

Abstract
Randomized clinical trials have established the efficacy of naltrexone for reducing quantity of alcohol consumption and incidence of relapse to heavy drinking. To evaluate putative treatment mechanisms, human laboratory studies have examined naltrexone's effects on alcohol responses and self‐administration during short‐term medication protocols. Results from these studies are inconsistent and have yet to be examined in aggregate. This meta‐analysis aimed to quantify naltrexone's effects on alcohol self‐administration and craving in the context of placebo‐controlled human laboratory trials. Potential moderators of medication effects were also examined. Meta‐analyses of alcohol self‐administration (k = 9, N = 490) and craving (k = 16, N = 748) confirmed that, under controlled experimental conditions, naltrexone reduces the quantity of consumption (Hedges' g = −.277, SE = .074, 95 percent CI = −.421, −.133, p < .001) and magnitude of self‐reported craving (g = −.286, SE = .066, 95 percent CI = −.416, −.156, p < .001) relative to placebo. Subgroup and moderation analyses found no evidence that effect sizes differed by study population (dependent versus non‐dependent drinkers), laboratory paradigm or duration of medication exposure. These results substantiate prior evidence for reductions in event‐level craving and consumption as potential treatment mediators, also establishing effect sizes to inform future human laboratory trials. From a clinical perspective, these results may provide additional evidence regarding naltrexone's efficacy in the context of acute or subacute dosing regimens.
Keywords: Alcohol use disorder, opioid antagonist, pharmacotherapy, relapse, subjective response
Introduction
Since the approval of naltrexone as a treatment for alcohol dependence by the U.S. Federal Drug Administration (FDA) in 1994, considerable evidence has accumulated to support its efficacy as a front‐line pharmacotherapy. Meta‐analyses support that, as an adjunct to psychosocial therapy, daily naltrexone reduces the number of drinking days, quantity of alcohol consumed and incidence of relapse to heavy drinking relative to placebo—albeit with modest effect sizes (Jonas et al., 2014; Maisel et al., 2013; Rösner et al., 2010). Meta‐analyses further indicate relatively greater efficacy of naltrexone versus acamprosate for reducing heavy drinking and craving (Maisel et al., 2013). Nonetheless, naltrexone remains significantly under‐utilized in clinical practice (Heilig et al., 2011; Heilig, 2015).
The earliest randomized controlled trials (RCTs) of naltrexone generated important evidence as to potential treatment mechanisms (O'Malley et al., 1992; Volpicelli et al., 1992; reviewed in Pettinati et al., 2006). In the seminal trial naltrexone reduced the incidence of relapse to heavy drinking, quantity of consumption and weekly reports of craving, but not the likelihood of an initial lapse (Volpicelli et al., 1992). The relative specificity of treatment effects to heavy drinking outcomes (as opposed to abstinence) has since been replicated across clinical trials (Pettinati et al., 2006) and meta‐analyses (Rösner et al., 2010). Early trials also suggested that patients treated with naltrexone reported fewer drinks consumed and diminished ‘high’ during initial lapses (Volpicelli et al., 1995), implicating reductions in event‐level consumption and hedonic effects of alcohol as putative biobehavioral mechanisms for naltrexone's efficacy (Pettinati et al., 2006; Sinha and O'Malley, 1999; Volpicelli et al., 1995).
Notably, these mechanisms are consistent with naltrexone's neuropharmacological profile as a competitive μ‐opioid receptor antagonist. Hedonic effects of alcohol are attributed in part to μ‐opioid receptor activation following alcohol‐induced release of endogenous opioids. Preclinical data suggest that μ‐opioid receptor activation indirectly promotes dopamine release in the nucleus accumbens via a pathway involving GABAergic and dopaminergic neurons in the ventral tegmental area (Heilig et al., 2011). Conversely, μ‐opioid blockade blunts alcohol‐related dopamine release in the nucleus accumbens (Benjamin et al., 1993; Di Chiara et al., 1996; Spanagel et al., 1992), presumably contributing to reductions in alcohol reward and self‐administration (SA) (Gonzales and Weiss, 1998). These findings complement clinical evidence concerning reductions in craving and event‐level consumption as potential mechanisms for naltrexone's efficacy (Pettinati et al., 2006; Sinclair, 2001).
Indirect support for these potential mechanisms comes from controlled human laboratory investigations, which first emerged shortly following the first naltrexone RCTs (Swift et al., 1994). Human laboratory models involving cue exposure and/or alcohol administration are well suited for testing biobehavioral mechanisms of medication efficacy (O'Malley et al., 2002; Plebani et al., 2012), and have been used to evaluate naltrexone's effects on craving, subjective responses and SA for over two decades. While several of these studies have reported naltrexone‐related reductions in laboratory measures of craving and/or consumption, results are mixed overall. A number of laboratory studies found no significant effect of naltrexone in reducing alcohol craving (e.g. de Wit et al., 1999; Doty et al., 1997; McGeary et al., 2006; Rohsenow et al., 2000) or SA (e.g. Anton et al., 2012). In other cases, medication effects were evident only in subgroup analyses (e.g. Drobes et al., 2003; Krishnan‐Sarin et al., 2007; Palfai et al., 1999). Potential explanations for these inconsistencies include differences in populations (e.g. dependent versus non‐dependent drinkers) and laboratory paradigms across studies. Additionally, considering that naltrexone's effects on clinical outcomes are modest in magnitude (Rösner et al., 2010), inadequate statistical power in small‐scale human laboratory studies could partly account for inconsistent findings.
The fact that naltrexone human laboratory studies utilize acute (single‐dose) or sub‐acute (e.g. several days) medication schedules has, among other reasons, rendered them ineligible for prior meta‐analyses, which have required a minimum treatment duration of 4–12 weeks (e.g. Jonas et al., 2014; Rösner et al., 2010). Meta‐analysis permits aggregation of results across studies, improving power and precision in effect size estimates. Evaluating human experimental results in a meta‐analytic framework would allow quantification of medication effects under human laboratory conditions, potentially clarifying inconsistent findings and informing effect size estimates for future studies. Verifying medication effects on laboratory craving and SA could also inform questions of clinical efficacy, including potential treatment mediators. Finally, given the exclusive use of acute or sub‐acute medication schedules in human laboratory trials, these findings are potentially relevant for the efficacy of short‐term opioid antagonist treatments for reducing alcohol craving and consumption (e.g. Niciu and Arias, 2013). The present study applied meta‐analysis to evaluate the aggregate effects of naltrexone versus placebo on measures of laboratory SA and craving, with the primary aims of quantifying medication effects under experimental conditions and testing potential moderators of medication effects.
Method
Search strategy and study selection
Study procedures were carried out in accordance with the Preferred Reporting Items for Systematic Reviews and Meta‐Analyses (PRISMA) criteria (Moher et al., 2009). Search procedures aimed to identify studies that met the following criteria: (1) reported a randomized trial of naltrexone versus placebo; (2) included a human laboratory paradigm involving cue exposure, alcohol administration or alcohol SA under controlled (i.e. experimenter‐observed) conditions; (3) included alcohol SA and/or craving outcomes; (4) published in a peer‐reviewed, English language journal; and (5) included data sufficient for calculating an effect size. The primary search method consisted of electronic database queries (PubMed, PsycInfo, EMBASE) using the following terms: (alcohol AND naltrexone) AND (laboratory OR craving OR ‘cue reactivity’ OR ‘cue‐elicited’ OR ‘cue exposure’ OR ‘alcohol administration’ OR urge OR ‘subjective response’). Secondary manual searches consisted of (1) reviewing references from each candidate study; and (2) examining naltrexone reviews and meta‐analyses. The search process was initially conducted in 2014 and repeated in 2015 prior to data analysis (no additional qualifying studies were identified).
Overall, 399 non‐duplicate abstracts were identified in primary searches, with six additional studies identified in secondary searches. Studies with potential to meet the inclusion criteria were retrieved for full review (n = 43). Of these, 23 were excluded because they involved secondary analysis of a previously published study (9), did not report quantitative results on SA or craving (5), reported insufficient data for calculating an effect size (3), or were otherwise deemed ineligible (6) (see Fig. 1). The minimum quality criteria for inclusion were (1) use of a randomized, placebo‐controlled design to evaluate medication effects; and (2) publication in a peer‐review journal. While unpublished data were not included for this reason, publication bias was evaluated using standard procedures (described below). Studies were also coded on several methodological features (e.g. random assignment, blinding) using a modified rating scheme (Downs & Black, 1998), resulting in a score of methodological quality ranging from 0 to 22.
Figure 1.
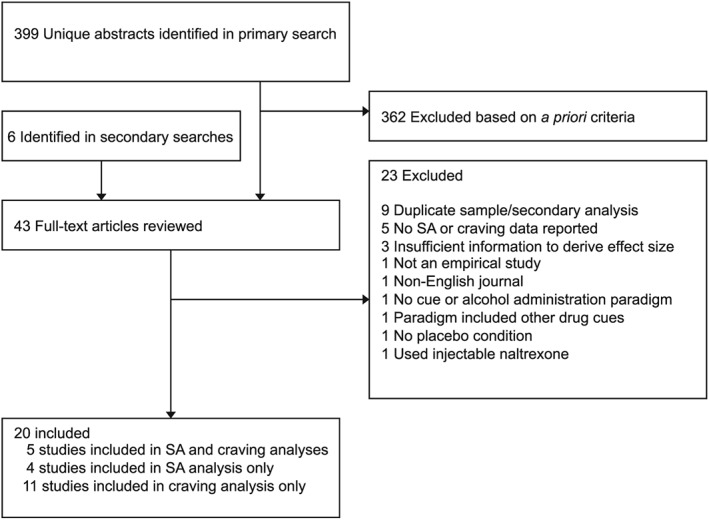
Study selection process
Table 1 presents the studies and primary outcome(s) included in the analyses. Twenty studies were retained for inclusion in one or both meta‐analyses. Of these, 9 contributed an effect to the SA analysis and 16 contributed an effect to the craving analysis (5 studies contributed effects for both). In some cases, studies reporting both SA and craving assessments were only included in one of the two analyses: SA data were excluded for one study because of the use of non‐alcoholic beer, and two studies omitted quantitative data on craving (see Supplemental Table A).
Table 1.
Studies included in meta‐analyses of laboratory self‐administration and craving.
|
Authors/ Year |
Paradigm | Population | Subgroups | N | Analysis | Medication days | Outcome measure analyzed | Craving time points analyzed |
|---|---|---|---|---|---|---|---|---|
| Anton et al., 2004 | ASA, in simulated bar with alternative reinforcer | Alcohol dependent | Delayed access versus immediate access | 40 | ASA | 7 | Number of weight‐adjusted drinks | |
| Anton et al., 2012 | ASA, in simulated bar with alternative reinforcer | Alcohol dependent | OPRM1 genotype | 83 | ASA | 7 | Number of weight‐adjusted drinks | |
| Davidson et al., 1996 | ASA in groups, in bar setting | Non‐dependent | 16 |
ASA, Craving |
8 | Mean BrAC; single item urge rating | Baseline, 60 minutes | |
| Davidson et al., 1999 | ASA in groups, in bar setting |
Non‐ dependent |
51 |
ASA, Craving |
7 | Number of standard drinks; AUQ | Baseline, end of drinking session (90 minutes) | |
| de Wit et al., 1999 | ASA in groups, in laboratory with choice paradigm (alcohol, placebo) | Non‐dependent |
13, 24 |
ASA, Craving |
1 | Number of weight‐adjusted drinks; DEQ ‘want’ | Peak change observed across time points | |
| Doty et al., 1997 | Alcohol challenge (0.25 g/kg ethanol or placebo), in groups, in laboratory | Non‐dependent |
Drinker status (light versus moderate) |
25 | Craving | 1 | DEQ ‘want’ | Peak change observed across time points |
| Drobes et al., 2003 a | ASA, in simulated bar with alternative reinforcer | Alcohol dependent and social drinkers | Alcohol dependence status | 147 | ASA | 8 | Number of weight‐adjusted drinks | |
| Drobes et al., 2004 a | Alcohol challenge (0.4 g/kg for men, 0.34 g/kg for women), in simulated bar | Alcohol dependent and social drinkers | Alcohol dependence status | 148 | Craving | 8 | Alcohol Craving Questionnaire‐Now (adapted) | Baseline and 10, 20, 40, 60 minutes |
| Krishnan‐Sarin et al., 2007 | ASA, in laboratory with alternative reinforcer | Alcohol dependent | Family history of alcohol dependence; medication dose (50 versus 100 mg) | 92 | ASA | 6 | Number of weight‐adjusted drinks | |
| Kruse et al., 2012 | ASA, in laboratory with alcohol cue exposure and manipulation of alcohol availability | Alcohol dependent | 58 |
ASA, Craving |
9 | Weight of beer consumed (g); Alcohol Craving Questionnaire (adapted) | Mean ratings across alcohol cue exposures | |
| McGeary et al., 2006 | Cue reactivity (water versus alcohol cue), individually, in laboratory | Mixed | OPRM1 and DRD4 genotype | 90 | Craving | 10 | Singe‐item urge rating | Mean ratings across all cue exposures |
| Miranda et al., 2014 | Cue reactivity (water versus alcohol cue), individually, in laboratory | Mixed | 22 | Craving | 10 | AUQ | Water cue versus mean ratings across two alcohol cues | |
| Monti et al., 1999 | Cue reactivity (water versus alcohol cue), individually, in laboratory | Alcohol dependent | 41 | Craving | 7 | Single‐item urge rating | Ratings following each of two alcohol cue exposures | |
| Na and Lee, 2002 | Alcohol challenge (0.5 ml/kg of 25 percent alcohol beverage), individually, in simulated bar | Non‐dependent | 15 | Craving | 7 | Single‐item urge rating | Baseline, 20 minutes and 60 minutes | |
| O'Malley et al., 2002 | ASA, in laboratory with alternative reinforcer | Alcohol dependent | 18, 16 |
ASA, Craving |
6 | Number of weight‐adjusted mini drinks; AUQ | Baseline and mean post‐alcohol rating (80, 110, 150 and 180 minutes) | |
| Ooteman et al., 2007 | Cue reactivity, in laboratory (alcohol‐cue only; pre‐ and post‐cue assessments both before and after medication) | Alcohol dependent | 75 | Craving | 21 | Single‐item urge rating | Change in peak craving during cue exposure relative to baseline | |
| Palfai et al., 1999 | Cue reactivity (water versus alcohol cue), in laboratory | Non‐dependent | High versus low alcohol expectancies | 36 | Craving | 1 | Single‐item urge rating | Water cue versus alcohol cue |
| Ray and Hutchison, 2007 | Alcohol challenge (successive BrACs of 20, 40 and 60 mg%), in laboratory | Non‐dependent | OPRM1 genotype | 40 | Craving | 3 | AUQ | Ratings at target BrACs: 20, 40 and 60 mg% |
| Ray et al., 2012 | Alcohol challenge (successive BrACs of 20, 40 and 60 mg%) in laboratory | Non‐dependent | OPRM1 genotype | 35 | Craving | 4 | AUQ | Mean ratings across baseline and 20, 40 and 60 mg% BrAC |
| Rohsenow et al., 2000 | Cue reactivity (no cue, juice, alcohol) in laboratory | Alcohol dependent | 56 | Craving | 1 | Single‐item urge rating | Baseline and ratings after each of two alcohol cue exposures |
ASA = Alcohol self‐administration. AUQ = Alcohol Urge Questionnaire. BrAC = Breath alcohol concentration. DEQ = Drug Effects Questionnaire.
Data abstraction and outcomes
Two coders (CH, JW) independently extracted effect size data and recorded characteristics from qualifying studies. Descriptive information included demographic factors (age, sex and baseline drinking quantity/frequency), naltrexone administration protocol (dosage, number of mediation days), diagnostic status (alcohol‐dependent or non‐dependent drinkers), design features (e.g. within or between subjects) and descriptions of the laboratory paradigm.
To generate effect size data, means and standard deviations (as reported in the manuscript) were used whenever possible. If means and standard errors were reported, standard errors were converted to standard deviations. When such information was unavailable, other metrics (e.g. t or F tests) were used when possible. In cases where group means were depicted graphically without sufficient text information, means and standard errors (or standard deviations) were extracted from figures by the two coders independently. Where minor discrepancies were observed because of measurement error, the two estimates were averaged for the analysis. Any other discrepancies across the coders in the data extracted from the text or figures were resolved by consensus through discussion.
Self‐administration
Measures of SA included number of standard drinks consumed (k = 1), number of participant‐standardized (i.e. adjusted for sex and body weight) laboratory drinks consumed (k = 6), breath alcohol concentration (BrAC) at the end of SA (k = 1) and weight of beverage consumed (k = 1). If studies reported more than one SA outcome, we coded the primary outcome. One study used a within‐subjects design without sufficient data to establish a pre‐to‐post correlation for the SA outcome; we imputed this value by estimating a moderate correlation (r = .50), similar to procedures reported in another meta‐analysis of naltrexone (Maisel et al., 2013).
Craving
Studies using a composite measure of self‐reported craving (e.g. the Alcohol Urge Questionnaire, AUQ), or a single‐item measure of craving (or synonymous construct; i.e. ‘urge,’ ‘wanting’) contributed to the craving analysis (k = 16). For studies assessing craving at multiple points during the session, effects were averaged across time points at the study level (Quinn and Fromme, 2011). If studies reported data on more than one craving measure (e.g. the AUQ and a visual analogue scale), or that different craving scales were used at different time points, we prioritized multi‐item scales over single item scales and prioritized assessments taken during cue exposure over retrospective assessments conducted following cue exposure. In studies using within‐subjects designs, we assumed the same pre‐post correlation noted above if pre‐post correlations were not reported. In addition to examining craving, we evaluated the possibility of analyzing other categories of subjective responses (e.g. stimulation, sedation). However, craving was the most commonly used subjective response outcome, and most studies reporting subjective response measures included craving. Given this issue, and that craving is considered a proximal determinant of consumption (Monti et al., 1999), we elected to restrict the analysis to craving as the sole self‐report outcome. Although a small number of otherwise eligible studies reporting other subjective effects were excluded as a result, focusing on craving allowed us to report on arguably the most clinically relevant outcome, while avoiding the potential problem of combining different subjective response domains in the analysis (King et al., 2011).
Statistical Analyses
Separate meta‐analyses were planned for SA and craving. Individual studies (k) served as the unit of analysis. Hedge's g, the standardized mean difference, served as the measure of effect size and was computed in Comprehensive Meta Analysis (Borenstein et al., 2005). In cases where reported data were insufficient to calculate g, we used alternate approaches (described above) to estimate Cohen's d, then converted values to g. Random effects models, which do not assume homogeneity of effects, were selected for all analyses. Effect sizes were coded such that a negative value indicates a reduction in consumption or craving in the naltrexone condition relative to placebo.
All qualifying studies included assignment to 50mg naltrexone (the standard clinical dose) and placebo. Two studies included a second dosage condition (100 mg, Krishnan‐Sarin et al., 2007; 25 mg, Doty et al., 1997). To maintain consistency across studies, data from alternate dosage conditions were omitted from analysis. Some studies reported results separately for participant subgroups (e.g. stratified on family history, diagnostic status, genotype). In these cases, effects for each subgroup were estimated and combined at the study level to ensure that each study contributed only one effect. To examine whether outliers could have substantially altered the summary effects, sensitivity analyses were conducted by re‐estimating the summary effect while removing each study in turn.
Heterogeneity between studies was assessed with the Q test and the I 2 statistic. Q denotes the ratio of observed variation to within‐study error; a significant Q statistic would imply the need to reject the null hypothesis that all studies share a common effect size (Borenstein et al., 2009). Because Q is sensitive to the number of studies (k), I 2 served as an additional index of heterogeneity. I 2 reflects the relative proportion of heterogeneity in observed effect sizes across studies that can be attributed to true heterogeneity, versus heterogeneity attributable to chance (i.e. because of within‐study error). Publication bias was evaluated using Rosenthal's fail‐safe N (Rosenthal, 1979). Because null effects may go unpublished, N estimates the number of additional null findings (assuming an effect size of 0 for such studies) that would be required before the overall effect would no longer reach statistical significance. The Trim and Fill method (Duval and Tweedie, 2000) was also applied as an additional estimate of publication bias.
Subgroup and moderator analyses
Within each meta‐analysis we planned subgroup analyses to compare effect sizes for alcohol‐dependent versus non‐dependent drinkers. These analyses were restricted to studies in which the entire sample consisted of either population (two studies were excluded from the craving analysis because they utilized mixed populations). One additional study recruited both dependent and non‐dependent drinkers, but reported SA and craving outcomes separately in each subgroup (Drobes et al., 2003; 2004). Results from this study were retained in the population analysis by allowing the study to contribute separate effects for dependent and non‐dependent drinkers.
Additional subgroup analyses were planned to compare medication effects based on methodological features of the laboratory paradigms. Among studies assessing medication effects on craving, some utilized alcohol cue exposure paradigms without alcohol administration, while others assessed craving following alcohol administration. Therefore, a subgroup analysis compared medication effects on craving in response to cue exposure (k = 6) versus acute alcohol exposure (k = 10). For the SA analysis, we planned a subgroup analysis to compare the most commonly used SA paradigm (O'Malley et al., 2002) (k = 5) to studies using alternate paradigms (k = 4). Subgroup analyses were conducted using a mixed effects model, such that a random effects model is used to combine studies within subgroup, and a fixed effect model is applied to yield the overall effect (Borenstein et al., 2009).
To estimate medication effects on craving prior to alcohol or cue exposure, a supplementary analysis examined medication effects on baseline craving. This analysis included all cue exposure and alcohol administration studies that reported data for baseline craving levels prior to cue or alcohol exposure (k = 7). Finally, to examine whether medication effects differed as a function of duration of medication exposure, we planned moderator analyses to examine whether number of dosage days moderated the effect size. Moderator analyses were conducted separately for SA and craving outcomes using meta‐regression. These analyses assumed a random effects model, with the moderating effect assessed based on the significance of the Z test statistic (Borenstein et al., 2009).
Results
Descriptive information
Descriptive details and a summary of findings from all studies are provided in Supplemental Table A. Studies qualifying for inclusion were published between 1996 and 2014. Individual sample sizes contributing to the effects of interest ranged from 13 to 148. With the exception of one study from South Korea and one from the Netherlands, qualifying studies were conducted in the United States. Quality ratings of the 20 studies ranged from 11 to 18 (mean = 13.9, standard deviation = 2.2). None of the included studies fulfilled all the quality criteria, with the least frequently met criteria being information on handling data for subjects lost to follow‐up, presence of source data and information on representativeness of the study sample. Of note, only one study provided justification of sample sizes on the basis of power calculations. Nonetheless, the most critical quality features, such as description of main outcomes, descriptive sample characteristics, randomization and intervention descriptions, blinding procedures, reporting of primary outcomes, reporting measures of variability and describing statistical methods, were present in almost all studies.
Self‐administration
The SA meta‐analysis comprised 490 participants. Four different SA paradigms were represented, the most common (5 studies) being a paradigm involving administration of ‘mini‐drinks’ (target blood alcohol concentration increment .015 g/dl per drink), first published by O'Malley et al. (2002). Other paradigms were reported by (Davidson et al., 1996; 1999, Kruse et al., 2012) and de Wit et al. (1999). Five studies included participants diagnosed with alcohol dependence, three studies included non‐dependent drinkers and one study included both types of participants.
Under a random effects model, the core SA analysis demonstrated a significant reduction in consumption in naltrexone versus placebo conditions (g = −.277, SE = .074, 95 percent CI = −.421, −.133, p < .001), without evidence of significant between‐study heterogeneity in effect sizes (Q (8) = 9.15, p = .33, I 2 = 12.57) (Fig. 2). The relatively low I 2 statistic indicates that most between‐study variance in effect sizes is likely attributable to random error. Sequential removal of each study left the point estimate relatively unchanged (range: g = −.311 to −.259, all ps < .005). The fail‐safe N was 33; therefore, the estimated number of null studies needed to render the overall effect non‐significant exceeded the number of studies in the meta‐analysis by a factor of 3.7. The summary estimate did not change appreciably using Trim and Fill values. In sum, the SA analysis supported a significant overall effect of naltrexone in reducing the quantity of ad libitum alcohol consumed under experimental conditions. Although small in magnitude, this effect appeared robust and reliable when aggregated across studies.
Figure 2.
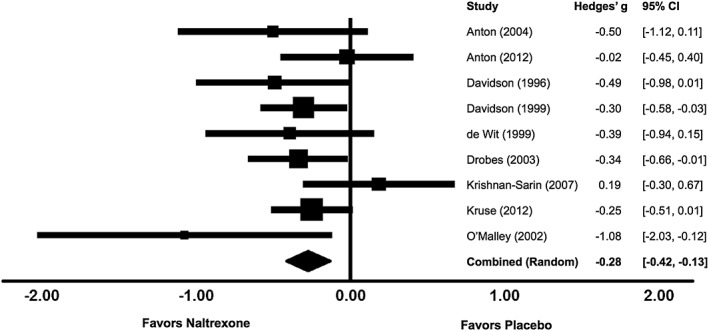
Effects of naltrexone versus placebo on laboratory alcohol self‐administration
Mixed effects subgroup analyses evaluated whether medication effects on SA differed by population or laboratory paradigm. There was no evidence that medication effects differed by population: naltrexone decreased consumption in both alcohol‐dependent (g = −.250, SE = .104, 95 percent CI = −.453, −.047, p = .016, k = 6) and non‐dependent drinkers (g = −.320, SE = .122, 95 percent CI = −.559, −.081, p = .009, k = 4), without evidence of heterogeneity in effects across subgroups (Q between (1) = .19, p = .66). The subgroup analysis of study paradigm found comparable effect sizes among studies using the ‘mini‐drink’ paradigm (g = −.222, SE = .119, 95 percent CI = −.456, .012, p = .063, k = 5) versus alternate paradigms (g = −.321, SE = .104, 95 percent CI = −.525, −.117, p = .002, k = 4). Although the confidence interval for the ‘mini‐drink’ studies contained zero, the assumption of homogeneity could not be rejected (Q between (1) = .39, p = .53). Although the mini‐drink studies showed a somewhat smaller overall effect size relative to studies utilizing other paradigms, the single largest effect came from the first published study of the mini‐drink paradigm (O'Malley et al., 2002). Finally, we tested the moderating effect of medication days using meta‐regression. There was no evidence that number of medication days was associated with differences in effect size (B = .00, 95 percent CI = −.07–.07, Z = .09, p = .93).
Craving
The craving analysis included 748 participants. Under a random effects model, there was a significant overall effect of naltrexone in reducing craving (g = −.286, SE = .066, 95 percent CI = −.416, −.156, p < .001, Fig. 3). In contrast to the SA analysis, the assumption of homogeneity did not hold for craving (Q (15) = 25.37, p = .045, I 2 = 40.88), suggesting variability in the magnitude of the effect across studies. Sequential removal of each study showed that the effect remained significant in all cases (range: g = −.244 to −.322, all ps < .001). The fail‐safe N was 128 (8 times the number of included studies), suggesting a low likelihood of publication bias, and the summary estimate remained virtually unchanged using Trim and Fill values.
Figure 3.
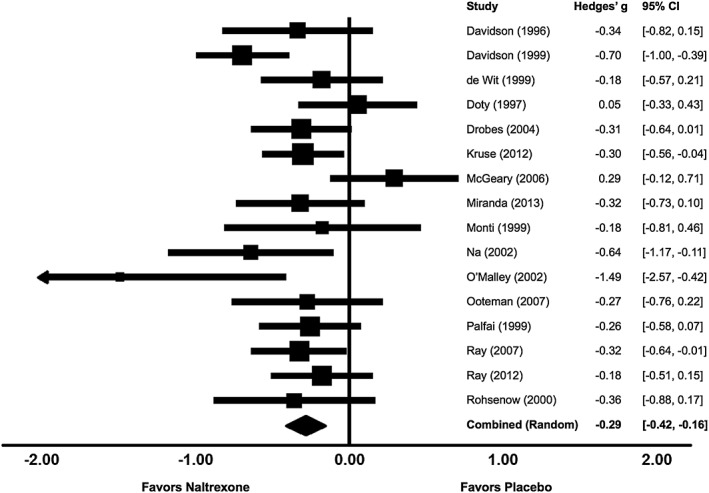
Effects of naltrexone versus placebo on laboratory alcohol craving
A subgroup analysis suggested somewhat larger reductions in craving during alcohol administration (k = 10, g = −.342, SE = .080, 95 percent CI = −.498, −.185, p < .001) versus cue exposure without alcohol consumed (k = 6, g = −.173 SE = .114, 95 percent CI = −.397, .051, p = .129); however, the overall difference did not reach significance (Q between (1) = 1.47, p = .23). The subgroup analysis for study population found no evidence of differences in the extent of craving reduction among alcohol‐dependent (k = 6, g = −.322, SE = .110, 95 percent CI = −.538, −.107, p = .003) and non‐dependent drinkers (k = 9, g = −.323, SE = .077, 95 percent CI = −.474, −.173, p < .001, Q between (1) = .00, p = .99). Also, there was no evidence that number of medication days was associated with the magnitude of reduction in craving (B = −.005, 95 percent CI = −.026–.017, Z = −.41, p = .68). The supplementary analysis on baseline craving levels (i.e. prior to alcohol administration or alcohol cue exposure) found a significant reduction in baseline craving (k = 7, g = −.321, SE = .083, 95 percent CI = −.483, −.158, p < .001), indicating that reductions in craving preceded cue exposure or alcohol administration.
Discussion
Naltrexone human laboratory studies have been important for evaluating potential mechanisms of medication efficacy, but quantitative synthesis of these findings has not been reported previously. Importantly, reductions in event‐level craving and drinking quantity are considered primary biobehavioral mechanisms for treatment effects (Heilig et al., 2011; Pettinati et al., 2006). In evaluating the aggregate effects of naltrexone versus placebo on laboratory measures of SA and craving, this study represents the first meta‐analysis to quantify medication effects on alcohol‐related outcomes exclusively in a human laboratory context.
The SA analysis confirmed that short‐term treatment with naltrexone reduces the quantity of ad libitum consumption under controlled experimental conditions. This effect was modest (g = −.277) but reliable, with minimal heterogeneity observed across studies. By comparison, effect sizes for heavy drinking or drinking quantity outcomes across randomized clinical trials generally range between g = .10 and .20 (Jonas et al., 2014; Maisel et al., 2013; Rösner et al., 2010). Thus, a modest effect on laboratory SA is consistent with what is known about naltrexone's effects on self‐reported drinking quantity during longer periods of treatment. Importantly, the current findings confirm that naltrexone reduces consumption using data collected at the event level—an observation also reported in the context of ecological momentary assessment (EMA) research (Miranda et al., 2014). Moreover, in contrast to RCT and EMA studies—which rely on self‐reports subject to recall and/or reporting biases—this analysis confirms a significant overall medication effect on objective indicators of consumption.
The craving meta‐analysis showed that naltrexone significantly reduced acute craving, with the overall effect again being modest (g = −.286). In the context of clinical trials, naltrexone effects on craving outcomes have generally been less consistent relative to heavy drinking outcomes. A prior meta‐analysis found no differences in craving between naltrexone and placebo groups during treatment periods lasting up to three months (Srisurapanont and Jarusuraisin, 2005). In a more recent meta‐analysis, results from 26 RCTs suggested a small but significant medication effect (g = .14) on craving (Maisel et al., 2013). Relatively smaller (or non‐significant) medication effects in RCT meta‐analyses may reflect the use of retrospective craving reports that span days or weeks. On the other hand, the apparently reliable effect for craving in the current analysis is consistent with the notion that evoking craving in laboratory settings, including with in vivo exposure to alcohol or alcohol cues, might provide a more sensitive signal of medication effects (Sinha and O'Malley, 1999). This finding could also be viewed as consistent with the expectation for larger effect sizes when phenotype assessment occurs under controlled experimental conditions (Ray and Heilig, 2013).
Whereas the assumption of homogeneity in effect sizes could not be rejected in the SA analysis, the craving analysis suggested significant heterogeneity in effect sizes. However, subgroup and moderation analysis did not provide clear indication as to potential sources of this heterogeneity. Stratification analyses supported significant craving reductions both at baseline and following exposure to alcohol or alcohol cues. Although medication effects appeared relatively stronger in the context of alcohol administration relative to cue exposure, the difference in effect sizes did not reach statistical significance. Medication effects on craving also did not differ between dependent and non‐dependent samples. Overall, these results provide further support that naltrexone blunts acute craving both prior to and in response to alcohol cues, and among dependent and non‐dependent drinkers. Meta‐analytic evidence for significant medication effects on acute craving is particularly notable given the recent addition of craving as a diagnostic criterion for alcohol use disorder in DSM‐5 (Murphy et al., 2014).
Studies subsumed in this analysis clearly differ from naltrexone RCTs in several respects, as evidenced by their exclusion from prior meta‐analyses. All were conducted under artificial conditions (although some used simulated bar settings), and most were conducted without a social context. Importantly, laboratory SA paradigms have historically resulted in relatively low peak BrAC levels, which may reflect the use of drink cutoffs, the influence of experimenters, or other factors (Zimmermann et al., 2013). Although the artificial laboratory context could be viewed as limiting generalizability of these results, testing medication effects under controlled experimental conditions is an important complement to RCTs (O'Malley et al., 2002; Plebani et al., 2012). For example, the current analyses involve objective measures of alcohol consumption and in‐the‐moment craving assessments, neither of which are typical of RCTs, thus complementing results from prior meta‐analyses. Studies in this review also diverge from RCTs in the exclusive use of acute or sub‐acute medication schedules. In this respect, the present results are potentially relevant for applications of short‐term opioid antagonist therapy to reduce acute alcohol craving and consumption (Niciu and Arias, 2013). Also noteworthy is that these analyses found no evidence that number of dosage days moderated effect sizes for craving or SA. However, the moderation analyses are qualified by the relatively small number of studies and the restricted range of medication exposure.
While naltrexone RCTs are generally limited to alcohol‐dependent subjects, studies in this review included both non‐dependent and dependent drinkers. Differences in study populations allowed for stratification analyses, which suggested no significant difference in medication effect sizes across populations. This finding has clinical implications and complements prior evidence for naltrexone's efficacy in non‐dependent heavy drinkers (O'Malley et al., 2015). Also notable is that the SA studies did not involve treatment‐seeking participants, meaning that reductions in consumption presumably occurred absent current motivation to limit drinking (although some SA studies imposed a financial incentive for limiting consumption). Overall, the fact that human laboratory studies depart significantly from traditional RCTs, yet yield comparable medication effects, suggests generalizability of medication effects across settings.
Several limitations of the current study should be considered. While meta‐analyses of naltrexone RCTs have included representation from several countries (Rösner et al., 2010; Srisurapanont and Jarusuraisin, 2005), the present analysis is limited mostly to U.S. studies. As noted, a potential limitation is that the paradigms represented in this study have reduced external validity relative to treatment trials. An additional limitation was the exclusion of a minority of studies that assessed subjective responses without assessing craving. Although other domains of subjective responses—in particular, reductions in perceived stimulation (King et al., 1997; Swift et al., 1994)—are important potential treatment mediators, the fact that most studies measured craving allowed us to focus on arguably the most clinically relevant construct, while limiting concerns about mixing different subjective response domains across studies. An additional qualification is that few studies involved dosage manipulations, precluding examination of dose‐related effects. Also, while some studies reported larger medication effects as a function of specific experimental conditions or population subgroups—potentially pointing to predictors of treatment response—these moderators were not examined frequently enough to allow for investigation at the meta‐analytic level. Finally, the present analyses did not include unpublished studies; however, estimates of publication bias suggested a low likelihood that inclusion of unpublished null findings would render the overall effects non‐significant.
Human laboratory paradigms are proposed as a critical bridge between preclinical studies and large‐scale medication trials (McKee, 2009; Perkins and Lerman, 2011; Ray et al., 2010), and are expected to play an important role in treatment development (Litten et al., 2015). The advantages of laboratory paradigms include the ability to quantify objective measures of behavior, examine genetic moderators under controlled conditions and isolate theoretical mediators of pharmacotherapy effects (Perkins and Lerman, 2011; Plebani et al., 2012; Ray et al., 2010). In principle, lower heterogeneity in effect sizes can be expected in the context of controlled laboratory trials; of note, measures of heterogeneity in this analysis appeared relatively lower than those reported in other meta‐analyses (Maisel et al., 2013; Rösner et al., 2010). The additional possibility of larger effect sizes under experimental conditions is an additional argument for laboratory‐based medication screening trials, which can provide a cost‐efficient means of medication screening (McKee, 2009; Perkins and Lerman, 2011).
Notwithstanding the advantages of human laboratory paradigms for medication screening, the current study highlights methodological considerations for future work. Most notably, the current study establishes that naltrexone human laboratory trials have largely been underpowered to detect significant medication effects on par with the aggregate effect sizes observed here. Illustrating this point, the apparent robustness of medication effects when aggregated across studies is in contrast to the conclusions drawn from individual studies. Among study‐level effect sizes generated for these analyses, 8 of 9 SA effects suggested relatively lower consumption in naltrexone versus placebo conditions, with only 3 of these effects being statistically significant. Similarly, 14 of 16 craving effects indicated relatively lower craving in naltrexone versus placebo conditions, with only 5 of these effects being statistically significant. Notably, only one study clearly reported that a priori power analyses had been conducted. Given that a primary rationale for human laboratory trials is to detect an initial medication signal before proceeding to larger RCTs, mitigating Type II error is important to avoid premature exclusion of candidate medications. The present results imply that future human laboratory studies should be powered to detect small‐to‐medium medication effects—which may nonetheless translate to clinically relevant outcomes at the population level (Roerecke et al., 2015). These considerations argue for larger sample sizes in human laboratory medication trials. Alternatively, effect sizes could be prioritized over traditional significance testing when determining the threshold for an initial medication signal.
An additional consideration is that studies in this review did not utilize standardized reporting conventions to facilitate systematic review. Use of standardized reporting schemes in future human laboratory studies (e.g. http://handbook.cochrane.org/) would facilitate transparent reporting and estimation of potential biases. Overall, further application of human laboratory methods in medication screening trials will ultimately facilitate additional meta‐analytic studies to quantify medication effects, allowing clearer inferences about the significance and magnitude of pharmacotherapy effects on intermediate therapeutic targets.
Funding and Disclosures
The authors acknowledge support from the Canadian Institutes of Health Research (MSH‐130189, MFE‐140817), the Canada Research Chairs Program, and the Ontario Mental Health Foundation. JR has received grants, personal fees and other (membership Nalmefene medical board until 2014) from Lundbeck, outside the submitted work. The other authors declare no conflicts of interest.
Authors Contribution
CH developed the study, conducted the literature searches, organized data coding and data abstraction, conducted primary analyses and drafted the manuscript. CH acknowledges access to the data. JW conducted study coding, effect size abstraction and calculation, and statistical analyses. AS assisted with identifying relevant studies and coding studies on methodological features. JR provided statistical and conceptual expertise and a critical review of the manuscript. All authors reviewed the final manuscript.
Supporting information
Supporting info item
Hendershot, C. S. , Wardell, J. D. , Samokhvalov, A. V. , and Rehm, J. (2017) Effects of naltrexone on alcohol self‐administration and craving: meta‐analysis of human laboratory studies. Addiction Biology, 22: 1515–1527. doi: 10.1111/adb.12425.
References
*Denotes studies included in meta‐analysis
- * Anton RF, Drobes DJ, Voronin K, Durazo‐Avizu R, Moak D (2004) Naltrexone effects on alcohol consumption in a clinical laboratory paradigm: temporal effects of drinking. Psychopharmacology (Berl) 173:32–40. [DOI] [PubMed] [Google Scholar]
- * Anton RF, Voronin KK, Randall PK, Myrick H, Tiffany A (2012) Naltrexone modification of drinking effects in a subacute treatment and bar‐lab paradigm: influence of OPRM1 and dopamine transporter (SLC6A3) genes. Alcohol Clin Exp Res 36:2000–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin D, Grant ER, Pohorecky LA (1993) Naltrexone reverses ethanol‐induced dopamine release in the nucleus accumbens in awake, freely moving rats. Brain Res 621:137–140. [DOI] [PubMed] [Google Scholar]
- Borenstein M, Hedges L, Higgins J, Rothstein H (2005) Comprehensive Meta‐Analysis, Version 2. Biostat: Englewood, NJ. [Google Scholar]
- Borenstein M, Hedges LV, Higgins JPT, Rothstein HR (2009) Introduction to Meta‐Analysis. Chichester, UK: John Wiley & Sons. [Google Scholar]
- * Davidson D, Palfai T, Bird C, Swift R (1999) Effects of naltrexone on alcohol self‐administration in heavy drinkers. Alcohol Clin Exp Res 23:195–203. [PubMed] [Google Scholar]
- * Davidson D, Swift R, Fitz E (1996) Naltrexone increases the latency to drink alcohol in social drinkers. Alcohol Clin Exp Res 20:732–739. [DOI] [PubMed] [Google Scholar]
- * de Wit H, Svenson J, York A (1999) Non‐specific effect of naltrexone on ethanol consumption in social drinkers. Psychopharmacology (Berl) 146:33–41. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Acquas E, Tanda G (1996) Ethanol as a neurochemical surrogate of conventional reinforcers: the dopamine‐opioid link. Alcohol 13:13–17. [DOI] [PubMed] [Google Scholar]
- * Doty P, Kirk JM, Cramblett MJ, de Wit H (1997) Behavioral responses to ethanol in light and moderate social drinkers following naltrexone pretreatment. Drug Alcohol Depend 47:109–116. [DOI] [PubMed] [Google Scholar]
- Downs SH, Black N (1998) The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non‐randomised studies of health care interventions. J Epidemiol Community Health 1998;52:377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * Drobes DJ, Anton RF, Thomas SE (2003) A clinical laboratory paradigm for evaluating medication effects on alcohol consumption: naltrexone and nalmefene. Neuropsychopharmacology 28:755–764. [DOI] [PubMed] [Google Scholar]
- * Drobes DJ, Anton RF, Thomas SE, Voronin K (2004) Effects of naltrexone and nalmefene on subjective response to alcohol among non‐treatment seeking alcoholics and social drinkers. Alcohol Clin Exp Res 28:1362–1370. [DOI] [PubMed] [Google Scholar]
- Duval S, Tweedie R (2000) Trim and fill: a simple funnel‐plot‐based method of testing and adjusting for publication bias in meta‐analysis. Biometrics 56:455–463. [DOI] [PubMed] [Google Scholar]
- Gonzales RA, Weiss F (1998) Suppression of ethanol‐reinforced behavior by naltrexone is associated with attenuation of the ethanol‐induced increase in dialysate dopamine levels in the nucleus accumbens. J Neurosci 18:10663–10671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M (2015) The Thirteenth Step: Addiction in the Age of Brain Science. Columbia University Press: New York. [Google Scholar]
- Heilig M, Goldman D, Berrettini W, O'Brien CP (2011) Pharmacogenetic approaches to the treatment of alcohol addiction. Nat Rev Neurosci 12:670–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas DE, Amick HR, Feltner C, Bobashev G, Thomas K, Wines R, Kim MM, Shanahan ES, Gass CE, Rowe CJ, Garbutt JC (2014) Pharmacotherapy for adults with alcohol use disorders in outpatient settings: a systematic review and meta‐analysis. JAMA 311:1889–1900. [DOI] [PubMed] [Google Scholar]
- King AC, Roche DJ, Rueger SY (2011) Subjective responses to alcohol: A paradigm shift may be brewing. Alcohol Clin Exp Res 35:1726–1728. [DOI] [PubMed] [Google Scholar]
- King AC, Volpicelli JR, Frazer A, O'Brien CP (1997) Effect of naltrexone on subjective alcohol response in subjects at high and low risk for future alcohol dependence. Psychopharmacology (Berl) 129:15–22. [DOI] [PubMed] [Google Scholar]
- * Krishnan‐Sarin S, Krystal JH, Shi J, Pittman B, O'Malley SS (2007) Family history of alcoholism influences naltrexone‐induced reduction in alcohol drinking. Biol Psychiatry 62:694–697. [DOI] [PubMed] [Google Scholar]
- * Kruse MI, Radnovich AJ, Kalapatapu RK, Mehdiyoun N, Chambers RA, Davidson D (2012) Effects of alcohol availability, access to alcohol, and naltrexone on self‐reported craving and patterns of drinking in response to an alcohol‐cue availability procedure. J Stud Alcohol Drugs 73:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litten RZ, Ryan ML, Falk DE, Reilly M, Fertig JB, Koob GF (2015) Heterogeneity of alcohol use disorder: understanding mechanisms to advance personalized treatment. Alcohol Clin Exp Res 39:579–584. [DOI] [PubMed] [Google Scholar]
- Maisel NC, Blodgett JC, Wilbourne PL, Humphreys K, Finney JW (2013) Meta‐analysis of naltrexone and acamprosate for treating alcohol use disorders: when are these medications most helpful? Addiction 108:275–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * McGeary JE, Monti PM, Rohsenow DJ, Tidey J, Swift R, Miranda RJ (2006) Genetic moderators of naltrexone's effects on alcohol cue reactivity. Alcohol Clin Exp Res 30:1288–1296. [DOI] [PubMed] [Google Scholar]
- McKee SA (2009) Developing human laboratory models of smoking lapse behavior for medication screening. Addict Biol 14:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * Miranda R, Ray L, Blanchard A, Reynolds EK, Monti PM, Chun T, Justus A, Swift RM, Tidey J, Gwaltney CJ, Ramirez J (2014) Effects of naltrexone on adolescent alcohol cue reactivity and sensitivity: an initial randomized trial. Addict Biol 19:941–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group (2009) Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Med 151:264–269 [PMC free article] [PubMed] [Google Scholar]
- * Monti PM, Rohsenow DJ, Hutchison KE, Swift RM, Mueller TI, Colby SM, Brown RA, Gulliver SB, Gordon A, Abrams DB (1999) Naltrexone's effect on cue‐elicited craving among alcoholics in treatment. Alcohol Clin Exp Res 23:1386–1394. [PubMed] [Google Scholar]
- Murphy CM, Stojek MK, Few LR, Rothbaum AO, Mackillop J (2014) Craving as an alcohol use disorder symptom in DSM‐5: an empirical examination in a treatment‐seeking sample. Exp Clin Psychopharmacol 22:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * Na C, Lee Y‐S (2002) Alcohol urge and plasma beta‐endorphin change after alcohol challenge with naltrexone pretreatment in social drinkers. Prog Neuropsychopharmacol Biol Psychiatry 26:663–670. [DOI] [PubMed] [Google Scholar]
- Niciu MJ, Arias AJ (2013) Targeted opioid receptor antagonists in the treatment of alcohol use disorders. CNS Drugs 27:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley SS, Corbin WR, Leeman RF, DeMartini KS, Fucito LM, Ikomi J, Romano DM, Wu R, Toll BA, Sher KJ, Gueorguieva R, Kranzler HR (2015) Reduction of alcohol drinking in young adults by naltrexone: a double‐blind, placebo‐controlled, randomized clinical trial of efficacy and safety. J Clin Psychiatry 76:e207–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B (1992) Naltrexone and coping skills therapy for alcohol dependence. A controlled study. Arch Gen Psychiatry 49:881–887. [DOI] [PubMed] [Google Scholar]
- * O'Malley SS, Krishnan‐Sarin S, Farren C, Sinha R, Kreek MJ (2002) Naltrexone decreases craving and alcohol self‐administration in alcohol‐dependent subjects and activates the hypothalamo‐pituitary‐adrenocortical axis. Psychopharmacology (Berl) 160:19–29. [DOI] [PubMed] [Google Scholar]
- * Ooteman W, Koeter MWJ, Verheul R, Schippers GM, van den Brink W (2007) The effect of naltrexone and acamprosate on cue‐induced craving, autonomic nervous system and neuroendocrine reactions to alcohol‐related cues in alcoholics. Eur Neuropsychopharmacol 17:558–566. [DOI] [PubMed] [Google Scholar]
- * Palfai T, Davidson D, Swift R (1999) Influence of naltrexone on cue‐elicited craving among hazardous drinkers: the moderational role of positive outcome expectancies. Exp Clin Psychopharmacol 7:266–273. [DOI] [PubMed] [Google Scholar]
- Perkins KA, Lerman C (2011) Early human screening of medications to treat drug addiction: novel paradigms and the relevance of pharmacogenetics. Clin Pharmacol Ther 89:460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettinati HM, O'Brien CP, Rabinowitz AR, Wortman SP, Oslin DW, Kampman KM, Dackis CA (2006) The status of naltrexone in the treatment of alcohol dependence: specific effects on heavy drinking. J Clin Psychopharmacol 26:610–625. [DOI] [PubMed] [Google Scholar]
- Plebani JG, Ray LA, Morean ME, Corbin WR, MacKillop J, Amlung M, King AC (2012) Human laboratory paradigms in alcohol research. Alcohol Clin Exp Res 36:972–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn PD, Fromme K (2011) Subjective response to alcohol challenge: a quantitative review. Alcohol Clin Exp Res 35:1759–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * Ray LA, Bujarski S, Chin PF, Miotto K (2012) Pharmacogenetics of naltrexone in Asian Americans: a randomized placebo‐controlled laboratory study. Neuropsychopharmacol 37:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, Heilig MA (2013) Subjective responses to alcohol as an endophenotype: implications for alcoholism etiology and treatment development In: MacKillop J, Munafo M. eds. Genetic Influences on Addiction, pp 97–120. MIT Press: Cambridge, MA. [Google Scholar]
- * Ray LA, Hutchison KE (2007) Effects of naltrexone on alcohol sensitivity and genetic moderators of medication response: a double‐blind placebo‐controlled study. Arch Gen Psychiatry 64:1069–1077. [DOI] [PubMed] [Google Scholar]
- Ray LA, Hutchison KE, Tartter M (2010) Application of human laboratory models to pharmacotherapy development for alcohol dependence. Curr Pharm Des 16:2149–2158. [DOI] [PubMed] [Google Scholar]
- Roerecke M, Sørensen P, Laramée P, Rahhali N, Rehm J (2015) Clinical relevance of nalmefene versus placebo in alcohol treatment: reduction in mortality risk. J Psychopharmacol 29:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- * Rohsenow DJ, Monti PM, Hutchison KE, Swift RM, Colby SM, Kaplan GB (2000) Naltrexone's effects on reactivity to alcohol cues among alcoholic men. J Abnorm Psychol 109:738–742. [PubMed] [Google Scholar]
- Rosenthal R (1979) The file drawer problem and tolerance for null results. Psychol Bull 86:638–641. [Google Scholar]
- Rösner S, Hackl‐Herrwerth A, Leucht S, Vecchi S, Srisurapanont M, Soyka M (2010) Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev CD001867 Doi: 10.1002/14651858.CD001867.pub2. [DOI] [PubMed]
- Sinclair JD (2001) Evidence about the use of naltrexone and for different ways of using it in the treatment of alcoholism. Alcohol Alcohol 36:2–10. [DOI] [PubMed] [Google Scholar]
- Sinha R, O'Malley SS (1999) Craving for alcohol: findings from the clinic and the laboratory. Alcohol Alcohol 34:223–230. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Herz A, Shippenberg TS (1992) Opposing tonically active endogenous opioid systems modulate the mesolimbic dopaminergic pathway. Proc Natl Acad Sci U S A 89:2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srisurapanont M, Jarusuraisin N (2005) Naltrexone for the treatment of alcoholism: a meta‐analysis of randomized controlled trials. Int J Neuropsychopharm 8:267–280. [DOI] [PubMed] [Google Scholar]
- Swift RM, Whelihan W, Kuznetsov O, Buongiorno G, Hsuing H (1994) Naltrexone‐induced alterations in human ethanol intoxication. Am J Psychiatry 151:1463–1467. [DOI] [PubMed] [Google Scholar]
- Volpicelli JR, Alterman AI, Hayashida M, O'Brien CP (1992) Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry 49:876–880. [DOI] [PubMed] [Google Scholar]
- Volpicelli JR, Watson NT, King AC, Sherman CE, O'Brien CP (1995) Effect of naltrexone on alcohol “high” in alcoholics. Am J Psychiatry 152:613–615. [DOI] [PubMed] [Google Scholar]
- Zimmermann US, O'Connor S, Ramchandani VA (2013) Modeling alcohol self‐administration in the human laboratory. Curr Top Behav Neurosci 13:315–353. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item