

Abstract
Parkinson’s disease is associated with altered neural activity in the motor cortex. Chronic high-frequency deep brain stimulation (DBS) of the subthalamic nucleus (STN) is effective in suppressing parkinsonian motor symptoms and modulates cortical activity. However, the anatomical pathways responsible for STN DBS-mediated cortical modulation remain unclear. Cortical evoked potentials (cEP) generated by STN DBS reflect the response of cortex to subcortical stimulation, and the goal of this study was to determine the neural origin of STN DBS-generated cEP using a two-step approach. First, we recorded cEP over ipsilateral primary motor cortex during different frequencies of STN DBS in awake healthy and unilateral 6-OHDA-lesioned parkinsonian rats. Second, we used a detailed, biophysically based model of the thalamocortical network to deconstruct the neural origin of the recorded cEP. The in vivo cEP included short (R1)-, intermediate (R2)-, and long-latency (R3) responses. Model-based cortical responses to simulated STN DBS matched remarkably well the in vivo responses. The short-latency response was generated by antidromic activation of layer 5 pyramidal neurons, whereas recurrent activation of layer 5 pyramidal neurons via excitatory axon collaterals reproduced the intermediate-latency response. The long-latency response was generated by polysynaptic activation of layer 2/3 pyramidal neurons via the cortico-thalamic-cortical pathway. Antidromic activation of the hyperdirect pathway and subsequent intracortical and cortico-thalamo-cortical synaptic interactions were sufficient to generate cortical potential evoked by STN DBS, and orthodromic activation through basal ganglia-thalamus-cortex pathways was not required. These results demonstrate the utility of cEP to determine the neural elements activated by STN DBS that might modulate cortical activity and contribute to the suppression of parkinsonian symptoms.
NEW & NOTEWORTHY Subthalamic nucleus (STN) deep brain stimulation (DBS) is increasingly used to treat Parkinson’s disease (PD). Cortical potentials evoked by STN DBS in patients with PD exhibit consistent short-latency (1–3 ms), intermediate-latency (5–15 ms), and long-latency (18–25 ms) responses. The short-latency response occurs as a result of antidromic activation of the hyperdirect pathway comprising corticosubthalamic axons. However, the neural origins of intermediate- and long-latency responses remain elusive, and the dominant view is that these are produced through the orthodromic pathway (basal ganglia-thalamus-cortex). By combining in vivo electrophysiology with computational modeling, we demonstrate that antidromic activation of the cortico-thalamic-cortical pathway is sufficient to generate the intermediate- and long-latency cortical responses to STN DBS.
Keywords: antidromic activation, cortical evoked potentials, deep brain stimulation, hyperdirect pathway, motor cortex, Parkinson’s disease, subthalamic nucleus, thalamocortical model
INTRODUCTION
Parkinson’s disease (PD) is associated with changes in neural activity, including altered firing rates, increased burst discharges, and enhanced beta oscillatory activity/phase-amplitude coupling across the cortex-basal ganglia-thalamus network (Bergman et al. 1994; de Hemptinne et al. 2013; Filion and Tremblay 1991; Goldberg et al. 2002; Levy et al. 2002; Whitmer et al. 2012). High-frequency deep brain stimulation (DBS) of the subthalamic nucleus (STN) is effective in suppressing motor symptoms of PD and also modulates cortical activity (de Hemptinne et al. 2015; Johnson et al. 2015; Li et al. 2007, 2012). However, the anatomical pathways responsible for cortical modulation by STN DBS remain unclear and could include antidromic activation of corticofugal fibers (hyperdirect pathway) and/or orthodromic activation through basal ganglia-thalamus-cortex pathways (Li et al. 2014). Establishing the anatomical pathways responsible for cortical modulation during STN DBS provides a foundation for better understanding of therapeutic mechanisms and thereby optimizing DBS electrode placement and parameter selection.
Cortical evoked potentials (cEP) generated by STN DBS reflect the response of cortex to subcortical stimulation (Baker et al. 2002). Low-frequency (≤20 Hz) STN DBS evokes a short-latency (R1), an intermediate-latency (R2), and a long-latency (R3) response in the cortex of persons with PD (Walker et al. 2012). Current hypotheses suggest that the R1 response is due to antidromic activation of the hyperdirect pathway, whereas the R3 response results from orthodromic propagation through basal ganglia-thalamus-cortex pathways (Alhourani et al. 2015; Devergnas and Wichmann 2011; Udupa and Chen 2015). Furthermore, recent experimental studies in rodents suggest that selective activation of layer 5 (L5) neurons in the primary motor cortex (M1) is sufficient to ameliorate PD motor symptoms (Gradinaru et al. 2009; Sanders and Jaeger 2016). Thus determining the neural origin of cEPs will illuminate the pathways responsible for cortical modulation during STN DBS.
The objective of this study was to determine the neural origin of cEPs generated by STN DBS by using a two-step approach. First, we recorded cEPs over ipsilateral motor cortex during STN DBS at different frequencies in awake healthy and parkinsonian rats. Second, we implemented an anatomically based, lamina-specific biophysical model of the cortical column (Traub et al. 2005) and simulated DBS in the model to identify the neural origin of the recorded cEP. The results demonstrate that antidromic activation of L5 pyramidal neurons and subsequent intracortical and cortico-thalamo-cortical synaptic interactions were sufficient to generate cortical potentials evoked by STN DBS, and orthodromic propagation through basal ganglia-thalamus-cortex pathway was not required.
METHODS
Experiments were conducted in 15 adult female rats including both Long-Evans (n = 10) and Sprague-Dawley (n = 5) strains. Of the 15 rats, 10 rats (11 hemispheres) had at least one electrode tip reaching STN, and only data from those 10 rats were included in the analyses. These rats were part of a cohort also used in other experiments to investigate the effects of STN DBS to generate or reduce motor deficits. All surgical and experimental procedures were approved by the Duke University Institutional Animal Care and Use Committee.
Surgical procedure.
Rats were implanted unilaterally with stimulating microelectrodes in the STN and bilaterally with stainless steel screws over the motor cortex, except in one rat that had bilateral STN stimulating microelectrodes. Surgical procedures are described in detail elsewhere (McConnell et al. 2012). Briefly, stereotactic surgery was performed under 2–4% isoflurane or 3–7% sevoflurane anesthesia using aseptic technique. A low-impedance (10 kΩ) platinum-iridium four-electrode stimulation array (2 × 2; MicroProbes, Gaithersburg, MD) with interelectrode spacing of 300 µm and wire diameter of 75 µm was implanted into the STN using stereotactic coordinates [anterior (A), −3.6 mm; lateral (L), 2.6 mm; ventral (V), −6.8 to −7.2 mm relative to bregma; Paxinos and Watson 2006), and acute, single-channel intraoperative recordings of single- and multiunit neural activity were used to guide electrode placement. Stainless steel screws (1-mm diameter) were inserted through the skull to abut the dura over the ipsilateral and contralateral motor cortices (A, 2.5 mm; L, ±2.5 mm relative to bregma). Rats were implanted unilaterally with a cannula in the medial forebrain bundle (MFB; A, −2.0 mm; L, 2.0 mm; V, −7.0 mm). All implanted electrodes and the cannula were secured using dental acrylic attached to additional stainless steel screws anchored to the skull. Postoperatively, rats were monitored closely and recovered for at least 1 wk before any experimental testing.
6-Hydroxydopamine lesion.
To compare cEPs in healthy and parkinsonian rats, we rendered a subset of rats (n = 3) parkinsonian after completing experimental sessions in the healthy state. 6-Hydroxydopamine (6-OHDA) was infused into the MFB through the implanted cannula to induce unilateral degeneration of the dopaminergic neurons in substantia nigra pars compacta (SNc). We injected desipramine (5 mg/kg) and pargyline (50 mg/kg) 30 min before 6-OHDA lesion to protect other types of neurons from the neurotoxic effects of 6-OHDA. Before infusion, 5 mg of 6-OHDA (Sigma-Aldrich) were dissolved in 2 ml of ice-cold saline with 0.2% ascorbic acid, and 10 µl of 6-OHDA at a rate of 2 µl/min were infused through the cannula using a Hamilton syringe. Rats recovered for at least 1 wk before any experimental testing. The effectiveness of the unilateral 6-OHDA lesion was assessed using methamphetamine-induced circling. Lesioned rats with at least 90% loss of dopaminergic neurons in SNc exhibit a turning rate of ≥3 turns/min ipsilateral to the lesion following administration of methamphetamine (So et al. 2012b). One rat did not exhibit sufficient motor deficits during the circling test after the first lesion, and that rat received a second 6-OHDA injection.
Stimulation and recording setup.
Neural recordings were conducted in awake and freely moving rats inside a Faraday cage. Each trial included three epochs: 1-min prestimulation baseline, 1-min stimulation, and 1-min poststimulation baseline. Four stimulation frequencies (4.5, 9, 50, and 130 Hz) were applied in random order, and trials were repeated for each rat across multiple (2–4) days. We chose two low frequencies of 4.5 and 9 Hz with interpulse intervals (IPI; 222.2 and 111.1 ms, respectively) sufficiently long to enable recording of the long-latency component of the cEP. A frequency of 50 Hz enabled recording of only the R1 and R2 responses, because the IPI was shorter than the latency of the R3 component (~25 ms), and similarly, 130 Hz enabled recording of only the R1 component of the cEP.
Stimulation to the STN was performed with charge-balanced, symmetric biphasic pulses with a duration of 90 µs/phase. There were no thresholds defined for the cEP responses. Rather, before any experimental testing, all possible bipolar stimulating pairs were tested across a range of stimulation amplitudes (typically 10–100 µA) during 130-Hz STN stimulation while rats were in a circular chamber. The amplitude and bipolar stimulating pair that elicited sustained motor responses, including contralateral tuning, increased motor activity, and lack of side effects (e.g., involuntary muscle contractions of the limb and neck) were identified for each rat. This bipolar electrode pair and amplitude, unique for each rat, were then used for the entire study (see appendix, Table A1). Stimulation amplitudes ranged between 30 and 70 µA across rats, and each stimulation epoch included an amplitude ramp-up over 5 s. Stimulation patterns were generated using custom scripts (MATLAB) and applied through an isolated voltage-to-current converter (Analog Stimulus Isolator model 2200; A-M Systems) and a custom alternating current (AC) coupler.
cEPs from motor cortex were recorded from the cortical screw electrodes using a multichannel acquisition processor (MAP) system (Plexon, Dallas, TX). cEP were bandpass filtered (0.7 Hz–2 kHz, 2 and 4 poles, respectively) and amplified 5,000 times before digital sampling at 20 kHz.
Analysis of neural data.
Our sampling frequency of 20 kHz and low-pass cutoff of 2 kHz allowed recordings in the presence of stimulation artifact. For each rat and each stimulation frequency, we computed the stimulus pulse-triggered average of the filtered ipsilateral motor cortex response per trial, and then the responses were averaged across trials (Fig. 1). Data from the first and last 5 s of the stimulation epoch were excluded from analysis due to the ramping of stimulation amplitude.
Fig. 1.

Ipsilateral M1 electrocorticographic (ECoG) recordings in a rat at 4 different frequencies of subthalamic nucleus (STN) deep brain stimulation (DBS): 4.5, 9, 50, and 130 Hz. A1–A4: raw ECoG traces over 1 trial. Each trial consisted of 3 epochs: 1-min prestimulation, 1-min stimulation, and 1-min poststimulation. Bar indicates STN DBS “on” period. A5–A8: pulse-triggered response shown across all DBS pulses. A9–A12: pulse-triggered response averaged across all DBS pulses.
We conducted three experiments in a subset of rats to distinguish neural responses from stimulation artifact: neural recordings with stimulation polarity reversed, neural recordings during stimulation while rats were under 2–3% isoflurane or 4–4.5% sevoflurane anesthesia, and neural recordings with stimulation polarity reversed during anesthesia. The recordings under anesthesia served the purpose to differentiate response components that occurred as a result of direct excitation by the DBS pulse in contrast to those that arose as a result of synaptic mechanisms.
We quantified the peak amplitude and latency of the different components of the cEP (R1, R2, R3). All results are means ± SD. Pearson's correlation coefficient was used to quantify the similarity of amplitudes/latencies between all components of cEP responses measured before and after 6-OHDA lesion only in the three rats that were rendered parkinsonian after recordings were completed in the healthy state. The following statistical tests were undertaken to infer effects of STN DBS frequency on R1 peak amplitude/latency. First, we tested the assumption of normality of R1 amplitude and latency at each STN DBS frequency using the Shapiro-Wilk test. R1 peak amplitude and latency data at 130 Hz were not normally distributed. Therefore, statistical inferences were made on the effect of stimulation frequency on R1 peak amplitude and latency using Friedman’s one-way analysis of variance (ANOVA). When the omnibus test statistic revealed significance at P < 0.05, we performed the Dunn-Bonferroni test for post hoc paired comparisons between individual frequencies (Dunn 1964). All statistical analyses were performed using IBM SPSS Statistics software.
Histology.
At the conclusion of experimental measurements, rats were deeply anesthetized with pentobarbital sodium (100 mg/kg ip) and euthanized via intracardiac perfusion with 10% formalin. Brains were extracted and postfixed overnight at 4°C. Electrodes were removed, and the brain was placed in 30% sucrose solution for 2 days and then sectioned coronally at 50-µm thickness. Sections were stained with cytochrome oxidase to locate the electrode tracks (see appendix, Fig. A1A). We were not able to perform histology on some rats, and in those rats, the inference of STN electrode placement was based on contact testing. Rats with at least one of the bipolar pair lying within STN elicit a sustained motor response to high-frequency STN DBS (So et al. 2012b). Electrode placement in 8/10 rats was confirmed through postmortem histology and was inferred through contact testing in the remaining 2 rats. Tyrosine hydroxylase staining was used to evaluate the effectiveness of lesion of SNc dopaminergic neurons (see appendix, Fig. A1B). All three rats had >90% degeneration of tyrosine hydroxylase-positive neurons based on postmortem histology.
Computational model.
There are two potential pathways by which STN DBS can generate cortical activation (Devergnas and Wichmann 2011; Li et al. 2014; Miocinovic 2014; Walker and Guthrie 2013): 1) antidromic activation of the hyperdirect pathway (L5 pyramidal neurons) and subsequent intracortical and thalamocortical interactions and/or 2) orthodromic propagation through the STN-globus pallidus interna (GPi)-thalamus (TH) pathway. On the basis of our prior modeling experience, we did not expect that the STN-GPi-TH pathway would activate the cortex (Kumaravelu et al. 2016; So et al. 2012a). Therefore, we used a detailed biophysical model of the thalamocortical (TC) network that simulated intracortical interactions following activation of L5 pyramidal neurons by STN DBS to determine the neural origin of cEPs (Gleeson et al. 2010; Traub et al. 2005).
The TC network model included 356 model neurons, each with 50–137 compartments. The cortex and thalamus comprised 12 and 2 different populations of neurons, respectively, characterized according to their firing properties. The cortical neurons were arranged in a columnar fashion consisting of six layers (Fig. 2A). L2/3 included 100 regular spiking (RS) pyramidal neurons, 5 fast rhythmic bursting pyramidal neurons, 18 fast-spiking interneurons, and 9 low-threshold spiking interneurons; L4 included 24 RS spiny stellate cells; L5 included 80 intrinsic bursting tufted pyramidal cells and 20 RS pyramidal neurons; L6 included 50 RS nontufted pyramidal neurons, 20 fast-spiking interneurons, and 10 low-threshold spiking interneurons; and L1 mostly consisted of tufted dendrites arising from the L5 pyramidal neurons. The thalamic population consisted of 10 thalamocortical relay (TCR) cells and 10 reticular nuclei cells. Network interactions in the model were through both electrical (gap junctions) and chemical synaptic (AMPA, NMDA, and GABAA) connections (Fig. 2B). The biophysical properties of the cells and the network properties including synaptic connection strengths, type, density, delay, etc., are described in detail in the original publication (Traub et al. 2005) and were largely based on in vivo and in vitro data from rats. Model parameters were not altered in any way to fit any of the in vivo STN DBS-generated cEP responses. The population response to STN DBS was quantified in the presence of the intrinsic gamma oscillatory activity present in the original model.
Fig. 2.

Computational model design. A: schematic of the cortical column showing the different populations of cells across layers 1–6. Each color denotes a specific cell type in the column characterized on the basis of firing behavior. Depth and width of the cortical column were ~2,000 and ~400 µm, respectively. RS, regular spiking pyramidal neurons; FRB, fast rhythmic bursting pyramidal neurons; LTS, low-threshold spiking interneurons; IB, intrinsic bursting tufted pyramidal cells; TCR, thalamocortical relay cells; nRT, reticular thalamic nuclei cells. B: intracolumnar synaptic connectivity map showing both excitatory (glutamate: NMDA and AMPA) and inhibitory (GABAA) connections (Traub et al. 2005). E and I, excitatory and inhibitory neurons, respectively.
STN DBS was simulated in the model by injecting intracellular monophasic current pulses into L5 axon terminals with a magnitude of 0.5 nA and a pulse width of 0.3 ms such that each pulse produced one spike. We tested four different stimulation frequencies of 4.5, 9, 50, and 130 Hz in the model as in the experiments. Furthermore, we tested the effects of DBS amplitude on cEP by stimulating different proportions of L5 axons (20–80%) in steps of 20% at a frequency of 4.5 Hz. We chose to activate axon terminals of L5 neurons because axons in the hyperdirect pathway are believed to originate from L5 of the cortex (Mathai and Smith 2011), terminating axons have low thresholds to extracellular stimulation (Baldissera et al. 1972; Jankowska et al. 1975), and activation of the hyperdirect pathway has been implicated as a potential therapeutic mechanism of STN DBS (Gradinaru et al. 2009; Sanders and Jaeger 2016). We chose to activate directly L5 axon terminals without accounting for the very short conduction time to cortical L5. The latency of the R1 response can be divided into conduction from STN to L5 and conduction from L5 to the surface of the cortex. The model accounts for the conduction time that arises due to the second component but not the first, because we did not explicitly model the axonal projections from L5 to STN, i.e., the hyperdirect pathway. The conduction velocity of axons activated by STN DBS in rodents is ~5 m/s (Kita and Kita 2012), the distance from STN to L5 is ~5 mm, and thus the time taken for the activity to propagate from the hyperdirect collaterals to L5 would be ~1 ms. Similarly, the axons of the hyperdirect pathway in humans have rapid conduction velocities (~60 m/s; Walker et al. 2012).
Extracellular field potentials at the surface of the cortical column, intended to represent the experimentally recorded cEP, were computed from the transmembrane currents in each compartment (j) of each model cortical neuron (i), , using the equation
where ri,j is the distance of each cortical neural element from the recording electrode and σe is the isotropic and homogeneous conductivity of the extracellular medium (0.3 S/m) (Ranck 1963). We computed the stimulus pulse-triggered average of the model-generated responses to obtain simulated cEP responses similar to those measured experimentally. The simulated cEP was then smoothed using a second-order Butterworth bandpass filter with a passband of 0.5–300 Hz. We calculated poststimulus time histograms (PSTH) of spike times from neurons in specific layers with a bin width of 1 ms. To determine the neural origin of cEPs, we selectively removed synaptic connections between neurons in the same and different layers.
Simulations were implemented in NEURON 7.4 with equations solved using the backward Euler method with a time step of 0.025 ms and a total simulation time of 20 s (Hines and Carnevale 2001). The network simulation was parallelized by dividing the total number of neurons across 10 processors in a round-robin fashion and by setting up communication of spike times between processors through the Message Passing Interface (MPI) protocol (Hines and Carnevale 2008; Hines et al. 2008). The code for the network model required to replicate the model-based cEP response to STN DBS is available on ModelDB.
RESULTS
We measured the cEPs generated by STN DBS at four different frequencies (4.5, 9, 50, and 130 Hz) in awake rats and determined the neural origin of the recorded signal using a computational model of a cortical column to simulate the cEP.
Effect of STN DBS frequencies on cEP.
One of the challenges to interpreting cEP is distinguishing the short-latency R1 response from the stimulation artifact. We recorded cEP responses to STN DBS using two different stimulation polarities, which inverted the polarity of the artifact but not the polarity of the neural response. Stimulation pulse-triggered averages of the cEP with two different stimulation polarities at 4.5 Hz are shown in Fig. 3, which distinguishes the R1 response from stimulation artifacts.
Fig. 3.
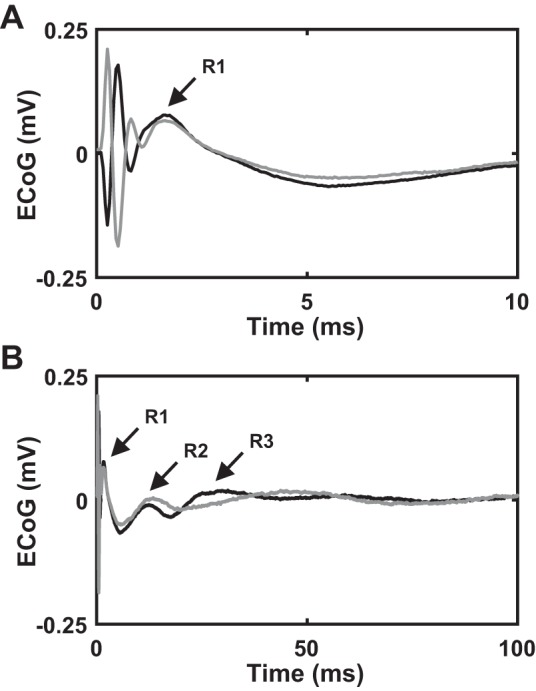
Reversing the polarity of stimulation to distinguish stimulation artifact from the short-latency evoked response. Stimulation settings for electrocorticographic (ECoG) recordings: charge-balanced, symmetric biphasic pulses with a duration of 90 µs/phase, amplitude of 40 µA, and frequency of 4.5 Hz. A and B: cortical evoked potential (cEP) responses to 2 different stimulation polarities superimposed on each other (n = 1). A is same as B, except it shows cEP data at a shorter timescale to enable visualization of the R1 response and stimulation artifact. Polarity of the short-latency (R1), intermediate-latency (R2), and long-latency (R3) components does not flip, unlike the stimulation artifacts. Gray trace represents cEP response to anodic-first biphasic subthalamic nucleus (STN) deep brain stimulation (DBS), whereas black trace represents cEP response to the polarity-flipped (cathodic first) biphasic STN DBS.
The cEP generated by STN DBS in healthy awake rats included a R1 response, a R2 response, and a R3 response. Furthermore, we chose to group rats on the basis of latency of components in the evoked response, i.e., rats exhibiting R1, R2, and R3 at identical timing were grouped together. This resulted in grouping cEP responses across six rats in one category and across four rats in the other.
In six rats, low-frequency STN DBS evoked the R1, R2, and R3 components (Fig. 4, A1 and A2), whereas 50- and 130-Hz STN DBS evoked the R1 response only due to the shorter IPIs (Fig. 4, A3 and A4). Although the 20-ms IPI at 50 Hz was longer than the latency of R2 response (~8.5 ms), the R2 response was somewhat variable at 50 Hz (Fig. 4A3). Amplitude and latency of cEP responses (R1, R2, and R3) to different STN DBS frequencies across these six rats are shown in Table 1. R1 peak amplitude and latency data at 130 Hz were not normally distributed (P < 0.05; Table 2). The amplitude of the R1 response decreased with increasing frequency (Fig. 4B1), and there were significant effects of STN DBS frequency on R1 peak amplitude [P = 0.011, Friedman’s ANOVA, χ2(3) = 11.160]. Post hoc analysis revealed a difference between 130 and 9 Hz (P = 0.009, significance value adjusted by Bonferroni correction for multiple comparisons). Although the amplitude of the R1 response to 130-Hz STN DBS decreased substantially compared with that to 4.5-Hz STN DBS, as well (Fig. 4B1), the result did not reach significance (P = 0.086). The R1 response latency increased with increasing frequency and was longest at 130 Hz (Fig. 4B4), and there were significant effects of STN DBS frequency on R1 response latency [P = 0.002, Friedman’s ANOVA, χ2(3) = 15]. Post hoc analysis revealed a difference in R1 response latency at 130-Hz compared with 4.5-Hz STN DBS (P = 0.001, significance value adjusted by Bonferroni correction for multiple comparisons). The amplitudes or latencies of R2 and R3 responses did not vary between 4.5 and 9 Hz (Fig. 4, B2, B3, B5, B6). The peak amplitude of the R1, R2 and R3 responses were remarkably stable over time with minimal waxing and waning during the 60-s stimulation epoch (Fig. 5). This effect was consistent across all six rats at all four STN DBS frequencies.
Fig. 4.

A: effect of subthalamic nucleus (STN) deep brain stimulation (DBS) frequency on the cortical evoked potential (cEP) response in healthy awake rats (n = 6). A1: 4.5 Hz. A2: 9 Hz. A3: 50 Hz. A4: 130 Hz. Stimulation parameters for electrocorticographic (ECoG) recordings: charge-balanced, symmetric biphasic pulses with a duration of 90 µs/phase and an amplitude range of 30–75 µA. STN DBS at low frequencies (4.5 and 9 Hz) evoked R1, R2, and R3 responses. High-frequency STN DBS (50 and 130 Hz) evoked an R1 response. Shading shows the range of cEP responses across rats, whereas black trace shows the mean cEP response across rats. B: quantification of cEP response features (peak amplitude and latency of peak amplitude) across STN DBS frequencies. On each box, the central mark indicates the median and bottom and top edges of box indicate the 25th and 75th percentiles, respectively. Whiskers extend to 1.5 times the interquartile range, and plus sign indicates outliers. B1, B2, and B3: peak amplitude of R1, R2, and R3 responses at different STN DBS frequencies. Absolute values were used in the case of R2 peak amplitude. Note a decrease in R1 peak amplitude at 130 Hz compared with lower frequencies. Friedman’s ANOVA identified effects of stimulation frequency on R1 peak amplitude (P = 0.011). B4, B5, and B6: latency of peak amplitude of R1, R2, and R3 responses at different STN DBS frequencies. Again, note an increase in the latency of peak amplitude of R1 response at higher compared with lower frequencies. Friedman’s ANOVA identified effects of stimulation frequency on the latency of R1 response (P = 0.002). *P < 0.05, significant difference between DBS frequencies (Dunn-Bonferroni method).
Table 1.
Peak amplitude and latency of peak amplitude of in vivo cEP response components R1, R2, and R3 in subset of 6 rats
| Peak Amplitude, mV |
Latency, ms |
||||
|---|---|---|---|---|---|
| STN DBS Frequency, Hz | Mean ± SD | Median | Mean ± SD | Median | |
| R1 | 4.5 | 0.12 ± 0.06 | 0.11 | 1.35 ± 0.07 | 1.34 |
| 9 | 0.12 ± 0.06 | 0.11 | 1.38 ± 0.06 | 1.35 | |
| 50 | 0.10 ± 0.04 | 0.11 | 1.65 ± 0.15 | 1.64 | |
| 130 | 0.03 ± 0.02 | 0.03 | 2.44 ± 0.46 | 2.73 | |
| R2 | 4.5 | 0.04 ± 0.02 | 0.05 | 8.33 ± 0.73 | 8.53 |
| 9 | 0.03 ± 0.01 | 0.04 | 8.63 ± 0.93 | 8.92 | |
| R3 | 4.5 | 0.04 ± 0.02 | 0.03 | 27.77 ± 2.42 | 28.03 |
| 9 | 0.04 ± 0.02 | 0.03 | 28.87 ± 1.67 | 29.17 | |
cEP, cortical evoked potential; STN, subthalamic nucleus; DBS, deep brain stimulation.
Table 2.
Assumption of normality of R1 peak amplitude and latency of peak amplitude at each STN DBS frequency
| STN DBS Frequency, Hz | Peak Amplitude, mV | Latency, ms | |
|---|---|---|---|
| R1 | 4.5 | W = 0.883 P = 0.281 | W = 0.958 P = 0.807 |
| 9 | W = 0.880 P = 0.271 | W = 0.832 P = 0.111 | |
| 50 | W = 0.914 P = 0.462 | W = 0.923 P = 0.531 | |
| 130 | W = 0.683 P = 0.006 | W = 0.744 P = 0.026 |
Data are P value and Shapiro-Wilk test statistic (W) at each subthalamic nucleus (STN) deep brain stimulation (DBS) frequency in rats (n = 6). Population is normally distributed if P > 0.05 and W > 0.8.
Fig. 5.

Pulse-triggered cortical evoked potentials (cEPs) in 6 healthy, awake rats during a 60-s epoch of 4.5-Hz subthalamic nucleus (STN) deep brain stimulation (DBS). A–F: waxing and waning of different components (R1, R2, R3) of the cEP during the 60-s stimulation period. Each panel shows data from 1 of the 6 rats. A1–F1 plot the pulse-triggered average response, A2–F2 plot the pulse-triggered response, and A3–F3 plot the peak amplitudes of R1 (blue trace), R2 (black trace), and R3 (red trace) responses. Color bars in A2–F2 represent the magnitude of the pulse-triggered cEP response. Note the remarkable stability of all 3 cEP response components (R1, R2, and R3) across all 6 rats. ECoG, electrocorticographic recordings.
We explored the possibility of reconstructing the cEP response to high-frequency (130 Hz) stimulation by vector addition of time-shifted cEP responses to low-frequency (4.5 Hz) STN DBS (Fig. 6, A and B). The cEP response to 130-Hz STN DBS generated by this technique resembled closely the original 130-Hz response recorded in vivo (Pearson’s correlation coefficient R = 0.9636, P < 0.001, n = 1 rat; Fig. 6C). The reconstructed cEP also accounted for the DBS frequency-dependent effects on R1, and the peak amplitude of the synthesized response was close to the peak amplitude of the original 130-Hz R1 response (Fig. 6D). This implies that the R2 and R3 responses to 130-Hz STN DBS are similar to those of low-frequency STN DBS, although the short IPI precluded visualization of these response components, and suggests that the reduced magnitude of the R1 response during high-frequency STN DBS might be due to the interaction of the R1 response with the R2 and R3 components. Another factor that can contribute to the reduced R1 peak amplitude during high-frequency STN DBS is propagation failure of antidromically spikes during high-frequency stimulation. Antidromic spike propagation in axons of the hyperdirect pathway was faithful only for low stimulation frequencies (Li et al. 2012).
Fig. 6.

Generation of cortical evoked potential (cEP) response to high-frequency (130 Hz) stimulation from the response to low-frequency (4.5 Hz) subthalamic nucleus (STN) deep brain stimulation (DBS). A: series of time-shifted pulse-triggered average response to 4.5-Hz stimulation with a delay period corresponding to the interpulse interval (IPI) of 130 Hz (i.e., 7.7 ms). B: vector addition of the time-shifted cEP response shown in A. C: pulse-triggered average response obtained from B (black trace) superimposed on cEP response to 130-Hz STN DBS recorded in vivo (gray trace). D: comparison of R1 peak amplitude between original cEP response to 130-Hz STN DBS recorded in vivo and the cEP response reconstructed from 4.5-Hz response using the technique described above (n = 5). On each box, the central mark indicates the median and bottom and top edges of box indicate the 25th and 75th percentiles, respectively. Whiskers extend to 1.5 times the interquartile range, and plus sign indicates outliers. Note the R1 peak amplitudes were not substantially different between the original and the reconstructed responses. This implies the decrease in the magnitude of R1 response during high-frequency stimulation might be due to the interaction of the R1 component with the R2 and R3 responses.
The major features of the cEP in healthy awake rats matched well the cEP recorded over the sensory-motor area (C4) of the ipsilateral cortex in humans with PD in response to STN DBS (Walker et al. 2012) (Fig. 7). The human cEP included R1, R2, and R3 components similar to those observed in rats (Fig. 7), although the R2 and R3 components occurred at somewhat shorter latencies in humans (R2, 5.7 ± 1.1 ms; R3, 22.2 ± 1.8 ms) than in rats (R2, 8.33 ± 0.73 ms; R3, 27.77 ± 2.42 ms). The amplitude of responses cannot be compared between the two studies because the responses in human were recorded from the scalp (EEG), while our recordings in the rat were from the surface of the dura (electrocorticography, ECoG). Furthermore, the effects of STN DBS frequency on R1 peak amplitude observed in rats mirrored those reported for humans (Walker et al. 2012).
Fig. 7.

Comparison of subthalamic nucleus (STN) deep brain stimulation (DBS)-induced cortical evoked potential (cEP) response between species: rat (A) and human (B). A: cEP response to 4.5-Hz STN DBS in healthy, awake rats. B: cEP response over the sensory-motor region of the cortex (C4) during 20-Hz STN DBS in a patient with Parkinson’s disease (recordings are from a study by Walker et al. 2012). Note the cEP response from both rats and human comprised R1, R2, and R3 components, although the R2 and R3 responses occurred at somewhat shorter latencies in human (R2: 5.7 ± 1.1 ms; R3: 22.2 ± 1.8 ms) than in rats (R2: 8.33 ± 0.73 ms; R3: 27.77 ± 2.42 ms). Furthermore, the cEP in rats was averaged across 6 rats, whereas the human cEP is from a single patient. cEP responses from each of these 6 rats are shown in Fig. 5.
In a subset of healthy awake rats (n = 4 rats, 5 hemispheres), the latencies of the R2 component of the cEP evoked by STN DBS differed somewhat from the response described above for the 6 rats, although postmortem histology or contact testing confirmed electrode positioning within the STN in all 10 animals. In these four rats, low-frequency STN DBS evoked the R1, R2, and R3 components, and 50- and 130-Hz STN DBS evoked the R1 and R2 and the R1 responses, respectively (Fig. 8). However, the R2 phase of the cEP in this subset occurred at a longer latency (13.56 ± 0.85 ms) compared with the R2 phase of the cEPs exhibited by the other subset of six rats (8.33 ± 0.73 ms) (Figs. 8B5 and 4B5). The amplitude and latency of cEP responses (R1, R2, and R3) to different STN DBS frequencies evoked in this subset of rats are shown in Table 3. The amplitude of the R1 response decreased with increasing frequency in this subset, as well (Fig. 8B1), and there were significant effects of STN DBS frequency on R1 peak amplitude [P = 0.026, Friedman’s ANOVA, χ2(3) = 9.3]. Post hoc analysis revealed a difference between 130 and 9 Hz (P = 0.037, significance adjusted by Bonferroni correction for multiple comparisons). Similarly, the latency of the R1 response increased with increasing frequency (Fig. 8B4), although the results did not reach significance [P = 0.50, Friedman’s ANOVA, χ2(3) = 7.8].
Fig. 8.

A: effect of subthalamic nucleus (STN) deep brain stimulation (DBS) frequency on the heterogeneous cortical evoked potential (cEP) response found in a subset of healthy, awake rats (n = 4 rats, 5 hemispheres). Stimulation parameters for electrocorticographic (ECoG) recordings: charge-balanced, symmetric biphasic pulses with a duration of 90 µs/phase and an amplitude range of 50–70 µA. A1: 4.5 Hz. A2: 9 Hz. A3: 50 Hz. A4: 130 Hz. STN DBS at low frequencies evoked R1, R2, and R3 responses. STN DBS at 50 Hz evoked R1 and R2 responses, whereas STN DBS at 130 Hz evoked only an R1 response. Shading shows the range of cEP responses across several rats, whereas black trace shows the mean cEP response across rats. This cEP differed from the response shown in Fig. 4, although postmortem histology or contact testing confirmed the stimulating electrode to be within STN. B: quantification of cEP features (peak amplitude and latency of peak amplitude) across STN DBS frequencies. On each box, the central mark indicates the median and bottom and top edges of box indicate the 25th and 75th percentiles, respectively. Whiskers extend to 1.5 times the interquartile range. B1, B2, and B3: peak amplitude of R1, R2, and R3 responses at different STN DBS frequencies. Note a decrease in R1 peak amplitude at 130 Hz compared with lower frequencies. Friedman’s ANOVA identified effects of stimulation frequency on R1 peak amplitude (P = 0.026). B4, B5, and B6: latency of peak amplitude of R1, R2, and R3 responses at different STN DBS frequencies. Note an increase in the latency of peak amplitude of R1 response at higher compared with lower frequencies, although the results were not statistically significant (P = 0.050, Friedman’s ANOVA). *P < 0.05, significant difference between DBS frequencies (Dunn-Bonferroni method).
Table 3.
Peak amplitude and latency of peak amplitude of in vivo cEP response components R1, R2 and R3 in subset of 4 rats
| Peak Amplitude, mV |
Latency, ms |
||||
|---|---|---|---|---|---|
| STN DBS Frequency, Hz | Mean ± SD | Median | Mean ± SD | Median | |
| R1 | 4.5 | 0.09 ± 0.07 | 0.10 | 1.41 ± 0.14 | 1.36 |
| 9 | 0.09 ± 0.07 | 0.07 | 1.46 ± 0.14 | 1.38 | |
| 50 | 0.05 ± 0.04 | 0.05 | 1.72 ± 0.25 | 1.56 | |
| 130 | 0.03 ± 0.003 | 0.03 | 2.35 ± 0.47 | 2.35 | |
| R2 | 4.5 | 0.01 ± 0.01 | 0.01 | 13.56 ± 0.85 | 13.53 |
| 9 | 0.01 ± 0.01 | 0.01 | 13.07 ± 0.99 | 12.79 | |
| 50 | 0.02 ± 0.01 | 0.02 | 15.04 ± 1.24 | 15.32 | |
| R3 | 4.5 | 0.02 ± 0.01 | 0.02 | 25.34 ± 0.94 | 24.88 |
| 9 | 0.02 ± 0.01 | 0.01 | 24.62 ± 1.18 | 24.91 | |
cEP, cortical evoked potential; STN, subthalamic nucleus; DBS, deep brain stimulation.
Effect of 6-OHDA lesion on cEP.
We lesioned the SNc in a subset of three rats using 6-OHDA and performed recordings during 9-Hz STN DBS to determine the effects of dopaminergic degeneration on the ipsilateral cEP. Figure 9 shows that the STN DBS-induced cEPs were quite similar before and after 6-OHDA lesion (Pearson’s correlation coefficients for each rat: R = 0.9061, Fig. 9A; R = 0.9092, Fig. 9B; R = 0.7031, Fig. 9C; P < 0.001). This suggests that the neural elements and anatomical pathways responsible for the cEP responses were largely independent of SNc dopaminergic modulation.
Fig. 9.
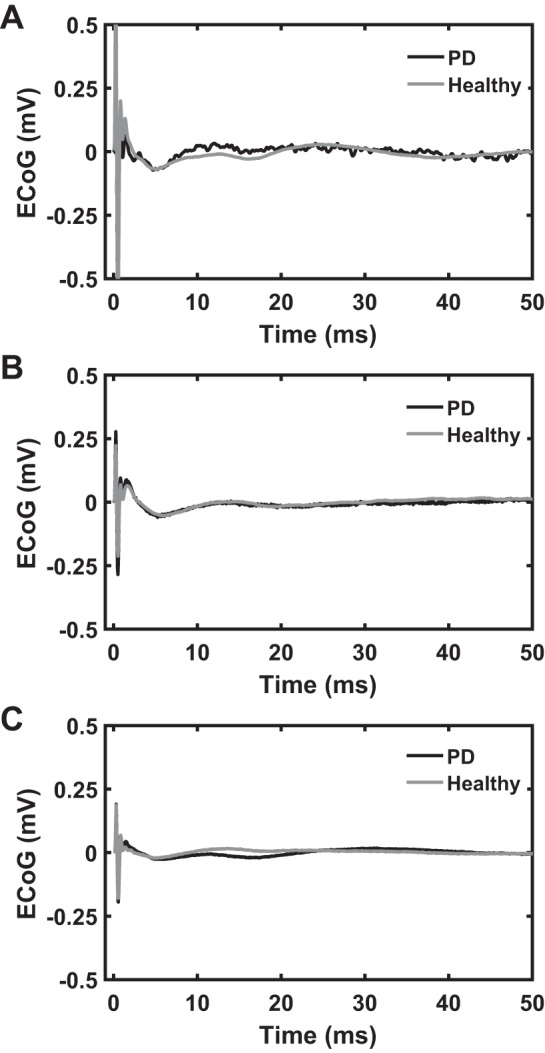
Effect of 6-hydroxydopamine (6-OHDA) lesion on cortical evoked potential (cEP) response to subthalamic nucleus (STN) deep brain stimulation (DBS) in awake rats (n = 3). Stimulation settings for electrocorticographic (ECoG) recordings: charge-balanced, symmetric biphasic pulses with a duration of 90 µs/phase, amplitude range of 50–70 µA, and frequency of 9 Hz. A–C: there is no substantial difference in cEP response in the parkinsonian state (black trace) compared with the healthy condition (gray trace). Each panel shows cEP response during healthy and 6-OHDA-lesioned states from 1 of the 3 rats.
Effect of volatile anesthetics on cEP.
The R1 response apparently resulted from direct excitation by the DBS pulse, because the latency (~1.4 ms) was too short for trans-synaptic activation. Similarly, the R3 response (~28 ms) apparently arose as a result of polysynaptic transmission, because the latency was too long for it to be due to direct excitation. However, the R2 response might be due to direct activation of small-diameter (slowly conducting) axons, might involve some synaptic transmission, or might be a combination of both. Therefore, we recorded cEPs in a subset of healthy rats (n = 3) first in awake conditions and then under anesthesia (Fig. 10A). The R2 component during 50-Hz STN DBS was completely suppressed following administration of anesthesia compared with the awake condition, whereas the magnitude of R1 response was modestly decreased compared with awake recordings (Fig. 10B). The excitability of corticospinal axons (L5) tends to decrease under volatile anesthetics due to the interaction of the anesthetic agent with ionic currents (especially Na+ current) at the nodes of Ranvier (Burke et al. 2000), and this is likely responsible for the reduction of R1 peak amplitude under anesthesia. However, activity that arises as a result of postsynaptic activation tends to be strongly suppressed by anesthesia (Berg‐Johnsen and Langmoen 1992), and the complete loss of R2 suggests that the generation of R2 involves synaptic transmission rather than direct excitation of small-diameter, slowly conducting axons.
Fig. 10.

A1–A3: cortical evoked potentials (cEP) under anesthesia and awake conditions in response to subthalamic nucleus (STN) deep brain stimulation (DBS) (n = 3). Stimulation settings for electrocorticographic (ECoG) recordings: charge-balanced, symmetric biphasic pulses with a duration of 90 µs/phase, amplitude range of 50–70 µA, and frequency of 50 Hz. Each panel shows cEP response during awake and anesthesia conditions from 1 of the 3 rats. The R2 response is completely suppressed following administration of anesthesia (isoflurane or sevoflurane), whereas the magnitude of R1 response decreased compared with that during the awake condition. B: quantification of R1 and R2 peak amplitudes under anesthesia and awake conditions. Note a decrease in R1 peak amplitude and a complete suppression of R2 response during anesthesia compared with the awake condition across all 3 rats.
Model-based deconstruction of the cEP.
We used a computational model of the cortical column to deconstruct the specific synaptic pathways responsible for generating the recorded cEP response, especially responses R2 and R3. We simulated STN DBS in the model by activating the axon terminals of L5 pyramidal neurons at four different frequencies (4.5, 9, 50, and 130 Hz; Fig. 11A). Furthermore, we simulated the effects of variations in DBS intensity by activating different proportions of the L5 axons (Fig. 11B). Figure 11B shows that the magnitudes of the later cEP phases (R2 and R3) increased and became more prominent at a higher intensity (60% of L5 axons activated) compared with lower intensity (20% of L5 axons activated). The model-generated cEPs matched well with the in vivo cEP response shown in Fig. 4, although there was polarity inversion of the response components. We analyzed the contribution of different neural structures (dendrites, soma, and axon) of model neurons to the model-based cEP response by computing ECoG using membrane currents from only the relevant structure, i.e., , , or . The dendrites and axonal elements of model neurons had a much greater contribution to the model-based cEP response than the soma (see appendix, Fig. A2).
Fig. 11.

A: model-based cortical evoked potential (cEP) response to different subthalamic nucleus (STN) deep brain stimulation (DBS) frequencies with activation of 60% of layer 5 (L5) axons. A1: 4.5 Hz. A2: 9 Hz. A3: 50 Hz. A4: 130 Hz. Model-based cEP response matched exceeding well with the in vivo response shown in Fig. 4. Note the reversal in polarity of cEP phases in model-based response compared with the in vivo cEP response shown in Fig. 4. B: effect of 4.5-Hz STN DBS intensity simulated by modulating the proportion of L5 pyramidal neuron axon activation. B1: 20%. B2: 40%. B3: 60%. B4: 80%. Note the magnitude of all phases (R1, R2, R3) increased and became more prominent at higher levels of activation. ECoG, electrocorticographic recordings.
Activation of model L5 pyramidal neurons was coincident with the R1 response at all STN DBS frequencies (Fig. 12). Recurrent activation of L5 pyramidal neurons was coincident with the R2 response, whereas the R3 phase was coincident with the activation of model L2/3 pyramidal neurons at 4.5 Hz (Fig. 12). The activation of L5 pyramidal neurons (R1) was the result of direct excitation due to the DBS pulse, whereas synaptic transmission led to the reactivation of L5 (R2) and activation of L2/3 (R3) pyramidal neurons (Fig. 12). We determined the synaptic pathways that resulted in the activation of L5 and L2/3 pyramidal neurons using the latency of neural activation in different cortical layers, as well as the thalamus (Fig. 12B). Activation of L5 pyramidal neurons by the DBS pulse resulted in recurrent activation of L5 pyramidal neurons, which led to postsynaptic activation of L6 pyramidal cells, which in turn generated a postsynaptic response in the TCR cells of the thalamus (Fig. 12, B4, B5, and B7). The TCR cells activated the L2/3 pyramidal cells via the L4 spiny stellate cells (Fig. 12, B1 and B3). On the basis of the latencies of model neuron responses, we conclude that monosynaptic activation of L5 pyramidal neurons via the L5→L5 pathway and polysynaptic activation of L2/3 pyramidal neurons via the L5→L5→L6→TH→L4→L2/3 pathway resulted in R2 and R3 responses, respectively (Fig. 12).
Fig. 12.

Model-based identification of the neural origin of cortical evoked potential (cEP) response to subthalamic nucleus (STN) deep brain stimulation (DBS). A and B: electrocorticographic (ECoG) recordings (A) and poststimulus time histograms (B) of underlying layer-specific neural activity evoked by 4.5-Hz STN DBS of 60% of layer 5 (L5) pyramidal cell axons in the computational model. B1: L2/3 pyramidal neurons. B2: L2/3 interneurons. B3: L4 spiny stellate cells. B4: L5 pyramidal neurons. B5: L6 pyramidal neurons. B6: L6 interneurons. B7: thalamocortical relay cells. B8: reticular thalamic nuclei cells. Activation of model L5 pyramidal neurons was coincident with R1 phase. Recurrent activation of L5 pyramidal neurons was coincident with R2 phase, whereas the timing of R3 phase was coincident with the activation of the model L2/3 pyramidal neurons. On the basis of the latency of neural activity in different layers, it can be concluded that the polysynaptic activation of L2/3 pyramidal neurons is through the L5→L5→L6→thalamus→L4→L2/3 pathway.
We deactivated synaptic connections individually and quantified the model neuron response to L5 axon stimulation. The magnitude of the R2 response decreased substantially following deactivation of recurrent L5 connections (Fig. 13A). Furthermore, deactivation of any synaptic connection in the L5→L5→L6→TH→L4→L2/3 pathway resulted in suppression of L2/3 pyramidal neuron activity and the R3 response (Fig. 13, A–E). Hence, the monosynaptic L5→L5 pathway and polysynaptic L5→L5→L6→TH→L4→L2/3 pathway were necessary for generation of the R2 and R3 responses following a DBS pulse. However, this does not negate the effect of other pathways in shaping the R2 and R3 responses. As an alternative confirmation of the role of these pathways in the generation of R2 and R3 responses, we deactivated all synaptic connections in the model except the connections in the L5→L5→L6→TH→L4→L2/3 pathway, and this reduced model was sufficient to produce all three cEP response components (R1, R2, and R3; Fig. 14).
Fig. 13.

Model-based deconstruction of subthalamic nucleus (STN) deep brain stimulation (DBS)-induced cortical evoked potential (cEP) by disconnecting individual synaptic connections in the layer 5 (L5)→L5→L6→thalamus (TH)→L4→L2/3 pathway. A–E: cEP responses (A1–E1) and poststimulus time histograms (A2–E2) to 4.5-Hz, 60% STN DBS following disconnection of L5→L5 recurrent excitatory connection (A), L5→L6 excitatory connection (B), L6→TH excitatory connection (C), TH→L4 excitatory connection (D), and L4→L2/3 excitatory connection (E). In A, note the suppression in activities of L5 and L2/3 pyramidal neurons and subsequent R2 and R3 responses (gray trace) compared with control (black trace). In B–E, note the suppression of L2/3 pyramidal neuron activity and subsequent R3 response (gray trace) following disruption of any synaptic connection in the L5→L6→TH→L4→L2/3 pathway compared with control condition (black trace). Gray traces show the cEP response following disconnection of a synaptic connection in the L5→L5→L6→TH→L4→L2/3 pathway, whereas black traces show the cEP response to 4.5-Hz, 60% STN DBS with all synaptic connections intact (control). ECoG, electrocorticographic recordings.
Fig. 14.

Model-based deconstruction of subthalamic nucleus (STN) deep brain stimulation (DBS)-induced cortical evoked potential (cEP) by evaluating the robustness of signal propagation through the layer 5 (L5)→L5→L6→thalamus (TH)→L4→L2/3 pathway. A1 and A2: cEP response and underlying spiking activity, respectively, to 4.5-Hz STN DBS of 60% L5 pyramidal cell axons generated by the cortical microcircuit with intact synaptic connections. B1 and B2: cEP response and underlying spiking activity, respectively, to 4.5-Hz STN DBS generated by a reduced cortical microcircuit with all synaptic connections disconnected except the L5→L5→L6→TH→L4→L2/3 pathway. The reduced microcircuit with only an intact L5→L5→L6→TH→L4→L2/3 polysynaptic pathway was sufficient to activate L5 and L2/3 pyramidal neurons following STN DBS-evoked stimulation of L5 pyramidal axons and subsequently generate the R1, R2, and R3 responses. ECoG, electrocorticographic recordings; Ex, excitatory neurons in L2/3 and L6.
DISCUSSION
We quantified the cEPs evoked by different frequencies of STN DBS using electrocorticographic recordings from M1 in awake healthy and parkinsonian rats. We used a detailed model of the cortical column interconnected to the TH to determine the neural origin of the in vivo cEP responses. The model revealed that the R1 response was due to the direct antidromic activation of L5 pyramidal neuron axons, the R2 component was due to the recurrent activation of L5 pyramidal neurons, and polysynaptic activation of L2/3 pyramidal neurons generated the R3 component. Antidromic activation of L5 pyramidal neurons and subsequent intracortical and cortico-thalamo-cortical synaptic interactions were sufficient to generate the cEP, and orthodromic activation through basal ganglia-thalamus-cortex pathways was not necessary (Devergnas and Wichmann 2011).
Role of synaptic pathways in generating cEP response to STN DBS.
STN DBS generates a series of complex network interactions involving multiple synaptic pathways. The hypothesis on the origin of STN DBS-induced cEP is that antidromic activation of the hyperdirect pathway mediates the R1 component, whereas orthodromic propagation through STN→GPi→TH→cortex (CTX) results in the R3 component (Devergnas and Wichmann 2011). Li et al. (2007) used collision tests to demonstrate antidromic activation of hyperdirect pathway during STN DBS, and we confirmed that R1 does indeed arise from antidromic activation of L5 axons projecting to STN. However, the model revealed that subsequent intracortical and cortico-thalamo-cortical synaptic interactions, following activation of axon terminals of L5 pyramidal neurons, were sufficient to generate the R3 response. The hypothesis that STN→GPi→TH→CTX pathway is responsible for the R3 evoked response was based on two observations (Devergnas and Wichmann 2011): 1) low-frequency GPi DBS evokes a R3 response in the cortex, and 2) the timing of the GPi DBS-evoked R3 response is ~10 ms earlier than the STN DBS-evoked response. Hence, because both STN DBS and GPi DBS evoke an R3 response in the cortex, the thinking was that a common pathway should mediate such a response. The well-known common link from STN/GPi to CTX is the STN→GPi→TH→CTX pathway. Thus, if indeed the STN→GPi→TH→CTX and GPi→TH→CTX pathways were producing the R3 cortical response during STN and GPi DBS, respectively, then the timing of R3 response due to GPi DBS must always be earlier compared with that due to STN DBS. However, in a study by Limousin et al. (1998), R3 cEPs evoked by STN DBS had a shorter latency (18–20 ms) than those evoked by GPi stimulation (25.0–25.8 ms). Several independent studies in patients with dystonia confirmed the timing of the long-latency response to GPi DBS reported by Limousin et al. (Bhanpuri et al. 2014; Ni et al. 2018; Tisch et al. 2008). Furthermore, if the long-latency responses to STN and GPi DBS are caused by the same pathway, then the effects of transcranial magnetic stimulation (TMS) delivered post-STN or post-GPi DBS on motor cortical excitability should be identical. However, this was found not to be the case. TMS delivered at the time of the R3 response to STN DBS increased motor cortical excitability (Kuriakose et al. 2010; Udupa et al. 2016) measured through motor evoked potentials, whereas cortical excitability decreased during TMS at the time of the R3 response to GPi DBS (Ni et al. 2018). In addition, Baker et al. (2002) showed that the major features of cEP were not substantially different in a PD patient with prior pallidotomy compared with those from PD subjects with intact pallidum. Therefore, different pathways might be responsible for mediating the cEP responses to STN and GPi DBS.
The pallidothalamic (GPi→TH) synaptic connections in the orthodromic pathway are inhibitory. Although inhibitory connections can evoke spikes by postinhibitory rebound excitation, experimental studies have shown the requirement of high-frequency activation of the presynaptic pallidal neuron to trigger a precise postsynaptic response (Goldberg et al. 2013; Person and Perkel 2005; Rivlin-Etzion et al. 2008). Numerous computational models of the basal ganglia circuit have shown that the increased burst firing in the PD state is capable of triggering rebound excitation in the TH neurons, whereas nonburst pallidal activity seen during the healthy or PD plus STN DBS states does not evoke any response in the TH (Guo et al. 2008; Kumaravelu et al. 2016; Rubin and Terman 2004; So et al. 2012a). Although the hypothesis implicating the STN→GPi→TH→CTX pathway in generating the R3 component of the cEP is plausible, no direct evidence exists supporting such a theory.
Role of synaptic pathways in mediating therapeutic benefits of STN DBS.
Our results do not clarify whether the cortical elements responsible for producing the cEP response play a role in the therapeutic mechanisms of STN DBS. We did not quantify motor responses to STN DBS in parkinsonian rats, and the synaptic interactions following antidromic activation of the hyperdirect pathway may or may not contribute to the therapeutic benefits of STN DBS. However, two studies support a role for the hyperdirect pathway in the therapeutic mechanism of STN DBS. First, selective optical stimulation of M1 L5 projection neurons reversed Parkinsonian motor symptoms in the 6-OHDA-lesioned rodent model of PD (Gradinaru et al. 2009; Sanders and Jaeger 2016). Second, Li et al. (2012) observed a significant correlation between the probability of antidromic spikes originating from the hyperdirect pathway and parkinsonian symptoms in the 6-OHDA-lesioned rat model of PD. Nonetheless, it is not possible to pinpoint the role of specific synaptic pathways in reversing motor symptoms from these studies, because selective activation of the axons of the hyperdirect pathway will also likely result in activation of STN, striatum, zona incerta, pontine nucleus, superior colliculus, brain stem, and associative thalamic nuclei via hyperdirect collaterals (Kita and Kita 2012).
Influence of axon diameter on cEP.
Ours is the first study to quantify the R3 response to STN DBS in rodents. Two prior studies quantified the cEP response in rats to STN DBS at frequencies >60 Hz (Dejean et al. 2009; Li et al. 2007), and thus it was only possible to visualize the R1 and R2 responses. In one study, all recordings were done under anesthesia (Li et al. 2007), whereas the other was an awake preparation similar to ours (Dejean et al. 2009). One surprising aspect of our cEP recordings from rat was the extent to which they matched cEPs from human (Walker et al. 2012). STN DBS-induced cEPs in rats included R1, R2, and R3 components, similarly to the human cEP, although there was a difference in the latency of the R3 response. There are several possible reasons for this difference. First, network connectivity of cortical microcircuits in humans may differ from than that of rodents. Second, the extent of myelination and axon diameter might be different between the two species. Although gray matter axons are myelinated to a lesser degree than white matter axons, recent experimental evidence suggests a different level of myelination of axons within the gray matter (Micheva et al. 2016; Tomassy et al. 2014). The latency of the model-based R3 response (~29 ms) matched more closely that of rats (~28 ms) than humans (~22 ms). The model included only the small-diameter, unmyelinated intracortical axons, thus implicating such axons in generating the R3 response in rodents, whereas large-diameter, myelinated axons might influence the timing of the R3 response in humans.
The lack of large-diameter axons in the model is not a large drawback, because the generation of the R1 response in rats appeared to be due mainly to activation of small-diameter axons. The latency of the R1 response in rats was ~1.4 ms, and the depth of STN from the dura is ~7 mm. Thus the conduction velocity of the axons activated by DBS and subsequently responsible for the generation of R1 response would be ~5 m/s. With the use of the standard value of 6 m/s per micrometer (Hursh 1939; Swadlow and Waxman 1975), the diameter of axons with a conduction velocity of ~5 m/s would be ~0.8 µm, and numerous experimental studies have identified such axons in the rat pyramidal tract/hyperdirect pathway (Dunkerley and Duncan 1969; Kita and Kita 2012). However, in humans, conduction velocities in the range of 60 m/s are necessary for the R1 response to have a latency of ~1 ms (Walker et al. 2012). The diameter range of such axons would be ~10 µm, and axons with such diameters exist in the primate pyramidal tract (Firmin et al. 2014), although in limited numbers.
Although cEPs in both rats and humans comprise R1, R2, and R3 components, all the underlying cognitive/behavioral effects of STN DBS may not be identical between rats and humans. For example, STN DBS in rats impairs reactive inhibitory control (Baunez et al. 2007), whereas STN DBS in humans improves inhibitory control (Mirabella et al. 2012, 2013).
Heterogeneity in R2 response.
We observed some heterogeneity of in the latency of the R2 component across rats. In the study by Li et al. (2007) across many rats, the mean latency of R2 response reported was 11.5 ms, and this latency may be the mean of averaging latencies of 8.33 and 13.56 ms that we found across the two groups of rats. One potential limitation in the conclusion of the dominant R2 latency in rats is the relatively small number of rats employed in the study. Possible reasons for heterogeneous cEPs across rats might be differences in electrode locations within STN or over M1 and the behavioral state of the rat (e.g., high-voltage spindles, rest as opposed to walking). All our recording sessions were conducted with the rats freely moving in the Faraday cage, and this freely moving condition could result in alteration of cortical dynamics leading to the heterogeneity of cEP responses. For example, there was a significant difference in cortical oscillatory activity in rodents between rest as opposed to walking on a treadmill (Lehmkuhle et al. 2009).
Activation of slowly conducting, small-diameter axons of the hyperdirect pathway/internal capsule is a possible alternative origin of the R2 response to that predicted by the model (Walker and Guthrie 2013; Walker et al. 2012). However, this is unlikely to be the case. First, the range of R2 latencies was 8.3–13.6 ms, and this means the range of axon diameters necessary to generate the R2 response would be 0.09–0.14 µm. Such thin-diameter axons are rare in the rat hyperdirect pathway, and only 10% of hyperdirect collaterals comprise axons <0.2 µm in diameter (Kita and Kita 2012). Second, R2 responses were completely suppressed under anesthesia, suggesting that R2 was the result of trans-synaptic activation. In a prior study, the R2 response was not completely suppressed under anesthesia (Li et al. 2007), perhaps because of the lower dose of anesthesia (1–2.5% isoflurane). Finally, Li et al. (2012) recorded single units from M1-L5 pyramidal neurons and found increased firing at ~4 ms in those neurons that exhibited antidromic spikes at ~1 ms, similar to the L5 PSTH obtained from the model. This suggests the recurrent activation of L5 pyramidal neurons following antidromic activation generates the R2 response. However, the possibility of thin-diameter axons of the hyperdirect pathway contributing to the R2 response, and subsequently, the distribution of thin- to ultrathin-diameter axons in the rat hyperdirect pathway resulting in the heterogeneity of the R2 response latency, cannot be excluded.
We found the R2 response to be indiscernible in some rats at 50-Hz STN DBS. The difficulty in discerning the R2 component might have to do with both the amplitude and the duration of R2 response being much smaller and shorter compared with the those of the R1 and R3 components. Furthermore, TMS paired to R1 and R3 responses, but not to R2, results in motor cortex facilitation (Hanajima et al. 2004; Kuriakose et al. 2010; Udupa et al. 2016; Udupa and Chen 2015). Hence, the small amplitude and the short duration of the R2 component might have contributed to an inability to detect it clearly in some rats.
Reversed polarity of model-based cEP compared with in vivo response.
Although the polarities of phases in model-based cEP responses were inverted with respect to the rat experimental cEPs, they matched well with cEP in humans obtained over the frontal cortex (FP2) but were inverted to those over the primary motor cortex (C4) (Walker et al. 2012). A common phenomenon in cEP recordings is reversal in polarity in traversing from the midline-frontal to the parietal leads of the EEG (sensorimotor) ipsilateral to stimulation (MacKinnon et al. 2005; Walker et al. 2012). This suggests that descending axons from the frontal cortex to STN might be the origin of antidromic activation following STN DBS. Motor, sensory, and prefrontal cortices comprise the cortico-subthalamic projections in rats (Afsharpour 1985; Canteras et al. 1988, 1990). Traditionally, the primary motor cortex is believed to be mostly agranular (no L4), the premotor and supplementary motor areas to be dysgranular (thin L4), and the prefrontal and somatosensory cortices to include prominent L4 neurons. L4 is one of the major recipients of thalamocortical projections, a key connection required to generate the R3 response. However, the classical view of M1 in rodents being agranular was recently challenged with the finding of L4 in rodent M1 with input-output characteristics equivalent to those found in sensory cortex L4 (Yamawaki et al. 2014). The circuit predicted by the model as being responsible for generating the R3 response is known to exist in the sensorimotor cortex (Alitto and Usrey 2003; Lindenbach and Bishop 2013).
Application of cEP for tuning and control of DBS.
The neural circuitry responsible for generating the STN DBS-induced cEP response appears largely independent of any SNc dopaminergic modulation, because cEPs were largely similar before and after unilateral 6-OHDA lesion. This conclusion is consistent with the observation made in rats that the R1 and R2 responses recorded under anesthesia were independent of SNc degeneration (Li et al. 2007). Therefore, cEP might not serve as a tool to probe the alteration in network dynamics during PD. However, the cEP might have utility to quantify the extent of neural activation during STN DBS. cEP can be used as an indicator to quantify the cortical response to various STN DBS parameters such as amplitude, frequency, pulse width, polarity, etc. We quantified the model-based cEP response to different intensities of STN DBS amplitude. Lower intensities resulted in only the R1 response, whereas prominent R2 and R3 responses were seen at higher intensities. In line with this observation, the magnitudes of later cEP phases in PD patients were higher at high STN DBS intensities compared with low intensity (Kelley et al. 2018). Thus cEP can be used to quantify the effects of STN DBS amplitude. In a different study, Kent and colleagues (Kent and Grill 2013; Kent et al. 2015) recorded the thalamic evoked compound action potential generated by various thalamic DBS parameters, but the cEP may have benefits for closed-loop DBS applications. First, stimulation artifacts and amplifier saturation appear to be less of a problem when cEPs are recorded during STN DBS because of the distance from the stimulating electrode, and our results demonstrate high-fidelity recordings of short-latency (R1) components of the cEP. Furthermore, the cEP might be used to select the appropriate electrode contact(s) for therapeutic stimulation. STN DBS from the most effective contact consistently evoked R3 at 22.2 ± 1.2 ms (Kuriakose et al. 2010), and this was true in rats, as well, with electrodes within STN evoking a consistent R3 (see appendix, Fig. A3). Finally, Walker et al. (2012) demonstrated that the comparable cEP can be recorded with EEG electrodes, and thus such selection and tuning could be done without the necessity of implanting additional electrodes.
GRANTS
This work was supported by National Institute of Neurological Disorders and Stroke Grants R37 NS040894 and R01 NS079312 and by the Duke Compute Cluster.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.K., C.S.O., W.M.G. conceived and designed research; K.K., C.S.O., and C.E.B. performed experiments; K.K., and C.S.O. analyzed data; K.K., C.S.O., C.E.B., and W.M.G. interpreted results of experiments; K.K. prepared figures; K.K. drafted manuscript; K.K., C.S.O., C.E.B., and W.M.G. edited and revised manuscript; K.K., C.S.O., C.E.B., and W.M.G. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Emily Shannon, Jaydeep Sambangi, and Jiashu Li for help with histology. We also thank Aman Aberra, Nathan Titus, and Dr. Dennis Turner for helpful discussions that greatly improved the manuscript.
APPENDIX
Here we provide additional figures and a table to support the results. Table A1 provides stimulation amplitudes used in the rat experiments. Figure A1 shows results of postmortem histology, Fig. A2 shows the contribution of different neural elements to the model-based cEP response, and Fig. A3 depicts the cEP with stimulating electrode tips located in zona incerta.
Fig. A1.

Postmortem histology. A: example of postmortem histology showing stimulating electrode tracks (arrows) with tip reaching subthalamic nucleus (STN; dotted outline) in a cytochrome oxidase-stained slice. B: example of postmortem histology from a unilateral 6-hydroxydopamine (6-OHDA)-lesioned rat showing loss of substantia nigra pars compacta (SNc) dopaminergic neurons on one side and intact dopaminergic neurons on the other in a tyrosine hydroxylase-stained slice.
Fig. A2.

Contribution of different neural structures (dendrites, soma, axon) to the model-based cortical evoked potential (cEP) response. Model-based cEP responses to 60%, 4.5-Hz subthalamic nucleus deep brain stimulation were computed from currents from dendrites, soma, and axons (A1), only dendritic currents (A2), only somatic currents (A3), and only axonal currents (A4) of model cortical neurons. ECoG, electrocorticographic recordings.
Fig. A3.

Cortical evoked potential (cEP) responses from rats with electrode tips in zona incerta (ZI), confirmed through postmortem histology (n = 3). Stimulation settings for electrocorticographic (ECoG) recordings: charge-balanced, symmetric biphasic pulses with a duration of 90 µs/phase, amplitude range of 60–90 µA, and frequency of 9 Hz. A–C each represent cEP data from 1 of the 3 rats. A1–C1: cEP data shown at a shorter timescale to enable visualization of the R1 response. Note the presence of R1 response in all 3 rats, although the electrode tips were in ZI. This is because the ascending fibers of the ZI are believed to have the same origin as the hyperdirect pathway, i.e., L5 of the cortex (Mitrofanis and Mikuletic 1999). A2–C2: the rats did not exhibit clearly distinguishable R2 and R3 responses, similarly to those seen when electrodes were within STN. Compare responses shown with those in Fig. 5.
Table A1.
Rat experiment stimulation parameters
| Animal | Strain | Stimulation Amplitude, µA |
|---|---|---|
| 1 | Sprague-Dawley | Left hemisphere: 60 (1+3−) Right hemisphere: 70 (1+2−) |
| 2 | Sprague-Dawley | 30 (2+3−) |
| 3 | Sprague-Dawley | 40 (4+3−) |
| 4 | Sprague-Dawley | 35 (2+4−) |
| 5 | Long-Evans | 50 (4+2−) |
| 6 | Long-Evans | 70 (2+3−) |
| 7 | Long-Evans | 65 (3+4−) |
| 8 | Long-Evans | 75 (1+3−) |
| 9 | Long-Evans | 50 (4+3−) |
| 10 | Long-Evans | 70 (2+4−) |
Electrode contacts are given in parentheses following stimulation amplitudes.
REFERENCES
- Afsharpour S. Topographical projections of the cerebral cortex to the subthalamic nucleus. J Comp Neurol 236: 14–28, 1985. doi: 10.1002/cne.902360103. [DOI] [PubMed] [Google Scholar]
- Alhourani A, McDowell MM, Randazzo MJ, Wozny TA, Kondylis ED, Lipski WJ, Beck S, Karp JF, Ghuman AS, Richardson RM. Network effects of deep brain stimulation. J Neurophysiol 114: 2105–2117, 2015. doi: 10.1152/jn.00275.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitto HJ, Usrey WM. Corticothalamic feedback and sensory processing. Curr Opin Neurobiol 13: 440–445, 2003. doi: 10.1016/S0959-4388(03)00096-5. [DOI] [PubMed] [Google Scholar]
- Baker KB, Montgomery EB Jr, Rezai AR, Burgess R, Lüders HO. Subthalamic nucleus deep brain stimulus evoked potentials: physiological and therapeutic implications. Mov Disord 17: 969–983, 2002. doi: 10.1002/mds.10206. [DOI] [PubMed] [Google Scholar]
- Baldissera F, Lundberg A, Udo M. Stimulation of pre- and postsynaptic elements in the red nucleus. Exp Brain Res 15: 151–167, 1972. doi: 10.1007/BF00235579. [DOI] [PubMed] [Google Scholar]
- Baunez C, Christakou A, Chudasama Y, Forni C, Robbins TW. Bilateral high-frequency stimulation of the subthalamic nucleus on attentional performance: transient deleterious effects and enhanced motivation in both intact and parkinsonian rats. Eur J Neurosci 25: 1187–1194, 2007. doi: 10.1111/j.1460-9568.2007.05373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg-Johnsen J, Langmoen IA. The effect of isoflurane on excitatory synaptic transmission in the rat hippocampus. Acta Anaesthesiol Scand 36: 350–355, 1992. doi: 10.1111/j.1399-6576.1992.tb03480.x. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol 72: 507–520, 1994. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Bhanpuri NH, Bertucco M, Ferman D, Young SJ, Liker MA, Krieger MD, Sanger TD. Deep brain stimulation evoked potentials may relate to clinical benefit in childhood dystonia. Brain Stimulat 7: 718–726, 2014. doi: 10.1016/j.brs.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Burke D, Bartley K, Woodforth IJ, Yakoubi A, Stephen JP. The effects of a volatile anaesthetic on the excitability of human corticospinal axons. Brain 123: 992–1000, 2000. doi: 10.1093/brain/123.5.992. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Shammah-Lagnado SJ, Silva BA, Ricardo JA. Somatosensory inputs to the subthalamic nucleus: a combined retrograde and anterograde horseradish peroxidase study in the rat. Brain Res 458: 53–64, 1988. doi: 10.1016/0006-8993(88)90495-7. [DOI] [PubMed] [Google Scholar]
- Canteras NS, Shammah-Lagnado SJ, Silva BA, Ricardo JA. Afferent connections of the subthalamic nucleus: a combined retrograde and anterograde horseradish peroxidase study in the rat. Brain Res 513: 43–59, 1990. doi: 10.1016/0006-8993(90)91087-W. [DOI] [PubMed] [Google Scholar]
- de Hemptinne C, Ryapolova-Webb ES, Air EL, Garcia PA, Miller KJ, Ojemann JG, Ostrem JL, Galifianakis NB, Starr PA. Exaggerated phase-amplitude coupling in the primary motor cortex in Parkinson disease. Proc Natl Acad Sci USA 110: 4780–4785, 2013. doi: 10.1073/pnas.1214546110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hemptinne C, Swann NC, Ostrem JL, Ryapolova-Webb ES, San Luciano M, Galifianakis NB, Starr PA. Therapeutic deep brain stimulation reduces cortical phase-amplitude coupling in Parkinson’s disease. Nat Neurosci 18: 779–786, 2015. doi: 10.1038/nn.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejean C, Hyland B, Arbuthnott G. Cortical effects of subthalamic stimulation correlate with behavioral recovery from dopamine antagonist induced akinesia. Cereb Cortex 19: 1055–1063, 2009. doi: 10.1093/cercor/bhn149. [DOI] [PubMed] [Google Scholar]
- Devergnas A, Wichmann T. Cortical potentials evoked by deep brain stimulation in the subthalamic area. Front Syst Neurosci 5: 30, 2011. doi: 10.3389/fnsys.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkerley GB, Duncan D. A light and electron microscopic study of the normal and the degenerating corticospinal tract in the rat. J Comp Neurol 137: 155–183, 1969. doi: 10.1002/cne.901370204. [DOI] [PubMed] [Google Scholar]
- Dunn OJ. Multiple comparisons using rank sums. Technometrics 6: 241–252, 1964. doi: 10.1080/00401706.1964.10490181. [DOI] [Google Scholar]
- Filion M, Tremblay L. Abnormal spontaneous activity of globus pallidus neurons in monkeys with MPTP-induced parkinsonism. Brain Res 547: 140–144, 1991. 10.1016/0006-8993(91)90585-J. [DOI] [PubMed] [Google Scholar]
- Firmin L, Field P, Maier MA, Kraskov A, Kirkwood PA, Nakajima K, Lemon RN, Glickstein M. Axon diameters and conduction velocities in the macaque pyramidal tract. J Neurophysiol 112: 1229–1240, 2014. doi: 10.1152/jn.00720.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson P, Crook S, Cannon RC, Hines ML, Billings GO, Farinella M, Morse TM, Davison AP, Ray S, Bhalla US, Barnes SR, Dimitrova YD, Silver RA. NeuroML: a language for describing data driven models of neurons and networks with a high degree of biological detail. PLOS Comput Biol 6: e1000815, 2010. doi: 10.1371/journal.pcbi.1000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Boraud T, Maraton S, Haber SN, Vaadia E, Bergman H. Enhanced synchrony among primary motor cortex neurons in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson’s disease. J Neurosci 22: 4639–4653, 2002. doi: 10.1523/JNEUROSCI.22-11-04639.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JH, Farries MA, Fee MS. Basal ganglia output to the thalamus: still a paradox. Trends Neurosci 36: 695–705, 2013. doi: 10.1016/j.tins.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science 324: 354–359, 2009. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Rubin JE, McIntyre CC, Vitek JL, Terman D. Thalamocortical relay fidelity varies across subthalamic nucleus deep brain stimulation protocols in a data-driven computational model. J Neurophysiol 99: 1477–1492, 2008. doi: 10.1152/jn.01080.2007. [DOI] [PubMed] [Google Scholar]
- Hanajima R, Ashby P, Lozano AM, Lang AE, Chen R. Single pulse stimulation of the human subthalamic nucleus facilitates the motor cortex at short intervals. J Neurophysiol 92: 1937–1943, 2004. doi: 10.1152/jn.00239.2004. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. NEURON: a tool for neuroscientists. Neuroscientist 7: 123–135, 2001. doi: 10.1177/107385840100700207. [DOI] [PubMed] [Google Scholar]
- Hines ML, Carnevale NT. Translating network models to parallel hardware in NEURON. J Neurosci Methods 169: 425–455, 2008. doi: 10.1016/j.jneumeth.2007.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines ML, Eichner H, Schürmann F. Neuron splitting in compute-bound parallel network simulations enables runtime scaling with twice as many processors. J Comput Neurosci 25: 203–210, 2008. doi: 10.1007/s10827-007-0073-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hursh J. Conduction velocity and diameter of nerve fibers. Am J Physiol 127: 131–139, 1939. doi: 10.1152/ajplegacy.1939.127.1.131. [DOI] [Google Scholar]
- Jankowska E, Padel Y, Tanaka R. The mode of activation of pyramidal tract cells by intracortical stimuli. J Physiol 249: 617–636, 1975. doi: 10.1113/jphysiol.1975.sp011034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Xu W, Baker KB, Zhang J, Vitek JL. Modulation of motor cortex neuronal activity and motor behavior during subthalamic nucleus stimulation in the normal primate. J Neurophysiol 113: 2549–2554, 2015. doi: 10.1152/jn.00997.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley R, Flouty O, Emmons EB, Kim Y, Kingyon J, Wessel JR, Oya H, Greenlee JD, Narayanan NS. A human prefrontal-subthalamic circuit for cognitive control. Brain 141: 205–216, 2018. doi: 10.1093/brain/awx300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent AR, Grill WM. Neural origin of evoked potentials during thalamic deep brain stimulation. J Neurophysiol 110: 826–843, 2013. doi: 10.1152/jn.00074.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent AR, Swan BD, Brocker DT, Turner DA, Gross RE, Grill WM. Measurement of evoked potentials during thalamic deep brain stimulation. Brain Stimulat 8: 42–56, 2015. doi: 10.1016/j.brs.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita T, Kita H. The subthalamic nucleus is one of multiple innervation sites for long-range corticofugal axons: a single-axon tracing study in the rat. J Neurosci 32: 5990–5999, 2012. doi: 10.1523/JNEUROSCI.5717-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaravelu K, Brocker DT, Grill WM. A biophysical model of the cortex-basal ganglia-thalamus network in the 6-OHDA lesioned rat model of Parkinson’s disease. J Comput Neurosci 40: 207–229, 2016. doi: 10.1007/s10827-016-0593-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose R, Saha U, Castillo G, Udupa K, Ni Z, Gunraj C, Mazzella F, Hamani C, Lang AE, Moro E, Lozano AM, Hodaie M, Chen R. The nature and time course of cortical activation following subthalamic stimulation in Parkinson’s disease. Cereb Cortex 20: 1926–1936, 2010. doi: 10.1093/cercor/bhp269. [DOI] [PubMed] [Google Scholar]
- Lehmkuhle MJ, Bhangoo SS, Kipke DR. The electrocorticogram signal can be modulated with deep brain stimulation of the subthalamic nucleus in the hemiparkinsonian rat. J Neurophysiol 102: 1811–1820, 2009. doi: 10.1152/jn.90844.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy R, Ashby P, Hutchison WD, Lang AE, Lozano AM, Dostrovsky JO. Dependence of subthalamic nucleus oscillations on movement and dopamine in Parkinson’s disease. Brain 125: 1196–1209, 2002. doi: 10.1093/brain/awf128. [DOI] [PubMed] [Google Scholar]
- Li Q, Ke Y, Chan DC, Qian ZM, Yung KK, Ko H, Arbuthnott GW, Yung W-H. Therapeutic deep brain stimulation in Parkinsonian rats directly influences motor cortex. Neuron 76: 1030–1041, 2012. doi: 10.1016/j.neuron.2012.09.032. [DOI] [PubMed] [Google Scholar]
- Li Q, Qian Z-M, Arbuthnott GW, Ke Y, Yung WH. Cortical effects of deep brain stimulation: implications for pathogenesis and treatment of Parkinson disease. JAMA Neurol 71: 100–103, 2014. doi: 10.1001/jamaneurol.2013.4221. [DOI] [PubMed] [Google Scholar]
- Li S, Arbuthnott GW, Jutras MJ, Goldberg JA, Jaeger D. Resonant antidromic cortical circuit activation as a consequence of high-frequency subthalamic deep-brain stimulation. J Neurophysiol 98: 3525–3537, 2007. doi: 10.1152/jn.00808.2007. [DOI] [PubMed] [Google Scholar]
- Limousin P, Brown P, Marsden J, Defebvre L, Rothwell J. Evoked potentials from subthalamic nucleus, internal pallidum and thalamic stimulation in parkinsonian and postural tremor patients. J Physiol 509: 176P–177P, 1998. [Google Scholar]
- Lindenbach D, Bishop C. Critical involvement of the motor cortex in the pathophysiology and treatment of Parkinson’s disease. Neurosci Biobehav Rev 37: 2737–2750, 2013. doi: 10.1016/j.neubiorev.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon CD, Webb RM, Silberstein P, Tisch S, Asselman P, Limousin P, Rothwell JC. Stimulation through electrodes implanted near the subthalamic nucleus activates projections to motor areas of cerebral cortex in patients with Parkinson’s disease. Eur J Neurosci 21: 1394–1402, 2005. doi: 10.1111/j.1460-9568.2005.03952.x. [DOI] [PubMed] [Google Scholar]
- Mathai A, Smith Y. The corticostriatal and corticosubthalamic pathways: two entries, one target. So what? Front Syst Neurosci 5: 64, 2011. doi: 10.3389/fnsys.2011.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell GC, So RQ, Hilliard JD, Lopomo P, Grill WM. Effective deep brain stimulation suppresses low-frequency network oscillations in the basal ganglia by regularizing neural firing patterns. J Neurosci 32: 15657–15668, 2012. doi: 10.1523/JNEUROSCI.2824-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Wolman D, Mensh BD, Pax E, Buchanan J, Smith SJ, Bock DD. A large fraction of neocortical myelin ensheathes axons of local inhibitory neurons. eLife 5: e15784, 2016. doi: 10.7554/eLife.15784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miocinovic S. Beyond the basal ganglia: cortical effects of deep brain stimulation. JAMA Neurol 71: 8–10, 2014. doi: 10.1001/jamaneurol.2013.4643. [DOI] [PubMed] [Google Scholar]
- Mirabella G, Iaconelli S, Modugno N, Giannini G, Lena F, Cantore G. Stimulation of subthalamic nuclei restores a near normal planning strategy in Parkinson’s patients. PLoS One 8: e62793, 2013. doi: 10.1371/journal.pone.0062793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabella G, Iaconelli S, Romanelli P, Modugno N, Lena F, Manfredi M, Cantore G. Deep brain stimulation of subthalamic nuclei affects arm response inhibition in Parkinson’s patients. Cereb Cortex 22: 1124–1132, 2012. doi: 10.1093/cercor/bhr187. [DOI] [PubMed] [Google Scholar]
- Mitrofanis J, Mikuletic L. Organisation of the cortical projection to the zona incerta of the thalamus. J Comp Neurol 412: 173–185, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- Ni Z, Kim SJ, Phielipp N, Ghosh S, Udupa K, Gunraj CA, Saha U, Hodaie M, Kalia SK, Lozano AM, Lee DJ, Moro E, Fasano A, Hallett M, Lang AE, Chen R. Pallidal deep brain stimulation modulates cortical excitability and plasticity. Ann Neurol 83: 352–362, 2018. doi: 10.1002/ana.25156. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Amsterdam: Academic, 2006. https://trove.nla.gov.au/version/221529547. [Google Scholar]
- Person AL, Perkel DJ. Unitary IPSPs drive precise thalamic spiking in a circuit required for learning. Neuron 46: 129–140, 2005. doi: 10.1016/j.neuron.2004.12.057. [DOI] [PubMed] [Google Scholar]
- Ranck JB., Jr Specific impedance of rabbit cerebral cortex. Exp Neurol 7: 144–152, 1963. doi: 10.1016/S0014-4886(63)80005-9. [DOI] [PubMed] [Google Scholar]
- Rivlin-Etzion M, Marmor O, Saban G, Rosin B, Haber SN, Vaadia E, Prut Y, Bergman H. Low-pass filter properties of basal ganglia cortical muscle loops in the normal and MPTP primate model of parkinsonism. J Neurosci 28: 633–649, 2008. doi: 10.1523/JNEUROSCI.3388-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JE, Terman D. High frequency stimulation of the subthalamic nucleus eliminates pathological thalamic rhythmicity in a computational model. J Comput Neurosci 16: 211–235, 2004. doi: 10.1023/B:JCNS.0000025686.47117.67. [DOI] [PubMed] [Google Scholar]
- Sanders TH, Jaeger D. Optogenetic stimulation of cortico-subthalamic projections is sufficient to ameliorate bradykinesia in 6-ohda lesioned mice. Neurobiol Dis 95: 225–237, 2016. doi: 10.1016/j.nbd.2016.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So RQ, Kent AR, Grill WM. Relative contributions of local cell and passing fiber activation and silencing to changes in thalamic fidelity during deep brain stimulation and lesioning: a computational modeling study. J Comput Neurosci 32: 499–519, 2012a. doi: 10.1007/s10827-011-0366-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So RQ, McConnell GC, August AT, Grill WM. Characterizing effects of subthalamic nucleus deep brain stimulation on methamphetamine-induced circling behavior in hemi-Parkinsonian rats. IEEE Trans Neural Syst Rehabil Eng 20: 626–635, 2012b. doi: 10.1109/TNSRE.2012.2197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swadlow HA, Waxman SG. Observations on impulse conduction along central axons. Proc Natl Acad Sci USA 72: 5156–5159, 1975. doi: 10.1073/pnas.72.12.5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisch S, Rothwell JC, Zrinzo L, Bhatia KP, Hariz M, Limousin P. Cortical evoked potentials from pallidal stimulation in patients with primary generalized dystonia. Mov Disord 23: 265–273, 2008. doi: 10.1002/mds.21835. [DOI] [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen HH, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, Arlotta P. Distinct profiles of myelin distribution along single axons of pyramidal neurons in the neocortex. Science 344: 319–324, 2014. doi: 10.1126/science.1249766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Contreras D, Cunningham MO, Murray H, LeBeau FE, Roopun A, Bibbig A, Wilent WB, Higley MJ, Whittington MA. Single-column thalamocortical network model exhibiting gamma oscillations, sleep spindles, and epileptogenic bursts. J Neurophysiol 93: 2194–2232, 2005. doi: 10.1152/jn.00983.2004. [DOI] [PubMed] [Google Scholar]
- Udupa K, Bahl N, Ni Z, Gunraj C, Mazzella F, Moro E, Hodaie M, Lozano AM, Lang AE, Chen R. Cortical plasticity induction by pairing subthalamic nucleus deep-brain stimulation and primary motor cortical transcranial magnetic stimulation in Parkinson’s disease. J Neurosci 36: 396–404, 2016. doi: 10.1523/JNEUROSCI.2499-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udupa K, Chen R. The mechanisms of action of deep brain stimulation and ideas for the future development. Prog Neurobiol 133: 27–49, 2015. doi: 10.1016/j.pneurobio.2015.08.001. [DOI] [PubMed] [Google Scholar]
- Walker HC, Guthrie BL. Noninvasive measurement of cortical activity during effective brain stimulation for movement disorders. Future Neurol 8: 9–12, 2013. doi: 10.2217/fnl.12.87. [DOI] [Google Scholar]
- Walker HC, Huang H, Gonzalez CL, Bryant JE, Killen J, Cutter GR, Knowlton RC, Montgomery EB, Guthrie BL, Watts RL. Short latency activation of cortex during clinically effective subthalamic deep brain stimulation for Parkinson’s disease. Mov Disord 27: 864–873, 2012. doi: 10.1002/mds.25025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmer D, de Solages C, Hill B, Yu H, Henderson JM, Bronte-Stewart H. High frequency deep brain stimulation attenuates subthalamic and cortical rhythms in Parkinson’s disease. Front Hum Neurosci 6: 155, 2012. doi: 10.3389/fnhum.2012.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki N, Borges K, Suter BA, Harris KD, Shepherd GM. A genuine layer 4 in motor cortex with prototypical synaptic circuit connectivity. eLife 3: e05422, 2014. doi: 10.7554/eLife.05422. [DOI] [PMC free article] [PubMed] [Google Scholar]