

Abstract
We recently reported the case of a young patient with multisystem failure carrying a de novo mutation in SLC12A2, the gene encoding the Na-K-2Cl cotransporter-1 (NKCC1). Heterologous expression studies in nonepithelial cells failed to demonstrate dominant-negative effects. In this study, we examined expression of the mutant cotransporter in epithelial cells. Using Madin-Darby canine kidney (MDCK) cells grown on glass coverslips, permeabilized support, and Matrigel, we show that the fluorescently tagged mutant cotransporter is expressed in cytoplasm and at the apical membrane and affects epithelium integrity. Expression of the mutant transporter at the apical membrane also results in the mislocalization of some of the wild-type transporter to the apical membrane. This mistargeting is specific to NKCC1 as the Na+-K+-ATPase remains localized on the basolateral membrane. To assess transporter localization in vivo, we created a mouse model using CRISPR/cas9 that reproduces the 11 bp deletion in exon 22 of Slc12a2. Although the mice do not display an overt phenotype, we show that the colon and salivary gland expresses wild-type NKCC1 abundantly at the apical pole, confirming the data obtained in cultured epithelial cells. Enough cotransporter must remain, however, on the basolateral membrane to participate in saliva secretion, as no significant decrease in saliva production was observed in the mutant mice.
Keywords: colon, CRISPR mouse, epithelia, MDCK cells, Na-K-2Cl cotransport, salivary gland
INTRODUCTION
Epithelia serve many important physiological roles, including operating as a physical barrier between distinct compartments. This separation is especially relevant when one compartment is in contact with the outside environment. This is the case for airway, gastric, and intestinal epithelia, but also for sweat, salivary, lacrimal, and pancreatic glands that connect to the outside world through small ducts. Epithelia form a physical barrier by limiting the movement of pathogens (e.g., bacteria in the intestinal lumen) and other substances, while tightly regulating the movement of key solutes.
Directional solute transport, another key function of epithelia, depends on the integrity of epithelial cell polarity and the expression of specific solute transporters or channels on the apical and basolateral membranes. Solute and fluid transport through epithelia is critical for a variety of physiological processes, including kidney function and control of blood pressure (45, 47), processing and absorption of nutrients (44, 48) or fluid secretion (32) in the digestive tract, and production of cerebrospinal (4, 26) or inner ear (50) fluids. In fluid secreting epithelia, such as those of sweat, salivary, lacrimal, and pancreatic glands, but also airway and intestine, fluid movement is driven by the movement of Cl−. Cl− enters the epithelial cell at the basolateral (serosal) membrane through the Na-K-2Cl cotransporter, NKCC1 (16), and exits the cell through apical (luminal or mucosal) Cl− channels, such as the cystic fibrosis transmembrane regulator or CFTR (14, 41). Disruption of epithelial barrier integrity or transporter and channel expression typically results in loss of epithelial function.
There are many factors that participate in the development of a disease. Complex diseases (e.g., diabetes and hypertension) are thought to involve multiple genes as well as environmental factors and are found in large segments of the population. Monogenic diseases instead are typically rarer, unless a genetic advantage is provided to the carriers [e.g., sickle cell anemia (18), cystic fibrosis (15, 23)]. Many disease mechanisms have also been discovered by studying the molecular mechanisms of rare monogenic mutations in families or populations [e.g., Familial Hyperkalemia and Hypertension or FHHt (33), Agenesis of Corpus Callosum with Peripheral Neuropathy or ACCPN (22)]. Whole exome sequencing of patients with complex disorders now provides an opportunity to map rare diseases with gene variants.
Recently, we reported the case of a teenage girl suffering from multiorgan failure. She carries a de novo 11 bp deletion in exon 22 of SLC12A2 (solute carrier family 12, member 2), a related gene encoding the Na-K-2Cl cotransporter-1 (9). The deletion results in the truncation of the last 187 amino acid residues of the cytosolic carboxyl-terminal tail of the cotransporter. In a preliminary study, we showed that in heterologous expression systems, the mutant transporter was expressed at the plasma membrane, yet was nonfunctional, and was not exerting dominant-negative effects on bumetanide-sensitive K+ transport.
Because the carboxyl-terminal tail is involved in dimerization and trafficking of the cotransporter to the basolateral membrane of epithelial cells, here we sought to assess the polarity of NKCC1 expression in epithelial cells. We show that expressing the mutant transporter in cultured epithelial cells affects the integrity of the epithelium, and decreases expression of the endogenous wild-type (WT) cotransporter. We also show that the mutant transporter fails to locate to the basolateral membrane, but instead accumulates in the cytoplasm and the subapical and apical membrane. In addition, the mutant transporter targets some of wild-type transporter to the apical membrane.
Disruption of epithelial integrity is likely to affect key physiological processes. One of these processes is saliva production, which is mediated by acinar cells of the parotid, submandibular, and sublingual glands. In normal conditions, fluid secretion originates at the base of the gland and is mediated by Cl− movement through the epithelial cells, via basolateral NKCC1-mediated transport and apical Cl− channels. Cl− is later exchanged for bicarbonate in the ducts (37). It is known that disruption of NKCC1 (12) or CFTR (1) function in mice results in decreased saliva secretion. Here we show that NKCC1 localizes to the subapical and apical membrane of salivary epithelial cells in a mouse model of the patient’s mutation. Despite the mistargeting of some NKCC1 protein to the apical membrane, stimulated saliva secretion was not affected in the NKCC1 mutant mice.
MATERIALS AND METHODS
Reagents.
Prolong Gold Antifade reagent with DAPI was obtained from Life Technologies (Grand Island, NY). Matrigel was obtained from Corning, and ActinRed and ActinGreen ready probes were purchased from Invitrogen (Carlsbad, CA). Streptavidin agarose resin was purchased from Thermo Scientific (Waltham, MA). The following antibodies were used as primary antibodies: NKCC1 (T4) from the University of Iowa Developmental Studies Hybridoma Bank (DSHB: T4 monoclonal, 1:1,000); PDXL/Gp135 (DHSB: Podocalyxin catalog no. 3F2D8 monoclonal, 1:500) ; NKCC1 (Abcam, catalog no. ab59791, 1:2,000); zonula occludens-1 (ZO-1; DSHB: R26.4 monoclonal, 1:200); green fluorescent protein (GFP; Vanderbilt VAPR Core, 1C9A5 monoclonal; 1:1,000); Alexa-Fluor-488 tagged anti-GFP (ThermoFisher: catalog no. A-21311; 1:200); Ezrin (Millipore, Clone 4A5, catalog no. MAB3822-C; 1:200); anti-tdTomato (ThermoFisher, Clone RF5R; catalog no. MA5-15257, 1:200), β-actin (Sigma, AC-74 monoclonal, catalog no. A2228; 1:5,000), and anti c-Myc (ThermoFisher, clone 9E10; catalog no. MA1-980, 1:2,000).
cDNA clones.
The enhanced GFP (EGFP) and tdTomato (TdT) open reading frames were cloned at the extreme amino terminus of the 3.7 kb mouse NKCC1 cDNA. In the process, the first nine amino acid residues of NKCC1 were eliminated. Prior experiments performed in Xenopus laevis oocytes demonstrated no functional differences between wild-type NKCC1 and EGFP-NKCC1 constructs (17). The mouse cDNA carrying the DFX mutation was previously described (9). Epitope-tagged NKCC1-WT and NKCC1-DFX constructs were subcloned into pCDNA3 plasmid. To create the mutant NKCC1-G345R mutant, we first subcloned a 363 bp SacI-MfeI fragment from full-length mouse NKCC1 cDNA into a pBluescript vector with modified multiple cloning site (pBSK75). Using the Quikchange mutagenesis kit (Stratagene) and oligonucleotide primers 5′-CAATGGGTTCGTGAGAGGAGGAAGAGCGTACTATTTAATCTCTAG-3′ and reverse, the codon encoding Gly345, GGA, was modified into AGA to encode for arginine. After sequence verification, the mutated fragment was reintroduced into the NKCC1 cDNA to create the mutated cDNA clone.
Generation of stable Madin-Darby canine kidney cell lines.
NKCC1-WT and NKCC1-DFX cDNAs purified with Qiagen Mediprep kit and quantitated by absorbance at 260, 280, and 320 nm were transfected into Madin-Darby canine kidney (MDCK) cells using Lipofectamine 2000 (Life Technologies). Cells were then transferred in selection media (DMEM supplemented with 5% FBS, 1% penicillin and streptomycin, and 500 μg/ml of Geneticin) for 14 days. Stable cells lines were subsequently sorted by FACS. GFP-, tdTomato, and dually transfected cells were maintained in selection media and expanded.
Polarized epithelial cell culture.
Parental and transfected MDCK cells were grown on 12-mm (500 μl top/1 ml bottom) or 24-mm (1 ml top/2 ml bottom) polycarbonate Transwell filter inserts (0.4 μm pore size; Corning) in complete media (DMEM supplemented with 5% FBS, 1% penicillin and streptomycin). The media were changed every other day and the cells were considered polarized after 5 days.
Three-dimensional culture of MDCK cells on Matrigel.
Three-dimensional (3-D) cell cultures was performed as previously described (5). Briefly, four-well glass slides (Laboratory-TEK II) were coated with 85 μl of Matrigel solution at 4°C. Matrigel was allowed to solidify for 20 min at 37°C. Parental MDCK NKCC1-WT and NKCC1-DFX-expressing MDCK cells were resuspended in DMEM at 1 × 104 cells/ml. A volume of 20 μl of cells suspension was added to 200 μl of 2% Matrigel. This cell suspension was then layered on top of the solidified Matrigel and incubated at 37°C, 5% CO2 in a humidified incubator. Conditioned media were aspirated every 48 h and replaced with fresh 2% Matrigel in complete DMEM media. Transparent cysts with a hollow lumen were visible under bright-field microscope by day 4.
Selective cell surface biotinylation.
MDCK cells were grown on 24-mm (0.4-μm pore size) Transwell filters as described above. Cells were washed three times with ice-cold HBSS containing MgCl2 and CaCl2 (HBSS2+). Sulfo-NHS-LC-biotin (Pierce, Rockford, IL) was resuspended in ice-cold HBSS2+ at 1 mg/ml. The cold biotin solution was then added to either the apical side (1 ml) or the basolateral side (2 ml) of Transwell filters and incubated on ice in the cold room for 30 min. After the incubation period, cells were washed with ice-cold HBSS2+ and quenched with ice-cold 100 mM glycine in HBSS2+ and were washed again three times with ice-cold HBSS2+. Transwell filters were then cut and place in 1 ml of lysis buffer (125 mM NaCl, 200 mM HEPES pH 7.6, 1 mM EDTA, 1 mM EGTA 1% Triton X-100, 0.5% SDS, 10% glycerol and protease inhibitors) and incubated at 4°C on hematologic mixer for 30 min. Cell lysates were precleared by centrifugation at 14,000 rpm for 20 min. The resulting supernatant was immunoprecipitated with 50 μl of streptavidin agarose resin.
Western blot analysis.
For membrane and cytosolic fractionation; cells growing on six-well plates were first lysed with a hypotonic lysis buffer (10 mM Tris·HCl pH 7.5) supplemented with protease inhibitors (Roche Applied Science, Indianapolis, IN). Cell lysates were incubated at 4°C for 20 min with rotation, and then centrifuged at 14,000 rpm for 20 min. Supernatants containing the cytosolic fraction were transferred to new tubes and the heavy membranes containing pellets were resuspended in lysis buffer (125 mM NaCl, 200 mM HEPES pH 7.6, 1 mM EDTA, 1 mM EGTA 1% Triton X-100, 0.5% SDS, 10% glycerol and protease inhibitors). For whole cell lysate, cells were directly lysed in lysis buffer. Whole cell lysates, membrane and cytosolic fractions were passed through a minicolumn to capture cellular DNA. Equal amounts of protein (40 μg) of each fraction and whole cell lysates were subjected to 7.5% SDS-PAGE and the gel was transferred to a polyvinylidene difluoride (PVDF) membrane (ThermoFisher Scientific). The membrane was probed with mouse monoclonal anti-GFP antibody, followed by a horseradish peroxidase (HRP)-conjugated secondary anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA) and revealed using enhanced chemiluminescence (Perkin Elmer, Wellesley, MA). For oocytes, groups of 8–10 oocytes injected with wild-type or G345R mutant NKCC1 cRNA were lysed in 240–300 μl buffer containing 150 mM NaCl, 50 mM Tris pH 8.5, 2 mM EDTA, 0.1% SDS, 0.5% Na-deoxycholate, and 1% CHAPS. Equal volumes (25 μl) were added to 25 μl 4× sample buffer + β-mercaptoethanol, denatured at 75°C for 15 min, separated on a 4–20% pre-cast gradient gel (Bio-Rad, Hercules, CA), transferred to PVDF membrane, and probed with anti-c-myc antibody (ThermoFisher, Waltham, MA).
Immunoprecipitation.
Confluent MDCK cells were lysed with 1 ml of lysis buffer (125 mM NaCl, 200 mM HEPES pH 7.6, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 0.5% SDS, 10% glycerol) supplemented with protease inhibitors. Cell lysates (500 μg) were mixed with 10 μl of anti-GFP (GFP-NKCC1) or IgG antibodies and incubated in a 1.5-ml Eppendorff tube overnight at 4°C, under constant rotation using a Hematology/Chemistry mixer. The next day, 50 μl of protein-A Agarose beads/slurry were added and incubated at 4°C for 2 h with rotation. Immunoprecipitates were then centrifuged at 10,000 rpm for 1 min and washed five times with ice-cold PBS. Immunoprecipitated proteins were separated from the beads by adding 50 μl of 4× SDS sample buffer containing 500 mM DTT and heated at 75°C for 15 min. Precipitated proteins were separated by SDS-PAGE and blotted on PVDF membranes. Membranes were probed with mCherry (tdTomato) primary antibody followed by HRP-conjugated anti-mouse secondary antibody and revealed using enhanced chemiluminescence.
Sucrose gradient.
Confluent (4×) 10-cm dishes of MDCKWT or MDCKDFX cells were lysed in 1 ml of hypotonic lysis buffer (10 mM Tris·HCl, pH 7.5) supplemented with protease inhibitors. Cell lysates were then incubated on ice for 1 h and frequently vortexed. The 1 ml cell lysates were mixed with 1 ml of 5 M sucrose in PBS and placed at the bottom of a SW41 centrifuge tube, overlaid with 6 ml of 2 M sucrose and 3 ml of 0.25 M sucrose, and ultracentrifuged at 120,000 g for 16 h. Fourteen fractions of ~800 μl each were then collected from the top of the gradient. Aliquots (40 μl) of each fraction were mixed with 40 μl sample buffer containing 500 mM of DTT and denatured at 75°C for 20 min. Fractions were resolved on 10% polyacrylamide gel electrophoresis and analyzed by Western blotting.
Live cell imaging.
MDCKWT and MDCKDFX were cultured on MatTek Glass Bottom Microwell dishes until they reached 100% confluency. Cells were then washed twice with HBSS2+ and incubated in HBSS2+ containing 1 μg/ml of PureBlu Hoechst 33342 (Bio-Rad) and 100 μM of ER-Tracker Red (BODIPY TR, ThermoFisher) for 30 min at 37°C. Cells were then washed twice with HBSS2+, incubated in HBSS2+ containing 2.5% FBS, and imaged. All images were captured live on a Zeiss LSM 880 laser scanning confocal microscope. Samples were scanned using a ×63 oil objective. The images were exported as TIFF files using Zeiss ZEN Lite 2012 software.
Immunofluorescence.
MDCK cells expressing epitope-tagged NKCC1-WT and NKCC1-DFX were cultured on glass coverslips until they reached 100% confluency. Cells were fixed with cold (−20°C) methanol, washed, and mounted on microscope slides with ProLong Gold antifade reagent with DAPI (Invitrogen). Images were captured on a Zeiss LSM 880 laser scanning confocal microscope. Samples were scanned using a ×63 oil objective. Z-stack slices were set at 0.5-μm intervals. The images were exported as TIFF files using Zeiss ZEN Lite 2012 software. For 3-D cultures, MDCK cysts maintained in four-well LabTek chamber slides were fixed with freshly prepared 2% paraformaldehyde; this was followed by permeabilization in Immuno-Fluorescence Buffer (0.5% BSA, 0.1% Triton X-100, and 0.1% Tween 20 in HBSS2+) for 20 min followed by incubation in blocking buffer (5% BSA, 0.1% Triton X-100, and 0.1% Tween 20 in HBSS2+) for 2 h and incubation with primary antibody for 2 h. Both Alexa-Fluor-tagged anti-GFP and anti-Gp135 were added at the same time. Alternatively, Alexa-Fluor-tagged anti-GFP was added simultaneously with ActinRed Ready Probes reagent to visualize actin filaments. Slides were washed 3 × 20 min with HBSS2+ and incubated with fluorophore-conjugated (Cy3 or FITC) secondary antibodies for 1 h. Chambers were removed from the microscope slide and mounted according to the manufacturer’s protocol. Slides were imaged on the Zeiss LSM 880 as described above.
Transcription of complementary RNA.
DNA clones, inserted into the Xenopus laevis oocyte expression vector pBF and encoding the mouse NKCC1 or NKCC1 G345R mutant, were linearized by incubation at 37°C overnight with MluI (New England Biolabs, Beverly, MA), purified using a QIAquick PCR Purification Kit (Qiagen, Valencia, CA), and transcribed into cRNA using a mMESSAGE mMACHINE SP6 transcription kit (Life Technologies). Complementary RNA (cRNA) was then purified by precipitation with LiCl (transcription kit), washed with 70% ethanol, and resuspended in diethylpyrocarbonate-treated water. RNA quality and quantity was assessed using denaturing agarose gel electrophoresis and spectrophotometric methods.
Isolation and injection of Xenopus laevis oocytes.
Ovarian lobes were isolated from female Xenopus laevis frogs as previously described (6, 39) and in accordance with an approved Vanderbilt University Institutional Animal Care and Use Committee (IACUC) protocol. Individual oocytes were dissociated using collagenase D treatment (4 × 90 min in 5 mg collagenase D/ml Ca2+-free ND-80 medium, Sigma, St. Louis, MO). Oocytes were then maintained overnight in modified L15 solution [250 ml Leibovitz L15 Ringer (Invitrogen, Carlsbad, CA), 200 ml deionized water, 952 mg HEPES (acid form), and 44 mg/l gentamycin (Invitrogen); pH 7.4; 195 to 200 mosM] at 16°C. The following day, groups of 25 oocytes were injected with 50 nl containing 15 ng Na-K-2Cl cotransporter-1 (NKCC1 or mutant) cRNA and returned to the incubator. Western blot analysis and 86Rb tracer flux studies to measure Na-K-2Cl cotransport expression and function, respectively, were assessed 3 days postinjection.
86Rb uptake experiments.
K+ influx was measured through unidirectional 86Rb uptakes in groups of 20–25 oocytes in 35-mm dishes. Oocytes were washed once with 3 ml isosmotic saline (96 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, pH 7.4, 200 mosM) and preincubated for 15 min in 1 ml identical solution containing 1 mM ouabain. The solution was then aspirated and replaced with 1 ml isosmotic or hyperosmotic flux solution containing 5 μCi 86Rb. Two 5 μl samples of flux solution were taken at the beginning of each uptake period, placed in scintillation vials, and used as standards. After 1 h uptake, the radioactive solution was aspirated and the oocytes were washed four times with 3 ml ice-cold isosmotic or hyperosmotic solution. Single oocytes were then transferred into glass vials, lysed for 1 h with 200 μl 0.25 N NaOH, and neutralized with 100 μl glacial acetic acid. 86Rb activity was measured by β-scintillation counting. K+ influx is expressed in nanomoles K+ per oocyte per hour.
Generation of the NKCC1-DFX mouse.
Two attempts were made to generate the NKCC1-DFX allele. In the first attempt, a 20 bp sequence (AGAATACTATAGATGTCTGG, boxed in Fig. 12A) located in exon 22 of mouse Slc12a2, and followed by TGG (coding for the second of two adjacent tryptophan residues) as proto-spacer adjacent motif, was selected to create a target-specific guide RNA molecule. The sequence flanked by BbsI sites was ligated in pX330, a vector expressing the guide RNA under a strong U6 promoter and cas9 under a hybrid chicken β-actin (Cbh) promoter. The vector was injected alongside a 195 base repair oligonucleotide into 961 mouse embryos. The repair oligo contained 91 base homology arms, a codon substituting valine residue 1020 to phenylalanine, a unique ClaI restriction site, and a few additional third-base mutations to introduce two stop codons and prevent targeting of cas9 to the repair DNA (Fig. 11A). Out of 961 embryos injected, 687 were transferred to 30 pseudo-pregnant females to generate 110 pups. At weaning, genotyping was done by amplifying a 421 bp fragment followed by sequencing. Table 1 shows a large number of mutant alleles produced, but none of them carried the designed mutation. In our second and successful attempt, the 20 bp target sequence was shifted three residues for the protospacer adjacent motif (PAM) to encode the first tryptophan residue (GGAAGAATACTATAGATGTC, boxed in Fig. 12B), and the 163 repair oligonucleotide consisted of different size homology arms, 11 bases deleted as in the patient, and a third-base mutation to introduce a unique ClaI restriction site. Twenty pups were generated out of 213 embryos injected and 142 embryos transferred to 6 pseudo-pregnant females. Again, a large number of mutant alleles were generated (Table 1), including the designed DFX allele.
Fig. 11.

NKCC1-DFX does not affect the polarity of MDCK cells. A and B: MDCK cells stably transfected with NKCC1-WT (A) or NKCC1-DFX (B) were grown on coverslips for 5 days and immunostained with antibodies to GFP (GFP-NKCC1) and the α-subunit of the Na-K-ATPase. C–H: localization of Na-K-ATPase (red) and NKCC1 (green) in MDCK cells transfected with eGFP-NKCC1-WT (C–E) or eGFP-NKCC1-DFX (F–H), and grown in 3-D extracellular matrix. Scale bars, 10 μm or 20 μm. eGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
Table 1.
Outcome of CRISPR/cas9 injections to generate a NKCC1-DFX mouse
| Injection | Guide | Repair | DOB | No. Pups | WT | Hetero | Homo | Chimeras |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | (+) 195 b | 1/04/16 | 0 | 0 | 0 | 0 | 0 |
| 2 | 1 | (+) 195 b | 1/05/16 | 8 | 8 | 0 | 0 | 0 |
| 3 | 1 | (+) 195 b | 1/10/16 | 10 | 4 | 4 | 2 | 0 |
| 4 | 1 | (+) 195 b | 4/10/16 | 3 | 2 | 0 | 1 | 0 |
| 5 | 1 | (+) 195 b | 4/13/16 | 31 | 20 | 2 | 6 | 3 |
| 6 | 1 | (+) 195 b | 4/19/16 | 22 | 9 | 1 | 7 | 5 |
| 7 | 1 | (+) 195 b | 7/12/16 | 10 | 7 | 0 | 0 | 3 |
| 8 | 1 | (+) 195 b | 7/18/16 | 21 | 13 | 2 | 6 | 0 |
| 9 | 2 | (−) 163 b | 7/19/16 | 9 | 1 | 1 | 5 | 2 |
| 10 | 2 | (−) 163 b | 7/19/16 | 11 | 7 | 0 | 2* | 2 |
| Total | 125 | 71 (57%) | 10 (8%) | 29 (23%) | 15 (12%) |
Sequences of RNA guides are as follows: guide 1, AGAATACTATAGATGTCTGGTGG; guide 2, GGAAGAATACTATAGATGTCTGG (Protospacer Adjacent Motifs are underlined). Repair oligonucleotides are single stranded. The number of bases and strand used are indicated. Chimeras are defined by the presence of more than two separate alleles. They are observed when separate CRISPR events occur in multicell embryos. DOB, date of birth; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
One homozygote female carried the DFX mutation. The other allele had the following amino acid sequence: DSIGLVAFx.
Fig. 10.

Coexpression of NKCC1-WT and NKCC1-DFX causes the mistrafficking of the WT cotransporter to the apical membrane. A and B: MDCK cells stably cotransfected with TdT-NKCC1-WT and eGFP-NKCC1-WT (A) or TdT-NKCC1-WT and eGFP-NKCC1-DFX (B) were polarized on Transwell filters for 5 days. Cells were then fixed and counterstained with anti-GFP anti-TdT antibodies. Z-sections were captured at 5-μm intervals on the Zeiss LSM 880 microscope. Bars, 20 μm. C: whole cell lysates from dually transfected cells as in A and B were prepared. Protein lysates (400 μg) were immunoprecipitated with anti-GFP antibody and analyzed by Western blotting. D: whole cell lysates from MDCK cells expressing TdT-NKCC1-WT, eGFP-NKCC1-WT, eGFP-NKCC1-DFX or dually transfected as in A and B were analyzed by Western blotting. E: polarized MDCK coexpressing TdT-NKCC1-WT and eGFP-NKCC1-WT or TdT-NKCC1-WT and eGFP-NKCC1-DFX constructs were selectively biotinylated on the apical membrane or basolateral membrane. Cell lysates were prepared and equal amounts of protein were immunoprecipitated with streptavidin agarose beads and analyzed by Western blotting. AP, apical; BL, basolateral; eGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; TdT (TdTomato), tandem dimer red fluorescent protein; WT, wild type.
Mice and tissue preparation.
Tissues were prepared from 20-wk-old wild-type and NKCC1-DFX male and female mice. Animals were anesthetized by isoflurane inhalation and killed by decapitation. Animal protocols were reviewed and approved by the Vanderbilt University IACUC, and all experiments were performed in accordance with IACUC guidelines. Tissue was removed, washed in ice-cold PBS, and fixed in PBS containing 4% paraformaldehyde overnight. Tissue was then transferred to 30% sucrose in PBS and incubated overnight at 4°C. Tissue was subsequently embedded in OCT compound medium (Tissue-Tek, Japan) and stored at −80°C. Sections (10 μm) were prepared in a cryostat (Microm model HM505N, Walldorf, Germany) at −20°C and mounted on Superfrost Plus microscope slides (Fisher Scientific), dried at room temperature, and stored at −20°C.
Immunohistochemistry of mouse frozen tissue.
Sections were defrosted and equilibrated at room temperature for 10 min. Tissue sections were rehydrated by three consecutive 5-min washes in 100%, 95%, and 70% ethanol; followed by a 10-min wash in PBS. Tissue sections were permeabilized in 1% SDS solution or 0.1% Triton X-100 for 5 min at room temperature. Sections were washed 3 × 5 min in PBS. Sections were then treated with microwave-heated 10 mM sodium citrate containing 0.05% Tween 20 and allowed to cool down at room temperature for 1.5 h. Sections were washed again 2 × 5 min in PBS. Sections were blocked with blocking buffer (5% BSA + 1% goat serum + 0.5% Triton X-100) in PBS for 1 h. Indicated primary antibodies were resuspended in blocking buffer and incubated at room temperature for 1.5 h then transferred to 4°C for overnight incubation. The following day, sections were washed 3 × 10 min with PBS. Fluorophore-tagged secondary antibodies were resuspended in blocking buffer and incubated at room temperature for 2 h. Finally, sections were washed 3 × 10 min in PBS and mounted with ProLong Gold antifade reagent with DAPI and imaged on the Zeiss LSM 880 microscope.
Salivary secretion assay.
Twelve-week-old mice of both sexes and of wild-type and DFX genotypes were used to measure saliva secretion. Anesthesia was induced with 5% isoflurane and oxygen and maintained under a 2% isoflurane and 98% oxygen through a nose cone. Saliva secretion was stimulated by subcutaneous injection of 50 μl of pilocarpine solution (100 μM dissolved in isotonic saline). Saliva was collected for 15 min using a narrow (0.5 mm outside diameter) catheter attached to an Eppendorf tube connected to a vacuum. Weight and volume of collected saliva were measured after the 15 min collection time.
RESULTS
In previous studies, we demonstrated that addition of an epitope tag to the NH2-terminal tail of NKCC1 did not affect the trafficking and function of NKCC1 (17). This observation led to the transfection of MDCK cells with vectors that express GFP-tagged NKCC1 or GFP-tagged NKCC1-DFX (truncated transporter). The cells were chosen despite their relative resistance to transfection because they are able to form a well-organized polarized epithelial cell layer in culture. After selection in the presence of G418 and fluorescence-activated cell sorting, cells were plated on glass coverslips. A significant difference could be seen between cells transfected with wild-type and mutant cotransporters. Cells transfected with mutant NKCC1 were rounded, elongated, and showed disruption of well-organized honeycomb-like structures (Fig. 1, A and D). Similar data were observed with the tight junction marker ZO-1. Staining with ZO-1 also showed an increase in the number of MDCK cells junctions involving two and four cells as opposed to the typical tricellular junction (Fig. 1, B and E, and Fig. 2). Merged images indicate that NKCC1-DFX accumulates in the cytoplasm (Fig. 1, C and F) and is uncoupled from the lateral membranes (Fig. 1, C and F, X–Z views).
Fig. 1.

NKCC1-DFX is mistrafficked and affects the morphology of epithelial cells on glass substrate. MDCK cells stably transfected with eGFP-NKCC1-WT (A–C) and eGFP-NKCC1-DFX (D–F) were plated on glass coverslips, fixed, and stained with anti-GFP and ZO-1 antibodies. A: membrane expression of GFP-tagged NKCC1-WT showing expression at the membrane. X-Z view shows lateral membrane staining, but note that the cells are mostly flat. B: staining for tight junction protein ZO-1 in NKCC1-WT-transfected cells. C: merged GFP-NKCC1-WT and ZO-1 images show colocalization at the plasma membrane. D: expression of GFP-tagged NKCC1-DFX showing accumulation of the mutant cotransporter inside the cell. E: staining for tight junction protein ZO-1 in NKCC1-DFX-transfected cells shows elongated and round cells. F: merged GFP-NKCC1-DFX and ZO-1 images show that the cotransporter is uncoupled from the lateral membranes. eGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type; ZO-1: zonula occludens-1.
Fig. 2.
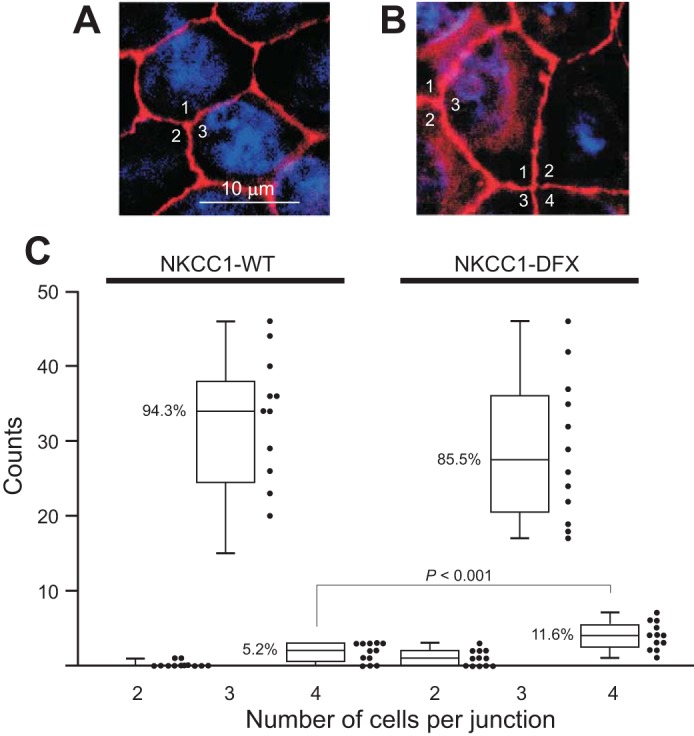
Junctions with 4 cells increase in NKCC1-DFX-expressing cells. A: picture showing multiple 3-cell junctions in MDCK cells transfected with wild-type NKCC1 cDNA. B: picture showing a 4-cell junction and multiple 3-cell junctions in MDCK cells transfected with mutant NKCC1-DFX cDNA. C: quantitation: 12 photographs each were analyzed and cells junctions were counted in MDCK cells expressing wild-type or mutant cotransporter. As shown by the box-and-whisker plot, the majority of junctions tie 3 cells (94.3% in wild-type and 85.5% in mutant cells). The number of 4-cell junctions more than doubles (5.2% to 11.5%) in the mutant transporters (12 images from 12 independent experiments and 406 junctions per condition). MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1.
Using c-myc and GFP epitopes tagged to the extreme NH2-domain of NKCC1-WT and NKCC1-DFX, we previously demonstrated that both wild-type and mutant proteins are readily expressed in oocytes and human cell lines and transported to the plasma membrane (9). Since the DFX mutation truncates 187 amino acids residues from the COOH domain of NKCC1, we used T4, a monoclonal antibody raised to the transporter COOH domain, to demonstrate that this antibody does not recognize the mutant transporter. Oocytes injected with cRNA complementary to NKCC1-WT and NKCC1-DFX were analyzed by immunoblotting, and we show that T4 antibody only recognized wild-type NKCC1 (Fig. 3A). Subcellular fractionation of membrane versus cytosolic compartments in MDCK cells revealed that both wild-type and mutant GFP-tagged transporters are associated with the membrane fractions, whereas the peripheral membrane protein marker ezrin was found in the cytosolic fraction (Fig. 3B). However, the cytosolic pool of NKCC1-DFX observed in Fig. 1 could not be detected in the cytosolic fraction (Fig. 3B, lane 6). Note that when the T4 antibody is used, the native transporter can be seen in untransfected cells and in cells transfected with wild-type mouse NKCC1 only. When NKCC1-DFX is transfected, the NKCC1 signal is markedly reduced (Fig. 3B, middle), indicating that the mutant transporter significantly affects expression of the wild-type transporter. NKCC1 has been shown to associate with detergent-resistant membranes or lipid rafts (19). Therefore we sought to analyze the distribution of NKCC1-DFX. Whole cell lysates were fractionated by ultracentrifugation on discontinuous sucrose gradients and the fractions were collected from the top of the tube. Analysis of each fraction by Western blotting indicates that wild-type NKCC1 is distributed in two pools (Fig. 3C). The low-density pool located at fractions 5 and 6 where caveolin-1-containing lipid rafts typically float (Fig. 3C, fraction: 5, 6, and 7) and a high-density pool toward the bottom of the gradient and which include heavy and intact plasma membrane fragments (Fig. 3C, fraction: 11, 12, 13, and 14). However, when MDCKDFX lysates are fractionated on the sucrose gradient, GFP signal can only be seen associated with the heavy and intact plasma membrane at the bottom of the tube, indicating that NKCC1-DFX cytosolic and raft fractions are too low to be detected by Western blotting (Fig. 3C, bottom). We next analyzed the cellular localization of NKCC1 by live cell microscopy. In cells transfected with the wild-type NKCC1 and incubated with an ER-Tracker reagent, the cotransporter mainly localized to the plasma membrane (Fig. 3, D and E). In the mutant transfected cells the mutant cotransporter colocalizes with the endoplasmic reticulum (ER) marker but the majority of the signal is cytosolic (Fig. 3, F and G). Similar data were obtained with the Golgi-Tracker (data not shown).
Fig. 3.

NKCC1-DFX is associated with membranes and trafficked out the ER. A: oocytes were injected with NKCC1-WT or NKCC1-DFX cRNA, lysed, and analyzed by Western blotting. T4 antibody, which targets the carboxyl-terminal domain of NKCC1, does not recognize the NKCC1-DFX mutant. Solid arrows, NKCC1 signal; open arrow, unspecific signal. B: subcellular fractionation of MDCK cells stably transfected with NKCC1-WT and NKCC1-DFX followed by immunoblotting of each fraction with GFP, T4, and ezrin antibodies. C: whole cell lysates of MDCK transfected with MDCK-WT and MDCK-DFX were fractionated on a discontinuous sucrose gradient. Aliquots (40 μl) of fractions (0.8 ml) collected from the top were separated by SDS-PAGE and analyzed by Western blotting. D–G: MDCK-WT (D and E) and MDCK-DFX (F and G) were cultured in MatTek Glass Bottom Microwell dishes. Cells were stained with PureBlu Hoechst 33342 and ER-Tracker Red and imaged live on the Zeiss LSM 880 confocal microscope. Bars, 20 μm. C, cytosolic compartment; ER, endoplasmic reticulum; GFP, green fluorescent protein; M, membrane compartment; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
To confirm the effect of NKCC1-DFX on the expression of endogenous NKCC1 as seen in Fig. 3B, we performed six independent immunoblotting analyses of parental MDCK, NKCC1-WT, and NKCC1-DFX transfected cells (Fig. 4A). Quantitative analysis indicates that in NKCC1-DFX transfected cells; endogenous NKCC1 expression is decreased by 60% (Fig. 4B).
Fig. 4.
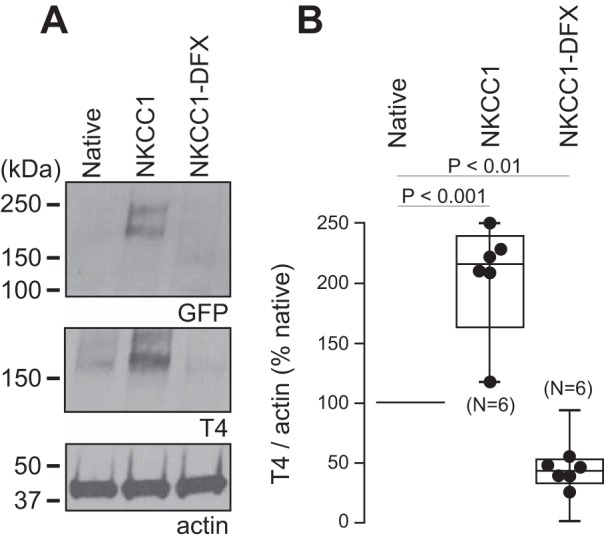
Expression of NKCC1-DFX results in decreased abundance of endogenous NKCC1. A: whole cell lysates of parental MDCK cells and MDCK cells stably transfected with NKCC1-WT or NKCC1-DFX were separated by SDS-PAGE followed by Western blotting with GFP, T4, and actin antibodies. B: box-and-whisker plot showing quantification of NKCC1 expression by densitometry analysis of 6 individual Western blotting experiments as in A (n = 6, ANOVA, P < 0.01). GFP, green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
When MDCK cells were grown on permeabilized support, in contrast to flat cells on glass coverslips, they formed an epithelium of tall cells that mimics a simple columnar epithelium. Confocal microscopy of MDCK cells expressing eGFP-NKCC1-WT showed intense lateral plasma membrane staining (Fig. 5A, X–Z and Y–Z views). Remarkably, in MDCK cells expressing the eGFP-NKCC1-DFX mutant, most of the signal is observed at the apical/subapical membrane of polarized cells as indicated by colocalization with the apical membrane marker Gp135/podocalyxin (Fig. 5B, X–Z and Y–Z views), indicating that trafficking to the plasma membrane as well as endocytosis and sorting to intracellular compartments is probably affected as well. Cell surface expression was also assessed using selective cell surface biotinylation of MDCK cells grown on permeabilized support. The biotin reagent was placed in the solution bathing either the apical (top chamber) or the basolateral (bottom chamber) membrane. As shown in the representative immunoblot (Fig. 5C), the NKCC1 signal is intense on the basolateral side and minimal on the apical side; whereas in NKCC1-DFX transfected cells, native NKCC1 signal is abundant on both apical and basolateral sides.
Fig. 5.

NKCC1-DFX mistrafficks from basolateral membrane to subapical pole and apical membrane. A and B: MDCK cells stably transfected with eGFP-NKCC1-WT (A) and eGFP-NKCC1-DFX (B) were polarized on Transwell filters for 5 days. Cells were then fixed and stained with anti-GFP and Gp135/podocalyxin. Z-sections were captured at 5-μm intervals on the Zeiss LSM 880 microscope. Bars, 20 μm. C: polarized MDCK cells expressing NKCC1-WT and NKCC1-DFX were selectively biotinylated on the apical membrane or basolateral membrane. Cell lysates were prepared, and equal amounts of protein were immunoprecipitated with streptavidin agarose beads and analyzed by Western blotting. AP, apical; BL, basolateral; eGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
This unexpected result was verified using cells grown in 3-D Matrigel. When cells are seeded at low density on Matrigel with 2% Matrigel in the culture medium, cells divided and created small organoids which developed with time into cysts. Figure 6A shows such a cyst with one cell layer separating an internal cavity from the extracellular milieu. Staining of the wild-type cotransporter was observed on the basolateral membranes, as anticipated for NKCC1. The situation was again different with NKCC1-DFX where some of the staining was observed on the apical membrane, although some basolateral staining could still be seen (Fig. 6B). Costaining with the apical membrane marker Gp135/podocalyxin indicates that in NKCC1-WT transfected cells, the cotransporter remained at the basal and lateral membranes (Fig. 6C), whereas in mutant transfected cells, a fraction of the cotransporter translocated to the apical membrane while the majority of the signal was dispersed throughout the cell (Fig. 6D). This indicates that NKCC1-DFX not only affects cotransporter trafficking to the plasma membrane but sorting to different endocytic compartments and recycling is affected as well.
Fig. 6.

NKCC1-DFX mistrafficks to the apical pole and apical membrane. A and B: MDCK cells stably transfected with eGFP-NKCC1-WT (A) or eGFP-NKCC1-DFX (B) were grown in 3-D Matrigel for 14 days. Cells were subsequently fixed and counterstained with anti-GFP antibody. Images were captured on the Zeiss LSM 880 microscope. C and D: MDCK cells stably transfected with eGFP-NKCC1-WT (C) and eGFP-NKCC1-DFX (D) were grown in 3-D Matrigel for 10 days and immunostained with anti-GFP (GFP-NKCC1) antibody, and Gp135/podocalyxin antibody to label the apical membranes. Bars, 10 μm. eGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
The formation of cysts in Matrigel was also affected by the mutant cotransporter. Under normal conditions (wild-type NKCC1), a large cavity (single lumen formation), as seen in Fig. 7, A–C, was observed in most cysts. Expression of NKCC1-DFX in MDCK cells gives rise to cysts with multiple lumens (Fig. 7, E–H). To validate this observation, we quantitated the number of cysts with multiple lumens in four independent experiments done with NKCC1-WT versus NKCC1-DFX transfected cells. In wild-type transfected cells, the incidence of cysts with multiple lumen after day 8 in culture was ~2–3%, and reached 45% in NKCC1-DFX transfected cells (Fig. 7D).
Fig. 7.

NKCC1-DFX increases the number of MDCK cysts with multiple lumen. MDCK cells stably transfected with eGFP-NKCC1-WT (A–C) and eGFP-NKCC1-DFX (E–H) were grown in 3-D Matrigel for 8–14 days and immunostained with anti-GFP (NKCC1) antibody, and ActinRed or Gp135/podocalyxin antibodies to label the apical membranes. Bars, 10 μm. D: quantification of multiluminal cysts in NKCC1-WT and NKCC1-DFX in 3-D Matrigel. Data are representative of 4 individual experiments where 12 cysts per experiment were analyzed (total, n = 48). eGFP, enhanced green fluorescent protein; MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type. Asterisks indicate individual lumen.
To assess whether the phenotype observed in NKCC1-DFX transfected cells was due to disruption of NKCC1 function or the presence of the mutant cotransporter, we used two MDCK cell lines that were previously shown to lack NKCC1 function (30). To better understand why these cells have a nonfunctional NKCC1, we extracted RNA from parental MDCK-N and mutants MDCK-LK-C1 and MDCK-LK-A3, PCR amplified segments of the cotransporter from the three different cell lines, and sequenced them. We found complete loss of exon 4 (96 bases, which conserved the frame), which encodes transmembrane domain TM2 and part of ICL1, in MDCK-LK-C1 cells and an amino acid substitution, G345R, in MDCK-LK-A3 cells (Fig. 8, A and B). Whole cell lysate was analyzed by Western blotting, confirming the expression of NKCC1 in all three cell lines (Fig. 8C). As previously reported by Payne et al. (38), MDCK-LK-C1 cell line expresses a lower-molecular-weight transporter (Fig. 8C, lane 3), whereas mutant NKCC1 expression in MDCK-LK-A3 cell line is slightly increased or highly glycosylated (Fig. 8C, lane 2). Mutation of Gly345, which constitutes the first residue of an α-helix, into charged arginine leads to a nonfunctional transporter when expressed in Xenopus laevis oocytes (Fig. 8D). When these two MDCK clones expressing nonfunctional NKCC1 were plated on Matrigel, they formed single lumen cysts as wild-type MDCK cells (Fig. 9, A–F). However, the sizes of the cysts were greatly reduced from an average of 1,560 pl for wild-type cysts versus 30–60 pl for mutant cysts (Fig. 9G). The size difference was due to the number and not the size of cells, as the sizes of the nuclei were roughly the same (see circles drawn around nuclei in Fig. 9, E and F). Note that the change in morphology in cells expressing NKCC1-DFX is clearly different from the change observed with MDCK cells lacking functional NKCC1 altogether (Fig. 9, D–F versus Fig. 7, E–H).
Fig. 8.

Sequence showing SLC12A2 mutations in MDCK-LK-C1 and MDCK-LK-A3 cells. Cells were mutagenized and functionally characterized for transport function in the past (30) but never characterized at the gene level. Canine-specific PCR primers were designed to amplify overlapping fragments of the dog cotransporter cDNA. A: cotransporter from MDCK-LK-C1 cells lacks entire exon 4 (96 bp), which encodes transmembrane domain 2 and a portion of intracellular loop 1. NKCC1 from MDCK-LK-A3 cells have a G>A substitution in exon 5, leading to amino acid residue 345 substitution from glycine to arginine. B: model of the TM2 and the beginning of ICL1 obtained from I-TASSER demonstrating the importance of the 345 location as it represents the first residue of an α-helix. C: detection of NKCC1 in MDCK-WT, MDCK-LK-A3, and MDCK-LK-C1 cells with T4 monoclonal antibody. D: NKCC1-G345R mutant (as MDCK-LK-A3) is expressed but nonfunctional in Xenopus laevis oocytes. Both cotransporters are observed with the c-myc epitope in NKCC1-injected oocytes, but not in water-injected oocytes. Bars represent means ± SE; n = 20–25 oocytes. Difference between black bars is highly significant: P < 0.001, ANOVA. MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1; SLC12A2, solute carrier family 12, member 2; WT, wild type.
Fig. 9.

NKCC1 regulates MDCK spheroid volume. A–C: MDCK-N (A) and low K+ resistant mutants MDCK-LK-C1 (B) and MDCK-LK-A3 (C) were grown in 3-D Matrigel for 8 days and immunostained with T4 anti-NKCC1 antibody (Red), and ActinGreen Ready Probes reagent to label the apical membrane. Confocal immunofluorescence micrographs of MDCK spheroids were imaged on the Zeiss LSM 880 microscope. Scale bars, 200 μm. D–F: the same cells were imaged at higher magnification. Circles are drawn around a few nuclei to indicate their sizes. Scale bars, 20 μm. G: MDCK-N and low K+ resistant mutants MDCK-LK-C1 and MDCK-LK-A3 spheroid diameters were determined with Zeiss ZEN 2012 software and volumes were calculated. MDCK, Madin-Darby canine kidney; NKCC1, Na+-K+-2Cl− cotransporter-1.
To test whether the mutant cotransporter affects the trafficking of the wild-type cotransporter, cotransfection experiments with tdTomato-NKCC1 and eGFP-NKCC1-DFX were performed. On permeabilized support, the “rounding,” which could also be referred to as “expansion of the apical membrane,” phenotype observed in cells overexpressing the mutant transporter type (Fig. 5B, X–Z view) was rescued, and colocalization was observed at the plasma membrane (Fig. 10B). In contrast to wild-type (Fig. 10A), most of the signal was observed on the apical side (Fig. 10B). Given that the working unit of NKCC1 is a dimer, we tested whether NKCC1-DFX can interact with wild-type NKCC1. GFP pulldowns from cells dually transfected with either TdT-NKCC1-WT and eGFP-NKCC1-WT or TdT-NKCC1-WT and eGFP-NKCC1-DFX showed that the mutant GFP-tagged cotransporter coprecipitates with tdTomato-tagged NKCC1-WT (Fig. 10C). Whole cell lysate analysis indicates that tdTomato antibody does not cross-react with GFP as only cells transfected with the tdTomato construct showed a band by Western blotting (Fig. 10D). Strong apical staining localization was also observed using side-specific cell surface biotinylation in MDCK cells grown on a permeabilized substrate (Fig. 10E).
Mutant NKCC1-DFX could affect the trafficking of the functional wild-type transporter through protein-protein interaction, as dimerization seems to be facilitated by the truncation of the particular portion of the transporter’s COOH terminus (9). Alternatively, the entire sorting machinery of the cell could be affected. To address this possibility, we examined the membrane localization of the α1-subunit of the Na+-K+-ATPase which is typically expressed on the basolateral membrane of polarized MDCK cells. Expression of NKCC1-DFX did not affect the localization of the α1-subunit of the Na+-K+-ATPase which was mainly localized to the basolateral membrane (Fig. 11). As observed before, NKCC1-DFX expression again led to many cysts with multiple lumens (data not shown). In this case, cells at the junction of these multiluminal cysts showed an atypical localization of the basolateral markers. Thus, NKCC1 is not necessary for cell polarity but may play a role in lumen formation.
To further assess the impact of the truncation mutation on epithelial function, we created a mouse model carrying the patient’s 11 bp deletion in Slc12a2. Using CRISPR/cas9 technology, we targeted exon 22 of Slc12a2 by selecting guide RNAs directing cas9 to break DNA upstream of the codon coding Trp1021 (Fig. 12A) or upstream of the preceding codon encoding Val1020 (Fig. 12B). Separate repair oligonucleotides were used to introduce a phenylalanine residue after Asp1019, followed by a stop codon. A large number of mutants (43%) were produced, indicating high efficiency of cas9 targeting for both designs (Table 1). Although a large number of animals (22%) exhibited events on both alleles, only 1 pup out of 47 (2%) demonstrated repair via homologous recombination. The designed mutation (DFX) was obtained with guide no. 2 and with the 163-base long repair oligonucleotide. The female mouse carrying the DFX allele also carried a distinct mutation in her second slc12a2 allele (Fig. 12D), making it a compound heterozygous NKCC1 mouse or a functional NKCC1-null mouse. This unique mouse was fortunately a female, as NKCC1-null males are infertile (7, 35). The mouse, which displayed the well-documented debilitating inner ear deficit phenotype (7, 13), produced progeny but did not nurture her pups. The line was successfully expanded by fostering newborn pups with surrogate mothers in separate cages.
Fig. 12.

Generation of the CRISPR/cas9 NKCC1-DFX mouse. A and B: two targeting designs of the Slc12a2 allele at exon 22 are shown. Boxed sequences on the forward strands were used to guide cas9. Guide sequences are followed by TGG as protospacer adjacent motif (PAM) sequences. Location of the DNA break sites is indicated by arrowheads. Design of DNA repair shows arms of recombination surrounding 13 bp mutated (A) or 3 bp mutated (B) sequences. In both cases a ClaI site was inserted to facilitate genotyping. C: chromatograph showing sequence of wild-type allele. D: chromatographs showing separation of the two mutant alleles in the progeny of the unique NKCC1-DFX female. Mutant allele 1 alongside a wild-type allele reveals exact mutations as designed in B. Mutant allele 2 alongside a wild-type allele reveals DNA repair through unspecific insertion/deletion. NKCC1, Na+-K+-2Cl− cotransporter-1; Slc12a2, solute carrier family 12, member 2.
NKCC1 expression was first assessed in brain lysates isolated from NKCC1WT/WT, NKCC1WT/DFX (or NKCC1-DFX), and NKCC1DFX/DFX [functional knockout (KO)] mice. We first demonstrated that contrary to T4 antibody, which does not bind to the mutant cotransporter (Fig. 3A), the Abcam antibody, which targets the amino-terminal tail of NKCC1, recognizes both wild-type and NKCC1-DFX (Fig. 13A). Using both antibodies, NKCC1 signal was observed in NKCC1-WT and NKCC1-DFX brain samples but not in the NKCC1DFX/DFX brain sample, indicating that in the absence of a wild-type NKCC1, the mutant protein is likely unstable and rapidly degraded (Fig. 13B). Using the T4 antibody we demonstrate reduced levels of NKCC1 in the NKCC1-DFX mice (Fig. 13, C and D), consistent with the presence of one wild-type allele.
Fig. 13.
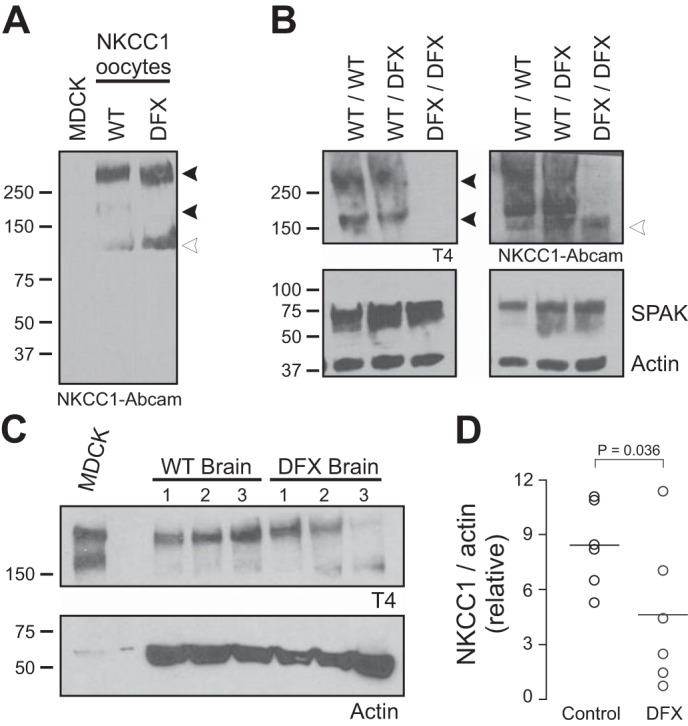
Expression of wild-type NKCC1 is reduced by ~50% in brain of NKCC1-DFX heterozygote mice. A: Western blot analysis of oocyte lysates from Fig. 2A with a NH2-terminal NKCC1 Abcam antibody showing expression of wild-type NKCC1 and NKCC1-DFX in respective lanes. B: Western blot analysis of the cotransporter in brains from control NKCC1WT/WT, mutant NKCC1WT/DFX, and NKCC1DFX/DFX mice. SPAK and actin antibodies were used to demonstrate equal loading. C: brain lysates from 3 mice of each genotype were collected and subjected to SDS-PAGE and probed with T4 and actin antibodies. D: quantitation of NKCC1 expression normalized to β-actin from samples from 6 mice. NKCC1 levels were significantly reduced in the NKCC1-DFX brain (n = 6, P < 0.05, t-test). NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type.
To assess the overall health of the mice, we weighed a cohort of 21 mice every 3 days for the first 3 wk and weekly thereafter until 12 wk of age. Sex and genotypes were not known for the first 21 days, but we did not distinguish two populations of weights. Figure 14 shows the growth curves of males and females of both genotypes. Whereas the weight of males increased faster than the weight of females, there was no difference between genotypes. This observation was consistent with the absence of an overt phenotype in the mutant mice compared with their wild-type littermates.
Fig. 14.
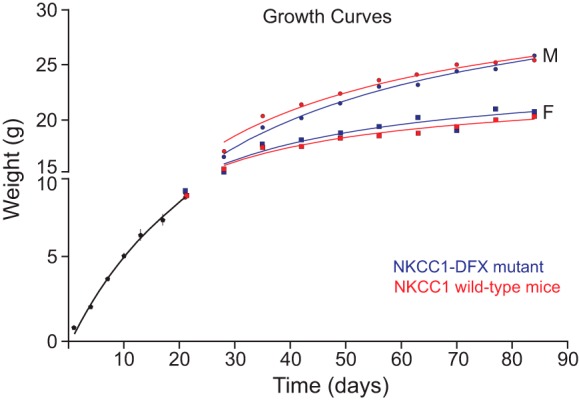
Growth curves demonstrate absence of general health phenotype of NKCC1-DFX mice. Mice were weighed every 3 days for 3 wk and weekly thereafter until 84 days old or 12 wk of age. Weights were separated by sex and genotype after postnatal day P21. Red curves, NKCC1 wild-type mice; blue curves, NKCC1-DFX mice. F, female; M, male; NKCC1, Na+-K+-2Cl− cotransporter-1.
To confirm the mistargeting of NKCC1 in the mouse model, we isolated the colons from wild-type, NKCC1-DFX, and functional KO (DFX/DFX) mice. First, using the Abcam antibody, we showed that NKCC1 located in the basolateral membranes of colonic epithelial cells in the wild-type mouse (Fig. 15, A, C, E, G). However, in the NKCC1-DFX mouse, the cotransporter appeared both in the basolateral membrane but also accumulated at the apical and subapical pole of the epithelial cells (Fig. 15, B, D, F, H). By using the T4 antibody, which does not recognize the mutant transporter, we showed that the wild-type cotransporter in wild-type mice mainly localizes to the basolateral membrane mouse colonic epithelial cells (Fig. 15I), whereas the signal is seen mainly at the subapical pole in the NKCC1 mutant mouse (Fig. 15J). As expected, no signal was detected in the NKCC1DFX/DFX (KO) mouse (Fig. 15, K and L). Combined, these data indicate that NKCC1-DFX causes mistrafficking of the wild-type transporter in colonic epithelial cells.
Fig. 15.

NKCC1 is mistrafficked to the apical/subapical membrane in NKCC1WT/DFX mouse colon. A and B: representative fluorescence micrographs showing NKCC1 (green) expression in colon sections from NKCC1WT/WT (A) and NKCC1WT/DFX (B) mice using the Abcam NH2-specific NKCC1 antibody. Scale bars, 20 μm. C and D: same panels as A and B with DAPI staining nuclei. E and F: high magnification of NKCC1WT/WT and NKCC1WT/DFX colon crypt regions. G and H: same panels as E and F with DAPI staining nuclei. I: T4 (carboxyl-terminus-specific) antibody was used to stain NKCC1WT/WT colon sections. J: T4 antibody was used to stain NKCC1WT/DFX colon sections. K and L: T4 antibody was used to stain NKCC1DFX/DFX (NKCC1-KO) colon sections. Scale bars, 20 μm. KO, knockout; NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type. Asterisks indicate NKCC1 signal at apical pole.
Given that the patient suffers from dry mouth and it was previously reported that the NKCC1 KO mouse displays a deficit in salivation (12), we wanted to test saliva secretion in the NKCC1-DFX mouse. First, we isolated salivary glands from wild-type and NKCC1-DFX mice and stained with T4 antibody. While a signal was observed on the basolateral membrane of salivary gland epithelial cells from both genotypes (Fig. 16), a strong signal was observed at the apical pole in the glands of NKCC1-DFX mice (Fig. 16, B, C, E, F), consistent with the data obtained with transfected polarized MDCK cells in culture and colon sections. Pilocarpine-stimulated saliva secretion was slightly decreased in NKCC1-DFX (100 μl/15 min) compared with NKCC1-WT (110 μl/15 min), but no significant difference was observed: P > 0.05, Student’s t-test, n = 4 per genotype (Fig. 16G).
Fig. 16.

NKCC1 is mistrafficked to the apical/subapical membrane of NKCC1WT/DFX mouse salivary gland. A–C: representative fluorescence micrographs showing NKCC1 (green) expression in salivary glands isolated from wild-type (A) and NKCC1WT/DFX mice (B and C). NKCC1 normally localizes to the basolateral membrane but in NKCC1WT/DFX mice, the cotransporter can be seen at the apical membrane. D–F: same panels with DAPI staining nuclei. Scale bars, 10 μm. G: amount of saliva collected from NKCC1-WT and NKCC1-DFX after pilocarpine injection (n = 4; P > 0.05 or nonsignificant). NKCC1, Na+-K+-2Cl− cotransporter-1; WT, wild type. Asterisks indicate NKCC1 signal at apical pole.
DISCUSSION
A 16-yr-old female patient carrying a de novo mutation in NKCC1 suffers from small intestinal dysmotility and dietary intolerance that led to total parenteral nutrition; autonomic bladder dysfunction that ultimately led to complete loss of organ function; decreased energy, fatigue, and exercise intolerance; orthostatic hypotension; chronic pain; pancreatic insufficiency, and other endocrine abnormalities (9). While inactivation of one allele does not typically result in a strong phenotype, if the gene product from the mutated allele is produced, it can exert dominant-negative effects and have pathogenic consequences. Using Xenopus laevis oocytes and fibroblasts isolated from the patient, we failed to detect any dominant negative effect of the mutant transporter over the wild-type transporter, although as anticipated, the truncated transporter was nonfunctional (9). The studies reported here revealed a different situation in epithelial cells.
When MDCK cells were transfected with NKCC1-DFX, we observed significant morphological changes such as rounding of the cells and appearance of multiple junctions on plastic substrate, appearance of multiple lumens in 3-D cultures, as well as mistrafficking of the mutant transporter to the subapical/apical membrane. This is in contrast to transfection of wild-type NKCC1, where cells retain their morphology and demonstrate staining of the basolateral membranes. We also found that in the presence of the truncated transporter, some of the wild-type transporter is also present at the apical membrane.
In cells overexpressing GFP-NKCC1-DFX, we also observed strong GFP signal accumulated inside the MDCK cells. COOH-terminal truncation of NKCC1 typically results in the accumulation/aggregation of the protein in the endoplasmic reticulum and absence of cell surface expression. Proper trafficking seems to necessitate the presence of a carboxyl-terminal sequence containing a dileucine motif (34). Costaining of NKCC1-DFX with ER marker revealed that, whereas some signal was colocalizing with the ER marker, the majority of the signal did not show ER localization. So where is the transporter located?
Several pieces of data indicate that the mutant transporter is associated with membranes. First, Western blot analysis of cytosolic and membrane fractions revealed expression of NKCC1-DFX associated with membranes. Second, the majority of the NKCC1-DFX signal following sucrose gradient ultracentrifugation was found in the pellet fraction. Third, when cells were cultured in polarized conditions on permeable filters or in 3-D Matrigel, some intense signal colocalizing with Gp135 was also observed at the apical pole. Note that we have never seen by Western blot analysis a GFP signal at 27 kDa, indicating that GFP is separated from the transporter and soluble in the cytosol. A likely location of the mutant transporter, aside the apical membrane, is subapical endosomal compartments. In polarized epithelial cells, there are two distinct early endosomes compartments: the apical early endosomes and the basolateral early endosomes. These two compartments do not communicate directly (21). However, both of these compartments can contribute cargo to the subapical compartment also known as the common endosome. The fact that NKCC1-DFX is seen at the basolateral membrane, the apical membrane and accumulates at the subapical domain indicates that multiple sorting signals are affected by this mutation. It is possible that NKCC1-DFX containing dimers are routed to the apical and basolateral membranes and from there trafficked to the common endosome or subapical compartment where the cotransporter has a tendency to accumulate (43). Alternatively, NKCC1-DFX containing dimers are trafficked from the Golgi to the common endosome/subapical compartment (where they accumulate) and occasionally targeted to the apical membrane or recycled to the basolateral membrane.
Cross-linking and Western blot experiments have shown that the structural unit of NKCC1 in cells and tissues is a homodimer (31). This observation is consistent with the crystal structure of bacterial Na+-dependent amino acid transporters, which belong to the same 12 transmembrane domain-containing amino acid, polyamine, organocation transporter superfamily. Resolution of Aquifex aeolicus LeuT transporter revealed the importance of transmembrane domains TM9 and TM12 in the formation of contacts between monomers (49). In addition to these transmembrane points of contact, the carboxyl-terminal tail of NKCC1 was shown to also serve as a point of interaction and provide specificity between monomers (36). The carboxyl-terminal regions involved are two relatively large (~130–150 aa) fragments (42) that were subsequently shown to form protein folds consisting of five parallel β-sheets separated by 3–4 α-helices (46). These domains have extensive domain contact, which are used in a “head to tail” arrangement between monomers (46). Interestingly, the patient mutation truncates the cotransporter at the very beginning of the second protein fold leaving the first domain intact which can then interact with the second domain of the wild-type subunit. This arrangement could stabilize the WT-DFX dimer and allows its expression at the plasma membrane. In support for this hypothesis was the demonstration through coimmunoprecipitation of an interaction between wild-type and mutant NKCC1.
Mistargeting of the NKCC1-DFX cotransporter to the apical pole was demonstrated in cultured MDCK cells and in colon and salivary gland epithelial cells from the NKCC1-DFX mouse model. In all cases the presence of a native wild-type protein likely stabilized the mutant transporter and allowed its expression at the plasma membrane, but why was the complex targeted to the apical membrane? Under normal conditions, NKCC1 is targeted to the basolateral membrane of epithelial cells (8, 24), whereas NKCC2, a related cotransporter expressed in the thick ascending limb of Henle, is targeted to the apical membrane (25). A study by Carmosino and colleagues (2) showed that NKCC1 possesses a sequence that targets the transporter to the basolateral membrane. This sequence is located within exon 21 of SLC12A2 (NKCC1), which is alternatively spliced in NKCC1 (40) and is absent in SLC12A1 (NKCC2). Exon 21 encodes a short 16 amino acid peptide containing two consecutive leucine residues that were shown to be critical for basolateral sorting. In contrast to typical dileucine motifs that are preceded by acidic residues, none of the residues surrounding the two leucines seemed to matter; including a stretch of four acidic residues located eight amino acids upstream (2). Our data, therefore, indicate that some context must exist around the presence of the two leucines, as exon 21 and these residues are present in the NKCC1-DFX mutant transporter.
Cyst formation in MDCK cells has been examined experimentally and has been modeled (11). Cysts start developing their lumen by the end of day 2 and although multiple lumens appeared in ~50% of the cysts, their frequency decreased over time. The cysts grow by cell division at a high rate from days 2–6 and then the rate of growth decreases. Modeling revealed that when cells divided with random orientation, the number of single lumen cysts decreased and the number of multilumen cysts increased (11). This was consistent with observations done with MDCK cells that had LGN, a G protein-signaling modulator that plays a role in spindle orientation during cell division, knocked down or targeted to the apical membrane (51). Decreased cell death within the lumen and delayed cell polarization also contributed to the decrease in single lumen cysts (29). Observation that NKCC1-DFX expression increases the frequency of multilumen cysts indicates that mistargeting of the transporter affects the formation of the cyst. Neither NKCC1-LK-C1 nor NKCC1-LK-A3 produces such mistargeting, and thus these cells form perfectly shaped single lumen cysts which fail to grow to their full sizes. This indicates that the size of the cyst is correlated with NKCC1 function, i.e., accumulation of fluid in the lumen. In the absence of fluid accumulation, there are likely signals that prevent cell division and expansion of cyst size. The occasional polarity reversal that we observed in the 3-D Matrigel cysts formed by MDCK cells expressing NKCC1-DFX is likely due to mixed signals that come from the cells that surround the multiple lumens. Indeed, when the cysts were perfectly formed with a single lumen, the polarity of the epithelium was unchanged as demonstrated by the presence of the Na+-K+-ATPase on the basolateral membrane. It is important to note that when MDCK cells overexpress NKCC1-DFX, while multilumen cysts are formed instead of one, the overall size of the cyst is not markedly decreased, definitely not small like the cysts formed by the NKCC1-deficient MDCK LK-C1 and MDCK LK-A3 cell lines. This observation indicates the presence of some functional NKCC1 able to mediate fluid secretion and cyst growth. If NKCC1 and NKCC1-DFX dimerize randomly, one anticipates one-fourth of dimers consisting of wild-type transporters. Whether DFX-containing dimers affect the traffic of normal dimers in unknown, but the size of the NKCC1-DFX expressing cysts suggests functional transporters on the basolateral membrane. Our staining experiments concur with this assessment.
Creation of the NKCC1-DFX mouse model allowed us to verify in vivo the mistargeting of NKCC1-DFX in epithelia. Because the T4 antibody does not recognizes the NKCC1-DFX transporter, the immunofluorescence signal we see in the NKCC1-DFX mouse with T4 comes from wild-type NKCC1. As anticipated based on our heterologous expression experiments in MDCK cells, we showed that wild-type NKCC1 is mistargeted to the apical pole of epithelial cells. We focused on colon and salivary gland, which represent two prototypical Cl− secreting epithelia with a basolateral Na-K-2Cl cotransporter and an apical Cl− channel (3, 10, 20, 27). Upon activation of apical Cl− conductance, i.e., through increased cAMP levels, intracellular Cl− levels decrease, leading to activation of the basolateral cotransporter (28). This activation replenishes Cl− in the cell, thereby allowing sustained Cl−-mediated fluid secretion. In the colon and salivary gland isolated from NKCC1-DFX mice, NKCC1 is mostly detected at the apical pole. Mistrafficking of the transporter should affect the level of saliva secretion. We did not, however, observe significant differences in the amount of saliva produced by the NKCC1-DFX mice, compared with wild-type mice. This might be due to enough transporter units left on the basolateral membrane, like with the MDCK cell cysts expressing NKCC1-DFX (see above). How many transporters are necessary for proper saliva secretion? While the NKCC1 knockout mouse (no functional transporters) display a deficit in saliva production (12), absence of NKCC1 does not completely abolish the secretion. In fact, the glands are still able to produce a quarter of the amount generated by glands from wild-type animals (12). If one-fourth of dimers are wild-type homodimers expressed in the basolateral membrane, there might be enough functional units to sustain saliva secretion.
The fact that the NKCC1-DFX mouse does not display an overt phenotype is interesting in light of the severe clinical presentations of the patient. Growth curves show similar growth patterns in NKCC1-DFX mice versus wild-type at least until 12 wk of age. It is important to note that the gastrointestinal and bladder dysfunctions occurred progressively in the patient and it is only recently that complete loss of organ function was detected. This rather slow timeline indicates that other factors are likely to be at play, such as inflammatory conditions or some other tissue injuries that the patient has experienced but our mice have not yet encountered as they live in a pathogen-free barrier facility. Alternatively, additional mutations in other genes might have predisposed the patient to the ill effects of NKCC1 mislocalization. Whether expression of a wild-type cotransporter is a prerequisite for the clinical presentation of the patient is still unknown. The female carrying the NKCC1-DFX allele together with another disrupted allele that was the product of cas9 injection clearly displayed the inner ear deficit phenotype characteristic of NKCC1 knockout mice (7, 13). This mouse does not have a wild-type allele which helps the mutant transporter to reach the plasma membrane. Thus, the knockout situation is clearly different from the NKCC1-DFX situation where the mutant transporter can exert dominant-negative effects on the wild-type cotransporter in epithelial cells.
GRANTS
This work was supported by NIH National Institute of General Medical Sciences Grant GM-118944 and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant DK-093501 to E. Delpire. Confocal imaging was performed in part through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource (supported by NIH National Cancer Institute Grant CA-68485, NIDDK Grants DK-20593, DK-58404, and DK-59637, and National Eye Institute Grant EY-08126).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
R.K. and E.D. conceived and designed research; R.K., S.O., and E.D. performed experiments; R.K., S.O., and E.D. analyzed data; R.K., S.O., and E.D. interpreted results of experiments; R.K. and E.D. prepared figures; R.K. and E.D. drafted manuscript; R.K., S.O., and E.D. edited and revised manuscript; R.K., S.O., and E.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Arohan Subramanya (University of Pittsburgh Medical Center) for indicating to us the possibility of a trafficking defect in epithelial cells. This idea prompted us to conduct this study.
REFERENCES
- 1.Best JA, Quinton PM. Salivary secretion assay for drug efficacy for cystic fibrosis in mice. Exp Physiol 90: 189–193, 2005. doi: 10.1113/expphysiol.2004.028720. [DOI] [PubMed] [Google Scholar]
- 2.Carmosino M, Giménez I, Caplan M, Forbush B. Exon loss accounts for differential sorting of Na-K-Cl cotransporters in polarized epithelial cells. Mol Biol Cell 19: 4341–4351, 2008. doi: 10.1091/mbc.e08-05-0478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cook DI, Young JA. Fluid and electrolyte secretion by salivary glands. In: Handbook of Physiology. The Gastrointestinal System. Salivary, Gastric, Pancreatic, and Hepatobiliary Secretion, edited by Forte JG, Schultz SG. Bethesda, MD: American Physiological Society, 1989, p. 1–23. doi: 10.1002/cphy.cp060301. [DOI] [Google Scholar]
- 4.Damkier HH, Brown PD, Praetorius J. Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev 93: 1847–1892, 2013. doi: 10.1152/physrev.00004.2013. [DOI] [PubMed] [Google Scholar]
- 5.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30: 256–268, 2003. doi: 10.1016/S1046-2023(03)00032-X. [DOI] [PubMed] [Google Scholar]
- 6.Delpire E, Gagnon KB, Ledford JJ, Wallace JM. Housing and husbandry of Xenopus laevis affect the quality of oocytes for heterologous expression studies. J Am Assoc Lab Anim Sci 50: 46–53, 2011. [PMC free article] [PubMed] [Google Scholar]
- 7.Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl co-transporter. Nat Genet 22: 192–195, 1999. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- 8.Delpire E, Rauchman MI, Beier DR, Hebert SC, Gullans SR. Molecular cloning and chromosome localization of a putative basolateral Na+-K+-2Cl− cotransporter from mouse inner medullary collecting duct (mIMCD-3) cells. J Biol Chem 269: 25677–25683, 1994. [PubMed] [Google Scholar]
- 9.Delpire E, Wolfe L, Flores B, Koumangoye R, Schornak CC, Omer S, Pusey B, Lau C, Markello T, Adams DR. A patient with multisystem dysfunction carries a truncation mutation in human SLC12A2, the gene encoding the Na-K-2Cl cotransporter, NKCC1. Cold Spring Harb Mol Case Stud 2: a001289, 2016. doi: 10.1101/mcs.a001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dharmsathaphorn K, Mandel KG, Masui H, McRoberts JA. Vasoactive intestinal polypeptide-induced chloride secretion by a colonic epithelial cell line. Direct participation of a basolaterally localized Na+,K+,Cl− cotransport system. J Clin Invest 75: 462–471, 1985. doi: 10.1172/JCI111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelberg JA, Datta A, Mostov KE, Hunt CA. MDCK cystogenesis driven by cell stabilization within computational analogues. PLOS Comput Biol 7: e1002030, 2011. doi: 10.1371/journal.pcbi.1002030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evans RL, Park K, Turner RJ, Watson GE, Nguyen HV, Dennett MR, Hand AR, Flagella M, Shull GE, Melvin JE. Severe impairment of salivation in Na+/K+/2Cl− cotransporter (NKCC1)-deficient mice. J Biol Chem 275: 26720–26726, 2000. doi: 10.1074/jbc.M003753200. [DOI] [PubMed] [Google Scholar]
- 13.Flagella M, Clarke LL, Miller ML, Erway LC, Giannella RA, Andringa A, Gawenis LR, Kramer J, Duffy JJ, Doetschman T, Lorenz JN, Yamoah EN, Cardell EL, Shull GE. Mice lacking the basolateral Na-K-2Cl cotransporter have impaired epithelial chloride secretion and are profoundly deaf. J Biol Chem 274: 26946–26955, 1999. doi: 10.1074/jbc.274.38.26946. [DOI] [PubMed] [Google Scholar]
- 14.Frizzell RA, Hanrahan JW. Physiology of epithelial chloride and fluid secretion. Cold Spring Harb Perspect Med 2: a009563, 2012. doi: 10.1101/cshperspect.a009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 266: 107–109, 1994. doi: 10.1126/science.7524148. [DOI] [PubMed] [Google Scholar]
- 16.Gagnon KB, Delpire E. Physiology of SLC12 transporters: lessons from inherited human genetic mutations and genetically engineered mouse knockouts. Am J Physiol Cell Physiol 304: C693–C714, 2013. doi: 10.1152/ajpcell.00350.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gagnon KB, England R, Delpire E. Volume sensitivity of cation-Cl− cotransporters is modulated by the interaction of two kinases: Ste20-related proline-alanine-rich kinase and WNK4. Am J Physiol Cell Physiol 290: C134–C142, 2006. doi: 10.1152/ajpcell.00037.2005. [DOI] [PubMed] [Google Scholar]
- 18.Gong L, Parikh S, Rosenthal PJ, Greenhouse B. Biochemical and immunological mechanisms by which sickle cell trait protects against malaria. Malar J 12: 317, 2013. doi: 10.1186/1475-2875-12-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hartmann AM, Blaesse P, Kranz T, Wenz M, Schindler J, Kaila K, Friauf E, Nothwang HG. Opposite effect of membrane raft perturbation on transport activity of KCC2 and NKCC1. J Neurochem 111: 321–331, 2009. doi: 10.1111/j.1471-4159.2009.06343.x. [DOI] [PubMed] [Google Scholar]
- 20.Heintze K, Stewart CP, Frizzell RA. Sodium-dependent chloride secretion across rabbit descending colon. Am J Gastro Intest Liver Physiol 244: G357–G365, 1983. doi: 10.1152/ajpgi.1983.244.4.G357. [DOI] [PubMed] [Google Scholar]
- 21.Hoekstra D, Tyteca D, van IJzendoorn SC. The subapical compartment: a traffic center in membrane polarity development. J Cell Sci 117: 2183–2192, 2004. doi: 10.1242/jcs.01217. [DOI] [PubMed] [Google Scholar]
- 22.Howard HC, Mount DB, Rochefort D, Byun N, Dupré N, Lu J, Fan X, Song L, Rivière JB, Prévost C, Horst J, Simonati A, Lemcke B, Welch R, England R, Zhan FQ, Mercado A, Siesser WB, George AL Jr, McDonald MP, Bouchard JP, Mathieu J, Delpire E, Rouleau GA. The K-Cl cotransporter KCC3 is mutant in a severe peripheral neuropathy associated with agenesis of the corpus callosumsum. Nat Genet 32: 384–392, 2002. doi: 10.1038/ng1002. [DOI] [PubMed] [Google Scholar]
- 23.Josefson D. CF gene may protect against typhoid fever. BMJ 316: 1481, 1998. doi: 10.1136/bmj.316.7143.1477j. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan MR, Plotkin MD, Brown D, Hebert SC, Delpire E. Expression of the mouse Na-K-2Cl cotransporter, mBSC2, in the terminal inner medullary collecting duct, the glomerular and extraglomerular mesangium, and the glomerular afferent arteriole. J Clin Invest 98: 723–730, 1996. doi: 10.1172/JCI118844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplan MR, Plotkin MD, Lee WS, Xu ZC, Lytton J, Hebert SC. Apical localization of the Na-K-Cl cotransporter, rBSC1, on rat thick ascending limbs. Kidney Int 49: 40–47, 1996. doi: 10.1038/ki.1996.6. [DOI] [PubMed] [Google Scholar]
- 26.Karimy JK, Zhang J, Carusillo Theriault B, Duran D, Gavankar C, Diluna ML, Delpire E, Medzhitov R, Gerzanich V, Simard JM, Kahle KT. Inflammation-dependent cerebrospinal fluid hypersecretion by the choroid plexus epithelium in posthemorrhagic hydrocephalus. Nat Med 23: 997–1003, 2017. doi: 10.1038/nm.4361. [DOI] [PubMed] [Google Scholar]
- 27.Kidokoro M, Nakamoto T, Mukaibo T, Kondo Y, Munemasa T, Imamura A, Masaki C, Hosokawa R. Na+-K+-2Cl− cotransporter-mediated fluid secretion increases under hypotonic osmolarity in the mouse submandibular salivary gland. Am J Physiol Renal Physiol 306: F1155–F1160, 2014. doi: 10.1152/ajprenal.00709.2012. [DOI] [PubMed] [Google Scholar]
- 28.Lytle C, McManus T. Coordinate modulation of Na-K-2Cl cotransport and K-Cl cotransport by cell volume and chloride. Am J Physiol Cell Physiol 283: C1422–C1431, 2002. doi: 10.1152/ajpcell.00130.2002. [DOI] [PubMed] [Google Scholar]
- 29.Martín-Belmonte F, Yu W, Rodríguez-Fraticelli AE, Ewald AJ, Werb Z, Alonso MA, Mostov K. Cell-polarity dynamics controls the mechanism of lumen formation in epithelial morphogenesis. Curr Biol 18: 507–513, 2008. doi: 10.1016/j.cub.2008.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McRoberts JA, Tran CT, Saier MH Jr. Characterization of low potassium-resistant mutants of the Madin-Darby canine kidney cell line with defects in NaCl/KCl symport. J Biol Chem 258: 12320–12326, 1983. [PubMed] [Google Scholar]
- 31.Moore-Hoon ML, Turner RJ. The structural unit of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is a homodimer. Biochemistry 39: 3718–3724, 2000. doi: 10.1021/bi992301v. [DOI] [PubMed] [Google Scholar]
- 32.Murek M, Kopic S, Geibel J. Evidence for intestinal chloride secretion. Exp Physiol 95: 471–478, 2010. doi: 10.1113/expphysiol.2009.049445. [DOI] [PubMed] [Google Scholar]
- 33.Murthy M, Kurz T, O’Shaughnessy KM. WNK signalling pathways in blood pressure regulation. Cell Mol Life Sci 74: 1261–1280, 2017. doi: 10.1007/s00018-016-2402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nezu A, Parvin MN, Turner RJ. A conserved hydrophobic tetrad near the C terminus of the secretory Na+-K+-2Cl− cotransporter (NKCC1) is required for its correct intracellular processing. J Biol Chem 284: 6869–6876, 2009. doi: 10.1074/jbc.M804302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pace AJ, Lee E, Athirakul K, Coffman TM, O’Brien DA, Koller BH. Failure of spermatogenesis in mouse lines deficient in the Na+-K+-2Cl− cotransporter. J Clin Invest 105: 441–450, 2000. doi: 10.1172/JCI8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Parvin MN, Gerelsaikhan T, Turner RJ. Regions in the cytosolic C-terminus of the secretory Na+-K+-2Cl− cotransporter NKCC1 are required for its homodimerization. Biochemistry 46: 9630–9637, 2007. doi: 10.1021/bi700881a. [DOI] [PubMed] [Google Scholar]
- 37.Patterson K, Catalán MA, Melvin JE, Yule DI, Crampin EJ, Sneyd J. A quantitative analysis of electrolyte exchange in the salivary duct. Am J Physiol Gastrointest Liver Physiol 303: G1153–G1163, 2012. doi: 10.1152/ajpgi.00364.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Payne JA, Ferrell C, Chung CY. Endogenous and exogenous Na-K-Cl cotransporter expression in a low K-resistant mutant MDCK cell line. Am J Physiol Cell Physiol 280: C1607–C1615, 2001. doi: 10.1152/ajpcell.2001.280.6.C1607. [DOI] [PubMed] [Google Scholar]
- 39.Ponce-Coria J, Markadieu N, Austin T, Flammang L, Rios K, Welling PA, Delpire E. A novel Ste20-related proline/alanine-rich kinase (SPAK)-independent pathway involving calcium-binding protein 39 (Cab39) and serine threonine kinase with no lysine member 4 (WNK4) in the activation of Na-K-Cl cotransporters. J Biol Chem 289: 17680–17688, 2014. doi: 10.1074/jbc.M113.540518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Randall J, Thorne T, Delpire E. Partial cloning and characterization of Slc12a2: the gene encoding the secretory Na+-K+-2Cl− cotransporter. Am J Physiol Cell Physiol 273: C1267–C1277, 1997. doi: 10.1152/ajpcell.1997.273.4.C1267. [DOI] [PubMed] [Google Scholar]
- 41.Saint-Criq V, Gray MA. Role of CFTR in epithelial physiology. Cell Mol Life Sci 74: 93–115, 2017. doi: 10.1007/s00018-016-2391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simard CF, Brunet GM, Daigle ND, Montminy V, Caron L, Isenring P. Self-interacting domains in the C terminus of a cation-Cl− cotransporter described for the first time. J Biol Chem 279: 40769–40777, 2004. doi: 10.1074/jbc.M406458200. [DOI] [PubMed] [Google Scholar]
- 43.Stoops EH, Caplan MJ. Trafficking to the apical and basolateral membranes in polarized epithelial cells. J Am Soc Nephrol 25: 1375–1386, 2014. doi: 10.1681/ASN.2013080883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thwaites DT, Anderson CM. H+-coupled nutrient, micronutrient and drug transporters in the mammalian small intestine. Exp Physiol 92: 603–619, 2007. doi: 10.1113/expphysiol.2005.029959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verouti SN, Boscardin E, Hummler E, Frateschi S. Regulation of blood pressure and renal function by NCC and ENaC: lessons from genetically engineered mice. Curr Opin Pharmacol 21: 60–72, 2015. doi: 10.1016/j.coph.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 46.Warmuth S, Zimmermann I, Dutzler R. X-ray structure of the C-terminal domain of a prokaryotic cation-chloride cotransporter. Structure 17: 538–546, 2009. doi: 10.1016/j.str.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 47.Welling PA, Chang YP, Delpire E, Wade JB. Multigene kinase network, kidney transport, and salt in essential hypertension. Kidney Int 77: 1063–1069, 2010. doi: 10.1038/ki.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright EM, Loo DD. Coupling between Na+, sugar, and water transport across the intestine. Ann NY Acad Sci 915: 54–66, 2000. doi: 10.1111/j.1749-6632.2000.tb05223.x. [DOI] [PubMed] [Google Scholar]
- 49.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437: 215–223, 2005. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 50.Zdebik AA, Wangemann P, Jentsch TJ. Potassium ion movement in the inner ear: insights from genetic disease and mouse models. Physiology (Bethesda) 24: 307–316, 2009. doi: 10.1152/physiol.00018.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng Z, Zhu H, Wan Q, Liu J, Xiao Z, Siderovski DP, Du Q. LGN regulates mitotic spindle orientation during epithelial morphogenesis. J Cell Biol 189: 275–288, 2010. doi: 10.1083/jcb.200910021. [DOI] [PMC free article] [PubMed] [Google Scholar]