

SUMMARY
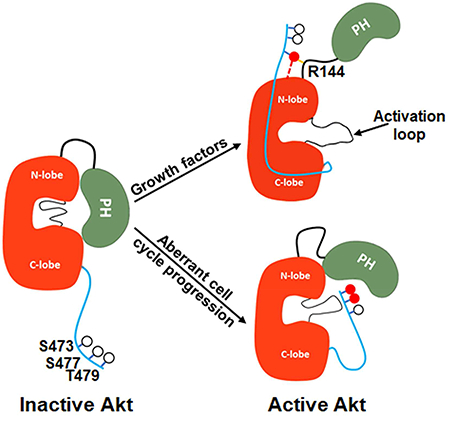
Akt is a critical protein kinase that drives cancer proliferation, modulates metabolism, and is activated by C-terminal phosphorylation. The current structural model for Akt activation by C-terminal phosphorylation has centered on intramolecular interactions between the C-terminal tail and the N-lobe of the kinase domain. Here, we employ expressed protein ligation to produce site-specifically phosphorylated forms of purified Akt1 that are well-suited for mechanistic analysis. Using biochemical, crystallographic, and cellular approaches, we determine that pSer473-Akt activation is driven by an intramolecular interaction between the C-tail and the pleckstrin homology (PH)-kinase domain linker that relieves PH domain-mediated Akt1 autoinhibition. Moreover, dual phosphorylation at Ser477/Thr479 activates Akt1 through a different allosteric mechanism via an apparent activation loop interaction that reduces autoinhibition by the PH domain and weakens PIP3 affinity. These results provide a new framework for understanding how Akt is controlled in cell signaling and suggest distinct functions for differentially modified Akt forms.
Graphical abstract
INTRODUCTION
The protein Ser/Thr kinase Akt1 (a.k.a. Akt, protein kinase B) and its two paralogs Akt2 and Akt3 have been intensively studied in cancer and metabolism (Manning and Toker, 2017). Akt1 is a key component of the phospholipid PIP3 (phosphatidyl inositol 3,4,5-triphosphate) signaling network (Fruman et al., 2017). In this network, the levels of PIP3 are controlled by the opposing actions of PI3-kinase and PTEN phosphatase (Fruman et al., 2017). Activation of PI3-kinase by growth factor receptor stimulation or mutation drives PIP2 to PIP3 conversion (Fruman et al., 2017). In the classical model of Akt1 regulation, phospholipid PIP3 recruits Akt1 to the plasma membrane (James et al., 1996) where it is acted upon by two protein kinases, mTORC2 and PDK1, which phosphorylate Akt1 on its C-terminus (Ser473) and activation loop (Thr308), respectively (Alessi et al., 1996; Alessi et al., 1997; Sarbassov et al., 2005). Dual phosphorylation of Akt1 leads to its enhanced kinase activity that targets protein substrates including GSK3 and Foxo1/3a (Manning and Toker, 2017). Akt1 activity can promote cell growth, block apoptosis, and mediate insulin response (Manning and Toker, 2017) and Akt1 inhibitors are in clinical development including ATP-site and allosteric site binding compounds (Crabb et al., 2017; Larsen et al., 2017).
Akt1-3 belong to the 60+ member AGC family of protein kinases (Leroux et al., 2017; Pearce et al., 2010) many of which are also regulated by activation loop and C-terminal phosphorylation. In some AGC enzymes, the C-terminal phosphorylation site appears to be naturally substituted with an acidic Asp residue and S473D has been reported to simulate the pSer473 state of Akt1 (Alessi et al., 1996; Pearce et al., 2010; Pearlman et al., 2011; Yang et al., 2002a; Yang et al., 2002b). Unlike most AGC kinases, Akt1-3 possess a pleckstrin homology (PH) domain that shows high affinity for PIP3 (Manning and Toker, 2017; Milburn et al., 2003). The Akt1-3 domain architecture includes an N-terminal PH domain followed by an unstructured linker, a conserved kinase domain, and a flexible C-terminal regulatory domain (Manning and Toker, 2017) (Fig. 1A).
Figure 1. Semisynthesis of site-specifically phosphorylated Akt1 constructs.

A) Semisynthesis strategy for C-terminally phosphorylated Akt1 proteins with highlighted phosphorylations (red balls) that are studied in this work. C-terminally truncated recombinant Akt1 containing PDK1-catalyzed pThr308 and a C-terminal intein generated thioester is ligated to N-Cys synthetic C-tail peptides without or with phosphorylations at residues Ser473, Ser477 and Thr479. B) Representative C-terminal tail synthetic peptide sequences containing distinct phosphorylation states. C) Coomassie stained SDSPAGE of selected purified pThr308 containing Akt1 semisynthetic proteins: M, molecular weight standards, lane 1, unligated C-terminally truncated Akt1 MESNA thioester, lane 2, full-length FL-Akt1-pThr308, lane 3, FL-Akt1-pThr308/pSer473, lane 4, FL-Akt1-pThr308/3p-Ser473,Ser477,Thr479. D) Western blot analysis of selected purified semisynthetic Akt1 forms: M, MW markers, lane 1, non-pThr308, non-C-terminally phosphorylated full length Akt1, lane 2, FL-Akt1-pThr308, lane 3, FL-Akt1-pSer473, lane 4, FL-Akt1-pThr308/pSer473.
The molecular mechanism of Akt1-3 regulation by C-terminal phosphorylation has been investigated previously (Alessi et al., 1996; Calleja et al., 2009; Yang et al., 2002a; Yang et al., 2002b). An X-ray crystal structure has been reported of an Akt1 form (aa144-480, Δ143-Akt1) lacking the PH and linker regions but possessing pThr308 and an Asp473 phospho-mimic in a ternary complex with a peptide substrate and nucleotide analog AMPPNP (Yang et al., 2002a). In this structure, the Akt1 C-terminal region interacts with the N-lobe of the kinase domain, apparently driving an active conformation. Consistent with this idea, an X-ray structure of the Akt1 kinase domain with a C-terminal deletion shows an inactive Akt1 conformation (Yang et al., 2002b). An X-ray structure of an Akt1 form containing the PH domain but lacking the C-terminus (aa1-443) in complex with allosteric inhibitor VIII displays an inactive kinase conformation involving an intramolecular interaction between the kinase and PH domains (Wu et al., 2010). It has been unclear how important the PH domain might be in Akt1 autoinhibition since C-terminal deletion itself appears to facilitate formation of the inactive state and the presence of VIII may have induced the PH-kinase domain interaction. It has recently been reported, however, that PIP3 can activate Akt by PH domain engagement (Ebner et al., 2017).
Beyond the well-established Ser473 phosphorylation, it has been discovered that Akt1 can be concurrently phosphorylated in cells on Ser477 and Thr479 by Cdk2/cyclinA2 (Liu et al., 2014). Ser477 and Thr479 phosphorylation have been proposed to turn on Akt1 in the cell nucleus and facilitate cell cycle progression. The mechanism of Akt1 activation by these non-canonical C-terminal phosphorylations, studied with Asp/Glu substitutions and peptide complementation experiments has been suggested to mimic that of Ser473 phosphorylation (Liu et al., 2014).
In addition, the influence of C-terminal phosphorylation on Thr308 phosphorylation in Akt1 has been uncertain. Based on cell-based experiments, it has been suggested that prior phosphorylation of Ser473 may stimulate phosphorylation of Thr308 (Guo et al., 2014), although this was not observed in other studies (Alessi et al., 1996; Jacinto et al., 2006).
A limitation in the analysis of Akt1 regulation has been the absence of purified Akt containing the desired phospho-modifications. pThr308 can be reliably installed using PDK1 (Gao et al., 2005) but purified Akt1 with specific C-terminal phosphorylations has not yet been reported. Here we use expressed protein ligation to generate the desired Akt1 phospho-forms. Expressed protein ligation is a method of protein semisynthesis involving the chemoselective reaction between a recombinant protein C-terminal thioester with a synthetic peptide containing an N-terminal Cys, resulting in a standard amide bond-containing protein backbone (Muir et al., 1998). With the requisite Akt1 phospho-forms in hand, we performed a combination of experiments that establish a new understanding of how Akt1 regulation occurs.
RESULTS
Production and characterization of full-length Akt1 semisynthetic proteins
Expressed protein ligation was performed by generating recombinant Akt1 protein (aa1-459 or aa144-459) carrying a C-terminal thioester and reacting it with N-Cys synthetic phospho-peptides (aa460-480) (Fig. 1). The recombinant Akt1 segments were expressed in SF9 insect cells resulting in Akt1-intein-CBD (chitin binding domain) fusion proteins (Bolduc et al., 2013). To install phosphorylation at Thr308, we co-expressed PDK1 and Akt1 while treating cells with phosphatase inhibitor (Fabbro et al., 1999; Kumar et al., 2001) or used recombinant PDK1 to phosphorylate the Akt1 thioester fragments in vitro. Both worked efficiently, netting what appeared by western blot to be stoichiometric pThr308 (Fig. 1). Note that by performing the PDK1 phosphorylations on the C-terminally truncated forms of Akt1, where Akt1 exists in an inactive state (Yang et al., 2002b) and therefore autophosphorylation is expected to be minimal, precise control of C-terminal phosphorylation of Akt1 is achieved. Akt1 lacking significant levels of phosphorylation at Thr308 (<5%) was obtained by omitting PDK1 transfection.
Chemoselective ligations of the aa460-480 N-Cys synthetic phospho-peptides to the requisite recombinant Akt1 MESNA thioester proceeded smoothly (>90%) and produced a full slate of pThr308 and non-pThr308 full-length semisynthetic Akt1s (aal-480) and a subset of aa144-480 Akt1 forms with various combinations of C-terminal phosphorylations or Asp473. It should be noted that since Akt1 contains a Cys460 at the ligation junction, the natural Akt1 amino acid sequence was preserved. Gel filtration and anion exchange chromatography of the semisynthetic Akt1s led to purified proteins and demonstrated that each is a monomer. In addition to coomassie staining of SDSPAGE, Western blot with anti-pSer473 and anti-pThr308 antibodies supported the appropriate phosphorylation status of the target proteins (Fig. 1D). We also showed that these proteins were phosphorylated at Thr450, a fold-stabilizing site, presumably incorporated during SF9 cell expression (Fig S1a).
Enzymatic analysis of full-length Akt1 semisynthetic proteins
To assess the catalytic properties of the semisynthetic full-length Akt1 forms, we adopted a radioactive assay which measures the incorporation of 32P into a GSK3 peptide substrate (Tarrant et al., 2012; Yang et al., 2002a; Zhang et al., 2006) and showed activity was linear with respect to time and Akt1 concentration in these assay conditions (Fig. S1b-e). We then measured the kcat and ATP Km values as well as the apparent GSK3 peptide Km values as shown in Fig. 2. Comparing the catalytic efficiencies (kcat/Km) for the pThr308 set of Akt1s (Fig. 2B) revealed that pThr308/pSer473 (A1) was about 500-fold greater than that of the non-C-terminally phosphorylated Akt1 (A2) and this resulted from a higher kcat and reduced Km for ATP, although the apparent peptide substrate Kms were similar. Of note, the Asp473 (A4), pSer477 (A7) and pSer479 (A8) forms showed quite low kcat/Kms that were within 3-fold of that of the non-C-terminally phosphorylated enzyme, A2 (Fig. 2B). The catalytic similarity of the Asp473 (A4) and the non-C-terminally phosphorylated (A2) forms relative to the much greater kinase activity of pSer473 (A1) was surprising because Asp473 has been suggested to be a phospho-mimetic in Akt1 (Alessi et al., 1996). We also assessed the catalytic effects of combinations of the C-terminal phosphorylations and noted that the triply phosphorylated C-tail form (A5) was slightly (~2-fold) more active than pSer473 (A1). Interestingly, Akt1 containing both non-canonical C-terminal phosphorylations (pSer477/pThr479 , A6) showed a robust kcat, comparable to that of A1 but an elevated Km for ATP and an overall 8-fold lower catalytic efficiency relative to A1. These results suggest that dual phosphorylation of Ser477 and Thr479 can synergize to promote Akt1 catalysis. In addition, we measured the sensitivity of A1 and A6 to the ATP-site Akt inhibitor GDC0068 (Blake et al., 2012). Interestingly, A1 displayed an 8-fold lower apparent Ki for GDC0068 relative to that of A6, indicative of a perturbed ATP binding pocket in A6 (see Fig. S1f,g).
Figure 2. Enzymatic characterization of semisynthetic Akt constructs by radiometric measurements.

A) Representative steady-state kinetic plots for v/[E] vs [ATP] with 20 mM GSK3 peptide substrate (left), and v/[E] vs [GSK3 peptide] with 20 mM ATP (right). Enzyme concentrations are 2 nM FL-Akt1-pThr308/pSer473; 4 nM FL-Akt1-pThr308/2p-Ser477,Thr479, and 500 nM non-phosphorylated FL-Akt1. The reactions were carried out at 300C for 10 min. B) Enzymatic parameters for the semisynthetic full length or Δ143 Akt1 proteins are shown ± standard error. The kcat and kcat/Km values were obtained from kinetic plots for v/[E] vs [ATP] (*), (n=2).
Akt1 forms lacking phosphorylation at Thr308 showed marked reductions in kinase activity (Fig. 2B) with 500-fold reduced catalytic efficiency when comparing the pSer473 form (A3) to A1 or about 1,500-fold when comparing completely unphosphorylated enzyme (A9) to A1 (Fig. 2B). Therefore, C-terminal phosphorylation and activation loop phosphorylation in Akt1 show independent and synergistic effects in the regulation of Akt1, as reported previously (Alessi et al., 1996).
To assess whether C-terminal phosphorylation of Akt1 could influence the rate of Thr308 phosphorylation by PDK1, we ran a time course that monitored the appearance of pThr308 by western blot (Fig. S2a, b). These experiments showed that pSer473 and to a lesser extent pSer477 and pThr479 enhance the rate of pThr308 appearance and combinations showed a maximal effect of ~5-fold (Fig.S2c). These results suggest that should C-terminal phosphorylation of the Akt1 precede Thr308 phosphorylation, it could promote PDK1-mediated activation. Fluorescence binding studies with PDK1 showed that its Kd for engaging the non-C-terminally phosphorylated Akt1 (0.2 μM) was actually about 2-fold lower than for binding to pSer473 Akt1 (Fig. S2d). These results imply that the basis for enhanced PDK1 action on C-terminally phosphorylated Akt1 stems from a more optimized orientation for catalysis rather than a tightening of affinity between the two kinases (Rettenmaier et al., 2014) (Fig. S2e).
Structural and enzymatic analysis of N-terminally deleted (Δ143) Akt1 forms
Given the major differences in catalytic efficiencies of A1 and A5 vs. A2 and A4, we pursued the X-ray crystal structure of the C-terminally phosphorylated forms in the context of the Akt1 catalytic domain (Δ143-Akt1) as S473D Δ143-Akt1 had been successfully crystallized previously (Yang et al., 2002a). Initial attempts to obtain high quality crystals of C-terminally phosphorylated Δ143-Akt1 in complex with GSK3 peptide substrate (GRPRTTSFAE) and AMPPNP were unsuccessful. To facilitate co-crystallization, we synthesized an ATP-peptide conjugate bisubstrate analog (Figure 3B and S3a-c) (Cheng et al., 2006) designed to inhibit Akt1. We showed that this GSK3 peptide-ATP conjugate could potently inhibit Akt1 kinase activity (apparent Ki 3.6 μM) and were able to determine the X-ray structure of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 in complex with the bisubstrate analog at 2.4 Å resolution (Fig. 3 A, B, C, Table S3, PDB ID 6BUU). To our surprise, this protein structure proved extremely similar to that of the previously reported Δ143-Akt1-pThr308/Asp473 (PDB 4EKK) (Fig. 3C) with the r.m.s.d. <0.58 Å between these structures (Figure 3C). In particular, the phosphate of pSer473 and the carboxylate of Asp473 made nearly identical hydrogen bonds to Gln218 in the N-terminal lobe of the kinase domain (Fig. 3C) (Calleja et al., 2009). The pThr479 in our structure was not observed and the pSer477 appeared to be surface exposed (Fig. S3d). This structural similarity between Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 and Δ143-Akt1-pThr308/Asp473 was unexpected since the catalytic efficiency of pSer473-Akt1 (A1) is >100-fold larger than that of the Asp473 form (A4).
Figure 3. X-ray structural analysis of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479.

A) Overall crystal structure of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 complexed with bisubstrate ATP-GSK3 (teal). B) A blow up of the region highlighted with a square box in Figure 3A showing chemical formula (top) and 3D structure (bottom) of bisubstrate ATP-GSK3. C) Structural comparison of the C-tail with S473D (orange, PDB: 4EKK) and pS473 (purple), interactions with Q218 are highlighted. D) Kinase assays of semisynthetic Akt1 proteins with pSer473 and Q218A and basic patch (142-KHR-144) AAA replacements. The kinase assays were carried out as described in Figure 2A and the enzymatic parameters are shown in Figure 2B. E) Alignment of the PH-kinase linkers (aa 135-164) from different human Akt paralogs and from Akt1 from different species with Uniprot IDs in parentheses and position of Arg144 highlighted in the red box.
These results prompted the comparison of the steady-state kinetic parameters of Δ143-Akt1-pThr308 constructs possessing pSer473 (A15), Asp473 (A16), 2p-Ser477,Thr479 (A17), and an unmodified C-terminus (A18) (Fig. 2B). We found that in the context of the isolated Akt1 catalytic domain, phospho-modifications were of little consequence and the kcat/Kms were within about 3-fold of each other (Fig. 2B). These results are consistent with the similarities of the Asp473 and pSer473 interactions in the Akt1 catalytic domain structures and led us to consider that the basis of Akt1 activation by C-terminal phosphorylation involved tail-interactions beyond the kinase domain.
The PH-Kinase Domain Linker and Akt Regulation
Based on the X-ray structure of Δl43-Akt1-pThr308/3p-Ser473,Ser477,Thr479 which showed that Arg144 was only about 6 Å from the phosphate of pSer473 (Fig. S3d), we speculated that pSer473 could interact with a fairly conserved basic patch of linker residues (Lys142-His143-Arg144) in the context of full-length Akt1. If so, such a pSer473-basic patch interaction might account for the 100-fold catalytic activity difference between the pSer473 (A1) and Asp473 (A4) Akt1 forms. With its small size, only two oxygens and one negative charge in its side chain, Asp473 is unable to interact with Gln218 and the basic patch concurrently in a fashion available to the dianionic trioxophospho-modified sidechain of pSer473. We thus prepared the Akt1 form A10 in which the basic patch aa142-144 is replaced with three Ala residues and also the Q218A Akt1 mutant All (Fig. 2B) to probe the interaction between pSer473 and Gln218. The catalytic efficiencies of A10 and All showed major reductions, 50-fold and l50-fold respectively, rendering the activities similar to that of the non-C-terminally phosphorylated form A2 (Fig. 2B and Fig. 3D). These results suggest that the proposed basic patch and Gln2l8 interactions with pSer473 are critical for Akt1 activation.
PIP3 interactions with semisynthetic Akts
Because of the importance of the C-terminal modifications in the full-length but not N-terminally deleted Akt1 proteins for catalytic activation, we suspected that intramolecular autoinhibition by the PH domain was antagonized by C-terminal phosphorylations. To further probe Akt1 activation mechanisms, we measured the apparent affinity of a soluble PIP3 analog (di-C6-PIP3) with Akt1 forms using a fluorescence anisotropy assay (Fig. S4a). This analysis demonstrated that the Kd values of soluble PIP3 for the pSer473 form (Al) is about 0.2 μM, nearly identical to that of non-C-terminally phosphorylated form A2 (Fig. S4c,d). Interestingly, PIP3 shows slightly tighter binding (Kd 0.09 μM) to the non-phosphorylated Akt1 form A9 (Fig. S4b). It has been reported that vesicle-containing PIP3 and PI(3,4)P2 can stimulate the catalytic activity of Akt in vitro (Ebner et al., 20l7). We confirmed high affinity soluble PI(3,4)P2 binding to the Akt1 PH domain (Fig. S5d). However, in our hands, soluble PIP3, soluble PI(3,4)P2, and vesicle embedded PIP3 did not stimulate the enzymatic activity of non-C-terminally phosphorylated Akt1 (A2) (Fig. S4e). One theoretical difficulty in analyzing PIP3’s effects on kinase activity is the potential for PIP3 to be sequestered by Mg which is added in large excess in kinase assays (Ebner et al., 20l7). We probed for the possibility of soluble PIP3 sequestration by Mg using fluorescent binding assays and found that Mg addition up to l0 mM only had a modest effect on PIP3 binding to the Akt1 PH domain, increasing the Kd~3-fold (Fig. S5b). Since the PIP3 concentration that we used was still far above this Kd level, we deduce that PIP3 occupancy of the Akt1 PH domain per se does not induce Akt1 activation.
Crystal structure of Δ122 C-terminally phosphorylated Akt1
We next determined the X-ray structure of an intermediate length Akt1 construct containing C-terminal phosphorylations (aal23-480, Δl22-Akt1-pThr308/3p-Ser473,Ser477,Thr479) at 2.l Å resolution that included a longer segment of the PH-kinase linker (Fig. 4, Table S3, PDB ID 6CO1). The structure of Δ122-Akt1-Thr308/3p-Ser473,Ser477,Thr479 is fairly similar to that of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 but shows additional electron density for the linker basic patch residues aa142-144 (Lys-His-Arg) (Fig. 4C, Figure S3h). In particular, the Arg144 sidechain makes a well-defined salt bridge with the pSer473 (Fig.4D, Figure S3h) and the backbone carbonyl of His143 forms a hydrogen bond with the sidechain of Ser475 (Fig. S3g). These results provide direct evidence in support of an important role for the basic patch residues aa142-144 binding to pSer473 in the tail.
Figure 4. Phospho-Ser473 interacts with Arg144 in the PH-kinase linker basic patch.

A) Cellular analysis of the effect of basic patch on the Akt phosphorylation and activity. Akt1/2 knock-out HCT116 cells (Ericson et al., 2010) were transfected with pcDNA3 plasmids expressing wild type (WT) and K142A, H143A, R144A (3A) full-length Akt1s. After serum starvation, the cells were stimulated with growth factors for the time indicated. The cells were lysed and analyzed by western blot with Akt antibodies (left panel) and Foxo1 or Foxo3a antibodies (right panel). N.: non-transfected and stimulated with growth factors for 10 min., (−): transfected with DNA plasmids but not stimulated with growth factors; n=3 forassays. B) Quantification of phosphorylation level of Akt Ser473 (blue for WT Akt1 and orange for 3A-Akt1) and Foxo1 Thr24/Foxo3a Thr32 (pink for WT Akt1 and green for 3A-Akt1) using Image J (n=3, SEM shown, p<0.01) of blots represented by Figure 4A. C) X-ray structure of D122-Akt1-pThr308/3p-Ser473,Ser477,Thr479complexed with bisubstrate analog and residues Gln218, pSer473 and Arg144 are highlighted. D) Zoom-in of the region highlighted with a square in Figure 4C shows the interaction of pSer473 with Gln218 and Arg144, the pSer473 to Arg144 H-bond is 3.0 Å
To further test this model, we generated the R144A point mutation in the context of pSer473Akt1 (A12) (Fig. 2B) and found that A12’s catalytic efficiency is ~50-fold below the wt version A1, similar to that of the Q218A (A11) and basic patch mutant (A10) forms (Fig. 2B). These results corroborate the X-ray crystallographic findings that the electrostatic interaction between the sidechains of Arg144 and pSer473 are key in Akt1 kinase activation.
Cellular analysis of the Akt1 linker basic patch mutant
To understand the physiological significance of the role of the linker aa142-144 basic patch and Arg144 in Akt1 activation, we investigated the role of these residues in cell transfection experiments. We employed human HCT116 colon cancer cells in which Akt1 and Akt2 were genetically deleted (Ericson et al., 2010) and confirmed that wt transfected full-length Akt1 confers response to growth factors as assessed by Foxo1a/3 phosphorylation (Fig. 4A,B) (Ericson et al., 2010). In contrast, transfected Akt1 possessing the basic patch 142-AAA-144 mutation showed diminished Foxo1a/3 phosphorylation (Fig. 4A,B). Interestingly, basic patch mutant Akt1 also displayed a loss in growth factor-mediated Ser473 phosphorylation relative to wt Akt1 (Fig. 4A, B). To determine whether the reduction in Ser473 phosphorylation of the basic patch AAA mutant Akt1 was somehow an indirect effect of its lower Akt1 catalytic activity, we investigated D274A Akt1 in which the active site base Asp274 sidechain is replaced (Ebner et al., 2017). We observed that D274A Akt1 showed robust Ser473 phosphorylation after cells were growth factor treated (Fig. S6a,b), however, Foxo1a/3 phosphorylation was reduced (Fig. S6a,b). Therefore, reduced Akt1 Ser473 phosphorylation in the setting of R144A is not simply related to a general reduction in kinase activity associated with R144A but was likely related to the specific loss of the salt bridge between Arg144 and pSer473. This salt bridge disruption presumably shifts the equilibrium of pSer473 Akt1 to a conformation that exposes pSer473, subjecting it to phosphatase cleavage (Fig. 4A). R144A Akt1 also showed diminished pSer473 in response to growth factors (Fig. S6c,d).
As Arg144 is part of the linker between the PH and kinase domains, we hypothesized that the linker length and/or conformation could be important in tuning the pSer473-basic patch interaction to counteract the PH-mediated autoinhibition of the kinase domain (Fig. 5A). Consequently, we designed an Akt1 form in which six Gly residues are inserted into the center of the PH-kinase linker of Akt1 (123-Gly-6xGly-Ser-124 Akt1) (Chen et al., 2017). By inserting the hexa-Gly segment, it was presumed that potential change in linker tension induced by pSer473-Arg144 and its tug on the PH domain would be relieved. In cellular experiments with transfected Gly6-Akt1, it was found that pFoxo1a/3 and pSer473 were both diminished relative to wt Akt1 transfected cells. These results support the notion that pSer473 may influence Akt1 PH-kinase linker tension to loosen PH domain-mediated autoinhibition. In contrast, with the hexa-Gly insertion, the conformational equilibria would favor the autoinhibited PH-kinase domain state even after phosphorylation of Ser473 in Akt1 (Fig. 5B-D) which would ultimately trigger hydrolysis of pSer473 by cellular phosphatases.
Figure 5. The molecular mechanism of Akt1 activation by C-terminal Ser473-phosphorylation.

A) Cartoon model for Akt1 activation induced by phospho-Ser473. Without C-terminal phosphorylation, Akt1 exists in an inactive conformation with autoinhibition involving a PH domain-kinase domain interaction. Phosphorylation of Ser473 by mTORC2 can activate Akt1 via the interaction of pSer473 with the N-lobe and Arg144 within the PH-kinase linker that relieves autoinhibition by the PH domain.B) Cellular assays for an Akt construct in which a hexa-Gly (6G) segment is inserted into the PH-kinase linker compared with wild type Akt1. Assays were carried out as described in Figure 4A. C) Quantification of the phosphorylation level of Ser473 (blue for WT Akt1 and purple for 6G-Akt) and Foxo1 Thr24/Foxo3a Thr32 (pink for WT Akt1 and yellow for 6G-Akt1) from three replicates (n=3, SEM shown, p<0.01). D) Cartoon depicted shows the model for the effect of a hexa-Gly insertion into the Akt1 PH-linker. The 6 Gly insertion is proposed to enhance linker flexibility stabilizing the intramolecular PH-kinase interaction even after C-terminal phosphorylation of Akt1 on Ser473.
Mechanism of Akt1 activation by Ser477/Thr479 dual phosphorylation
In contrast to the mutagenesis effects of the linker basic patch (A10) and Gln218 (A11) on the kinase activity of pSer473, the effects of these mutations (A13, A14) on pSer477, pThr479 Akt1 were minor (kcat/Kms of A6 vs. A13 and A14 are within~2-fold, Fig. 2B). These results suggest that the structural basis of Akt1 activation by dual phosphorylation of Ser477 and Thr479 is distinct from that of phosphorylation of Ser473. The soluble PIP3 affinity for pSer477,pThr479 Akt1 (A6) (Kd 0.8 μ M) is 4-fold greater than that of pSer473 (A1) or non-C-terminally phosphorylated (A2), revealing that the C-terminal phosphorylations at Ser477 and Thr479 induce weakening of the PIP3-PH interaction (Fig. 7A,B, Fig. S4). Taken together with the Gln218 and basic patch mutagenesis results, the weakening of PIP3 binding implies that the pSer477/pThr479 C-tail relieves PH domain-mediated Akt1 autoinhibiton through a distinct mechanism.
Figure 7. The dual phosphorylations at Ser477 and Thr479 activate Akt1 in a mechanism distinct from that of pSer473.

A) Binding assay of PIP3 with FL-Akt1-pThr308, pSer477,pThr479 (A6) measured with fluorescence anisotropy. Varying concentrations of Akt1 were mixed with 50 nM fluorescein-labeled soluble PIP3. Measurements (n=2) were fit to quadratic binding isotherms and Kd value shown ± SEM. B) Table for the affinity binding (Kd) values of soluble PIP3 with three semisynthetic FL-Akt1 proteins: A1, A2, and A6.C) Cartoon model for how phosphorylation of Ser477 and Thr479 of Akt1 can relieve autoinhibition by interaction with the activation loop and PH domain, partially displacing the PH domain from its interaction with the kinase domain.
PIP3’s reduced affinity for A6 relative to A1 led us to consider that the pSer477/pThr479 motif was serving as an intramolecular PIP3 mimetic. However, the catalytic activity of pSer477,pThr479 (A6) was minimally impacted by the addition of excess soluble PIP3 (Fig. S5a), arguing against this possibility. We deduced that dual Ser477 and Thr479 phosphorylation partially relieves Akt1 autoinhibition, not by engaging the classical PIP3 binding site on the PH domain, but by influencing the PH domain in a fashion that both weakens PIP3 binding and kinase domain binding. Such differential activation mechanisms of A6 relative to A1 are also consistent with the distinct sensitivities to the allosteric Akt inhibitor MK2206 (Fig. S1f,g).
To further probe the molecular mechanism of A6 activation, we generated a semisynthetic form of this protein, FL-472Bpa-Akt1-pThr308/2pSer477,Thr479, that replaces Phe472 with the analog benzoylphenylalanine (Bpa) (Chen et al., 2016; Herblin et al., 1987). Bpa is a photo-activatable amino acid that can covalently crosslink residues to map intramolecular protein interactions (Chen et al., 2016; Herblin et al., 1987). To enrich for C-terminal cross-linked peptide, we incorporated Lys-biotin in place of His468 (468KBio) and found that UV exposure of this Bpa-Akt1 form followed by LC-MS/MS analysis led to one cross-linked peptide with a significant Crossfinder score (>500) (Mueller-Planitz, 2015). This cross-linked peptide (Crossfinder score 1933) was composed of the modified C-terminal segment of Akt1 (aa466-480) and a short fragment of the activation loop (302-DGATMK-307) (Fig. 7, Fig. S7) and its formation was dependent on UV exposure (Tables S4, S5). Structural assignment of this unique cross-link was further confirmed using electron transfer dissociation (ETD)-MS/MS. Based on X-ray structure analysis, the activation loop conformation in the activated form of Akt1 and the PH domain in the autoinhibited structure of Akt1 clash and should be mutually exclusive in the catalytically active conformation of Akt1 (Figure S3e,f). Thus, if the pSer477/pThr479 tail sits near the activation loop in its catalytically competent conformation as indicated by the photo-cross-linking, this should displace the PH domain from the kinase domain and relieve autoinhibition. A cartoon model for this mechanism, that also reflects a presumed contact of the tail near the PH domain with its diminished PIP3 affinity, is illustrated in Fig. 7C.
DISCUSSION
A principal finding of these studies is that C-terminal phosphorylations on Akt1 can operate outside of the N-lobe of the kinase domain to activate Akt1 catalytic activity. These results contrast models from prior studies on Akt and other AGC kinases (Leroux et al., 2017; Pearce et al., 2010). Our results also do not support the idea that the PH domain engagement by a PIP3 molecule directly relieves PH-mediated autoinhibition as proposed by Ebner et al (2017). We think that our findings may differ from this recent study since we employed site-specifically modified Akt1 forms, a direct kinase assay that measures phospho-peptide product (rather than ADP formation), and optimal Mg concentration. Our findings indicate that PIP3’s activation of Akt in cells is likely due to the well-documented recruitment of Akt to PIP3 to membranes to facilitate PDK1 and mTORC2 catalyzed activation loop and C-terminal phosphorylation. Interestingly, the PIP3 affinity of pThr308 Akt1 is 2.5-fold weaker compared with the unmodified activation loop form of Akt1 (Fig. S4). This affinity difference can be rationalized based on a comparison of the unmodified PH-kinase and pThr308 kinase domain X-ray crystal structures where the activation loop phosphate would clash with the PIP3 binding cavity on the PH domain (Fig. S3e,f). Perhaps Thr308 phosphorylation can help eject Akt from membranes.
We find that C-terminal phosphorylation of Akt1 can enhance PDK1 phosphorylation of Akt1’s activation loop. Structurally, this may be explained by the presumed conformational change which brings the C-terminal PIFtide motif closer to Thr308 (Rettenmaier et al., 2014). From a functional perspective, these data suggest that C-terminal Akt phosphorylation can stimulate activation loop phosphorylation without the requirement of membrane interactions.
The discovery that the PH-kinase linker plays a direct and critical role in activation by Ser473 phosphorylation may help explain how PTMs on the linker of Akt isoforms including Ser phosphorylation, Cys modification, and Pro hydroxylation may influence Akt regulation (Bellacosa et al., 1998; Guo et al., 2016; Long et al., 2017; Wani et al., 2011). Such modifications may alter the availability of Arg144 for C-tail interaction through changes in the conformation or tension in the PH-kinase linker.
The distinct Akt mechanisms of activation for pSer477/pThr479 form (A6) compared with the pSer473 form (A1) are notable. This dichotomy of activation mechanisms differs from the predicted parsimonious model (Liu et al., 2014) in which the three nearby phosphorylation events all drive similar interactions. An initial clue that the pSer477 and pThr479 behaved differently from pSer473 in regulating Akt1 was based on the observation that single Ser477 or Thr479 phosphorylations were catalytically indistinguishable from unphosphorylated C-tail Akt1. In addition, the Km for ATP is markedly elevated in the pSer477/pThr479 Akt1 form (A6) compared with the pSer473 Akt1 form (A1) , and appears similar to that of the unphosphorylated C-tail form (A2). The high ATP Km for the pSer477/pThr479 Akt1 (A6) is unlikely to impact the Akt1 kinase rate in cells relative to pSer473 Akt1 (A1) given the millimolar intracellular ATP concentration. Nonetheless, the therapeutic implications may be important as this pSer477/pThr479 enzyme form (A6) showed an 8-fold higher Ki for the ATP-site inhibitor GDC0068 compared with the pSer473 Akt1 form (A1).
The strongest evidence for the different activation mechanisms of pSer477/pThr479 (A6) vs. pSer473 (A1) Akt1 comes from the results of the mutagenesis of Gln218 and the basic patch aa142-144 KHR. The simple hypothesis that pSer477/pThr479 tail acts as a structural PIP3 mimic in engaging the canonical PIP3 binding site on the Akt1 PH domain seems unlikely. Evidence against this is that PIP3 addition to the pSer477/pThr479 Akt1 form (A6) has little impact on kinase activity (Fig. S5a). Rather, the current data based on PIP3 affinity modulation and photo-cross-linking suggest that the pSer477/pThr479 tail allosterically activates Akt1 kinase activity by binding to the activation loop in proximity to the PH domain (Fig. 7C). In this model, the PH domain is displaced from the kinase domain, preventing autoinhibition. That A6 shows reduced PIP3 affinity is consistent with an environmental change for the PH domain in this conformation. Moreover, this weakened PIP3 affinity implies that A6 is well suited to phosphorylate nonmembrane associated protein substrates in the nucleus as previously proposed (Liu et al., 2014).
The two different “on” states of Akt1, pSer473 and pSer477/pThr479 are uncommon in the context of kinase regulation. Although there appear to be a number of sub-types of inactive kinase conformations (Huse and Kuriyan, 2002), it is generally accepted that there is a singular active kinase conformation that is responsible for phosphoryl transfer. There may be biological advantages to having more than one active kinase state. For example, perhaps an intermediate reactivity state would be better suited to tuning signal transduction flux. Alternatively, multiple active state conformations may show differences in protein substrate or site selectivity. Our results suggest that D473 in Akt1 is not a reliable mimic of pSer473 despite its use in cellular studies. We speculate that D473 may induce indirect effects in cells that lead to altered phenotypes. Perhaps D473 triggers other PTMs such as pThr308, pTyr474 (Conus et al., 2002), or pThr477/pThr479.
On the technical side, these studies further illustrate the value of expressed protein ligation for proteins produced with baculovirus/insect cell systems. This approach may allow for a broadening of understanding of other Akt PTMs (Conus et al., 2002; Fan et al., 2013; Mahajan et al., 2010; Yang et al., 2009) in future work.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal rabbit anti-pan Akt (clone 11E7) | Cell Signaling Technology | Cat. # 4685S |
| Monoclonal rabbit anti-pThr308 Akt (clone D25E6) | Cell Signaling Technology | Cat. # 13038S |
| Monoclonal rabbit anti-pSer473 Akt (clone 193H12) | Cell Signaling Technology | Cat. # 4058S |
| Monoclonal rabbit anti-pThr450 Akt (clone D5G4) | Cell Signaling Technology | Cat. # 12178S |
| Monoclonal rabbit anti-Foxo1 (clone C29H4) | Cell Signaling Technology | Cat. # 2880S |
| Monoclonal rabbit anti-Foxo3a (clone 75D8) | Cell Signaling Technology | Cat. # 2497S |
| Monoclonal rabbit anti-pT24 Foxo1/ pT32 Foxo3a | Cell Signaling Technology | Cat. # 9464S |
| Bacterial and Virus Strains | ||
| Escherichia coli, strain DH10Bac | Invitrogen | Cat. # 18297010 |
| Escherichia coli, strain Rosetta2 (DE3) | Novagen | Cat. # 71400 |
| pTXB1-Akt(1-121)-MxeIntein-CBD | This paper | N/A |
| pFastBac1-Akt(2-459)-MxeIntein-CBD | This paper | N/A |
| pFastBac1-Akt(144-459)-MxeIntein-CBD | This paper | N/A |
| pFastBac1-Akt(123-459)-MxeIntein-CBD | This paper | N/A |
| pFastBac1-Q218A, Akt(2-459)-MxeIntein-CBD | This paper | N/A |
| pFastBac1-K142A, H143A, R144A-Akt(2-459)-MxeIntein-CBD | This paper | N/A |
| pFastBac1-GST-PDK1 | This paper | N/A |
| pcDNA3-Flag-HA-K142A, H143A, R144A-Akt1 | This paper | N/A |
| pcDNA3-Flag-HA-R144A-Akt1 | This paper | N/A |
| pcDNA3-Flag-HA-6Glys-Akt1 | This paper | N/A |
| pcDNA3-Flag-HA-D274A-Akt1 | This paper | N/A |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| TEV protease | Daniel J. Leahy, Johns Hopkins University | N/A |
| Synthetic peptides used in this study | Mendeley data | http://dx.doi.org/10.17632/8d5hgmvhjm.1 |
| Fluorescein-NHS | Cat. # 76608-16-7 | |
| ATP-γ-S | Sigma Aldrich | Cat. # 10102342001 |
| Recombinant human insulin | Thermo Scientific | Cat. # 12585014 |
| Human IGF-1 | Cell Signaling Technology | Cat.# 8917SC |
| Human EGF | Millipore Sigma | Cat.# E9644 |
| 32P-ATP | Perkin Elmer | Cat.# NEG002Z2-50UC |
| Lipofectamine 3000 | Thermo Scientific | Cat. # L3000015 |
| Monomeric Avidin Agarose | Thermo Scientific | Cat. # 20228 |
| Pierce avidin | Thermo Scientific | Cat. # 21128 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Raw and analyzed data | This paper and Mendeley data | http://dx.doi.org/10.17632/8d5hgmvhjm.1 |
| Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 structure | This paper | PDB: 6BUU |
| D122-Akt1-pThr308/3p-Ser473,Ser477,Thr479 structure | This paper | PDB: 6CO1 |
| Experimental Models: Cell Lines | ||
| Sf21 | Invitrogen | Cat. # 11497-013 |
| Sf9 | Invitrogen | Cat. # 11496-015 |
| Akt1/2 knock out HCT116 | Ericson et al., 2010 | N/A |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Primers used in this study, see Table S1 | This paper | |
| Recombinant DNA | ||
| pcDNA3-Flag-HA-Akt1 | Hsieh et al., 2004 | Addgene # 9021 |
| pcDNA3-PDK1 | GenScript | Cat. # OHu59606 |
| Software and Algorithms | ||
| HKL-2000 | Otwinowski and Minor, 1997 | N/A |
| COOT | Emsley et al., 2010 | N/A |
| Prism 6 | GraphPad | http://www.sraphpad.com |
| Chimera 1.11rc | Pettersen et al., 2004 | http://www.csl.ucsf.edu/chimera |
| ImageJ (Fiji) | https://fiji.sc/ | |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Philip A. Cole (pacole@bwh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Akt1/2 knocked-out HCT116 cells (Ericson et al., 2010) were routinely maintained in McCoy’s 5A medium (Corning grow) supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco) at 37°C with 5% CO2.
METHOD DETAILS
Peptide synthesis
Peptides corresponding to residues 460-480 of Akt1 (CVDSERRPHFPQFSYSASGTA) were synthesized either on a Prelude or PS3 peptide automated synthesizer (Protein Technologies) using the Fmoc strategy and solid phase peptide synthesis and phospho-residues were mono-benzyl protected. In particular, residues were double coupled (1.5 h for standard amino acids and 3 h for phospho-amino acids) and Fmoc groups were removed with 20% (v/v) piperidine in DMF over three 10 min cycles. Biotinylated GSK3 peptide substrate corresponding to residues 14-27 of GSK3(RSGRARTSSFAEPGGK) was synthesized using N-ε-biotin-lysine Wang resin (Iris Biotech).
To synthesize fluorescein labeled peptides, the allyloxycarbonyl (Alloc) protecting group of N-ε-Alloc-lysine was orthogonally removed using 4.4 eq. palladium dissolved in 20:1:0.5-chloroform:dichloromethane:acetic acid for 3 hours. The resin was washed with 0.5% sodium diethyldithiocarbonate trihydrate in DMF, and then 5% 4-methylmorpholine (NMM) in DMF. Subsequently, 1.2 eq. NHS-fluorescein (Thermo Scientific) dissolved in 5% NMM in DMF was added to the resin, and the coupling reaction was left overnight.
Peptides were deblocked and cleaved from resin with trifluoroacetic acid: water: triisopropylsilane (95:2.5:2.5, v/v/v) for 3 hours, then precipitated with chilled diethyl ether and purified using reverse-phase C18 HPLC using a gradient of water:acetonitrile containing 0.1% trifluoroacetic acid. Pure fractions were combined and concentrated on a rotavap and then lyophilized. Peptide structures were confirmed using MALDI mass spectrometry and peptide concentrations were determined by amino acid analysis.
To generate the ATP-GSK3 peptide bisubstrate, the Alloc protecting group from Nβ-Alloc-L-2,3-diaminopropionic acid (Sigma) was orthogonally deprotected as described above and then 10 eq. of bromoacetic anhydride (dissolved in 5% NMM in DMF) was added to the resin and the coupling reaction was performed for 3 hours to afford bromo-GSK3 peptide. Bromo-GSK3 peptide was cleaved from the resin as described above and purified using reverse-phase C18 HPLC and lyophilized. Bromo-GSK3 peptide (5 mg) was reacted with 2 eq. of ATP-gamma-S solution (Li salt, Roche) in 250 μl of aqueous buffer containing 100 mM NH4HCO3, pH 8.0 overnight at room temperature. The conjugation product ATP-GSK3 peptide was purified using reverse-phase C18 HPLC using basic conditions (10-35% gradient of acetonitrile to water containing 10 mM NH4HCO3) and the correct structure confirmed by mass spectrometry. The concentration of the GSK3 peptide-ATP bisubstrate analog was determined using UV spectrometry based on the extinction coefficient of ATP.
Molecular Cloning and Mutagenesis
For the expression of Akt1 (aa1-459, 144-459 or 123-459)-MxeIntein-CBD fusion proteins in insect cells, the human Akt1 gene from pcDNA3-Flag-HA-Akt1 (Addgene, # 9021) was subcloned into a modified pFastBac1 plasmid containing the gyrase MxeIntein and chitin binding domain (CBD). For the expression of 6xHistidine tagged Akt1(123-459)-MxeIntein-CBD fusion protein in E. coli, the Akt1 gene was subcloned into a pTXB1 plasmid. To produce Akt mutants for insect cell expression or mammalian cell-based assays, the QuickChange PCR method (Agilent Technologies) was employed. To generate GST-PDK1, the gene encoding human full length PDK1 (aa1-556) from pcDNA3-PDK1 (Genescript, # OHu59606) was subcloned into pFastBac1 plasmid and then the glutathione S-transferase (GST) tag was N-terminally inserted using a 2-step QuickChange PCR method. All DNA primers used in this work are listed in Table S1. The open reading frames of all wt and mutant constructs were verified by DNA sequencing.
Expression and purification of GST-PDK1
A pFastBac1 baculovirus vector containing GST-PDK1 fusion protein (PDK1 aa 1456) was used to make bacmid and then baculovirus in Sf21 insect cells using standard methods (Bolduc et al., 2013). The resulting baculovirus was used to infect Sf9 insect cells with multiplicity of infection (M.O.I.) of 5.0. After growing in Sf9 III media (Gibco) for 48 hours at 27°C, the cells were pelleted (700 × g, 10 min, 4°C and then resuspended in 1/20th of the media used for culture, pelleted again (discarding the supernatant), and then flash frozen in liquid nitrogen and stored at −80°C. The Sf9 cells from 400 ml culture were resuspended in 20 ml lysis buffer (50 mM HEPES pH 7.5, 1 mM EDTA, 1 mM EGTA, 0.5% Triton X-100, 1 mM sodium orthovanadate, 10 mM sodium β-glycerophosphate, 50 mM NaF, 5 mM sodium pyrophosphate, 1 μM benzamidine, 0.1% BME, 0.27 M sucrose containing one protease inhibitor tablet (Roche)) (Dario Alessi, 1997) and lysed in a 40 ml Dounce homogenizer on ice. The lysate was centrifuged (17,500 × g, 40 min, 4°C) and the supernatant was mixed with 3 ml glutathione resin (Thermo Fisher Scientific) at 4°C. The resin was washed 10 times with storage buffer (50 mM HEPES pH 7.5, 0.1 mM EGTA, 0.1% BME, 0.27 M sucrose) to remove Triton X-100 and then eluted with 50 mM glutathione in storage buffer. The resulting eluate was further purified by size exclusion chromatography using a Superdex 200 column equilibrated with storage buffer and the fractions (>90% pure using SDS PAGE and coomassie blue) were combined and concentrated to 1 mg/ml, aliquoted and stored at −80°C.
Generation of semisynthetic Akt proteins
The baculovirus-insect cell expression system was employed to express the Akt1-Mxeintein-CBD fusion construct (Akt1 aa2-459, 144-459 and 123-459) similarly to that described for GST-PDK1. For protein production, Sf9 insect cells were infected with the respective baculovirus at M.O.I. of 5.0 and cultured in Sf9 III media for 48 hours at 27°C. To obtain in vivo Thr308 phosphorylation, the Akt1(aa2-459)-MxeIntein-CBD fusion construct was co-expressed with GST-PDK1 in Sf9 insect cells with M.O.I.s of 5.0 and 1.0 for the baculovirus containing Akt1 and GST-PDK1, respectively. After growing infected Sf9 cells for ~36 hours at 27°C, 25 nM okadaic acid (Cell signaling technology-CST) was added, and the cells permitted to grow for an additional 16 hours and then harvested as described above. In the absence of GST-PDK1 co-expression and okadaic acid, Akt1s produced appeared to be lacking phosphorylation at Thr308 (<5% by western blot). The cells from 200 ml culture were lysed in a 20 ml Dounce homogenizer in 10 ml lysis buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 10% Glycerol, 0.1% Triton X-100) containing one dissolved protease inhibitor tablet (Roche). The lysate was centrifuged (17,500 × g, 40 min, 4°C) and the supernatant was added to a 5 ml bed of fibrous cellulose (Sigma). After 1 hour of incubation at 4°C, the lysate was filtered from the cellulose and then bound to 5 ml chitin resin (NEB). The resin was then washed with 150 ml washing buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 0.1% Triton X-100), incubated overnight in cleavage buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 300 mM MESNA (sodium mercaptoethylsulfonate), 1mM phenylmethylsulfonylfluoride (PMSF), 10% glycerol) at room temperature (RT). The cleavage buffer containing Akt thioester protein was eluted from the resin, and concentrated by ultrafiltration using an Amicon 10 KDa molecular weight cutoff (MWCO) filter. The N-terminal Akt1 protein fragments containing a C-terminal thioester were shown to be >90% pure by SDSPAGE and reacted with the synthetic N-Cys containing C-terminal Akt peptides (CVDSERRPHFPQFSYSASGTA) containing variable phosphorylation (bolded in aa sequence) or Asp473 in ligation buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM TCEP, 0.5 mM PMSF, 10% glycerol) for 5 hours at RT and then maintained overnight at 4°C. The ligation conversions were assessed by coomassie-stained SDSPAGE and typically shown to be greater than 95%. The semisynthetic Akt1 proteins were purified by size exclusion chromatography on a Superdex 200 column (GE Healthcare) with Akt1 storage buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2 mM beta-mercaptoethanol, 0.5mM (PMSF), 0.5 mM sodium orthovanadate, 10% glycerol) where they were shown to be monomeric. The purified fractions (>95% by coomassie stained SDSPAGE) were combined, concentrated to ~5 mg/ml, aliquoted and then stored at −80°C.
For semisynthetic truncated Akt1 constructs (aa 144-480 and aa123-480), a modified protocol involving in vitro GST-PDK1-catalyzed Thr308 phosphorylation was used to improve the Akt1 protein expression level in Sf9 cells. The Akt1 thioester protein (aa 144-459 or aa 123-459) obtained from the MESNA cleavage step was concentrated to 1 ml and treated with 2 μM GST-PDK1, 2 mM ATP in buffer 50 mM HEPES pH 7.5, 150 mM NaCl, 50 mM MESNA, 20 mM MgClz, 10% glycerol, 0.2 mM sodium orthovanadate for 3 hours at room temperature. The reaction mixture was then loaded on glutathione resins to remove GST-PDK1 and then exchanged into the ligation buffer for the Akt1 protein ligation reaction as described above.
To obtain bacterially expressed Akt1 (aa 123-459), the pTXB1 plasmid containing 6xHistidine tagged Akt1 (aa 123-459)-MxeIntein-CBD fusion protein was expressed in E. coli Rosetta (DE3)/pLysS in 1L 2xYT media containing 100 μg/ml ampicillin and 10 μg/ml Chloramphenicol. The cultures were grown in shaker flasks at 37°C until OD600 = 0.5, then 1 ml 0.3 M IPTG was added to induce expression and the cultures were further incubated for 16 hours at 16°C. Cells were pelleted and then resuspended in 20 ml lysis buffer, and lyzed by french press. The lysate was loaded on 5 ml chitin resin and the Akt1 fusion protein was purified with chitin resin as described above. The Akt1 protein was cleaved off from the resin with the cleavage buffer containing 100 mM DTT at room temperature overnight.
Kinase assays
The kinase parameters of semisynthetic Akt constructs were determined by phosphorylating N-ε-biotin-lysine GSK3 peptide (RSGRARTSSFAEPGGK) in radiometric reactions as previously described (Qiu et al., 2009). Radiometric kinase assays were carried out in small plastic tubes in a 25 μl reaction mixture containing 50 mM HEPES pH 7.5, 10 mM MgCl2, 1 mM EGTA, 2 mM DTT, 1 mM sodium orthovanadate, 0.5 mg/ml BSA (kinase reaction buffer), 0.42 μCi 32P-γ-ATP and varying amounts of biotinylated GSK3 peptide substrate (0-20 μM) and ATP (0-2 mM). Reactions were initiated by the addition of Akt1 (0-500 nM) and were typically performed at 30°C for 10 minutes. The kinase reactions were quenched by adding 20 ml of 100 mM EDTA and then 10 ml of 10 mg/ml Pierce avidin (Thermo Scientific) was added to each sample and incubated for 20 minutes at room temperature. Samples were transferred to centrifugal 30 KDa MWCO filtration units (Nanosep 30K, PALL) and washed two times with 100 ml and one time with 120 ml of washing buffer (0.5 M sodium phosphate, 0.5 M NaCl, pH 8.5). The filtration units were placed in 5 ml scintillation fluid and then beta emission measured using a Beckman liquid scintillation counter (Beckman LS6500). Background levels without Akt1 or peptide substrate were in the range of 500-1000 cpm and with points including enzyme and substrate were more than 2-fold above background. All measurements were made at least twice and replicates were typically within 20%.
Anisotropy measurements
To determine binding affinity of Akt1 constructs with GST-PDK1, different concentrations of GST-PDK1 were mixed with 0.4 μM of semisynthetic Akt constructs C-terminally labeled with fluorescein in binding buffer (50 mM HEPES pH 7.5, 2 mM DTT, 10 mM MgCl2) and with or without 1 mM AMP-PNP and incubated at room temperature for 30 minutes. For binding assays of Akt1 constructs with PIP3, different amounts of Akt1 were mixed with 50 nM fluorescein-labeled soluble PIP3 (Cayman Chemical) in binding buffer (50 mM HEPES pH 7.5, 2 mM DTT, 0.05 mg/ml ovalbumin) and incubated at room temperature for 30 minutes. Fluorescent anisotropy spectra were recorded by Fluoromax 3 system (Horiba) at 23°C with three different replicates.
The Kd values were determined by fitting the data to the quadratic binding equation below:
b=Kd + X + Fixed
Y= Y0 - [(Y0 - Ymax)/(2*Fixed)] * [b - sqrt(b^2 - 4*X*Fixed)] (Seamon et al., 2015; Weiser et al., 2017)
With X is ligand concentration and Y is anisotropy unit.
All measurements were made at least twice and replicates were typically with 20%.
The competition binding assays in which fluorescein-labeled soluble PIP3 was displaced from Akt PH domain by unlabeled soluble PIP3 (di-C6) or PI(3,4)P2 (di-C8) were performed as previously described (Weiser et al., 2017). 50 nM of fluorescein-labeled PIP3 was incubated with 1μM Akt PH domain and varied concentrations of the corresponding competitors in binding buffer at room temperature for 30 minutes, then fluorescence anisotropy measurements were performed as described above. The data were fit with a sigmoidal curve to determine IC50 values. The obtained IC50 values were used to calculate affinity binding (Ki) using the equation:
where L50 is the concentration of free fluorescein-labeled PIP3 at 50% inhibition, P0 is the concentration of free Akt PH domain at 0% inhibition, and Kd is dissociation constant of the fluorescein-labeled PIP3-Akt PH complex (Weiser et al., 2017).
Activation assays
1 μM Akt was mixed with 10 nM GST-PDk1 in activation buffer 50 mM HEPES, pH 7.5, 2 mM DTT, 10 mM MgCl2 and incubated at 30°C, the reaction was triggered by adding 1 mM ATP. 10 μl of the reaction was collected at the indicated time points and quenched by the addition of 4xSDS-loading buffer. The SDS samples were loaded on SDSPAGE gel for Western Blot analysis and imaged with anti-pT308 or pan-Akt primary antibodies.
Western blots
After transfers of protein from SDSPAGE gels to membranes using an iBlot system, the membranes were blocked overnight in 50 mg/ml BSA in TBS-T buffer at 4°C. Membranes were incubated with anti-Akt (pan or phospho) primary antibodies (CST) at a 1:5000 dilution overnight at 4°C, washed 3 times of 10 minutes with TBS-T buffer, and the cycle of antibody incubation and washing was repeated with secondary HRP-linked antibody (CST). Membranes were developed with Amersham ECL Western blotting detection reagents (GE Healthcare) and imaged by a GeneSys imaging system.
Crystallization
Semisynthetic Akt1 (aa144-480) protein with pThr308 and C-terminal triple-phospho residues Ser473, Ser477, Thr479 (10 mg/ml) was mixed with 1 mM ATP GSK3 bisubstrate peptide and 1 mM MnCl2. Crystals were grown at 4°C with 20% PEG3000, 0.2 M ammonium sulfate, 0.1 M HEPES pH 7.5 by vapor diffusion in sitting drop 24-well plates with a 1:1 volume ratio of protein to crystallization buffer for 4 days.
Semisynthetic Akt1 (aa123–480) protein with pThr308 and C-terminal triple-phospho residues Ser473, Ser477, Thr479 (10 mg/ml) was mixed with 1 mM ATP-GSK3 bisubstrate peptide and 1 mM MnCl2 and crystallized at 4°C with 12.5% PEG3350, 0.2 M ammonium acetate, 0.1 M HEPES pH 7.5 by using sitting drop 24-well plates and a 1:1 volume ratio of protein to crystallization buffer for 1 week. All crystals were cryoprotected in the same precipitant buffers that they were grown with 20 % (v/v) of 50% PEG solution added, flash frozen and stored in liquid nitrogen until submitted to X-rays and data collection.
Data collection and structural determination
Diffraction data of Akt1(aa144-480) in complex with ATP-GSK3 bisubstrate peptide were collected on a FR-e Super-Bright Rigaku (Americas Corporation, The Woodlands, TX) copper rotating anode x-ray generator as the source with a DECTRIS Pilatus 3R 200K-A detector at 100 K. Data were indexed, integrated and scaled with HKL3000 (Otwinowski and Minor, 1997). The structure of Akt1 (144-480) in complex with ATP-GSK3 bisubstrate peptide was determined by molecular replacement using the program AMoRE and S473D Akt1(aa144-480) (PDB ID 4EKK) (Lin et al., 2012) as initial template (Navaza, 2001). The model was rebuilt manually with interactive rounds of Coot and refined using refmac5 from the CCP4 suite. The final structure, PDB ID6BUU, was refined to 2.4 Å (Murshudov et al., 1997; Winn et al., 2003).
Diffraction data of Akt1(aa 123-480) in complex with ATP-GSK3 bisubstrate peptide were collected at National Synchrotron Light Source II (Brookhaven, NY) beam line 17-ID1on a DECTRIS Eiger 6M detector. Data were reduced with fastdp, aimless (Kabsch, 2010a, b; Winter and McAuley, 2011). The Akt1 (aa123-480) in complex with ATP-GSK3 bisubstrate peptide was determined by molecular replacement using PDB 6BUU as template. The initial models were rebuilt and refined as described above for PDB 6BUU. The final structure of Akt1 (aa123-480), PDB 6CO1, was refined to 2.1 Å. Final models were validated using Coot (Emsley et al., 2010), Molprobity (Chen et al., 2010). Structure figures were prepared with PyMOL (DeLano; DeLano, 2002; Schrodinger, 2015). RMSD between models were calculated using the LSQ function in Coot.
Mammalian cell signaling analysis
Akt1/2 knocked-out HCT116 cells (Ericson et al., 2010) were routinely maintained in McCoy’s 5A medium (Corning grow) supplemented with 10% FBS and 1% penicillin/streptomycin (Gibco) at 37°C with 5% CO2. When the cells reached ~70% confluence in 6-well plates, the cells were transfected with 1.5 μg of pcDNA3.1-Flag-HA-Akt plasmids complexed with 3μl Lipofectamine 3000 and 3 μl P3000 reagent in Opti-MEM medium for 24 hrs at 37 °C and 5% CO2. When indicated, the cells were rinsed twice by PBS and serum-starved for 18hrs in McCoy’s 5A with 0.5% FBS and stimulated with 100 ng/ml of insulin (ThermoFisher Scientific), 60 ng/ml of EGF (CST) and 60 ng/ml human IGF-1 (CST) for variable times (5, 10, 20, and 40 minutes) at 37 °C and 5% CO2. The cells were lysed by adding 80 μl RIPA buffer (CST) containing 1× complete protease inhibitor tablet and 1 mM PMSF, and shaking for 30 minutes at 4°C. 30 μg of total protein (BCA assay) was loaded on SDSPAGE. Membrane transfer and Western blotting was carried out as described above with 1:1000 dilution for primary antibodies: Akt, phospho Akt, FOXO1, FOXO3a; 1:200 dilution for phospho FOXO1/3a, and 1:5000 dilution for anti-β-actin antibody.
UV-induced protein crosslinking
The di-pSer477/Thr479 Akt C-tail peptide with replacements of Phe472 by Benzoylphenylalanine (Bpa) and of His468 by Nε-biotinylated Lysine was synthesized and ligated to truncated pThr308-Akt thioester as described above. The UV-induced cross-linking reaction was performed with 60 μg of Akt diluted in buffer 50 mM Tris-HCl pH7.8, 10 mM DTT, and UV-irradiated at 365 nm for 3 hrs in a quartz reaction vessel with circulating water at 4°C and constant stirring. Next, the UV-irradiated Akt was treated with 1 μM bovine alkaline phosphatase (Sigma Aldrich) for 2 hrs at room temperature, followed by reduction with 5 mM TCEP in 50 mM NH4HCO3 for 30 minutes at 60°C, and alkylation with 10 mM 2-chloroacetamide for 30 minutes at room temperature in the dark. The Akt protein sample was desalted using Protein Desalting Spin Columns (Thermo Scientific), and digested with 2% (wt/wt) trypsin for 16 hrs at 37°C. The tryptic digestion was stopped by adding TFA to 0.1 % (v/v) final concentration, then desalted using C-18 spin columns (Thermo Scientific) and lyophilized. The dried peptides were reconstituted in PBS buffer, pH 7.4 for enrichment of biotinylated peptides using monomeric avidin agarose (Thermo Scientific). The biotinylated peptides were eluted from the avidin agarose with 2 mM d-Biotin in PBS buffer, and desalted using C-18 spin columns, lyophilized and stored until mass spectrometry analysis.
Nano LC-MS Analysis
The biotinylated peptides from crosslinking were reconstituted in 20% acetonitrile, 0.1% formic acid with 50 mM guanidine HCl and analyzed by nano LC-MS using a NanoAcquity UPLC system (Waters, Milford, MA) interfaced to a QExactive HF mass spectrometer (Thermofisher Scientific, San Jose, CA) (Ficarro et al., 2009). Peptides were injected onto a self packed pre-column (4 cm POROS 10 R2, Applied Biosystems, Framingham, MA) and gradient eluted (10–70% B in 40 minutes; A=0.2 M acetic acid in water, B=0.2 M acetic acid in acetonitrile) to an analytical column (30 um I.D., packed with 50 cm Monitor C18, Column Engineering, Ontario, CA) with integrated ESI emitter tip (spray voltage = 3 kV). The mass spectrometer was operated in data-dependent mode, where the 10 most abundant ions in each MS scan (120K resolution, 3E6 target, lock mass enabled=445.120025) were subjected to MS/MS (15K resolution, 2E5 target, 100 ms max fill time). Dynamic exclusion was enabled with a repeat count of 1 and an exclusion time of 30 seconds. Raw MS/MS data were converted to mzXML using multiplierz (Alexander et al., 2017; Parikh et al., 2009) and MS/MS spectra matched to peptide sequences with Crossfinder version 1.1 (Mueller-Planitz, 2015). The Crossfinder data are complied in Tables S4 and S5. Crossfinder hits were validated using mzStudio (Ficarro et al., 2017).
CE-MS Analysis
The biotinylated peptides from crosslinking were reconstituted in 50% acetonitrile, 1% formic acid with 100 mM ammonium acetate and analyzed by CE-MS using a ZipChip CE system and autosampler (908 Devices, Boston, MA) interfaced to an Orbitrap Lumos mass spectrometer (ThermoFisher Scientific). Peptide solutions were loaded for 30 seconds and separation performed at 500 V/cm for 10 minutes using an HR chip (22 cm separation channel) with a background electrolyte composed of 1% formic acid in 50% acetonitrile. Pressure assist was utilized and started at 1 minute. The mass spectrometer was programmed to perform continuous MS1 (15K resolution, 1E6 target) and targeted MS2 (quadrupole isolation, 2 Da isolation window, ETD dissociation with charge calibrated parameters, 250 ms max fill time, 5E5 target, FTMS detection with 15K resolution) scans.
Preparation of phospholipid vesicles
The DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) phospholipid small unilamellar vesicles with 5 mol % of bulk PIP3 (1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol-3′,4′,5′-trisphosphate) (Avanti Polar Lipids) were prepared as previously described (Chu et al., 2014). Briefly, 5 mg of phospholipid DOPC and 100 mg of bulk PIP3 were dissolved in 1 ml of chloroform in a 10-ml round bottom flask, and chloroform was removed by nitrogen flow to form a thin lipid film. Next, the film was hydrated in 50 mM HEPES, pH 7.5 for 1 h with vortexing every 10 minutes in between. The lipid mixture was subjected to 5 cycles of freezing with dry ice-methanol followed by thawing with cool tap water and then under warm (42°C) water for 30 seconds. The lipid solution was passed 20 times through a miniextruder with a 100-nm polycarbonate membrane (Avanti Polar Lipids). The resulting vesicles were collected and kept under argon at 4°C for the next steps.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters are reported in the figure legends and in the STAR Methods. All the biochemical assays were repeated at least twice. The western blots for cell transfection studies were repeated at least thrice. The bands were quantified using ImageJ software, and the error bars represent the SEM.
DATA AND SOFTWARE AVAILABILITY
Atomic coordinates and structure factors for the Akt1 (144-480) in complex with ATP-GSK3 bisubstrate peptide have been deposited in the protein data bank (PDB) under ID code 6BUU, and for Akt1(aa 123-480) in complex with ATP-GSK3 bisubstrate peptide under ID code 6CO1.
Raw and analyzed data for this paper have been deposited to Mendeley Data: http://dx.doi.org/10.17632/8d5hgmvhjm.1
Supplementary Material
Figure S1. Biochemical and pharmacological analysis of semisynthetic Akt1 proteins, Related to Figure 1. (a) Western blot analysis of phospho-Thr450 for insect cell expressed Akt proteins: FL-Akt1-pThr308/ non-p C tail Akt1 (lane 1); FL-Akt1-pThr308/pSer473 (lane 2); FL-Akt1-pThr308/pSer477,pThr479 (lane 3); N-tags, Akt(123-459) (lane 4); and E. coli expressed 6xHis, Akt(122-459) (lane 5) with total Akt antibody (top panel) and anti-pThr450 antibody (bottom panel). (b-e) Akt1 activity is linear vs time and enzyme concentration under the conditions of these kinase assays. Representative plots showing product/enzyme (P/[E]) concentration vs. time and velocity vs. Akt1 concentration. Assays were carried out with 20 μM GSK3 peptide and 200 μM ATP. (b) and (c) P/[E] vs time using FL-Akt1-pThr308/pSer473 (10nM) and Δ143-Akt1-pThr308/pSer473 (2.5 nM), respectively. (d) and (e) Activity vs enzyme concentration for FL-Akt1-pThr308/pSer473 and Δ143-Akt1-pThr308/pSer473, respectively. Experiments were performed on two separate occasions (n=2). (f) Kinase inhibition assays for FL-Akt1-pThr308/pSer473 (red) and FL-Akt1-pThr308/2pSer477,Thr479 (blue) with ATP-competitive inhibitor GDC-0068 (left) and allosteric inhibitor MK2206 (right). Inhibition assays were run with 250 μM ATP, 20 μM GSK3 peptide substrate and 10 mM MgCl2. The inhibitors were preincubated with enzyme for 10 minutes on ice, and the reaction then performed for 10 minutes, 30°C. (g) Inhibitor parameters obtained in kinase inhibition assays. Each measurement was performed in duplicate on at least two separate occasions.
Figure S2. C-tail phosphorylation effects on PDK1-catalyzed Akt1 Thr308 phosphorylation, Related to Figure 1. (a) Schematic for the in vitro phosphorylation of Akt1 by PDK1. For these reactions, semisynthetic Akt1 (1 μM) was mixed with 10 nM GST-PDK1 in 50 mM HEPES, pH 7.5, 2 mM DTT, 10 mM MgCl2 and incubated at 30°C, and initiated by adding 1 mM ATP. Aliquots (10 μl) of the reaction were collected at the indicated time points and quenched with SDS-loading buffer. Reaction samples were analyzed by Western Blot (b) with anti-pThr308 (left) or total anti-Akt (right) primary antibodies and densitometry was used to calculate the half-time to completion. (c) The table summarizes the half-times for each of the Akt1 forms. (d) The affinity of GST-PDK1 with non-p C tail (left, red) or pSer473 (right, blue) Akt proteins with site-specific C-terminal fluorescein labeling was measured using fluorescence anisotropy. 0.4 μM of fluorescein-labeled Akt1 mixed with various concentrations of GST-PDK1 in binding buffer (50 mM HEPES, pH 7.5, 2 mM DTT, 20 mM MgCl2, 55 mM NaCl and 1 mM AMP-PNP); mixtures were preincubated at room temperature for 30 minutes and then anisotropy measurements were made. Kd values were obtained using quadratic fits and shown ±SEM (n=2). (e) The cartoon depicts a model for the effect of C-terminal phosphorylation on PDK1-catalyzed Thr308 phosphorylation. The Akt C-tail containing a conserved HF motif (469-FPQF-472) that specifically binds to a functionally conserved allosteric site on the protein kinase PDK1 called the PIF pocket (Rettenmaier et al., 2014; Romano et al., 2009). The binding of pSer473 in the C-tail to Arg144 in the PH-kinase linker arranges the HF motif to orient the PDK1 kinase domain for Akt1 Thr308 phosphorylation.
Figure S3. X-ray structural analysis of semisynthetic phospho-Akt constructs., Related to Figures 3 and 4. (a) Superposition of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 (purple) on Δ143-Akt1-pThr308/Asp473 (orange, PDB: 4EKK) with related hydrogen bonds to Gln218 side chain and backbone although one of the pSer473 non-bridging oxygens is not engaged in interactions. (b-c) Three dimensional structures of GSK3 and AMP-PNP from 4EKK (b), and GSK3-ATP bisubstrate analog (c). (d) A blow up of the phospho C-tail of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 shows the distance of pSer473 about 6.0 Å from the phosphate of pSer473. (e-f) A superposition of Δ122-Akt1-pThr308/3p-Ser473, Ser477, Thr479 (Akt1 active form) on the autoinhibited form of Akt1 (aa 2-446) containing PH and kinase domains (PDB: 4EJN) (Ashwell et al., 2012) shows that the activation loop of the autoinhibited form (green) has an orientation distinct from that of the active form (black) (e) and a steric clash of the activation loop of the active form (black balls) with the PH domain (yellow) from the autoinhibited Akt1 form (f). (g-h) Zoom-in structure of the phospho C tail of Δ122-Akt1-pThr308/3p-Ser473, Ser477, Thr479 shows a hydrogen bond of His143 and Ser475 (g), a well-defined salt bridge of pSer473 and Arg144 aswell as the hydrogen bonds of pSer473 to Gln218 (h). The electron density map shown is 2Fo-Fc contoured at 1 sigma.
Figure S4. Binding and kinase assays of PIP3 and PI(3,4)P2 with semisynthetic Akt1 proteins, Related to Figure 4. (a) Schematic for the binding affinity measurements of fluorescein-labeled soluble PIP3 (di-C6) with semisynthetic Akt proteins. (b-d) Affinity of PIP3 for non-phosphorylated FL-Akt1 (b), FL-Akt1-pThr308/pSer473(c), and FL-Akt1-pThr308/non-p C tail (d) measured with fluorescence anisotropy. Varying concentrations of Akt1s were mixed with 50 nM fluorescein-labeled soluble PIP3. Measurements (n=2) were fit to quadratic binding isotherms and Kd values shown +/− standard error. (e) Kinase assays measuring the effect of bulk PIP3 within phospholipid DOPC vesicles (5 mol% of PIP3) and soluble PI(3,4)P2 (di-C8) on the activity of semisynthetic FL-Akt1 proteins with pThr308/non-p C tail or pThr308/pSer473 using 400 μM DOPC-PIP3 vesicles (~20 mM PIP3) or 20 μM soluble PI(3,4)P2 in kinase buffer containing 250 μM ATP, 20 μM GSK3 peptide, 10 mM MgCl2.
Figure S5. Characterization of the effects of PIP3 and Mg2+ on Akt1 activity, Related to Figure 4. (a) Kinase assay measuring the effect of adding various concentrations of PIP3 on the activity of semisynthetic FL-Akt1 proteins with pThr308/pSer477,pThr479or pThr308/non-p C tail. Error bars show technical duplicates. (b) Schematic for the binding affinity measurements of fluoresceinlabeled soluble PIP3 (di-C6) with isolated Akt PH domain. (c) Binding assays show increase in fluorescence anisotropy of fluorescein labeled PIP3 with increasing concentrations of Akt PH domain. Measurements were performed at various concentrations of MgCl2 to observe differences in Kd, which is reported for each condition. (d) Competition binding assays in which fluorescein-labeled PIP3 was displaced from Akt PH domain by soluble PIP3 (di-C6) (left) and PI(3,4)P2 (di-C8) (right). The obtained IC50 values were used to calculate Kd values as described in Star methods.
Figure S6. Cellular signaling assays for wild type (WT) Akt1 and its kinase-inactive mutant D274A (a,b), and WT and R144A Akt1 constructs (c,d), Related to Figures 4 and 5. Four pcDNA3 plasmids for WT, D274A, 3A and R144A were transfected into Akt1/2 knock out HCT116 cells, the cells were serum starved and then stimulated by growth factors insulin, EGF and hIGF1 for various time points (5 to 40 minutes). The cells were lysed and loaded on SDS-PAGE gel for WB analysis with antibodies for Akt (left panel in a, c) and its downstream substrates Foxo1 and Foxo3a (right panel in a, c). (b, d) Quantification of phosphorylation level of Akt1 Ser473 (blue, red in b, and blue, violet in d) and Foxo1 Thr24/Foxo3a Thr32 (pink, green in b, and pink, dark red in d) from at least three replicates of cellular assays (n≥3, SEM shown, p<0.01).
Figure S7. Photo-crosslinking and mass spectrometry analysis, Related to Figure 6. Full MS/MS spectra of RPKBioFPQBSYSASGTA (peptide 1) crosslinked to DGATMK (peptide 2) in the activation loop of Akt as described in Figure 6. B: Bpa group, KBio:Nε-bioinylated lysine.
Figure 6.

Dissecting the pSer477/pThr479 intramolecular interaction in Akt1 using photo-crosslinking. A) Schematic representation for the UV-activatable crosslinking of the Bpa group from the pSer477/pThr479 C tail with the activation loop. B) MS and C) MS/MS spectra of RPKbioFPQBSYSASGTA (peptide 1) crosslinked to DGATMK (peptide 2) in the activation loop of Akt1. (C) Ions of type b from peptide 1 and peptide 2 are highlighted in blue and green, respectively. Similarly, ions of type y from peptide 1 and peptide 2 are shown in red and yellow. Each product ion is also labeled with its parent sequence (“1” or “2”) followed by the charge state of the ion (i.e. 2+). For example, y1 (2/1+) corresponds to the y1 ion of peptide 2 singly charged. Specific crosslinked sequences and internal fragments are also indicated (See Figure S7 for the rest of m/z spectrum). −H2O, water loss; Kbio, biotinylated lysine; B, benzoylphenylalanine.
Highlights.
-
-
Site-specifically C-terminally phosphorylated purified Akt1 forms were analyzed
-
-
Phosphorylation of Ser473 of Akt1 activates by binding the PH-kinase domain linker
-
-
Dual Ser477/Thr479 phosphorylation stimulates Akt1 via activation loop interaction
ACKNOWLEDGMENTS
We thank the NIH grant CA74305 and FAMRI foundation (P.A.C.), and NIH grant CA062924 (S.B.G.) for financial support. We thank T.K. Harris, Y. Hwang, Z. Wang, B. Weiser, M. Wu and B. Zucconi-Bennett for advice and assistance. We thank B. Vogelstein for Akt knockout cell lines, and J. Powell for Foxo antibodies. This research used resources of the Advanced Photon Source, a Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility. The Life Science Biomedical Technology Research resource (LSBR) used here is supported by P41GM111244, and DOE KP1605010. As a National Synchrotron Light Source II facility resource at Brookhaven National Laboratory, work performed at the LSBR is supported in part by DE-SC0012704 (KC0401040).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, and Hemmings BA (1996). Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15, 6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, and Cohen P (1997). Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 7, 261–269. [DOI] [PubMed] [Google Scholar]
- Alexander WM, Ficarro SB, Adelmant G, and Marto JA (2017). multiplierz v2.0: A Python-based ecosystem for shared access and analysis of native mass spectrometry data. Proteomics 17.. [DOI] [PubMed] [Google Scholar]
- Ashwell MA, Lapierre JM, Brassard C, Bresciano K, Bull C, Cornell-Kennon S, Eathiraj S, France DS, Hall T, Hill J, et al. (2012). Discovery and optimization of a series of 3-(3-phenyl-3H-imidazo[4,5-b]pyridin-2-yl)pyridin-2-amines: orally bioavailable, selective, and potent ATP-independent Akt inhibitors. J Med Chem 55, 5291–5310. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, and Tsichlis P (1998). Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene 17, 313–325. [DOI] [PubMed] [Google Scholar]
- Blake JF, Xu R, Bencsik JR, Xiao D, Kalian NC, Schlachter S, Mitchell IS, Spencer KL, Banka AL, Wallace EM, et al. (2012). Discovery and preclinical pharmacology of a selective ATP-competitive Akt inhibitor (GDC-0068) for the treatment of human tumors. J Med Chem 55, 8110–8127. [DOI] [PubMed] [Google Scholar]
- Bolduc D, Rahdar M, Tu-Sekine B, Sivakumaren SC, Raben D, Amzel LM, Devreotes P, Gabelli SB, and Cole P (2013). Phosphorylation-mediated PTEN conformational closure and deactivation revealed with protein semisynthesis. Elife 2, e00691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calleja V, Laguerre M, Parker PJ, and Larijani B (2009). Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol 7, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Dempsey DR, Thomas SN, Hayward D, Bolduc DM, and Cole PA (2016). Molecular Features of Phosphatase and Tensin Homolog (PTEN) Regulation by C-terminal Phosphorylation. J Biol Chem 291, 14160–14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Jiang H, Xu W, Li X, Dempsey DR, Zhang X, Devreotes P, Wolberger C, Amzel LM, Gabelli SB, et al. (2017). A Tunable Brake for HECT Ubiquitin Ligases. Mol Cell 66, 345–357 e346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KY, Noble ME, Skamnaki V, Brown NR, Lowe ED, Kontogiannis L, Shen K, Cole PA, Siligardi G, and Johnson LN (2006). The role of the phospho-CDK2/cyclin A recruitment site in substrate recognition. J Biol Chem 281, 23167–23179. [DOI] [PubMed] [Google Scholar]
- Chu NK, Shabbir W, Bove-Fenderson E, Araman C, Lemmens-Gruber R, Harris DA, and Becker CF (2014). A C-terminal membrane anchor affects the interactions of prion proteins with lipid membranes. J Biol Chem 289, 30144–30160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conus NM, Hannan KM, Cristiano BE, Hemmings BA, and Pearson RB (2002). Direct identification of tyrosine 474 as a regulatory phosphorylation site for the Akt protein kinase. J Biol Chem 277, 38021–38028. [DOI] [PubMed] [Google Scholar]
- Crabb SJ, Birtle AJ, Martin K, Downs N, Ratcliffe I, Maishman T, Ellis M, Griffiths G, Thompson S, Ksiazek L, et al. (2017). ProCAID: a phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Invest New Drugs 35, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL The PyMOL Molecular Graphics System (2002). [Google Scholar]
- DeLano WL (2002). The PyMOL Molecular Graphics System (San Carlos, CA, USA, DeLano: Scientific; ). [Google Scholar]
- Ebner M, Lucic I, Leonard TA, and Yudushkin I (2017). PI(3,4,5)P3 Engagement Restricts Akt Activity to Cellular Membranes. Mol Cell 65, 416–431 e416. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson K, Gan C, Cheong I, Rago C, Samuels Y, Velculescu VE, Kinzler KW, Huso DL, Vogelstein B, and Papadopoulos N (2010). Genetic inactivation of AKT1, AKT2, and PDPK1 in human colorectal cancer cells clarifies their roles in tumor growth regulation. Proc Natl Acad Sci U S A 107, 2598–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbro D, Batt D, Rose P, Schacher B, Roberts TM, and Ferrari S (1999). Homogeneous purification of human recombinant GST-Akt/PKB from Sf9 cells. Protein Expr Purif 17, 83–88. [DOI] [PubMed] [Google Scholar]
- Fan CD, Lum MA, Xu C, Black JD, and Wang X (2013). Ubiquitin-dependent regulation of phospho-AKT dynamics by the ubiquitin E3 ligase, NEDD4–1, in the insulin-like growth factor-1 response. J Biol Chem 288, 1674–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarro SB, Alexander WM, and Marto JA (2017). mzStudio: A Dynamic Digital Canvas for User-Driven Interrogation of Mass Spectrometry Data. Proteomes 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficarro SB, Zhang Y, Lu Y, Moghimi AR, Askenazi M, Hyatt E, Smith ED, Boyer L, Schlaeger TM, Luckey CJ, et al. (2009). Improved electrospray ionization efficiency compensates for diminished chromatographic resolution and enables proteomics analysis of tyrosine signaling in embryonic stem cells. Anal Chem 81, 3440–3447. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, and Abraham RT (2017). The PI3K Pathway in Human Disease. Cell 170, 605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Yo P, and Harris TK (2005). Improved yields for baculovirus-mediated expression of human His(6)-PDK1 and His(6)-PKBbeta/Akt2 and characterization of phospho-specific isoforms for design of inhibitors that stabilize inactive conformations. Protein Expr Purif 43, 44–56. [DOI] [PubMed] [Google Scholar]
- Guo H, Gao M, Lu Y, Liang J, Lorenzi PL, Bai S, Hawke DH, Li J, Dogruluk T, Scott KL, et al. (2014). Coordinate phosphorylation of multiple residues on single AKT1 and AKT2 molecules. Oncogene 33, 3463–3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Chakraborty AA, Liu P, Gan W, Zheng X, Inuzuka H, Wang B, Zhang J, Zhang L, Yuan M, et al. (2016). pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 353, 929–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herblin WF, Kauer JC, and Tam SW (1987). Photoinactivation of the mu opioid receptor using a novel synthetic morphiceptin analog. Eur J Pharmacol 139, 273–279. [DOI] [PubMed] [Google Scholar]
- Hsieh AC, Bo R, Manola J, Vazquez F, Bare O, Khvorova A, Scaringe S, and Sellers WR (2004). A library of siRNA duplexes targeting the phosphoinositide 3-kinase pathway: determinants of gene silencing for use in cell-based screens Nucleic Acids Res 32, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, and Kuriyan J (2002). The conformational plasticity of protein kinases. Cell 109, 275–282. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, and Su B (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137. [DOI] [PubMed] [Google Scholar]
- James SR, Downes CP, Gigg R, Grove SJ, Holmes AB, and Alessi DR (1996). Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3,4,5-trisphosphate without subsequent activation. Biochem J 315 (Pt 3), 709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010a). Integration, scaling, space-group assignment and postrefinement. Acta Crystallogr D Biol Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W (2010b). Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar CC, Diao R, Yin Z, Liu Y, Samatar AA, Madison V, and Xiao L (2001). Expression, purification, characterization and homology modeling of active Akt/PKB, a key enzyme involved in cell survival signaling. Biochim Biophys Acta 1526, 257–268. [DOI] [PubMed] [Google Scholar]
- Larsen JT, Shanafelt TD, Leis JF, LaPlant B, Call T, Pettinger A, Hanson C, Erlichman C, Habermann TM, Reeder C, et al. (2017). Akt inhibitor MK-2206 in combination with bendamustine and rituximab in relapsed or refractory chronic lymphocytic leukemia: Results from the N1087 alliance study. Am J Hematol 92, 759–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux AE, Schulze JO, and Biondi RM (2017). AGC kinases, mechanisms of regulation and innovative drug development. Semin Cancer Biol 48, 1–17.. [DOI] [PubMed] [Google Scholar]
- Lin K, Lin J, Wu WI, Ballard J, Lee BB, Gloor SL, Vigers GP, Morales TH, Friedman LS, Skelton N, et al. (2012). An ATP-site on-off switch that restricts phosphatase accessibility of Akt. Sci Signal 5, ra37. [DOI] [PubMed] [Google Scholar]
- Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, et al. (2014). Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508, 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MJ, Parvez S, Zhao Y, Surya SL, Wang Y, Zhang S, and Aye Y (2017). Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat Chem Biol 13, 333–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Rivera C, et al. (2010). Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PLoS One 5, e9646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning BD, and Toker A (2017). AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, and Van Aalten DM (2003). Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J 375, 531–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Planitz F (2015). Crossfinder-assisted mapping of protein crosslinks formed by site-specifically incorporated crosslinkers. Bioinformatics 31, 2043–2045. [DOI] [PubMed] [Google Scholar]
- Muir TW, Sondhi D, and Cole PA (1998). Expressed protein ligation: a general method for protein engineering. Proc Natl Acad Sci U S A 95, 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Navaza J (2001). Implementation of molecular replacement in AMoRe. Acta Crystallogr D Biol Crystallogr 57, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of x-ray diffraction data collected in oscillation mode. Methods in Enzymology 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Parikh JR, Askenazi M, Ficarro SB, Cashorali T, Webber JT, Blank NC, Zhang Y, and Marto JA (2009). multiplierz: an extensible API based desktop environment for proteomics data analysis. BMC Bioinformatics 10, 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce LR, Komander D, and Alessi DR (2010). The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 11, 9–22. [DOI] [PubMed] [Google Scholar]
- Pearlman SM, Serber Z, and Ferrell JE Jr. (2011). A mechanism for the evolution of phosphorylation sites. Cell 147, 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Qiu C, Tarrant MK, Boronina T, Longo PA, Kavran JM, Cole RN, Cole PA, and Leahy DJ (2009). In vitro enzymatic characterization of near full length EGFR in activated and inhibited states. Biochemistry 48, 6624–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettenmaier TJ, Sadowsky JD, Thomsen ND, Chen SC, Doak AK, Arkin MR, and Wells JA (2014). A small-molecule mimic of a peptide docking motif inhibits the protein kinase PDK1. Proc Natl Acad Sci U S A 111, 18590–18595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Romano RA, Kannan N, Kornev AP, Allison CJ, and Taylor SS (2009). A chimeric mechanism for polyvalent trans-phosphorylation of PKA by PDK1. Protein Sci 18, 1486–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrödinger L (2015). The PyMOL Molecular Graphics System, Version 1.8 . [Google Scholar]
- Seamon KJ, Sun Z, Shlyakhtenko LS, Lyubchenko YL, and Stivers JT (2015). SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res 43, 6486–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrant MK, Rho HS, Xie Z, Jiang YL, Gross C, Culhane JC, Yan G, Qian J, Ichikawa Y, Matsuoka T, et al. (2012). Regulation of CK2 by phosphorylation and O-GlcNAcylation revealed by semisynthesis. Nat Chem Biol 8, 262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, Paek E, Tsang AW, and Furdui CM (2011). Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci U S A 108, 10550–10555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser BP, Stivers JT, and Cole PA (2017). Investigation of N-Terminal Phospho-Regulation of Uracil DNA Glycosylase Using Protein Semisynthesis. Biophys J 113, 393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Murshudov GN, and Papiz MZ (2003). Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol 374, 300–321. [DOI] [PubMed] [Google Scholar]
- Winter G, and McAuley KE (2011). Automated data collection for macromolecular crystallography. Methods 55, 81–93. [DOI] [PubMed] [Google Scholar]
- Wu WI, Voegtli WC, Sturgis HL, Dizon FP, Vigers GP, and Brandhuber BJ (2010). Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One 5, e12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Cron P, Good VM, Thompson V, Hemmings BA, and Barford D (2002a). Crystal structure of an activated Akt/protein kinase B ternary complex with GSK3-peptide and AMP-PNP. Nat Struct Biol 9, 940–944. [DOI] [PubMed] [Google Scholar]
- Yang J, Cron P, Thompson V, Good VM, Hess D, Hemmings BA, and Barford D (2002b). Molecular mechanism for the regulation of protein kinase B/Akt by hydrophobic motif phosphorylation. Mol Cell 9, 1227–1240. [DOI] [PubMed] [Google Scholar]
- Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, et al. (2009). The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang S, Yamane H, Wahl R, Ali A, Lofgren JA, and Kendall RL (2006). Kinetic mechanism of AKT/PKB enzyme family. J Biol Chem 281, 13949–13956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Biochemical and pharmacological analysis of semisynthetic Akt1 proteins, Related to Figure 1. (a) Western blot analysis of phospho-Thr450 for insect cell expressed Akt proteins: FL-Akt1-pThr308/ non-p C tail Akt1 (lane 1); FL-Akt1-pThr308/pSer473 (lane 2); FL-Akt1-pThr308/pSer477,pThr479 (lane 3); N-tags, Akt(123-459) (lane 4); and E. coli expressed 6xHis, Akt(122-459) (lane 5) with total Akt antibody (top panel) and anti-pThr450 antibody (bottom panel). (b-e) Akt1 activity is linear vs time and enzyme concentration under the conditions of these kinase assays. Representative plots showing product/enzyme (P/[E]) concentration vs. time and velocity vs. Akt1 concentration. Assays were carried out with 20 μM GSK3 peptide and 200 μM ATP. (b) and (c) P/[E] vs time using FL-Akt1-pThr308/pSer473 (10nM) and Δ143-Akt1-pThr308/pSer473 (2.5 nM), respectively. (d) and (e) Activity vs enzyme concentration for FL-Akt1-pThr308/pSer473 and Δ143-Akt1-pThr308/pSer473, respectively. Experiments were performed on two separate occasions (n=2). (f) Kinase inhibition assays for FL-Akt1-pThr308/pSer473 (red) and FL-Akt1-pThr308/2pSer477,Thr479 (blue) with ATP-competitive inhibitor GDC-0068 (left) and allosteric inhibitor MK2206 (right). Inhibition assays were run with 250 μM ATP, 20 μM GSK3 peptide substrate and 10 mM MgCl2. The inhibitors were preincubated with enzyme for 10 minutes on ice, and the reaction then performed for 10 minutes, 30°C. (g) Inhibitor parameters obtained in kinase inhibition assays. Each measurement was performed in duplicate on at least two separate occasions.
Figure S2. C-tail phosphorylation effects on PDK1-catalyzed Akt1 Thr308 phosphorylation, Related to Figure 1. (a) Schematic for the in vitro phosphorylation of Akt1 by PDK1. For these reactions, semisynthetic Akt1 (1 μM) was mixed with 10 nM GST-PDK1 in 50 mM HEPES, pH 7.5, 2 mM DTT, 10 mM MgCl2 and incubated at 30°C, and initiated by adding 1 mM ATP. Aliquots (10 μl) of the reaction were collected at the indicated time points and quenched with SDS-loading buffer. Reaction samples were analyzed by Western Blot (b) with anti-pThr308 (left) or total anti-Akt (right) primary antibodies and densitometry was used to calculate the half-time to completion. (c) The table summarizes the half-times for each of the Akt1 forms. (d) The affinity of GST-PDK1 with non-p C tail (left, red) or pSer473 (right, blue) Akt proteins with site-specific C-terminal fluorescein labeling was measured using fluorescence anisotropy. 0.4 μM of fluorescein-labeled Akt1 mixed with various concentrations of GST-PDK1 in binding buffer (50 mM HEPES, pH 7.5, 2 mM DTT, 20 mM MgCl2, 55 mM NaCl and 1 mM AMP-PNP); mixtures were preincubated at room temperature for 30 minutes and then anisotropy measurements were made. Kd values were obtained using quadratic fits and shown ±SEM (n=2). (e) The cartoon depicts a model for the effect of C-terminal phosphorylation on PDK1-catalyzed Thr308 phosphorylation. The Akt C-tail containing a conserved HF motif (469-FPQF-472) that specifically binds to a functionally conserved allosteric site on the protein kinase PDK1 called the PIF pocket (Rettenmaier et al., 2014; Romano et al., 2009). The binding of pSer473 in the C-tail to Arg144 in the PH-kinase linker arranges the HF motif to orient the PDK1 kinase domain for Akt1 Thr308 phosphorylation.
Figure S3. X-ray structural analysis of semisynthetic phospho-Akt constructs., Related to Figures 3 and 4. (a) Superposition of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 (purple) on Δ143-Akt1-pThr308/Asp473 (orange, PDB: 4EKK) with related hydrogen bonds to Gln218 side chain and backbone although one of the pSer473 non-bridging oxygens is not engaged in interactions. (b-c) Three dimensional structures of GSK3 and AMP-PNP from 4EKK (b), and GSK3-ATP bisubstrate analog (c). (d) A blow up of the phospho C-tail of Δ143-Akt1-pThr308/3p-Ser473,Ser477,Thr479 shows the distance of pSer473 about 6.0 Å from the phosphate of pSer473. (e-f) A superposition of Δ122-Akt1-pThr308/3p-Ser473, Ser477, Thr479 (Akt1 active form) on the autoinhibited form of Akt1 (aa 2-446) containing PH and kinase domains (PDB: 4EJN) (Ashwell et al., 2012) shows that the activation loop of the autoinhibited form (green) has an orientation distinct from that of the active form (black) (e) and a steric clash of the activation loop of the active form (black balls) with the PH domain (yellow) from the autoinhibited Akt1 form (f). (g-h) Zoom-in structure of the phospho C tail of Δ122-Akt1-pThr308/3p-Ser473, Ser477, Thr479 shows a hydrogen bond of His143 and Ser475 (g), a well-defined salt bridge of pSer473 and Arg144 aswell as the hydrogen bonds of pSer473 to Gln218 (h). The electron density map shown is 2Fo-Fc contoured at 1 sigma.
Figure S4. Binding and kinase assays of PIP3 and PI(3,4)P2 with semisynthetic Akt1 proteins, Related to Figure 4. (a) Schematic for the binding affinity measurements of fluorescein-labeled soluble PIP3 (di-C6) with semisynthetic Akt proteins. (b-d) Affinity of PIP3 for non-phosphorylated FL-Akt1 (b), FL-Akt1-pThr308/pSer473(c), and FL-Akt1-pThr308/non-p C tail (d) measured with fluorescence anisotropy. Varying concentrations of Akt1s were mixed with 50 nM fluorescein-labeled soluble PIP3. Measurements (n=2) were fit to quadratic binding isotherms and Kd values shown +/− standard error. (e) Kinase assays measuring the effect of bulk PIP3 within phospholipid DOPC vesicles (5 mol% of PIP3) and soluble PI(3,4)P2 (di-C8) on the activity of semisynthetic FL-Akt1 proteins with pThr308/non-p C tail or pThr308/pSer473 using 400 μM DOPC-PIP3 vesicles (~20 mM PIP3) or 20 μM soluble PI(3,4)P2 in kinase buffer containing 250 μM ATP, 20 μM GSK3 peptide, 10 mM MgCl2.
Figure S5. Characterization of the effects of PIP3 and Mg2+ on Akt1 activity, Related to Figure 4. (a) Kinase assay measuring the effect of adding various concentrations of PIP3 on the activity of semisynthetic FL-Akt1 proteins with pThr308/pSer477,pThr479or pThr308/non-p C tail. Error bars show technical duplicates. (b) Schematic for the binding affinity measurements of fluoresceinlabeled soluble PIP3 (di-C6) with isolated Akt PH domain. (c) Binding assays show increase in fluorescence anisotropy of fluorescein labeled PIP3 with increasing concentrations of Akt PH domain. Measurements were performed at various concentrations of MgCl2 to observe differences in Kd, which is reported for each condition. (d) Competition binding assays in which fluorescein-labeled PIP3 was displaced from Akt PH domain by soluble PIP3 (di-C6) (left) and PI(3,4)P2 (di-C8) (right). The obtained IC50 values were used to calculate Kd values as described in Star methods.
Figure S6. Cellular signaling assays for wild type (WT) Akt1 and its kinase-inactive mutant D274A (a,b), and WT and R144A Akt1 constructs (c,d), Related to Figures 4 and 5. Four pcDNA3 plasmids for WT, D274A, 3A and R144A were transfected into Akt1/2 knock out HCT116 cells, the cells were serum starved and then stimulated by growth factors insulin, EGF and hIGF1 for various time points (5 to 40 minutes). The cells were lysed and loaded on SDS-PAGE gel for WB analysis with antibodies for Akt (left panel in a, c) and its downstream substrates Foxo1 and Foxo3a (right panel in a, c). (b, d) Quantification of phosphorylation level of Akt1 Ser473 (blue, red in b, and blue, violet in d) and Foxo1 Thr24/Foxo3a Thr32 (pink, green in b, and pink, dark red in d) from at least three replicates of cellular assays (n≥3, SEM shown, p<0.01).
Figure S7. Photo-crosslinking and mass spectrometry analysis, Related to Figure 6. Full MS/MS spectra of RPKBioFPQBSYSASGTA (peptide 1) crosslinked to DGATMK (peptide 2) in the activation loop of Akt as described in Figure 6. B: Bpa group, KBio:Nε-bioinylated lysine.