein is 92% identical to the V. cholerae Lrp protein and is completely conserved in the helix-turn-helix motif (Lintner et al., 2008). In EMSA using fluorescently labeled crtS, Lrp was seen to bind crtS with an approximate KD of 5.5 ± 1.2 nM (Figure 3B). The addition of leucine altered the distribution of the Lrp-shifted species, indicating that crtS-binding is responsive to the presence of leucine (Supplementary Figure S3). Lrp was seen to bind equally well to Δ3′crtS and Δ5′crtS and slightly less well to Δ5′Δ3′crtS, indicating that it has multiple binding sites within crtS, but it appears that the site(s) within the 5′ terminal sequences are required for promoter repression (Supplementary Figure S4). A search for Lrp binding site within crtS using a SELEX derived consensus sequence (Cui et al., 1995) revealed a putative site with 12/15 matches covering 50 - 64 bp region. The first four bp of this putative Lrp binding site are lost upon truncation of the 5′ sequences, possibly explaining the loss of repression in Δ5′crtS. In addition to the 15 bp consensus, the three to five flanking bases also contribute to specific binding by Lrp (Cui et al., 1995), which are also missing in Δ5′crtS.
Lrp Is Required for Chr2 Replication-Licensing by crtS in E. coli and V. cholerae
In order to test the effect of Lrp on the replication enhancement function of crtS, the copy number of ori2-containing plasmids was measured in Δlrp strains. While in WT E. coli, the copy number of pori2 increased about threefold in the presence of pcrtS compared to the empty vector, no such increase was observed in the Δlrp strain (Figure 4A). This indicates that crtS fails to function as an enhancer of Chr2 replication in the absence of Lrp. Upon complementing with an Lrp-expressing plasmid, plrp, the copy number of pori2 increased about fourfold in the presence of PcrtS, whereas the vector copy number was unaffected, indicating that Lrp is essential for crtS function in E. coli. To test whether the Lrp was required solely to repress PcrtS, a promoter-defective mutant of crtS (crtS-10m, (Baek and Chattoraj, 2014)) was used in which two bases within the −10 element of the promoter were mutated. The mutation was previously shown to retain the replication enhancement function of crtS in WT E. coli while possessing low promoter activity. However, pcrtS-10m failed to increase pori2 copy number in the E. coli Δlrp strain (Supplementary Figure S5). Introduction of the plrp plasmid restored the function of pcrtS-10m. This indicates that keeping the promoter repressed may not be the sole and perhaps not the primary function of Lrp on crtS.
FIGURE 4.

Lrp is necessary for licensing of Chr2 replication by crtS. (A) Copy number of pori2 (a plasmid carrying the origin of Chr2, pTVC22) in E. coli WT (BR8706), Δlrp (CVC3260), and Δlrp/plrp (CVC3274/pJWD-2). The cells also contained a source of RctB (pTVC11) and additionally either a vector (white bars) or a crtS-containing plasmid (black bars). The copy numbers were normalized to the value in WT cells, set as 1. pori2 copy number increases threefold in the presence of pcrtS in WT but not in Δlrp cells. Complementing Δlrp cells with plrp increases pori2 copy number even more that that was seen in WT cells, suggesting that Lrp could be limiting in WT cells. Data represent mean ± SEM from three biological replicates. (B) Histograms of ori2 foci number per cell in WT V. cholerae (CVC3058) with either the vector control (pTrc99A) or plrp, and in V. cholerae Δlrp cells (CVC3286) with complementing plrp and after curing plrp. Representative microscopy images from each strain are shown above the histograms. Note that the number of cells without any ori2 foci increases in the absence of lrp, indicating that Lrp is crucial for Chr2 replication in V. cholerae. Data represent mean ± SEM from at least 1000 cells imaged from three biological replicates.
Lrp is not essential for viability of E. coli or V. cholerae (Lintner et al., 2008; Srivastava et al., 2011; Fu et al., 2013). To test the requirement of Lrp for Chr2 replication in V. cholerae, the lrp gene was deleted in a strain where fluorescently tagged ori2 foci could be visualized. The deletion was initially made in the presence of a plasmid supplying Lrp (pJWD-2). Upon deletion of chromosomal lrp and curing of plrp, the percentage of cells without an ori2 focus increased dramatically (from 7 to 80%) (Figure 4B). This indicates that although lrp gene is not essential, the protein contributes dramatically to Chr2 replication. The contribution seems greater in the defined medium used for microscopy, where the growth was slower than in LB (Supplementary Figure S6). In fact, the Δlrp strain never appears to enter logarithmic growth in the microscopy medium. At least in E. coli, an Lrp-associated minimal medium growth defect results largely from effects in nitrogen assimilation (Paul et al., 2007; van Heeswijk et al., 2013). The requirement of Lrp in Chr2 replication/cell growth thus exhibits media-dependency.
Interestingly, in cells where the complementing plrp plasmid was not cured, Chr2 copy number was higher than when the cells had the empty vector (Figure 4B). This outcome was obtained in the WT strain containing plrp as well, where 45% of cells possessed two or more ori2 foci as compared to 30% when cells contained the empty vector, suggesting that Lrp may normally be limiting for Chr2 replication. A test of whether Lrp functions solely via crtS to increase Chr2 replication was performed using a previously isolated ΔcrtS strain (Baek and Chattoraj, 2014). This ΔcrtS mutant possesses a mutation in rctB, which makes it a hyper-initiator (in WT V. cholerae) and apparently can compensate for the Chr2 replication defect that the absence of crtS confers. In this ΔcrtS strain, plrp failed to increase Chr2 replication (Supplementary Figure S7) and the distribution of ori2 foci in ΔcrtS with plrp, resembled that of the vector control, indicating that Lrp functions via crtS in licensing Chr2 replication.
Lrp Enhances RctB Binding to crtS
How could Lrp stimulate crtS function? One possibility is that Lrp modulates the interaction of crtS with the rate-limiting factor for Chr2 replication, which is known to be RctB (Pal et al., 2005; Duigou et al., 2006). This hypothesis was tested in vivo and in vitro. In WT E. coli, RctB was able to repress the derepressed PcrtS activity from Δ5′crtS by about 50% (Figure 2) but was unable to do so in the isogenic Δlrp strain (Figure 5A). These results suggest that RctB binding to crtS is Lrp dependent. To test this hypothesis, we performed an EMSA of crtS DNA with both RctB and Lrp. RctB was previously shown to bind crtS only when the site is supercoiled (Baek and Chattoraj, 2014). However, by changing the buffer condition, it was possible to detect RctB binding to linear crtS fragments (Figure 5B). Lrp alone also bound to the same fragment but with more affinity. When both Lrp and RctB were present, a super-shifted band was seen, indicating that both proteins are bound to crtS simultaneously. Presence of RctB and Lrp in the super-shifted band was confirmed by mass spectrometry.
FIGURE 5.
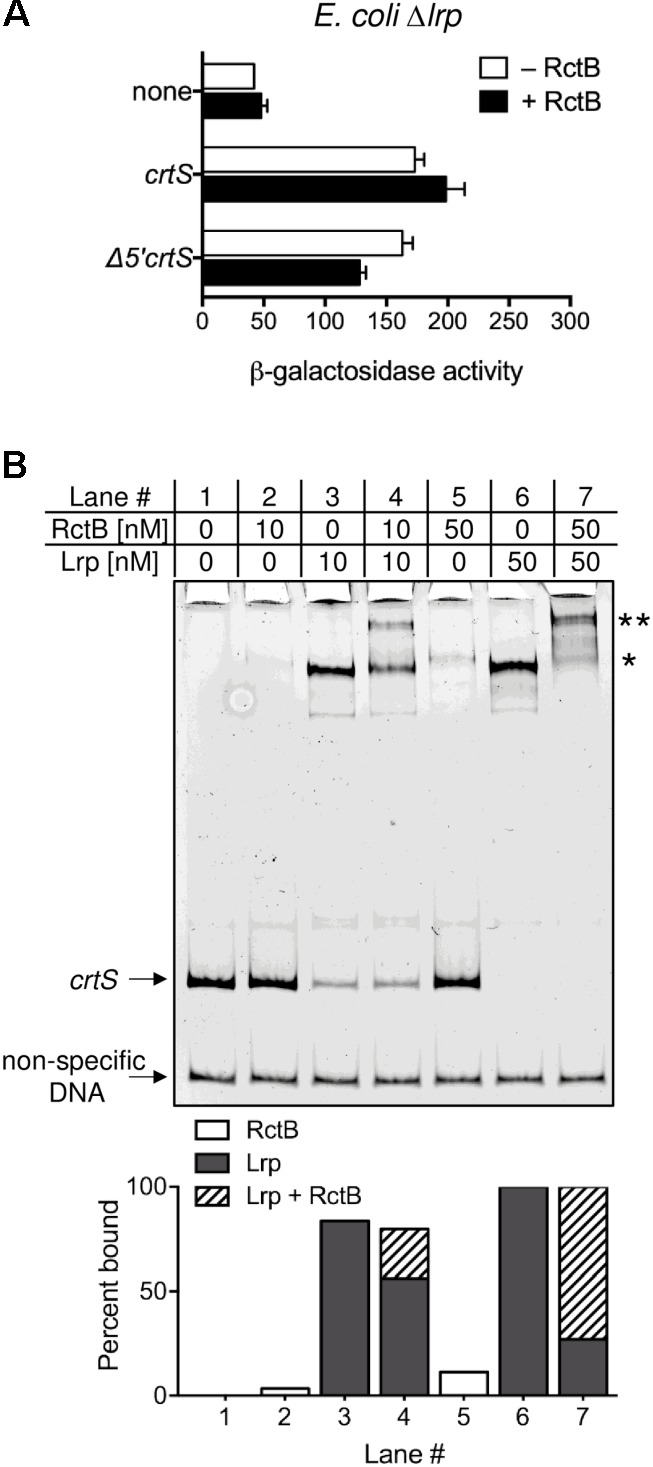
Lrp enhances RctB binding to crtS. (A) Promoter activity of crtS (from PcrtS) was measured as in Figure 2 in E. coli Δlrp (CVC3259) carrying either the empty vector (none, pMLB1109), or the same vector carrying crtS (pBJH235) or its 5′ truncated derivative Δ5′crtS (pPC067). Cells also had either a source of RctB (pRR24) or the corresponding empty vector (pPC020). Note that RctB, which was partially effective in repressing PcrtS in WT, fails to repress the promoter in Δlrp. Data represent mean ± SEM from three biological replicates. (B) EMSA of 5′-FAM labeled crtS DNA (upper arrow) and non-specific DNA (lower arrow) with Lrp and RctB. Both Lrp (lanes three and six) and RctB (lanes two and five) were individually seen to bind crtS specifically. Note that the major Lrp bound band (∗) is super-shifted in the presence of RctB (∗∗). The intensity of the super-shifted band is much higher than the RctB bound band, indicating that RctB binds better to Lrp bound crtS. Shown below are percentages of probe bound to RctB alone (white columns), Lrp alone (gray columns) and, both Lrp and RctB (hatched columns).
Leucine does not affect the binding of RctB to Lrp-bound crtS significantly (Supplementary Figure S8). While the total DNA bound by Lrp alone and by Lrp+RctB remained nearly the same, the intensity of the super-shifted band increased at the expense of the species bound to Lrp alone. Apparently, RctB binds with higher affinity to Lrp-bound crtS than to naked crtS. This is quantified by measuring RctB binding to either crtS or Lrp-bound crtS (Supplementary Figure S9A). The affinity of RctB to Lrp-bound crtS is nearly 10-fold higher than that to naked crtS (Supplementary Figure S9B). Lrp could also enhance the binding of RctB to Δ5′Δ3′crtS fragment (Supplementary Figure S10). This is not surprising, considering that Δ5′Δ3′crtS is functional in E. coli in multicopy, suggesting that RctB and Lrp can favorably interact on the Δ5′Δ3′crtS fragment, where at least one Lrp binding site also exists (Supplementary Figure S4). Lrp thus could enhance Chr2 replication by enhancing RctB binding to crtS.
Discussion
Requirement for Lrp in crtS Function
The Chr1 encoded crtS-mediated licensing of Chr2 replication is so far the only known mechanism by which the replication of one chromosome regulates the timing of the replication of the other. Here we report that the licensing function of crtS depends on the global transcription regulator, Lrp. Although many general DNA binding proteins, such as IHF, HU, Fis, and SeqA, are known to participate in DNA replication, this is the first evidence for Lrp participation, a protein sensitive to the environment and, in particular, to the intracellular concentration of leucine and other amino acids (Hart and Blumenthal, 2011). Growth phase control of DNA replication initiation is a little-studied aspect of cell cycle in bacteria, although starvation induced nucleotide alarmone (p)ppGpp has been known to inhibit new rounds of replication initiation for some time (Chiaramello and Zyskind, 1990). In the E. coli chromosome, the growth-phase regulated Fis protein signals to oriC to turn off DNA replication as the bacteria enter stationary phase (Cassler et al., 1995). So far, no Fis involvement in the origin of Chr2 replication has been detected in V. cholerae. The involvement of Lrp in Chr2 replication mirrors the involvement of Fis in sensitizing the chromosome to changes in cell physiology. The involvement of Lrp also makes crtS more comparable to DARS2 (DnaA reactivation site) of the E. coli chromosome (Kasho et al., 2014). Both crtS and DARS2 are involved in initiator remodeling, and both bind the cognate initiator and an additional factor, Lrp and Fis, respectively. The role of Lrp in crtS function thus could be analogous to Fis in DARS2 function.
We find that increasing Lrp concentration increases Chr2 replication (Figure 4B). Lrp concentration increases in stationary phase and upon other stresses to the cell (Landgraf et al., 1996). This suggests that Lrp could be utilized to promote Chr2 replication preferentially under stressful conditions. It is not possible to specify how the cells benefit from this preferential replication since functions of most of the genes in Chr2 are not known. It is known, however, that many more Chr2 genes are expressed during intestinal growth than during liquid culture, the basis of which is yet to be understood (Xu et al., 2003). One function of Lrp at crtS may be to maintain parity of chromosome numbers in stationary phase. In rich medium, Chr1 is maintained at two-fold higher copy number than Chr2 (Srivastava and Chattoraj, 2007; Stokke et al., 2011). When cells reach stationary phase both chromosomes have one copy each. To make this adjustment Chr2 must replicate an additional round after Chr1 replication has ceased. Increased Lrp concentrations during entry to stationary phase could help achieve this parity by stimulating Chr2 replication via crtS.
Lrp has been reported to control more genes in E. coli than any other global transcriptional regulators (Kroner et al., 2018; Shimada et al., 2018). A deletion of lrp in E. coli, however, is easily tolerated. In contrast, a deletion of lrp in V. cholerae is obtained only in the presence of a complementing plasmid and the deleted strain shows significant growth defect (Srivastava et al., 2011). Tn-seq analysis also showed fewer hits in lrp compared to many other targets considered “non-essential” in V. cholerae (Fu et al., 2013). The requirement of Lrp in crtS function, and hence in Chr2 replication, may explain why in V. cholerae Lrp is critical. Whole genome sequencing of Δlrp strains cured of complementing plasmids in this study did not reveal any suppressor mutations in 2/2 cases. At least in our growth conditions (in LB), the Δlrp strains appear to be viable, although slow-growing, whereas growth is more severely affected in the poorer synthetic medium used for microscopy (Supplementary Figure S6). V. cholerae possesses three hypothetical genes with significant identity (>35%) to Lrp (Supplementary Table S4), in addition to the widely distributed local regulator AsnC (Caspi et al., 2016; Unoarumhi et al., 2016). It is possible that in the absence of Lrp, some of its functions could be compensated for by these paralogs. If any paralogs exist in E. coli, they do not seem to substitute for Lrp. In E. coli the protein seems to be essential for crtS function (Figure 4A).
The Importance of the Less-Conserved 5′ Region of crtS
An intriguing feature of crtS is that its 5′ region, although less conserved than the remainder of the site, is crucial for its replication enhancement function. On the other hand, that same function is unaffected by deletion of the downstream sequences, which are better conserved. The conservation of non-essential region suggests that crtS serves additional functions that are not yet recognized. Variant forms of Lrp or its orthologs in different species may account for the relatively poor conservation of the 5′ region. If so, this likely involves the differences at the amino termini of the different Vibrio Lrp orthologs (Hart et al., 2011; Unoarumhi et al., 2016). Although crtS sequences from different Vibrio species are able to increase the copy number of orthologous pori2, the failure of certain crtS sequences to function with a few other pori2, could be due to differences in their cognate Lrp proteins (Kemter et al., 2018).
The 5′ region of crtS provides Lrp binding sites required for promoter repression as well as the enhancement of replication initiation. The nature of the relationship of the two functions to each other remains to be clarified, but they appear to be anti–correlated: truncation that resulted in increased promoter expression reduces the efficiency with which crtS can license Chr2 replication. It is possible that occupancy by RNA polymerase interferes with RctB binding to crtS. The presence of Lrp could thus aid RctB binding to crtS by preventing RNA polymerase from binding to the promoter. Lrp usually forms an octameric ring composed of two tetramers, upon which DNA is wrapped, causing significant bending to the DNA (de los Rios and Perona, 2007). It is possible that the bases on crtS preferred by RctB are made more accessible by bound Lrp, or that constructive protein-protein contacts are made between Lrp and RctB.
The low PcrtS activity under our laboratory conditions measured with a transcriptional fusion to lacZ was also evident from previous RNA-Seq analyses (Figure 2) (Baek and Chattoraj, 2014; Papenfort et al., 2015). RNA-Seq reads in V. cholerae at low and high cell densities did not reveal any measurable transcripts originating from PcrtS. Unless some conditions are found that activate the promoter naturally, the presence of the promoter might well be incidental to the Lrp requirement in crtS function (Alice and Crosa, 2012; Lin et al., 2007). Uncoupling the replication enhancement and promoter repressor function of Lrp by mutating the −10 box of PcrtS did not relieve the site from Lrp dependence (Supplementary Figure S5). This indicates that reduction of the promoter activity cannot be the only role of Lrp. To the extent analyzed, increasing RctB binding appears to be the main function of Lrp. In the immediate future, we seek to delineate the details of interactions among crtS, RctB and Lrp with the ultimate aim of understanding how they help RctB to license Chr2 replication and regulate that essential function.
Author Contributions
PC, RR, and DC designed the study and wrote the manuscript. PC and RR performed the experiments. All authors read and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Sankar Adhya for advice regarding PETG, Harris Bernstein for pTrc99A, Robert Blumenthal for pJWD-2, Nadim Majdalani for E. coli Δlrp from the Keio collection, Marie-Eve Val and Didier Mazel for V. cholerae MCH1, CW for V. cholerae Δlrp and Lisa Jenkins at the Collaborative Protein Technology Resource (CCR, NIH) for mass spectrometry analysis. We also grateful to Michael Yarmolinsky for thorough review of the manuscript and thoughtful comments. Finally, we thank the reviewers for their helpful comments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02103/full#supplementary-material
References
- Alice A. F., Crosa J. H. (2012). The TonB3 system in the human pathogen Vibrio vulnificus is under the control of the global regulators Lrp and cyclic AMP receptor protein. J. Bacteriol. 194 1897–1911. 10.1128/JB.06614-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., et al. (2006). Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:20060008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek J. H., Chattoraj D. K. (2014). Chromosome I controls chromosome II replication in Vibrio cholerae. PLoS Genet. 10:e1004184. 10.1371/journal.pgen.1004184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo J. M., Matthews R. G. (1994). The leucine-responsive regulatory protein, a global regulator of metabolism in Escherichia coli. Microbiol. Rev. 58 466–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi R., Billington R., Ferrer L., Foerster H., Fulcher C. A., Keseler I. M., et al. (2016). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 44 D471–D480. 10.1093/nar/gkv1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassler M. R., Grimwade J. E., Leonard A. C. (1995). Cell cycle-specific changes in nucleoprotein complexes at a chromosomal replication origin. EMBO J. 14 5833–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaramello A. E., Zyskind J. W. (1990). Coupling of DNA replication to growth rate in Escherichia coli: a possible role for guanosine tetraphosphate. J. Bacteriol. 172 2013–2019. 10.1128/jb.172.4.2013-2019.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho B. K., Barrett C. L., Knight E. M., Park Y. S., Palsson B. O. (2008). Genome-scale reconstruction of the Lrp regulatory network in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 105 19462–19467. 10.1073/pnas.0807227105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J. M., Brenner S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14 1188–1190. 10.1101/gr.849004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y., Wang Q., Stormo G. D., Calvo J. M. (1995). A consensus sequence for binding of Lrp to DNA. J. Bacteriol. 177 4872–4880. 10.1128/jb.177.17.4872-4880.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das N., Chattoraj D. K. (2004). Origin pairing (‘handcuffing’) and unpairing in the control of P1 plasmid replication. Mol. Microbiol. 54 836–849. 10.1111/j.1365-2958.2004.04322.x [DOI] [PubMed] [Google Scholar]
- Datsenko K. A., Wanner B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97 6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Rios S., Perona J. J. (2007). Structure of the Escherichia coli leucine-responsive regulatory protein Lrp reveals a novel octameric assembly. J. Mol. Biol. 366 1589–1602. 10.1016/j.jmb.2006.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duigou S., Knudsen K. G., Skovgaard O., Egan E. S., Lobner-Olesen A., Waldor M. K. (2006). Independent control of replication initiation of the two Vibrio cholerae chromosomes by DnaA and RctB. J. Bacteriol. 188 6419–6424. 10.1128/JB.00565-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan E. S., Fogel M. A., Waldor M. K. (2005). Divided genomes: negotiating the cell cycle in prokaryotes with multiple chromosomes. Mol. Microbiol. 56 1129–1138. 10.1111/j.1365-2958.2005.04622.x [DOI] [PubMed] [Google Scholar]
- Ernsting B. R., Denninger J. W., Blumenthal R. M., Matthews R. G. (1993). Regulation of the gltBDF operon of Escherichia coli: how is a leucine-insensitive operon regulated by the leucine-responsive regulatory protein? J. Bacteriol. 175 7160–7169. 10.1128/jb.175.22.7160-7169.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., Waldor M. K., Mekalanos J. J. (2013). Tn-Seq analysis of Vibrio cholerae intestinal colonization reveals a role for T6SS-mediated antibacterial activity in the host. Cell Host Microbe 14 652–663. 10.1016/j.chom.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding A., Weickert M. J., Tokeson J. P., Garges S., Adhya S. (1991). A mutation defining ultrainduction of the Escherichia coli gal operon. J. Bacteriol. 173 6294–6296. 10.1128/jb.173.19.6294-6296.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart B. R., Blumenthal R. M. (2011). Unexpected coregulator range for the global regulator Lrp of Escherichia coli and Proteus mirabilis. J. Bacteriol. 193 1054–1064. 10.1128/JB.01183-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart B. R., Mishra P. K., Lintner R. E., Hinerman J. M., Herr A. B., Blumenthal R. M. (2011). Recognition of DNA by the helix-turn-helix global regulatory protein Lrp is modulated by the amino terminus. J. Bacteriol. 193 3794–3803. 10.1128/JB.00191-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidelberg J. F., Eisen J. A., Nelson W. C., Clayton R. A., Gwinn M. L., Dodson R. J., et al. (2000). DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406 477–483. 10.1038/35020000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha J. K., Li M., Ghirlando R., Miller Jenkins L. M., Wlodawer A., Chattoraj D. (2017). The DnaK chaperone uses different mechanisms to promote and inhibit replication of Vibrio cholerae chromosome 2. mBio 8:e00427-17. 10.1128/mBio.00427-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasho K., Fujimitsu K., Matoba T., Oshima T., Katayama T. (2014). Timely binding of IHF and Fis to DARS2 regulates ATP-DnaA production and replication initiation. Nucleic Acids Res. 42 13134–13149. 10.1093/nar/gku1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemter F. S., Messerschmidt S. J., Schallopp N., Sobetzko P., Lang E., Bunk B., et al. (2018). Synchronous termination of replication of the two chromosomes is an evolutionary selected feature in Vibrionaceae. PLoS Genet. 14:e1007251. 10.1371/journal.pgen.1007251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner G. M., Wolfe M. B., Freddolino P. (2018). Escherichia coli Lrp regulates one-third of the genome via direct, cooperative, and indirect routes. bioRxiv [Preprint]. 10.1101/276808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landgraf J. R., Wu J., Calvo J. M. (1996). Effects of nutrition and growth rate on Lrp levels in Escherichia coli. J. Bacteriol. 178 6930–6936. 10.1128/jb.178.23.6930-6936.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W., Kovacikova G., Skorupski K. (2007). The quorum sensing regulator HapR downregulates the expression of the virulence gene transcription factor AphA in Vibrio cholerae by antagonizing Lrp- and VpsR-mediated activation. Mol. Microbiol. 64 953–967. 10.1111/j.1365-2958.2007.05693.x [DOI] [PubMed] [Google Scholar]
- Lintner R. E., Mishra P. K., Srivastava P., Martinez-Vaz B. M., Khodursky A. B., Blumenthal R. M. (2008). Limited functional conservation of a global regulator among related bacterial genera: Lrp in Escherichia, Proteus and Vibrio. BMC Microbiol. 8:60. 10.1186/1471-2180-8-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. H. (1992). A Short Course in Bacterial Genetics. A Laboratory Manual and Handbook for Escherichia coli and Related Bacteria. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Pal D., Venkova-Canova T., Srivastava P., Chattoraj D. K. (2005). Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J. Bacteriol. 187 7167–7175. 10.1128/JB.187.21.7167-7175.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papenfort K., Forstner K. U., Cong J. P., Sharma C. M., Bassler B. L. (2015). Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 112 E766–E775. 10.1073/pnas.1500203112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul L., Mishra P. K., Blumenthal R. M., Matthews R. G. (2007). Integration of regulatory signals through involvement of multiple global regulators: control of the Escherichia coli gltBDF operon by Lrp, IHF, Crp, and ArgR. BMC Microbiol. 7:2. 10.1186/1471-2180-7-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R., Ciaccia P. N., Filsuf T. A., Jha J. K., Chattoraj D. K. (2018). Chromosome 1 licenses chromosome 2 replication in Vibrio cholerae by doubling the crtS gene dosage. PLoS Genet. 14:e1007426. 10.1371/journal.pgen.1007426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer J., Jovanovic G., Kotta-Loizou I., Buck M. (2016). Single-step method for beta-galactosidase assays in Escherichia coli using a 96-well microplate reader. Anal. Biochem. 503 56–57. 10.1016/j.ab.2016.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Ogasawara H., Ishihama A. (2018). Single-target regulators form a minor group of transcription factors in Escherichia coli K-12. Nucleic Acids Res. 46 3921–3936. 10.1093/nar/gky138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava D., Harris R. C., Waters C. M. (2011). Integration of cyclic di-GMP and quorum sensing in the control of vpsT and aphA in Vibrio cholerae. J. Bacteriol. 193 6331–6341. 10.1128/JB.05167-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava P., Chattoraj D. K. (2007). Selective chromosome amplification in Vibrio cholerae. Mol. Microbiol. 66 1016–1028. 10.1111/j.1365-2958.2007.05973.x [DOI] [PubMed] [Google Scholar]
- Stokke C., Waldminghaus T., Skarstad K. (2011). Replication patterns and organization of replication forks in Vibrio cholerae. Microbiology 157 695–708. 10.1099/mic.0.045112-0 [DOI] [PubMed] [Google Scholar]
- Unoarumhi Y., Blumenthal R. M., Matson J. S. (2016). Evolution of a global regulator: Lrp in four orders of gamma-Proteobacteria. BMC Evol. Biol. 16:111. 10.1186/s12862-016-0685-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Val M. E., Marbouty M., de Lemos Martins F., Kennedy S. P., Kemble H., Bland M. J., et al. (2016). A checkpoint control orchestrates the replication of the two chromosomes of Vibrio cholerae. Sci. Adv. 2:e1501914. 10.1126/sciadv.1501914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Val M. E., Skovgaard O., Ducos-Galand M., Bland M. J., Mazel D. (2012). Genome engineering in Vibrio cholerae: a feasible approach to address biological issues. PLoS Genet. 8:e1002472. 10.1371/journal.pgen.1002472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeswijk W. C., Westerhoff H. V., Boogerd F. C. (2013). Nitrogen assimilation in Escherichia coli: putting molecular data into a systems perspective. Microbiol. Mol. Biol. Rev. 77 628–695. 10.1128/MMBR.00025-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Dziejman M., Mekalanos J. J. (2003). Determination of the transcriptome of Vibrio cholerae during intraintestinal growth and midexponential phase in vitro. Proc. Natl. Acad. Sci. U.S.A. 100 1286–1291. 10.1073/pnas.0337479100 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.