

Abstract
Pioglitazone is effective in improving insulin resistance and liver histology in patients with nonalcoholic steatohepatitis (NASH). Because dysfunctional mitochondrial metabolism is a central feature of NASH, we hypothesized that an important target of pioglitazone would be alleviating mitochondrial oxidative dysfunction. To this end, we studied hepatic mitochondrial metabolism in mice fed high-fructose high-transfat diet (TFD) supplemented with pioglitazone for 20 wk, using nuclear magnetic resonance-based 13C isotopomer analysis. Pioglitazone improved whole body and adipose insulin sensitivity in TFD-fed mice. Furthermore, pioglitazone reduced intrahepatic triglyceride content and fed plasma ketones and hepatic TCA cycle flux, anaplerosis, and pyruvate cycling in mice with NASH. This was associated with a marked reduction in most intrahepatic diacylglycerol classes and, to a lesser extent, some ceramide species (C22:1, C23:0). Considering the cross-talk between mitochondrial function and branched-chain amino acid (BCAA) metabolism, pioglitazone’s impact on plasma BCAA profile was determined in a cohort of human subjects. Pioglitazone improved the plasma BCAA concentration profile in patients with NASH. This appeared to be related to an improvement in BCAA degradation in multiple tissues. These results provide evidence that pioglitazone-induced changes in NASH are related to improvements in hepatic mitochondrial oxidative dysfunction and changes in whole body BCAA metabolism.
Keywords: insulin resistance, lipidomics, liver metabolism, mitochondria, nonalcoholic fatty liver disease
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver condition in obese patients with prediabetes or type 2 diabetes mellitus (T2DM) (3, 31). It is characterized by accumulation of liver fat (LFAT; >5.5%), hepatic insulin resistance, and dysregulated mitochondrial metabolism (36). Up to 40% of patients with NAFLD will progress to a more severe form of disease, nonalcoholic steatohepatitis (NASH), characterized by inflammation and hepatocellular injury with or without fibrosis (1, 6). Dysfunctional hepatic mitochondrial metabolism is a central feature of NAFLD, characterized by sustained induction of hepatic tricarboxylic acid (TCA) cycle flux and accumulation of lipotoxic intermediates of incomplete fat oxidation (e.g., long-chain acylcarnitines, diacylglycerols, and ceramides) (29, 34, 38). This metabolic milieu in the liver contributes to what we term mitochondrial oxidative dysfunction and promotes high rates of reactive oxygen species (ROS) production and inflammation (33).
Pioglitazone, a thiazolidinedione (TZD), reduces intrahepatic triglycerides (IHTG) and improves long-term metabolic and histological parameters in human patients (2, 7, 32). Pioglitazone’s beneficial effects on the liver are believed to occur primarily through peroxisome proliferator-activated receptor-γ (PPARγ)-mediated improvements in adipose tissue insulin sensitivity and the resultant reduction in free fatty acid delivery to the liver (8, 10–12). However, this alone does not fully explain its mode of action in NAFLD, and several pieces of evidence also suggest a direct action of pioglitazone on the liver. Pioglitazone reduces endoplasmic reticulum stress and suppresses inflammatory cytokine signaling in insulin-resistant mouse liver (19, 41). Furthermore, in isolated hepatocytes, pioglitazone may ameliorate mitochondrial respiratory chain activity (9). Consistent with this finding, pioglitazone reduces the observed elevated gluconeogenic flux observed in patients with T2DM (5, 13). Furthermore, studies have also shown that pioglitazone treatment lowers plasma branched-chain amino acids (BCAAs) (17, 18), whose levels are robustly correlated with insulin resistance and are predictive of T2DM onset (28, 39). This is significant, considering the emerging hypothesis that BCAAs can modulate mitochondrial lipid metabolism during insulin resistance (14, 24, 25).
We hypothesized that pioglitazone’s mechanism of action involves the alleviation of dysfunctional hepatic mitochondrial metabolism during NAFLD, specifically by reducing the accumulation of lipotoxic intermediates in the liver and thus relieving the metabolic burden on mitochondrial TCA cycle flux. Our data support this hypothesis, and our findings indicate that pioglitazone treatment alleviates high-fructose/high-transfat diet-induced lipotoxicity and improves mitochondrial oxidative flux in the liver. Concurrently, pioglitazone also improved whole body BCAA metabolism, which could have a significant impact for improving peripheral and hepatic insulin sensitivity.
RESEARCH DESIGN AND METHODS
Animal studies.
Animal studies were approved by the Institutional Animal Care and Use Committee at the University of Florida. C57BL/6J mice (Jackson Laboratories, Bar Harbor ME), were randomly assigned either a control diet (C; 10% kcal fat; Research Diets, no. D09100304) or a high-fructose high-transfat diet (TFD; 40% kcal fat, 20% kcal fructose, 2% cholesterol; Research Diets, no. D09100301) for 24 wk to induce NASH, as previously reported (4, 29). Following 4 wk of TFD feeding, a subgroup of mice was randomly assigned to a custom TFD diet supplemented with pioglitazone (0.01%) (PIO; Research Diets, no. D15032301). The 0.01% pioglitazone enrichment was designed to achieve a daily pioglitazone intake of 10 mg/kg body wt in mice. Metabolic measurements were conducted following a total of 20 wk on PIO diet, compared with their age-matched TFD and control counterparts.
Stable isotope infusions and metabolic analysis in mice.
After 24 wk on the diets, ~100 µl of blood was collected during the fed condition from the tail vein for analysis of plasma pioglitazone and other metabolites. Mice were then implanted with a jugular vein catheter for stable isotope infusions. Following 4–5 days of recovery and an overnight fast, mice were infused with [13C3]propionate (Cambridge Isotopes, Andover, MA) and [3,4-13C2]glucose (Omicron Biochemical, South Bend, IN) to evaluate hepatic mitochondrial metabolism and endogenous glucose production (EGP), respectively (29, 34). Following 90 min of stable isotope infusions, mice were euthanized, and plasma and tissue samples were collected and stored at −80°C for analysis.
Human study design.
To determine the impact of pioglitazone on BCAAs, we studied 50 human subjects who participated in a randomized control trial of pioglitazone in managing NAFLD and from whom plasma was available for the analysis detailed below (7). The study was approved by the Institutional Review Board of the University of Texas Health Science Center at San Antonio, and all patients signed a written, informed consent form before any procedures were done. All the selected patients were diagnosed with prediabetes or type 2 diabetes mellitus (T2DM) and had biopsy-proven NASH. Patients with any liver disease other than NASH (i.e., hepatitis B or C, autoimmune hepatitis, hemochromatosis, Wilson’s disease, or drug-induced hepatitis), type 1 diabetes, a history of clinically significant renal, pulmonary, or heart disease or a history of high alcohol intake (≥30 g/day in men or ≥20 g/day in women) were excluded from the study. After completion of the initial screening and baseline metabolic measurements, patients were prescribed a hypocaloric diet (−500 kcal) and were randomized to pioglitazone (Actos, Takeda Pharmaceuticals) 45mg daily or placebo for 18 mo. The following clinical and metabolic assessments were conducted at baseline and were repeated after 18 mo of pioglitazone (or placebo) therapy: 1) fasting plasma glucose, fasting plasma insulin, free fatty acids (FFA), and adiponectin concentrations; 2) total body fat (TBF) content measured by DEXA; 3) intrahepatic triglycerides (IHTG) measured by localized proton nuclear magnetic resonance spectroscopy; 4) insulin-induced suppression of EGP and of plasma FFA concentration, and the rate of glucose disappearance (Rd) measured during a low-dose (10 mlU·m−2·min−1) and high-dose (80 mlU·m−2·min−1) euglycemic-hyperinsulinemic clamp, respectively, as described previously (7).
Analysis of plasma BCAAs by gas chromatography-mass spectrometry.
Human plasma BCAA concentrations were determined in baseline samples before start of the insulin infusion and in samples from the last 30 min of the low-dose and high-dose insulin clamps. This was repeated at baseline and after 18 mo of pioglitazone treatment. Plasma BCAA concentrations were measured by isotope dilution with gas chromatography-mass spectrometry (GC-MS). For this, a known concentration of internal standard (hydrolyzed [U-13C,15N]-algae mixture) was added to 50 µl of the plasma. Plasma samples were deproteinized with cold acetone, and the supernatant was dried under nitrogen gas before conversion to the amino acids to their respective tert-butyldimethylsilyl (TBDMS) derivatives (37). The BCAA derivatives were separated on a HP-5MS column (30 m × 0.25 mm × 0.25 μm, Agilent) and fragmented under electron ionization (HP 5973N Mass Selective Detector, Agilent). The unlabeled BCAAs were compared with their respective internal standards to calculate their plasma concentrations.
EGP and hepatic mitochondrial TCA cycle metabolism by 13C-NMR in mice.
Glucose in mice's plasma was converted to the 1,2-isopropylidene glucofuranose derivative (monoacetone glucose) before 13C isotopomer analysis. Following 13C-NMR, the 13C-multiplet peak areas in monoacetone glucose were analyzed using 1-D NMR software ACD/Laboratories, 9.0, before metabolic analysis as reported previously (34). EGP was determined for the stable isotope dilution of [3,4-13C2]glucose in mouse plasma. Rates of mitochondrial anaplerosis, pyruvate cycling and TCA cycle flux were determined from the relative enrichments of the carbon-2 multiplets in glucose, following their normalization to EGP (29, 34).
Lipidomic analysis of mouse liver by high-resolution liquid chromatography-tandem mass spectrometry.
Approximately 15–20 mg of powdered liver tissue was used to determine the relative concentrations of various lipid classes, including triacylglycerols (TGs), diacylglycerol (DAGs), ceramides (Cer), lysophosphatidylcholine (LPC), phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylglycerol (PG) by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Liver tissue was homogenized with ceramic beads in chloroform-methanol (2:1, vol/vol) for Folch extraction of lipids, following addition of Cer (d18:1/17:0) and d5-DG internal standard mixture I (Avanti Lipids, Alabaster, AL). Metabolites extracted were quantified by peak area comparison with their respective or a representative internal standard. Ionization was performed with heated electrospray ionization probe (HESI II), and mass spectra were acquired using a Q-Exactive Orbitrap (Thermo Scientific). Mass spectra were acquired in full scan mode using data-dependent top5 analysis (ddMS2-top5) in both positive and negative polarity. MZmine 2.0 (30) was employed for feature processing as described previously (21). Lipids were identified using LipidMatch (22). LipidMatch default fragmentation criteria using a 5-ppm tolerance for matching experimental fragment masses to in silico fragment masses were used: for DAGs, as ammoniated adducts neutral losses of both fatty acyl chains had to be observed; for Cers, as protonated adducts both the loss of water and the loss of two waters and the fatty acyl fragment were required (e.g., m/z 264.2684 for d18:1 containing Cers); and for Cers, as formate adducts the loss of formate and the loss of water and the backbone (e.g., m/z 280.2646 for Cer containing 16:0 in the sn2 position) were required. Another 23 types (different adducts and classes) of Cer included in the LipidMatch libraries were also queried, including glucosyl Cer and Cer phosphates. After identification, lipids were quantified using LipidMatch Quant, which uses the closest eluting standard representative of a lipid class for relative quantification of each respective feature. Both LipidMatch and LipidMatch Quant are available at http://secim.ufl.edu/secim-tools/.
Analysis of plasma ketones in mice by GC-MS.
Fed and fasted plasma ketone concentrations were analyzed by stable isotope dilution using GC-MS. Briefly, a known concentration of internal standard (3-[13C2]hydroxybutyrate; Cambridge Isotopes, Andover, MA) was added to 10–20 µl of plasma. The samples were deprotenized with 500 µl of cold acetone, and the supernatant was dried under nitrogen gas, followed by convertion of ketones to their TBDMS derivatives, before separation on a HP-5MS column (30 m × 0.25 mm × 0.25 μm, Agilent) under electron impact ionization (HP 5973N Mass Selective Detector, Agilent). The unlabeled 3-hydroxybutyrate was compared with its internal standards to calculate its plasma concentration.
Pioglitazone concentration in mice plasma.
Fed plasma was collected from the tail vein. Fed and fasted plasma pioglitazone concentrations were measured by a novel LC-MS/MS assay described previously (20).
Western blotting.
Approximately 50 mg of frozen tissues was homogenized in RIPA buffer (Cell Signaling Technology, Danvers, MA) containing protease inhibitors (Roche Diagnostics, Indianapolis, IN). Following SDS-PAGE, proteins were transferred onto to a nitrocellulose membrane (ProtranTM, Whatman/GE Healthcare, Piscataway, NJ) and incubated with primary antibodies (Cell Signaling Technology).
Gene expression analysis.
Frozen tissues (liver, muscle, and adipose) were ground to fine powder in liquid nitrogen. mRNA was extracted from the tissues, and 25 ng of cDNA was amplified by quantitative real-time PCR (CFX Real Time System, Bio-Rad, C1000 Touch Thermal Cycler), as reported previously (37). The comparative threshold method was used to determine relative mRNA levels with cyclophilin as the internal control.
Biochemical measurements.
Fed and fasted FFA concentrations were determined using an analytical kit (Wako chemicals, Richmond, VA). Fed and fasted plasma insulin was measured by enzyme-linked immunoassay using the mouse Insulin ELISA kit (Crystal Chem, Downers Grove, IL). Plasma and liver TG concentrations were determined using an analytical kit from Sigma (St. Louis, MO).
Statistical analysis.
Data are presented as means ± SE, and differences between groups were analyzed using ANOVA. Pairwise mean comparisons were done using an unpaired t-test, and differences were considered significant at P ≤ 0.05 and as trends at P ≤ 0.10. Multivariate analysis was conducted on the various classes of lipids, using partial least squares discriminant analysis (PLS-DA; in MetaboAnalyst 3.0), to maximize the separation between groups of observations and to understand which variables were responsible for the observed separation. A quality of prediction (Q2) value greater than 0.5 and an index of reproducibility of the PLS-DA model (R2) value greater than 0.6 were considered good for the PLS-DA analysis of all the classes of lipids. Correlation analyses between BCAAs and measurements of insulin sensitivity in human subjects (hepatic and plasma FFA suppression by insulin and Rd) were conducted using Pearson’s correlation in Stata 11.1 software (StataCorp, College Station, TX).
RESULTS
Pioglitazone administration improved whole body insulin sensitivity and adipose tissue metabolism in mice with NASH.
Plasma concentration of pioglitazone during the fed conditions (3,492 ± 154 ng/ml) were 15-fold higher than the fasting values (203 ± 18 ng/ml). The plasma metabolic and hormonal profile following pioglitazone administration in mice with NASH (Table 1) illustrates improvement in whole body insulin sensitivity. Treatment with pioglitazone lowered fasting plasma glucose levels in mice with NASH (TFD: 109 ± 8 vs. PIO: 88 ± 4 mg/dl, P = 0.04). Whereas the fasting plasma insulin levels remained similar between TFD-fed mice and those administered pioglitazone, there was a significant decrease in fed insulin levels in mice administered pioglitazone in their diets (TFD: 1.5 ± 0.2 vs. PIO: 0.9 ± 0.1 ng/ml, P = 0.03). There was also a decrease in fed plasma FFA (TFD: 0.36 ± 0.04 vs. PIO: 0.23 ± 0.03 mM, P = 0.04) and ketones (TFD: 134 ± 22 vs. PIO: 85 ± 3 µM, P = 0.07) in the mice administered pioglitazone. The fed-to-fasted differences in plasma FFA and ketone levels following pioglitazone administration, relative to the TFD fed mice and control mice indicate increased sensitivity to insulin stimulation following pioglitazone treatment.
Table 1.
Metabolic characteristics of C57BL/6J mice fed either control (C), high-fructose/high-transfat (TFD), or TFD diet supplemented with pioglitazone (PIO)
| C | TFD | PIO | P | |
|---|---|---|---|---|
| Body weight, g | 36.3 ± 1.0 | 38.9 ± 1.3 | 36.5 ± 0.5‡ | 0.131 |
| Plasma glucose, (fasting) mg/dl | 88 ± 5 | 109 ± 8* | 88 ± 4‡ | 0.036 |
| Plasma insulin (fed), ng/ml | 1.1 ± 0.2 | 1.5 ± 0.2 | 0.9 ± 0.1‡ | 0.127 |
| Plasma insulin (fasting), ng/ml | 0.20 ± 0.03 | 0.17 ± 0.02 | 0.16 ± 0.01 | 0.220 |
| Plasma FFA (fed), mM | 0.23 ± 0.04 | 0.36 ± 0.04 | 0.23 ± 0.03‡ | 0.046 |
| Plasma FFA (fasting), mM | 0.60 ± 0.05 | 0.45 ± 0.06 | 0.43 ± 0.03† | 0.068 |
| Plasma ketones (fed), µM | 82 ± 8 | 134 ± 22* | 85 ± 3 | 0.034 |
| Plasma ketones (fasting), µM | 732 ± 16 | 506 ± 30* | 501 ± 47† | <0.001 |
| Plasma triglyceride (fasting), mg/ml | 1.0 ± 0.02 | 1.3 ± 0.2 | 1.6 ± 0.1† | 0.022 |
| Plasma adiponectin (fasting), µg/ml | 8.6 ± 0.2 | 5.8 ± 0.3* | 8.7 ± 0.5‡ | <0.001 |
| Plasma pioglitazone (fed), ng/ml | — | — | 3492 ± 154 | |
| Plasma pioglitazone (fasting), ng/ml | — | — | 203 ± 18 | |
| Liver weight, g | 1.4 ± 0.1 | 3.1 ± 0.3* | 2.5 ± 0.1† | <0.001 |
| Liver triglyceride, mg/g liver | 126 ± 17 | 322 ± 19* | 275 ± 24† | <0.001 |
| Liver triglyceride, mg/whole liver | 174 ± 33 | 994 ± 106* | 751 ± 87† | <0.001 |
Values are means ± SE (n = 5–7 per group). FFA, free fatty acids.
P ≤ 0.05 C vs. TFD;
P ≤ 0.05 TFD vs. PIO;
P ≤ 0.05 C vs. PIO.
Concurrently, pioglitazone-treated mice had higher plasma adiponectin levels relative to their TFD fed counterparts (TFD: 5.8 ± 0.3 vs. PIO: 8.7 ± 0.5 µg/ml, P < 0.01). Moreover, adipose tissue gene expression analysis demonstrated increased Ppara and Pparg activity and decreased activity of Tnfa, Il6, and Ucp2 following pioglitazone administration (Fig. 1). Insulin signaling in adipose tissue also tended to improve in response to pioglitazone administration, as reflected by the enhanced phosphorylation of Akt Thr308 relative to TFD feeding. Taken together, these results suggest that pioglitazone action improves adipose tissue insulin action and whole body insulin sensitivity.
Fig. 1.

Pioglitazone restores adipose tissue gene expression in mice with nonalcoholic steatohepatitis (NASH). Changes in adipose tissue genes involved in fatty acid metabolism and inflammation: (A) Ppara, (B) Pparg, (C) Il6, (D) Tnfa, and (E) Ucp2, with pioglitazone treatment points to its beneficial effects on adipose tissue insulin sensitivity and function. Data are represented as means ± SE (n = 4–5). *P ≤ 0.05 between control (C) and high-transfat/high-fructose fed (TFD); #P ≤ 0.05 between TFD and TFD + pioglitazone (PIO); $P ≤ 0.05 between C and PIO. mice; PIO, mice fed TFD diet supplemented with pioglitazone.
Pioglitazone administration decreased hepatic mitochondrial oxidative flux in mice with NASH.
Although pioglitazone’s beneficial effects on adipose tissue and peripheral insulin sensitivity are relatively well documented (8, 10), its effect on hepatic mitochondrial metabolism remains unclear. On the basis of the fact that chronic overactivity of hepatic oxidative metabolism, especially through the TCA cycle, is a central feature of NAFLD (23, 29, 34, 38), we tested pioglitazone’s ability to alleviate mitochondrial oxidative burden. The reduction in fasting blood glucose concentrations with pioglitazone (Table 1) was associated with a trend toward a lower rate of EGP (Fig. 2A) and a significant decrease in nutrient flux through the hepatic mitochondrial TCA cycle (Fig. 2B; TFD: 9.1 ± 1.2 vs. PIO: 5.3 ± 0.4 µmol/min, P < 0.01). Furthermore, there were also concomitant lowering of total mitochondrial anaplerosis (Fig. 2C; TFD: 26.0 ± 4.7 vs. PIO: 15.6 ± 0.9 µmol/min, P = 0.03) and pyruvate cycling (Fig. 2D; TFD: 20.0 ± 4.1 vs. PIO: 10.2 ± 0.8 µmol/min, P = 0.02) in response to pioglitazone. Taken together, these data provide evidence of an overall pioglitazone-induced reduction in the activity of mitochondrial TCA cycle metabolism. However, the high rates of total anaplerosis, which reflects the activity of pyruvate carboxylase enzyme and is closely coupled to phosphoenolpyruvate carboxykinase and cataplerosis, was tightly correlated to hepatic TCA cycle flux (Fig. 2E). This correlation is a robust reflection of the close association between hepatic TCA cycle and gluconeogenesis (29, 34, 38). Thus, pioglitazone’s glucose-lowering effects could be partly mediated through reduced gluconeogenesis fueled by the hepatic TCA cycle.
Fig. 2.

Pioglitazone administration downregulates hepatic mitochondrial TCA cycle activity in mice with NASH. (A) endogenous glucose production, (B) TCA cycle flux, (C) anaplerosis, and (D) pyruvate cycling, determined using 13C-NMR-based isotopomer analysis. TCA cycle flux, anaplerosis, and pyruvate cycling were all downregulated in mice fed a TFD diet supplemented with pioglitazone. E: the robust relationship between hepatic mitochondrial TCA cycle flux and mitochondrial anaplerosis also suggests that pioglitazone could be improving gluconeogenesis by modulating hepatic TCA cycle function. Values are means ± SE (n = 7–8 per group).
Pioglitazone administration decreased hepatic TG content and improved the lipidomic profile of the liver in mice with NASH.
As expected, pioglitazone administration reduced liver TG accumulation in the liver of mice fed the TFD diet (Table 1). We then determined whether the reduction in total TG accumulation following pioglitazone also translated to an improvement in the lipidomic profile of multiple lipid classes in the liver, utilizing high-resolution LC-MS/MS based metabolomics. Along with total hepatic lipid content, various classes of lipids, including TAGs and DAGs, showed significant reductions in pioglitazone-administered mice (Fig. 3). To maximize the separation between groups of observations and understand which variables are responsible for the separation, we conducted PLS-DA analysis with the cut-off for the quality of prediction (Q2) and the reproducibility of the model (R2) indees for all the classes of lipids at Q2 >0.5 and R2 >0.6. The pioglitazone-induced change in the lipidomic profile is illustrated through the clear separations in the PLS-DA score plots of the total lipids (Fig. 3B), TGs (Fig. 3D), DAGs (Fig. 3F), and Cer (Fig. 3H). These separations were indeed further evident through the targeted metabolomics analysis, which demonstrated significant reductions in several specific TGs, DAGs, and Cer species following pioglitazone treatment (Fig. 4, A–C). Taken together, with the significant reduction in mitochondrial TCA cycle flux, pioglitazone-induced changes in lipidomic profiles clearly illustrate increased efficiency of the mitochondrial fat oxidation machinery. Pioglitazone administration in mice was also accompanied by a significant lowering of Srebp1c gene expression, compared with the TFD-fed mice with NASH (expression relative to control mice ± SE: TFD, 2.48 ± 0.26; PIO, 1.18 ± 0.21). Taken together, with the improvements in lipidomic profiles, the lower expression of Srebp1c in the pioglitazone-administered livers could be evidence of improvements in lipid storage mechanisms. Pioglitazone administration also resulted in statistically significant changes in lipid intermediates from other classes, including LPCs, PCs, PEs, and PGs. While these changes clearly illustrate the ability of pioglitazone to alter lipid profile, the interpretation of the directionality of these changes and their clinical relevance, is beyond the scope of this paper but presents an attractive avenue for future studies.
Fig. 3.

Pioglitazone administration decreases multiple lipid classes in liver of mice with NASH. Hepatic content of each lipid class and their respective partial least squares discriminant analysis (PLS-DA) score plots for the 3 groups of mice (C, TFD, and PIO) are presented for total lipids (A and B), total triglycerides (C and D), total diacylglycerols (E and F), and total ceramides (G and H). *P ≤ 0.05 between C and TFD; #P ≤ 0.05 between TFD and PIO; $P ≤ 0.05 between C and PIO.
Fig. 4.

High-resolution LC-MS/MS-based lipidomic profiling of (A) triglycerides, (B) diacylglycerols, and (C) ceramides in the liver demonstrate significant changes in several of these lipid intermediates in the liver in response to pioglitazone administration in mice with NASH. Values for each bar are expressed as mean fold change from the control mice average ± SE (n = 6–8 per group). *P ≤ 0.05 between C and TFD; #P ≤ 0.05 between TFD and PIO; $P ≤ 0.05 between C and PIO.
Pioglitazone improved plasma BCAA profiles in human subjects with NASH.
There is growing evidence pointing to significant cross-talk between lipid metabolism and changes in BCAA metabolism during insulin resistance, obesity, and their comorbidities (24–26). Considering this, we investigated whether pioglitazone treatment also resulted in a parallel improvement in plasma amino acid profiles in human subjects with NASH under basal conditions as well as under low vs. high hyperinsulinemic-euglycemic clamp conditions. Baseline metabolic characteristics of human subjects randomized to either pioglitazone or placebo treatment are presented in Table 2. As can be observed, no important differences were observed between the groups at baseline. Pioglitazone administration for 18 mo resulted in lower levels of BCAAs compared with placebo under both basal fasting conditions and during the euglycemic-hyperinsulinemic clamp (Fig. 5, A–C). Of note, the change in suppression of all the three BCAAs after 18 mo of pioglitazone treatment, determined under a high-dose insulin clamp, correlated well with the corresponding change in suppression of plasma FFA and with whole body insulin-stimulated glucose uptake (Rd) (Fig. 5, D–I; P ≤ 0.10 and P ≤ 0.01, respectively). These results point to a dynamic interaction between pioglitazone-driven changes in the adipose tissue and skeletal muscle and BCAA metabolism. More importantly, these results also suggest that the beneficial effects of pioglitazone treatment extend beyond the normal paradigm of improving lipid accumulation in the liver.
Table 2.
Baseline metabolic characteristics of human subjects randomized to either placebo or pioglitazone treatment
| Placebo (n = 23) |
Pioglitazone (n = 27) |
P | |
|---|---|---|---|
| Age, yr | 54 (9) | 54 (8) | 0.98 |
| Sex (male), n (%) | 74% | 70% | 0.78 |
| Type 2 diabetes mellitus, n (%) | 70% | 63% | 0.62 |
| Body mass index, kg/m2 | 34.3 (4.7) | 33.8 (4.9) | 0.68 |
| Total body fat by DEXA, % | 34 (8) | 32 (7) | 0.60 |
| Liver fat, % | 16 (9) | 18 (9) | 0.65 |
| Fasting plasma glucose, mg/dl | 128 (31) | 128 (33) | 0.98 |
| A1c, % | 6.5 (0.9) | 6.6 (1.1) | 0.68 |
| Fasting plasma insulin, μU/ml | 19 (11) | 14 (10) | 0.07 |
| FFA, mmol/l | 0.57 (0.19) | 0.49 (0.17) | 0.12 |
| Aspartate aminotransferase, U/l | 51 (28) | 57 (31) | 0.46 |
| Alanine aminotransferase, U/l | 76 (45) | 76 (46) | 0.99 |
| Adiponectin, μg/ml | 8.2 (5.4) | 9.1 (4.8) | 0.53 |
| Suppression of FFA by insulin, % | 40 (19) | 46 (21) | 0.34 |
| Suppression of EGP by insulin, % | −0.4 (0.2) | −0.4 (0.2) | 0.77 |
| Rd, mg kgLBM−1 min−1 | 4.8 (2.7) | 6.5 (3.5) | 0.05 |
DEXA, dual-energy X-ray absorptiometry; EGP, endogenous glucose production; Rd, insulin-stimulated glucose disposal. Continuous variables presented as means (SD).
Fig. 5.

Impact of pioglitazone on plasma branched-chain amino acid (BCAA) metabolism in human subjects with NASH. Pioglitazone lowered plasma BCAA levels following 18 mo of treatment, as illustrated under low- and high-dose euglycemic insulin clamp for (A) leucine, (B) isoleucine, and (C) valine. The change in suppression of BCAAs following pioglitazone treatment, plotted against the change in suppression of plasma free fatty acid (FFA) levels (measured under a high-dose insulin clamp) following pioglitazone treatment revealed significant relationships for (D) leucine, (E) isoleucine, and (F) valine. The change in suppression of BCAAs following pioglitazone administration, plotted against the change in suppression of glucose disposal rate (Rd; an index of muscle insulin sensitivity measured under a high-dose euglycemic insulin clamp) following pioglitazone treatment revealed significant relationships for (G) leucine, (H) isoleucine, and (I) valine.
Pioglitazone normalized the gene expressions of enzymes involved in BCAA degradation in multiple tissues.
In a modest effort to determine whether pioglitazone could influence the activity of enzymes involved in the degradation of BCAAs, we determined the gene expression patterns of Bckdha and Bcat2 in multiple tissues from mice administered pioglitazone in their diets. Interestingly, in all three tissues, liver, skeletal muscle, and adipose tissue, the expressions of both Bckdha and Bcat2 were induced by pioglitazone administration compared with their TFD-fed counterparts (Fig. 6). These results suggest that pioglitazone can normalize dysfunctional BCAA degradation. This, in turn, could be responsible for the improvements in plasma BCAA profile observed in human subjects with NASH.
Fig. 6.
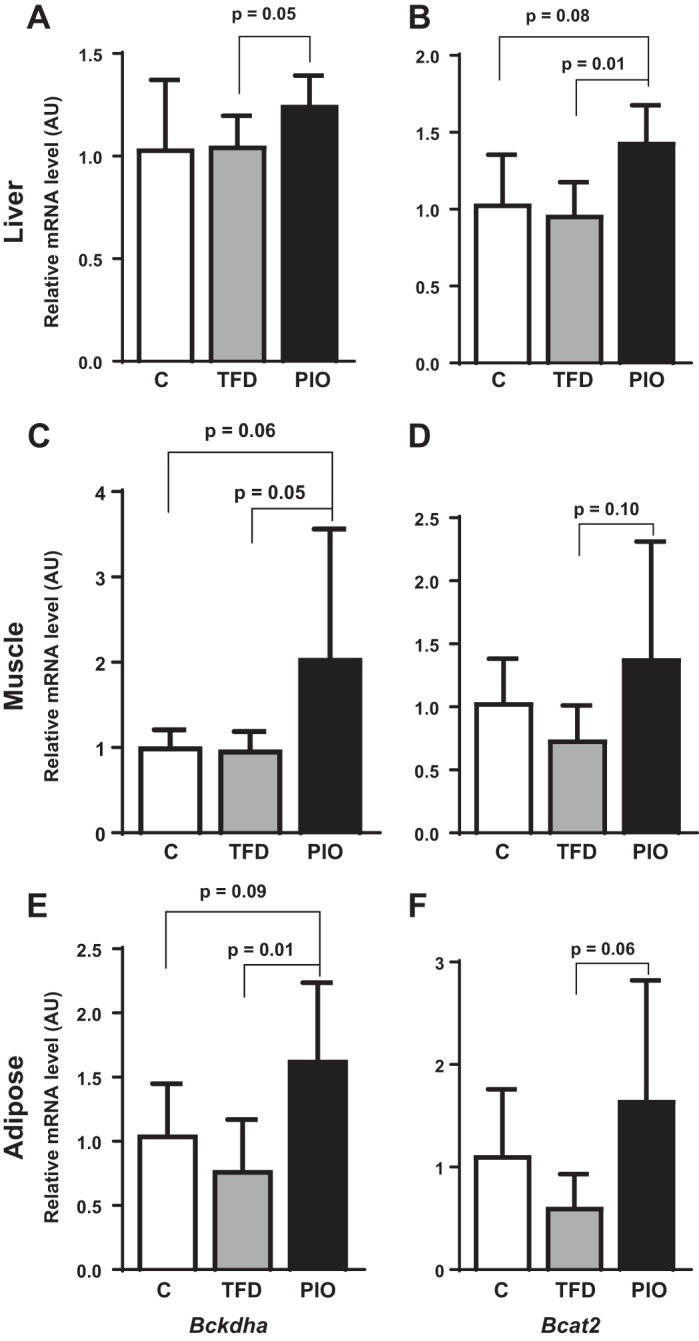
Impact of pioglitazone administration on BCAA degradation enzymes Bckdha and Bcat2, respectively, in (A and B) liver, (C and D) muscle, and (E and F) adipose tissue of mice fed either C, TFD, or PIO diets.
DISCUSSION
The therapeutic management of NAFLD with pioglitazone has been gaining prominence with the recent evidence of the long-term ability of pioglitazone to improve hepatic and peripheral insulin sensitivity, hepatic steatosis, and liver histology in patients with NASH (7). Despite these findings, it has never been tested whether these improvements in liver histology and hepatic steatosis with pioglitazone correlate with changes in mitochondrial metabolism and/or alleviation of the lipotoxic lipid profile, usually associated with NAFLD. Utilizing a mouse model of hepatic insulin resistance and NASH (4, 29), we show that the beneficial metabolic effects of pioglitazone in the liver are indeed mediated, at least in part, by enhancing mitochondrial TCA cycle metabolism with a concomitant improvement in the lipidomic profile. Furthermore, we also identified that pioglitazone’s therapeutic value extends beyond the normal paradigm of improving whole body lipid metabolism, also alleviating dysfunctional BCAA metabolism (18). These results suggest that pioglitazone targets insulin resistance across multiple tissues and mechanisms in NASH.
Pioglitazone is a PPARγ agonist, thought to act primarily through improving adipose tissue function and in turn whole body insulin sensitivity (6, 8, 36). Indeed, pioglitazone’s well known beneficial effects on adipose tissue and whole body insulin sensitivity were replicated in our mice model of NASH following pioglitazone treatment for 16 wk. Pioglitazone administration to TFD-fed mice resulted in a range of beneficial metabolic effects, including lowering of fed plasma insulin and FFA levels, normalization of plasma adiponectin levels, and improvements in adipose tissue gene expression of Ppara, Pparg, Tnfa, Il6, and Ucp2, together with modest improvements is phosphorylation of Akt in adipose tissue. These effects of pioglitazone point not only to improvements in adipose tissue insulin sensitivity, but also on the impact of pioglitazone to improve whole body insulin resistance, as previously documented in in vivo mouse models and in humans (7, 11, 12).
We then focused our investigations to identify how pioglitazone impacts hepatic metabolism, specifically mitochondrial TCA cycle, and accumulation of lipotoxic metabolites in the liver. Dysfunctional mitochondrial lipid metabolism is a central feature of NAFLD (23, 34, 36, 38). Mitochondria in the liver integrates several critical pathways of energy metabolism (e.g., β-oxidation, TCA cycle, and mitochondrial respiration), which fuels glucose production and lipid synthesis. Our laboratory and others’ have provided evidence that hepatic insulin resistance and inflammation closely mirror alterations in mitochondrial energy metabolism in both rodents and humans with NAFLD (23, 33, 34). During NAFLD, FFA overload and dysfunctional mitochondrial energy metabolism also result in the accumulation of lipotoxic intermediates (e.g., ceramides, DAGs) and oxidative stress (29). We first tested whether pioglitazone administration along with TFD diet, could remodel mitochondrial TCA cycle metabolism. Compared with mice with NASH, pioglitazone administration resulted in a significant downregulation of hepatic TCA cycle fluxes. It would be logical to assume that the lower activity of the hepatic TCA cycle could be a response to the lower rate of FFA delivery to the liver (7, 10, 12), a consequence of improved adipose tissue insulin sensitivity.
Pioglitazone’s ability to lower hepatic mitochondrial metabolism could have significant implications in slowing down the progression of liver disease from simple steatosis to NASH. An emerging body of evidence both in mouse models and in human subjects point to a hepatic mitochondrial remodeling resulting in a chronic induction of oxidative metabolism during simple steatosis and NASH (16, 23, 29, 34, 38). The induction of mitochondrial oxidative metabolism could be specific to the stage of the liver disease, for example lower induction in NASH compared with simple steatosis, highlighting an inflexibility of the liver mitochondria to efficiently adapt to lipid burden (23). Irrespective of this, chronic and sustained induction of mitochondrial oxidative metabolism could sustain inflammatory pathways, ROS production, and oxidative stress (33). Thus, we believe that a further therapeutic induction of mitochondrial oxidative metabolism with the goal of alleviating lipid burden on the hepatocyte, under conditions of NAFLD, will only worsen mitochondrial function and oxidative stress. Consequently, pioglitazone’s ability to modulate and lower hepatic TCA cycle activity could be beneficial in optimizing the mitochondrial oxidative machinery and relieving oxidative stress on the hepatocyte.
Pioglitazone’s impact on mitochondrial TCA cycle function could be put into perspective by considering its impact on improving the lipidomic profile in the liver. Apart from the significant lowering of the total hepatic lipid content, pioglitazone also decreased accumulation of several species of Cer and DAGs in comparison to mice with NASH. This significant effect of pioglitazone is illustrated by 1) the reduction in total hepatic content of these metabolites, 2) the PLSDA score plots of individual metabolite classes, demonstrating clear separation between the pioglitazone-administered group and mice with NASH, and 3) the targeted lipidomic analysis of TGs, DAGs, and Cer highlighting significant lowering of several individual species in response to pioglitazone administration. Taken together with the downregulation of hepatic TCA cycle metabolism, the improved lipidomic profile after pioglitazone administration points to an overall improvement in the efficiency of hepatic mitochondrial fat oxidation and lipid storage mechanisms. The substantial lowering of fed plasma ketones and upregulation of Srebp1c, a master regulator of lipid storage, following pioglitazone administration also attest to a significant improvement in the mitochondrial fat oxidation and lipid storage machinery, respectively.
The cross-talk between BCAAs and mitochondrial lipid metabolism is an emerging area of interest in the etiology of insulin resistance and NAFLD (24, 25, 37). We wanted to determine whether the pleiotropic effects of pioglitazone action extend into modulating BCAA metabolism. First, we determined the ability of pioglitazone to modulate plasma BCAAs in human subjects with NASH. Indeed, 18 mo of pioglitazone treatment lowered plasma BCAA levels under fasting basal conditions and also under low- and high-dose euglycemic-hyperinsulinemic clamps. Although these results cannot be taken as proof for a specific effect of pioglitazone on BCAA metabolism, we identified significant correlations between the change in suppression of BCAAs under a high-dose insulin clamp after pioglitazone treatment and a similar change in suppression of FFA. A significant correlation was also found between the change in suppression of BCAAs under a high-dose insulin clamp after pioglitazone treatment and similar change in glucose Rd. These observations point to the potential impact of pioglitazone on adipose tissue and muscle, respectively, in modulating plasma BCAA levels. These pioglitazone-mediated effects could also indirectly result in improvements in hepatic insulin sensitivity. To further determine whether pioglitazone’s tissue-specific actions is mediated through modulating BCAA catabolism, we determined tissue-specific gene expression of Bckdha and Bcat2 in liver, muscle, and adipose tissue of mice supplemented with pioglitazone compared with their counterparts with NASH. There was an overall induction of both Bckdha and Bcat2 in all the three tissues in response to pioglitazone administration in mice. Considering the fact that defects in BCAAs degradation are central to insulin resistance (15, 27, 35, 40), improvement in BCAA catabolism is another significant beneficial effect of pioglitazone treatment in the setting of NAFLD.
In summary, the results above offer unique insights to understanding the mechanism of action of pioglitazone. In a well-established animal model of NAFLD, in addition to well-established effects on adipose tissue and peripheral insulin sensitivity, pioglitazone had an important effect on hepatic TCA cycle flux, anaplerosis, and pyruvate cycling and was associated with a marked reduction in hepatic steatosis and toxic lipid metabolites (i.e., DAGs). In human subjects, changes induced by the thiazolidinedione on insulin resistance were associated with positive changes in BCAA metabolism, a novel observation that opens a new avenue for their role in NAFLD. Taken together, the data above indicate that pioglitazone may exert a broad spectrum of metabolic effects in NASH that clearly deserves future exploration.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant (RO1-DK-112865; to N. E. Sunny), the University of Florida Research Opportunity Seed Fund Award (00089467; to N. E. Sunny), the Southeast Center for Integrated Metabolomics; NIDDK Grant U24-DK-097209, the Burroughs Wellcome Fund (to K. Cusi), the American Diabetes Association (1-08-CR-08; to K. Cusi), and the National Institutes of Health and National Center for Research Resources Clinical and Translational Science Award to the University of Florida (UL1-TR-00064).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.K., F.B., K.C., and N.E.S. conceived and designed research; S.K., K.A., J.G., P.A., W.-Y.L., D.J., and N.E.S. performed experiments; S.K., F.B., J.P.K., T.J.G., and N.E.S. analyzed data; S.K., F.B., J.P.K., R.A.Y., R.F.F., T.J.G., K.C., and N.E.S. interpreted results of experiments; S.K., F.B., and N.E.S. prepared figures; S.K. and N.E.S. drafted manuscript; S.K., F.B., J.P.K., R.A.Y., R.F.F., T.J.G., K.C., and N.E.S. edited and revised manuscript; S.K., K.C., and N.E.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to James R. Rocca and the Advanced Magnetic Resonance Imaging and Spectroscopy Facility at the University of Florida, Gainesville, for assistance with carbon-13-based NMR isotopomer analysis. Ariana Vergara at the University of Florida provided valuable assistance with biochemical and gene expression assays.
REFERENCES
- 1.Ahmed A, Wong RJ, Harrison SA. Nonalcoholic fatty liver disease review: diagnosis, treatment, and outcomes. Clin Gastroenterol Hepatol 13: 2062–2070, 2015. doi: 10.1016/j.cgh.2015.07.029. [DOI] [PubMed] [Google Scholar]
- 2.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, Berria R, Ma JZ, Dwivedi S, Havranek R, Fincke C, DeFronzo R, Bannayan GA, Schenker S, Cusi K. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 355: 2297–2307, 2006. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 3.Bril F, Cusi K. Nonalcoholic fatty liver disease: the new complication of type 2 diabetes mellitus. Endocrinol Metab Clin North Am 45: 765–781, 2016. doi: 10.1016/j.ecl.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 4.Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, Athanacio J, Villescaz C, Ghosh SS, Heilig JS, Lowe C, Roth JD. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 305: G483–G495, 2013. doi: 10.1152/ajpgi.00079.2013. [DOI] [PubMed] [Google Scholar]
- 5.Coletta DK, Sriwijitkamol A, Wajcberg E, Tantiwong P, Li M, Prentki M, Madiraju M, Jenkinson CP, Cersosimo E, Musi N, Defronzo RA. Pioglitazone stimulates AMP-activated protein kinase signalling and increases the expression of genes involved in adiponectin signalling, mitochondrial function and fat oxidation in human skeletal muscle in vivo: a randomised trial. Diabetologia 52: 723–732, 2009. doi: 10.1007/s00125-008-1256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cusi K. Treatment of patients with type 2 diabetes and non-alcoholic fatty liver disease: current approaches and future directions. Diabetologia 59: 1112–1120, 2016. doi: 10.1007/s00125-016-3952-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, Tio F, Hardies J, Darland C, Musi N, Webb A, Portillo-Sanchez P. Long-term pioglitazone treatment for patients With nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med 165: 305–315, 2016. doi: 10.7326/M15-1774. [DOI] [PubMed] [Google Scholar]
- 8.de Souza CJ, Eckhardt M, Gagen K, Dong M, Chen W, Laurent D, Burkey BF. Effects of pioglitazone on adipose tissue remodeling within the setting of obesity and insulin resistance. Diabetes 50: 1863–1871, 2001. doi: 10.2337/diabetes.50.8.1863. [DOI] [PubMed] [Google Scholar]
- 9.García-Ruiz I, Solís-Muñoz P, Fernández-Moreira D, Muñoz-Yagüe T, Solís-Herruzo JA. Pioglitazone leads to an inactivation and disassembly of complex I of the mitochondrial respiratory chain. BMC Biol 11: 88, 2013. doi: 10.1186/1741-7007-11-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gastaldelli A, Casolaro A, Ciociaro D, Frascerra S, Nannipieri M, Buzzigoli E, Ferrannini E. Decreased whole body lipolysis as a mechanism of the lipid-lowering effect of pioglitazone in type 2 diabetic patients. Am J Physiol Endocrinol Metab 297: E225–E230, 2009. doi: 10.1152/ajpendo.90960.2008. [DOI] [PubMed] [Google Scholar]
- 11.Gastaldelli A, Harrison S, Belfort-Aguiar R, Hardies J, Balas B, Schenker S, Cusi K. Pioglitazone in the treatment of NASH: the role of adiponectin. Aliment Pharmacol Ther 32: 769–775, 2010. doi: 10.1111/j.1365-2036.2010.04405.x. [DOI] [PubMed] [Google Scholar]
- 12.Gastaldelli A, Harrison SA, Belfort-Aguilar R, Hardies LJ, Balas B, Schenker S, Cusi K. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology 50: 1087–1093, 2009. doi: 10.1002/hep.23116. [DOI] [PubMed] [Google Scholar]
- 13.Gastaldelli A, Miyazaki Y, Mahankali A, Berria R, Pettiti M, Buzzigoli E, Ferrannini E, DeFronzo RA. The effect of pioglitazone on the liver: role of adiponectin. Diabetes Care 29: 2275–2281, 2006. doi: 10.2337/dc05-2445. [DOI] [PubMed] [Google Scholar]
- 14.Green CR, Wallace M, Divakaruni AS, Phillips SA, Murphy AN, Ciaraldi TP, Metallo CM. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat Chem Biol 12: 15–21, 2016. doi: 10.1038/nchembio.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem 285: 11348–11356, 2010. doi: 10.1074/jbc.M109.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iozzo P, Bucci M, Roivainen A, Någren K, Järvisalo MJ, Kiss J, Guiducci L, Fielding B, Naum AG, Borra R, Virtanen K, Savunen T, Salvadori PA, Ferrannini E, Knuuti J, Nuutila P. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 139: 846–856, 2010. doi: 10.1053/j.gastro.2010.05.039. [DOI] [PubMed] [Google Scholar]
- 17.Iwasa M, Ishihara T, Mifuji-Moroka R, Fujita N, Kobayashi Y, Hasegawa H, Iwata K, Kaito M, Takei Y. Elevation of branched-chain amino acid levels in diabetes and NAFL and changes with antidiabetic drug treatment. Obes Res Clin Pract 9: 293–297, 2015. doi: 10.1016/j.orcp.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 18.Kakazu E, Kondo Y, Ninomiya M, Kimura O, Nagasaki F, Ueno Y, Shimosegawa T. The influence of pioglitazone on the plasma amino acid profile in patients with nonalcoholic steatohepatitis (NASH). Hepatol Int 7: 577–585, 2013. doi: 10.1007/s12072-012-9395-y. [DOI] [PubMed] [Google Scholar]
- 19.Kanatani Y, Usui I, Ishizuka K, Bukhari A, Fujisaka S, Urakaze M, Haruta T, Kishimoto T, Naka T, Kobayashi M. Effects of pioglitazone on suppressor of cytokine signaling 3 expression: potential mechanisms for its effects on insulin sensitivity and adiponectin expression. Diabetes 56: 795–803, 2007. doi: 10.2337/db06-1039. [DOI] [PubMed] [Google Scholar]
- 20.Kawaguchi-Suzuki M, Bril F, Sanchez PP, Cusi K, Frye RF. A validated liquid chromatography tandem mass spectrometry method for simultaneous determination of pioglitazone, hydroxypioglitazone, and ketopioglitazone in human plasma and its application to a clinical study. J Chromatogr B Analyt Technol Biomed Life Sci 969: 219–223, 2014. doi: 10.1016/j.jchromb.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 21.Koelmel JP, Kroeger NM, Gill EL, Ulmer CZ, Bowden JA, Patterson RE, Yost RA, Garrett TJ. Expanding lipidome coverage using LC-MS/MS data-dependent acquisition with automated exclusion list generation. J Am Soc Mass Spectrom 28: 908–917, 2017. doi: 10.1007/s13361-017-1608-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koelmel JP, Kroeger NM, Ulmer CZ, Bowden JA, Patterson RE, Cochran JA, Beecher CWW, Garrett TJ, Yost RA. LipidMatch: an automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinformatics 18: 331, 2017. doi: 10.1186/s12859-017-1744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, Schlensak M, Roden M. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 21: 739–746, 2015. doi: 10.1016/j.cmet.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 24.Kujala UM, Peltonen M, Laine MK, Kaprio J, Heinonen OJ, Sundvall J, Eriksson JG, Jula A, Sarna S, Kainulainen H. Branched-chain amino acid levels are related with surrogates of disturbed lipid metabolism among older men. Front Med (Lausanne) 3: 57, 2016. doi: 10.3389/fmed.2016.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lerin C, Goldfine AB, Boes T, Liu M, Kasif S, Dreyfuss JM, De Sousa-Coelho AL, Daher G, Manoli I, Sysol JR, Isganaitis E, Jessen N, Goodyear LJ, Beebe K, Gall W, Venditti CP, Patti ME. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab 5: 926–936, 2016. doi: 10.1016/j.molmet.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, Zhou B, Cao Y, Ritterhoff J, Gu H, Raftery D, Sun H, Tian R. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab 25: 374–385, 2017. doi: 10.1016/j.cmet.2016.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol 10: 723–736, 2014. doi: 10.1038/nrendo.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, Haqq AM, Shah SH, Arlotto M, Slentz CA, Rochon J, Gallup D, Ilkayeva O, Wenner BR, Yancy WS Jr, Eisenson H, Musante G, Surwit RS, Millington DS, Butler MD, Svetkey LP. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab 9: 311–326, 2009. doi: 10.1016/j.cmet.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patterson RE, Kalavalapalli S, Williams CM, Nautiyal M, Mathew JT, Martinez J, Reinhard MK, McDougall DJ, Rocca JR, Yost RA, Cusi K, Garrett TJ, Sunny NE. Lipotoxicity in steatohepatitis occurs despite an increase in tricarboxylic acid cycle activity. Am J Physiol Endocrinol Metab 310: E484–E494, 2016. doi: 10.1152/ajpendo.00492.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pluskal T, Castillo S, Villar-Briones A, Oresic M. MZmine 2: modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11: 395, 2010. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA 313: 2263–2273, 2015. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 32.Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR; NASH CRN . Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 362: 1675–1685, 2010. doi: 10.1056/NEJMoa0907929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, Sunny NE, He T, Nair LA, Livingston KA, Fu X, Merritt ME, Sherry AD, Malloy CR, Shelton JM, Lambert J, Parks EJ, Corbin I, Magnuson MA, Browning JD, Burgess SC. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest 126: 1605, 2016. doi: 10.1172/JCI86695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Satapati S, Sunny NE, Kucejova B, Fu X, He TT, Méndez-Lucas A, Shelton JM, Perales JC, Browning JD, Burgess SC. Elevated TCA cycle function in the pathology of diet-induced hepatic insulin resistance and fatty liver. J Lipid Res 53: 1080–1092, 2012. doi: 10.1194/jlr.M023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab 293: E1552–E1563, 2007. doi: 10.1152/ajpendo.00134.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sunny NE, Bril F, Cusi K. Mitochondrial adaptation in nonalcoholic fatty liver disease: novel mechanisms and treatment strategies. Trends Endocrinol Metab 28: 250–260, 2017. doi: 10.1016/j.tem.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 37.Sunny NE, Kalavalapalli S, Bril F, Garrett TJ, Nautiyal M, Mathew JT, Williams CM, Cusi K. Cross-talk between branched-chain amino acids and hepatic mitochondria is compromised in nonalcoholic fatty liver disease. Am J Physiol Endocrinol Metab 309: E311–E319, 2015. doi: 10.1152/ajpendo.00161.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sunny NE, Parks EJ, Browning JD, Burgess SC. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 14: 804–810, 2011. doi: 10.1016/j.cmet.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, O’Donnell CJ, Carr SA, Mootha VK, Florez JC, Souza A, Melander O, Clish CB, Gerszten RE. Metabolite profiles and the risk of developing diabetes. Nat Med 17: 448–453, 2011. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wiklund P, Zhang X, Pekkala S, Autio R, Kong L, Yang Y, Keinänen-Kiukaanniemi S, Alen M, Cheng S. Insulin resistance is associated with altered amino acid metabolism and adipose tissue dysfunction in normoglycemic women. Sci Rep 6: 24540, 2016. doi: 10.1038/srep24540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshiuchi K, Kaneto H, Matsuoka TA, Kasami R, Kohno K, Iwawaki T, Nakatani Y, Yamasaki Y, Shimomura I, Matsuhisa M. Pioglitazone reduces ER stress in the liver: direct monitoring of in vivo ER stress using ER stress-activated indicator transgenic mice. Endocr J 56: 1103–1111, 2009. doi: 10.1507/endocrj.K09E-140. [DOI] [PubMed] [Google Scholar]