

Abstract
With-no-lysine kinase 4 (WNK4) and kidney-specific (KS)-WNK1 regulate ROMK (Kir1.1) channels in a variety of cell models. We now explore the role of WNK4 and KS-WNK1 in regulating ROMK in the native distal convoluted tubule (DCT)/connecting tubule (CNT) by measuring tertiapin-Q (TPNQ; ROMK inhibitor)-sensitive K+ currents with whole cell recording. TPNQ-sensitive K+ currents in DCT2/CNT of KS-WNK1−/− and WNK4−/− mice were significantly smaller than that of WT mice. In contrast, the basolateral K+ channels (a Kir4.1/5.1 heterotetramer) in the DCT were not inhibited. Moreover, WNK4−/− mice were hypokalemic, while KS-WNK1−/− mice had normal plasma K+ levels. High K+ (HK) intake significantly increased TPNQ-sensitive K+ currents in DCT2/CNT of WT and WNK4−/− mice but not in KS-WNK1−/− mice. However, TPNQ-sensitive K+ currents in the cortical collecting duct (CCD) were normal not only under control conditions but also significantly increased in response to HK in KS-WNK1−/− mice. This suggests that the deletion of KS-WNK1-induced inhibition of ROMK occurs only in the DCT2/CNT. Renal clearance study further demonstrated that the deletion of KS-WNK1 did not affect the renal ability of K+ excretion under control conditions and during increasing K+ intake. Also, HK intake did not cause hyperkalemia in KS-WNK1−/− mice. We conclude that KS-WNK1 but not WNK4 is required for HK intake-induced stimulation of ROMK activity in DCT2/CNT. However, KS-WNK1 is not essential for HK-induced stimulation of ROMK in the CCD, and the lack of KS-WNK1 does not affect net renal K+ excretion.
Keywords: hyperkalemia, hypokalemia, K+ excretion, Kir1.1, Kir4.1
INTRODUCTION
With-no-lysine kinases (WNKs) are serine/threonine protein kinases (11) and WNK1, WNK3, and WNK4 are expressed in the aldosterone-sensitive distal nephrons (ASDNs) (10, 11, 13). A large body of evidence indicates that the WNK family plays an important role in the regulation of ROMK channels (3, 9, 12, 13, 22, 26). The WNK1 gene gives rise to two major isoforms: L-WNK1 (long-WNK1), the ubiquitous and catalytically active isoform, and KS-WNK1, a kinase-deficient isoform expressed only in the distal nephron (11). Overexpression of L-WNK1, L-WNK13, and L-WNK14 inhibits ROMK activity by stimulating the clathrin-dependent endocytosis of the channel (9). Intersectin (a scaffold protein) and the clathrin adaptor ARH (autosomal recessive hypercholesterolemia) have been reported to be involved in WNK4 or L-WNK1-mediated endocytosis of ROMK (4, 7). Although KS-WNK1 lacks kinase activity and does not block ROMK channels (2, 26), it antagonizes the inhibitory effect of L-WNK1 on ROMK, thereby indirectly activating the K+ channel.
Although experiments performed in cell models support the role of WNK4 and KS-WNK1 in the regulation of ROMK (7, 15, 22), it is still not tested whether WNK4 and KS-WNK1 regulate ROMK channels in vivo. It is generally accepted that ROMK channels expressed in ASDN play a role in mediating K+ excretion under control conditions while HK intake-induced stimulation of renal K+ excretion is achieved by the activation of both ROMK and Ca2+-activated big-conductance K+ channels (BK) in the ASDN (5, 8, 16, 18, 21). The ASDN includes the late part of distal convoluted tubule (DCT2), connecting tubule (CNT), and collecting duct. A previous study has demonstrated that ENaC activity in the DCT2/CNT segment was significantly higher than in the CCD under control conditions (19). Because ENaC activity provides the driving force for K+ excretion (21), a high ENaC activity suggests that the DCT2 should play an important role in mediating renal K+ excretion by ROMK, which is also highly expressed in the apical membrane (25). Moreover, both WNK4 and KS-WNK1 are highly expressed in the DCT2/CNT (20, 24). Thus, the aim of the present study is to explore the role of WNK4 and KS-WNK1 in the regulation of ROMK channels in the DCT2/CNT and in mediating the effect of HK intake on ROMK channels.
METHODS
Animals and tubule preparation.
C57BL/6J mice (either sex, 8–10 wk old), which have the same genetic background as those of WNK4−/−, KS-WNK1−/−, and KS-WNK1AQP2/AQP2 knockout (KO) mice, were purchased from the Jackson Laboratory (Bar Harbor, ME) and were used as wild-type (WT) control. These genetically modified mice were obtained from Dr. J. Hadchouel (Hospital Tenon, Paris, France). KS-WNK1−/− and KS-WNK1AQP2/AQP2 KO mice, in which KS-WNK1 is deleted in principal cells (PC), were bred in New York Medical College for the experiments. The mice were fed with normal K+ diet (1% KCl) or HK diet (5% KCl) for 7 days and had free access to water. The normal K+ diet (TD.90228) and HK diet (TD.110866) were purchased from Harlan Laboratory (Madison, WI). Mice were euthanized by CO2 inhalation plus cervical dislocation. After mice were euthanized, we opened the abdomen to expose the left kidney and perfused the left kidney with 2 ml L-15 medium (Life Technology) that contained Type 2 collagenase (250 units/ml). The collagenase-perfused kidney was then removed for further dissection. The renal cortex was separated and cut into small pieces for additional incubation in collagenase-containing L-15 media for 30–50 min at 37°C. The tissue was then washed three times with fresh L-15 medium and transferred to an ice-cold chamber for dissection.
We followed the method described previously to prepare the DCT/CNT for the experiments (29, 30). Figure 1 shows images of isolated distal nephron segments showing the location of early distal convoluted tubule (DCT1), late DCT (DCT2), CNT, and cortical collecting duct (CCD) in WT (Fig. 1A), KS-WNK1−/−(Fig. 1B), and WNK4−/− mice (Fig. 1C). Figure 1D is a tubule scheme indicating the position of isolated distal tubules in the whole nephron segments. The isolated DCT tubules were placed on a small cover glass coated with polylysine, and the cover glass was placed on a chamber mounted on an inverted microscope. The protocol for using mice was approved by New York Medical College independent Animal Use Committee.
Fig. 1.

Images of isolated distal nephron segments show the location of early distal convoluted tubule (DCT1), late DCT (DCT2), connecting tubule (CNT), and cortical collecting duct (CCD). in WT (A), kidney-specific-with-no-lysine kinase-null (KS-WNK1−/−) mice (B), and WNK4−/− mice (C). A tubule scheme indicates the position of isolated distal tubules in the whole nephron segments (D) Arrow indicates the starting position of the tubule where the patch-clamp experiments are performed in the corresponding segment.
Patch-clamp experiments.
The basolateral K+ conductance was measured in the early part of the DCT (DCT1), which was defined by the first 100-μm length of the DCT after glomerular attachment, while ROMK channel currents were measured at the last 100 μm of the DCT2 before CNT attachment. DCT2 is morphologically different from the CNT (the diameter of the CNT is smaller than that of the DCT2) (Fig. 1A). However, it is hard to determine the exact transition between DCT2 and CNT (especially in KS-WNK1−/− and WNK4−/− mice) because of possible tubule remodeling. Thus, we refer that TPNQ-sensitive K+ (ROMK) currents were measured at split-open DCT2/CNT for the study. For the measurement of whole cell K+ currents with the perforated patch recording, we used an Axon 200A amplifier. The tip of the pipette was filled with pipette solution containing 140 mM KCl, 2 mM MgATP, 1 mM EGTA, and 10 mM HEPES (pH 7.4) for K+ currents. The pipette was then back-filled with amphotericin B (20 μg/0.1 ml) containing the pipette solution. The bath solution contains symmetrical 140 mM KCl solution as used in the pipette for measuring whole cell K+ currents. After formation of a high-resistance seal (>2 GΩ), the membrane capacitance was monitored until the whole cell patch configuration was formed. The currents were low-pass filtered at 1 kHz, digitized by an Axon interface with a 4-kHz sampling rate (Digidata 1440A). Data were analyzed using the pClamp software system 9.0 (Axon).
Procedures for renal clearance.
Animals were anesthetized by 2–4% isoflurane through an inhaling mask. The mice were placed on a small heated blanket to maintain body temperature at 37°C. The trachea was cannulated to clear any mucus that may have been produced during the experiment. A carotid artery was catheterized with polyethylene (PE)-10 tubing for blood collection, and the jugular vein was also cannulated for intravenous infusion. The bladder was exposed and catheterized via a suprapubic incision with a 10-cm piece of PE-10 tubing for urine collections. After completion of the surgery, 0.3% body weight of isotonic saline was given intravenously (0.3 ml every 1–1.5 h) to replace surgical fluid losses and to maintain hemodynamics. Urine collections were made during a 30-min period, and four collections were performed for each experiment. After the renal clearance experiment, the mice were euthanized by intravenous somnasol. Plasma sodium or potassium concentrations were measured using a dual-channel flame photometer with internal lithium standard (Cole-Parmer Instrument, Vernon Hills, IL).
Material and statistical analysis.
We obtained hydrochlorothiazide (HCTZ) and tertiapin-Q (TPNQ) from Sigma Chemical (St. Louis, MO). Data were analyzed using Studentʼs t-test for comparisons between two groups or one-way or two-way ANOVA for comparisons among more than two groups. P < 0.05 was considered statistically significant. Data are presented as means ± SE.
RESULTS
We first measured TPNQ (400 nM)-sensitive K+ currents (ROMK) at −40 mV in the DCT2/CNT of WT, WNK4−/−, and KS-WNK1−/− mice on a normal K+ diet (1%). Results from five to seven experiments were summarized in Fig. 2 showing TPNQ-sensitive K+ currents in all three groups (WT: 960 ± 50 pA; WNK4−/−: 370 ± 50 pA; KS-WNK1−/−: 450 ± 40 pA). ROMK currents in WNK4−/− and KS-WNK1−/− mice were significantly lower than that of WT. To determine whether the deletion of WNK4 or KS-WNK1 may also inhibit the basolateral Kir4.1 in the DCT, we next used the whole cell recording to measure the basolateral K+ conductance in the DCT1. Because the only type K+ channel expressed in the DCT1 is a Kir4.1/Kir5.1 heterotetramer in the basolateral membrane (29, 30), the whole cell K+ conductance in the DCT1 represents the basolateral K+ channel activity. The right group of bars in Fig. 2 also summarizes the results obtained from six experiments in which Ba2+-sensitive basolateral K+ currents were measured with whole cell recording at −60 mV in the DCT1. It is apparent that the basolateral K+ conductance in DCT1 of WNK4−/− mice was significantly higher (1,850 ± 50 pA) than in WT mice (1,340 ± 75 pA), while it was similar in KS-WNK1−/− mice (1300 ± 65 pA). Thus, the deletion of WNK4 or KS-WNK1 inhibits ROMK channels in the DCT2/CNT, while the deletion of WNK4 leads to increased basolateral K+ channel activity in DCT1.
Fig. 2.
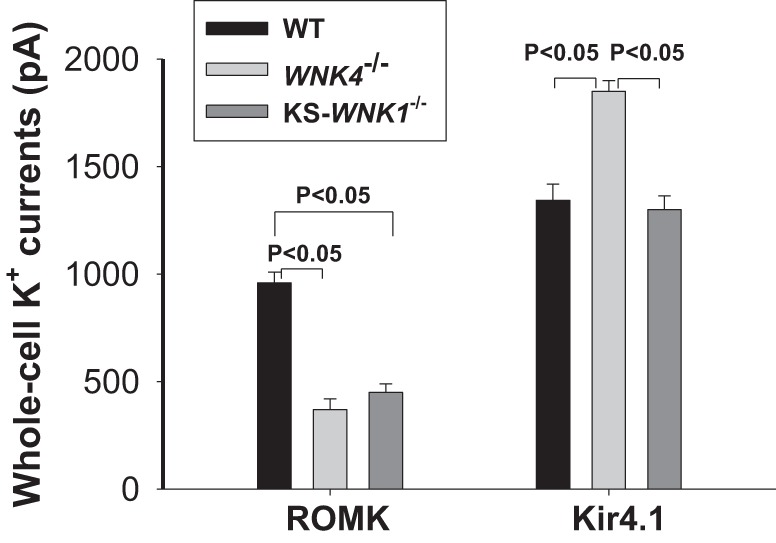
Deletion of with-no-lysine kinase 4 (WNK4) or kidney-specific-with-no-lysine kinase-null (KS-WNK1) inhibits ROMK but not Kir4.1. A bar graph summarizes results of experiments in which whole cell recording was used to measure tertiapin-Q (TPNQ)-sensitive K+ currents (ROMK) in DCT2/CNT and Ba2+-sensitive K+ currents (Kir4.1) in the basolateral membrane of DCT1 in WT, KS-WNK1−/−, and WNK4−/− mice. The results were obtained from five to seven experiments (tubules). A symmetrical solution containing 140 mM KCl, 2 mM MgATP, 1 mM EGTA, and 10 mM HEPES (pH 7.4) was used for the pipette and bath. The membrane potential was either clamped at −40 mV for the measurement of TPNQ-sensitive K+ currents or at −60 mV for the measurement of Ba2+-sensitive K+ currents.
We next examined the effect of HK intake on ROMK channel activity in the DCT2/CNT of WT mice. Figure 3A is a recording showing TPNQ-sensitive K+ currents in the DCT2/CNT at −40 mV. Figure 3B is a scatter graph summarizing the results from six experiments in which the whole cell TPNQ-sensitive K+ currents were measured at −40 mV in the DCT2/CNT of WT mice on normal K+ (NK, 1% KCl) or HK diet (5% KCl) for 7 days. As previously shown (28), HK intake increased the TPNQ-sensitive K+ currents in the DCT2/CNT (1870 ± 73 pA with HK and 960 ± 50 pA with NK). Figure 3C is a table showing that the administration of a HK diet for 7 days increased plasma K+ level from 3.8 ± 0.1 mM to 4.2 ± 0.1 mM.
Fig. 3.

HK intake stimulates ROMK in late distal convoluted tubule (DCT2)/connecting tubule (CNT). A: whole-cell recording shows tertiapin-Q (TPNQ)-sensitive K+ currents at −40 mV in DCT2/CNT of WT mice on normal K+ (1%) and a high K+ (5%), respectively. Arrows indicate addition of 400 nM TPNQ to the bath. B: scatterplot summarizes the experiments in which TPNQ-sensitive K+ currents were measured at −40 mV with whole cell recording. The results were obtained from six experiments (tubules). C: table shows plasma K+ concentration of WT mice on 1% K+ and 5% K+ diets for 7 days (n = 6 mice). *Significant difference between normal K+ (NK, 1% K+) and high K+ (HK, 5% K+) diets (P < 0.05).
We next examined whether the deletion of WNK4 affects HK intake-induced stimulation of ROMK channels in the DCT2/CNT by measuring TPNQ-sensitive K+ currents at −40 mV. Figure 4A is a set of two typical whole cell recordings showing TPNQ-sensitive K+ currents in the DCT2/CNT of WNK4−/− mice on NK and HK diets, respectively. Results from six similar experiments are summarized in Fig. 4B. We observed that the TPNQ-sensitive K+ currents increased from 370 ± 50 pA under control conditions to 1,010 ± 84 pA (HK) in the DCT2/CNT of WNK4−/− mice. Thus, the response of ROMK channels in the DCT2/CNT to dietary K+ intake in WNK4−/− mice was significantly blunted compared with WT mice, although the deletion of WNK4 did not completely abolish the stimulatory effect of HK intake on ROMK (Fig. 3C). We have also measured the plasma K+ concentrations in WNK4−/− mice (8–10 wk) on a NK diet and on a HK diet (Fig. 4D). We confirmed that WNK4−/− mice were hypokalemic (3.2 ± 0.1 mM) (1). Moreover, HK intake failed to correct hypokalemia (3.2 ± 0.1 mM). A previous study by Castañeda-Bueno et al. (1) demonstrated that WNK4−/− mice were hypokalemic and that HK intake for 4 days could correct hypokalemia, although plasma K+ tended to be still slightly lower under these conditions. The discrepancy between two findings may be induced by different experimental conditions, including different K+ content of normal diet, sex differences, and age differences. In Castañeda-Bueno’s experiment, the mice were fed with 1.2% K+ diet as a normal K+ diet, while we used 1% K+ diet as a normal K+ diet. Consequently, the plasma K+ level of WNK4−/− mice in their experiments (3.46 ± 0.1) was higher than the present one. Second, in their experiments only male mice were used, while we used both male and female mice. Finally, in Castañeda-Bueno’s experiment, the mice were older (12–16 wk) than those used in the present study (8–10 wk). We used 8- to 10-wk-old mice to perform the patch-clamp experiments because the dissection of the DCT was very difficult in the mice older than 12 wk. Thus, we also measured plasma K+ in the mice with similar age. We cannot exclude the possibility that these younger WNK4−/− mice may have less dietary K+ intake than those 12- to 16-wk-old WNK4−/− mice. Thus, even the HK diet could not completely restore the normal plasma K+ level in WNK4−/− mice in our experimental conditions. This suggests that WNK4 is essential for maintaining K+ homeostasis, although the deletion of WNK4 does not affect HK-induced stimulation of ROMK channels in the DCT2/CNT.
Fig. 4.

HK-induced stimulation of ROMK in late distal convoluted tubule (DCT2)/connecting tubule (CNT) is preserved in kidney-specific-with-no-lysine kinase-null (WNK4−/−) mice. A: whole cell recording shows tertiapin-Q (TPNQ)-sensitive K+ currents at −40 mV in of WNK4−/− mice on normal potassium (NK) and high potassium (HK). Arrows indicate the addition of 400 nM TPNQ to the bath. B: scatterplot summarizes the experiments in which TPNQ-sensitive K+ currents were measured at −40 mV with whole cell recording. Results were obtained from six experiments (tubules). C: line graph shows the effect of dietary K+ intake on TPNQ-sensitive K+ currents in the DCT of wild-type (WT) and WNK4−/− mice. The # symbol indicates the difference between WT and WNK4−/− mice is significant using two-way ANOVA. D: table shows plasma K+ concentrations of WNK4−/− mice on NK and HK diets for 7 days (n = 5 mice).
Having found that the HK-induced stimulation of ROMK is well preserved in the DCT2/CNT of WNK4−/− mice, we next examined the role of KS-WNK1 in mediating the effect of HK intake on ROMK in the DCT2/CNT. Figure 5A demonstrates a set of two typical whole cell recordings showing TPNQ-sensitive K+ currents in the DCT2/CNT of KS-WNK1−/− mice on NK and HK diets, respectively. In contrast to WNK4−/− mice, deletion of KS-WNK1 abolished the effect of HK intake on ROMK channel activity. Results from six similar experiments are summarized in Fig. 5B, showing that TPNQ-sensitive K+ currents in the DCT2/CNT of KS-WNK1−/− mice on a HK diet (520 ± 40 pA at −40 mV) were similar to that of mice on a NK diet (450 ± 40 pA at −40 mV). This finding suggests that KS-WNK1 plays an essential role in mediating the effect of HK intake on ROMK channels in the DCT2/CNT. However, although the deletion of KS-WNK1 abolished the effect of HK intake on ROMK channel activity in DCT2/CNT, plasma K+ concentrations of KS-WNK1−/− mice were normal under control conditions (3.8 ± 0.1 mM). Moreover, KS-WNK1−/− mice had no hyperkalemia during increasing K+ intake (4.0 ± 0.1 mM) (Fig. 5C). This suggests that the deletion of KS-WNK1 did not affect K+ homeostasis or renal K+ excretion in response to HK intake.
Fig. 5.

Deletion of kidney-specific-with-no-lysine kinase (KS-WNK1) abolishes the effect of high potassium (HK) on ROMK in late distal convoluted tubule (DCT2)/connecting tubule (CNT). A: whole cell recording shows tertiapin-Q (TPNQ)-sensitive K+ currents at −40 mV in DCT2/CNT of kidney-specific-with-no-lysine kinase-null (KS-WNK1−/−) mice on normal K+ (NK) and HK. Arrows indicate the addition of 400 nM TPNQ to the bath. B: scatterplot summarizes the experiments in which TPNQ-sensitive K+ currents were measured at −40 mV with whole cell recording. Results were obtained from six experiments (tubules). C: table shows plasma K+ concentration of KS-WNK1−/− mice on NK and HK diets for 7 days (n = 6 mice).
To test the possibility that the deletion of KS-WNK1 did not affect the overall renal K+ excretion, we used the renal clearance method to examine the effect of HCTZ (25 mg·1−1·kg body wt−1) on urinary K+ excretion (EK) in WT and KS-WNK1−/− mice on a NK or HK for 7 days. The mice were perfused with 0.3 ml isotonic saline (containing 140 mM Na+ and 5 mM K+) during 1 h after surgery to replace surgical fluid losses. Then, six urine collections were performed (30-min interval and two before HCTZ and four after HCTZ). Results from six experiments are summarized in Fig. 6A, showing that the application of HCTZ modestly but significantly increased renal K+ excretion in WT (control: 0.56 ± 0.05; HCTZ: 1.09 ± 0.1 μeq·min−1·100 g body wt−1) and in KS-WNK1−/− mice on a NK diet (control: 0.44 ± 0.03; HCTZ: 0.92 ± 0.10 μeq·min−1·100 g body wt−1). HK intake increased basal level of EK to 1.64 ± 0.2 μeq·min−1·100 g body wt−1. HCTZ did not significantly change EK in the mice on a HK diet (1.74 ± 0.25 μeq·min−1·100 g body wt−1). Moreover, a similar pattern was observed in KS-WNK1−/− mice on a HK diet and EK was 1.77 ± 0.14 μeq·min−1·100 g body wt−1 (before HCTZ) and 1.9 ± 0.25 μeq·min−1·100 g body wt−1 (after HCTZ). Thus, deletion of KS-WNK1 had no effect on the net renal K+ excretion under control conditions and after an increased dietary K+ intake. We also confirmed that the deletion of KS-WNK1 stimulated NCC activity since HCTZ-induced natriuresis was larger in KS-WNK1−/− mice (from 1.25 ± 0.1 to 4.0 ± 0.2 μeq·min−1·100 g body wt−1) than in WT mice (from 0.84 ± 0.13 to 2.5 ± 0.2 μeq·min−1·100 g body wt−1) (Fig. 6B) (6, 17). Moreover, we confirmed our previous finding that HK intake decreased HCTZ-induced natriuresis in WT mice (from 1.65 ± 0.21 to 2.6 ± 0.3 μeq·min−1·100 g body wt−1) (Fig. 6B) (27). However, HCTZ-induced natriuresis in KS-WNK1−/− mice on HK diet (from 1.35 ± 0.2 to 3.9 ± 0.2 μeq·min−1·100 g body wt−1) was still larger than in WT mice, suggesting that the deletion of KS-WNK1 compromised the inhibitory effect of HK intake on NCC. Thus, the deletion of KS-WNK1 affects the effect of HK intake on NCC, but it did not affect the renal K+ excretion.
Fig. 6.

Renal K+ excretion is normal in kidney-specific-with-no-lysine kinase-null (KS-WNK1−/−) mice. A: bar graph shows the results of experiments in which the effect of a single dose hydrochlorothiazide (HCTZ) on urinary K+ excretion (EK) within 120 min was measured with renal clearance method in wild-type (WT) or KS-WNK1−/− mice on normal potassium (NK) (1% K+) and high potassium (HK) (5% K+) diets, respectively. B: bar graph shows the results (n = 6 mice for each group) of experiments in which the effect of single dose HCTZ on urinary Na+ excretion (ENa) within 120 min was measured with renal clearance method in WT or KS-WNK1−/− mice on 1% and 5% K+ diets, respectively. *Significant difference as determined by Studentʼs t test (P < 0.05). #Significant difference as determined by Studentʼs t test and two-way ANOVA (P < 0.05).
Since the deletion of KS-WNK1 did not affect the net renal K+ excretion, we suspect that KS-WNK1-dependent regulation of ROMK may mainly occur in DCT2/CNT but not in the late portion of ASDN. Thus, we next measured TPNQ-sensitive K+ currents in the CCD with whole cell recording. Figure 7A summarizes results from six experiments showing that the depletion of KS-WNK1 did not abolish the effect of HK intake on ROMK channels in the CCD. HK intake increased TPNQ-sensitive K+ currents from 1,100 ± 110 pA to 2,060 ± 130 pA in the CCD of KS-WNK1−/− mice. This suggests that HK-induced stimulation of ROMK channel requires the involvement of KS-WNK1 only in the DCT2/CNT but not in the CCD. This hypothesis was further tested by measuring TPNQ-sensitive K+ currents in the CCD of KS-WNK1AQP2/AQP2 KO mice in which KS-WNK1 was deleted in PC (Fig. 7A). Indeed, HK intake was able to stimulate ROMK channels in the CCD (control diet: 540 ± 40; HK: 1,130 ± 70 pA; n = 5). Lower ROMK activity in the CCD of KS-WNK1AQP2/AQP2 KO mice than in KS-WNK1−/− mice may be due to the possibility that ROMK activity is normal in the DCT2/CNT of KS-WNK1AQP2/AQP2 KO mice. This possibility is confirmed by measurement of ROMK currents in the DCT2/CNT of KS-WNK1AQP2/AQP2 KO mice on NK and HK, respectively. Results are summarized in Fig. 7B showing that HK intake stimulated ROMK channels in DCT2/CNT of KS-WNK1AQP2/AQP2 KO mice from 900 ± 40 (NK) to 1,890 ± 50 pA (n = 5). Thus, KS-WNK1 is essential for the effect of HK intake on ROMK channels in the DCT2/CNT, but it is not required in PC.
Fig. 7.

Deletion of kidney-specific-with-no-lysine kinase 1 (KS-WNK1) does not affect high potassium (HK)-induced stimulation of ROMK in the cortical collecting duct (CCD). A: bar graph summarizes results of experiments in which whole cell recording was used to measure tertiapin-Q (TPNQ)-sensitive K+ currents (ROMK) in the CCD of KS-WNK1−/− and KS-WNK1AQP2/AQP2 KO mice on 1% and 5% K+ diets, respectively (n = 6 tubules). B: bar graph summarizes results of experiments in which whole cell recording was used to measure TPNQ-sensitive K+ currents (ROMK) in DCT2/CNT of KS-WNK1AQP2/AQP2 KO mice on 1% and 5% K+ diets, respectively (n = 5 tubules). A symmetrical solution containing 140 mM KCl, 2 mM MgATP, 1 mM EGTA, and 10 mM HEPES (pH 7.4) was used for the pipette and bath. The membrane potential was clamped at −40 mV.
DISCUSSION
In the present study, we have demonstrated that ROMK channel activity in the DCT2/CNT of WNK4−/− mice was downregulated. WNK4 has been shown to inhibit ROMK channels in a variety of cells transfected with ROMK and WNK4 (7, 9, 15). In the mice expressing the pseudohypoaldosteronism type II (PHAII) mutant WNK4 (TgWNK4PHAII mice), ROMK channel activity was significantly lower in the DCT2/CNT than the WT control, suggesting the role of WNK4 in inhibiting ROMK in vivo (28). However, the finding that TPNQ-sensitive K+ currents, an indication of ROMK channel activity, were decreased rather than increased in WNK4−/− mice argues the role of WNK4 in inhibiting ROMK in vivo. We speculate that the phenotype of low ROMK channel activity in TgWNK4PHAII mice may be induced by a strong pathophysiological role of WNK4PHAII mutant in the inhibition of ROMK while endogenous WNK4 copies may play only a minor role in regulating ROMK channels in the WNK4PHAII mouse model. However, in consideration of the fact that multiple factors, including WNK4, L-WNK1, KS-WNK1, WNK3, c-Src, and superoxide, have been shown to regulate ROMK channels (5, 8), it is difficult to accurately evaluate any single factor in regulating ROMK channels in in vivo animal models because of the interaction among all factors. Thus, it is conceivable that the role of WNK4 in regulating ROMK channels observed in cell models in which only one or two factors are investigated may be modified or changed in in vivo conditions. Thus, we speculate that the discrepancy between cell models and animal models may be due to the fact that WNK4 is not the only factor controlling ROMK channel activity in the native DCT2/CNT under physiological conditions. It is conceivable that ROMK channel activity is regulated not only by WNK4 but also L-WNK1 and KS-WNK1 in in vivo conditions. This notion is supported by our present study showing the role of KS-WNK1 in regulating ROMK channels in the DCT2/CNT. Thus, in the absence of WNK4, WNK family members other than WNK4 or more dominant physiological pathways may compensate for the function of WNK4 in regulating ROMK channels in the DCT2/CNT.
Also, the observation that HK intake was still able to stimulate ROMK channels in the DCT2/CNT of WNK4−/− mice suggests that WNK4 may not be essential for HK intake-induced stimulation of ROMK channels in vivo. However, the finding that WNK4−/− mice were hypokalemic under control conditions and even during increasing dietary K+ intake strongly suggests that WNK4 is essential for maintaining K+ homeostasis. However, we believe that the role of WNK4 in regulating renal K+ excretion and K+ homeostasis is mainly achieved by controlling NCC activity rather than by regulating ROMK. Previous studies demonstrated that WNK4 plays a unique role in the regulation of NCC in response to changes in plasma K+ concentrations at physiological ranges and that the deletion of WNK4 decreased NCC expression and activity (1, 23). Consequently, the deletion of WNK4 is expected to increase Na+ and fluid delivery to the late portion of ASDN and to enhance Na+ absorption, thereby causing K+ wasting and hypokalemia. Also, we suspect that hypokalemia may be responsible for low ROMK activity in the DCT2/CNT of WNK4−/− mice because hypokalemia is known to be a dominant factor inhibiting ROMK channel activity (14). Thus, although WNK4 may not be essential for the regulation of ROMK channels in DCT2/CNT, WNK4 plays a key role in the regulation of renal K+ excretion and K+ homeostasis by regulating NCC activity.
As in WNK4−/− mice, ROMK channel activity in DCT2/CNT of KS-WNK1−/− mice was also decreased. However, the low ROMK channel activity was not related to plasma K+ level because KS-WNK1−/− mice were normokalemic. Thus, the decreased ROMK channel activity in DCT2/CNT of KS-WNK1−/− mice was most likely due to the lack of stimulatory effect of KS-WNK1 on ROMK. Previous studies have demonstrated that KS-WNK1 can stimulate ROMK channel activity by antagonizing the L-WNK1-induced inhibition (12, 26). Since L-WNK1 has been shown to be expressed in the DCT (24), it is possible that the lack of KS-WNK1 may indirectly enhance the inhibitory effect of L-WNK1 on ROMK, thereby decreasing ROMK channel activity in DCT2/CNT. KS-WNK1 has been suggested to play a role in mediating the effect of HK intake on ROMK channels since a HK intake increases the expression of KS-WNK1, which could, thus attenuate the inhibitory effect of L-WNK1 on ROMK channels (12, 26). On the other hand, low K+ intake has been reported to decrease the expression of KS-WNK1, thereby enhancing L-WNK1-mediated inhibition of ROMK channels (12). Our present experiments support the role of KS-WNK1 in mediating the effect of HK intake on ROMK at least in the DCT2/CNT, since HK intake fails to increase ROMK channel activity in the DCT2/CNT of KS-WNK1−/− mice. Thus, we speculate that the lack of L-WNK1/KS-WNK1 interaction may be responsible for the absence of HK-induced stimulation of ROMK in DCT2/CNT of KS-WNK1−/− mice.
Although the HK-induced stimulation of ROMK in the DCT2/CNT was compromised in KS-WNK1−/− mice, HK intake did not induce hyperkalemia. This strongly suggests that the deletion of KS-WNK1 had no significant effect on overall renal K+ excretion and K+ homeostasis. This notion is supported by renal clearance experiments showing that the renal K+ excretion in KS-WNK1−/− mice was similar to those of WT mice on either normal K+ or a HK diet, although NCC activity was higher in KS-WNK1−/− mice than WT. DCT2/CNT should play a significant role in renal K+ excretion under control conditions due to high ENaC and ROMK activity (19, 28). However, the fact that KS-WNK1−/− mice were normokalemic under control conditions and had no hyperkalemia during increasing dietary K+ intake suggests the possibility that K+ excretion may be shifted from DCT2/CNT to the late portion of ASDN by mechanisms, including the stimulation of ROMK- and BK-dependent K+ secretion. Although it has been reported that K+ secretion in the isolated CCD of KS-WNK1−/− mice was decreased (2), the observation that HK intake still stimulated ROMK channel in the CCD of KS-WNK1−/− mice suggests that HK intake induced stimulation of ROMK and K+ secretion in the CCD is, at least in part, preserved in KS-WNK1−/− mice. The finding that the deletion of KS-WNK1 inhibits ROMK channels and impairs HK-induced stimulation of ROMK only in the DCT2/CNT, but not in the CCD, suggests the possibility that KS-WNK1 may not play a key role in the regulation of ROMK in the collecting duct. This notion is also supported by the observation that HK stimulated ROMK in PC of KS-WNK1AQP2/AQP2 KO mice. The reason for the lack of KS-WNK1-dependent regulation of ROMK in the CCD may be due to the fact that KS-WNK1 was mainly expressed in the DCT/CNT rather than in CCD (24). Alternatively, the role of KS-WMK1 in regulating ROMK is different between the DCT2/CNT and the collecting duct. In addition, we speculate that KS-WNK1/L-WNK1 interactions may play a role only in regulating ROMK channel activity in the DCT but may not play a dominant role in the regulation of NCC under physiological conditions. This notion is supported by the report that WNK4 activity, but not L-WNK1, was sensitive to changes in intracellular Cl− concentrations in a physiologically relevant range (23). This suggests the possibility that WNK4 may play a more important role in regulating NCC than L-WNK1. Thus, KS-WNK1-mediated modulation of L-WNK1 in the DCT should mainly affect ROMK activity in the DCT without changing NCC activity.
The observation that the basolateral K+ channel activity in the DCT of KS-WNK1−/− mice was the same as that of WT mice suggests that KS-WNK1 is not involved in the regulation of the basolateral Kir4.1 channels in the DCT. Although the basolateral K+ channel activity in the DCT of WNK4−/− mice was increased, it is unlikely that WNK4 inhibits Kir4.1 in vivo since WNK4 had no effect on Kir4.1 channels in HEK-293 cells transfected with Kir4.1/5.1 (W.-H. Wang, unpublished observations). Since WNK4−/− mice were hypokalemic, it is most likely that low plasma K+ is responsible for increasing the basolateral K+ conductance (27). In summary, WNK4 is not required for mediating the effect of HK intake on ROMK channels in the DCT2/CNT. However, WNK4 is essential for maintaining K+ homeostasis. We also conclude that KS-WNK1 regulates ROMK channels and mediates the stimulatory effect of HK intake on ROMK only in the DCT2/CNT but not in the CCD. Moreover, the deletion of KS-WNK1 has no net effect on the overall renal K+ excretion.
GRANTS
This work was supported by National Institutes of Health Grant DK-54983.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.H.E., J.H., J.T., and W.-H.W. conceived and designed research; P.W., Z.-X.G., X.-T.S., and W.-H.W. performed experiments; P.W., Z.-X.G., X.-T.S., and W.-H.W. analyzed data; P.W., Z.-X.G., D.H.E., J.H., J.T., and W.-H.W. interpreted results of experiments; P.W., Z.-X.G., and W.-H.W. prepared figures; P.W., Z.-X.G., D.H.E., J.H., J.T., and W.-H.W. edited and revised manuscript; P.W., Z.-X.G., X.-T.S., D.H.E., J.H., J.T., and W.-H.W. approved final version of manuscript; W.-H.W. drafted manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Marc Paulais for technical support during the course of the experiments.
REFERENCES
- 1.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+:Cl− cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA 109: 7929–7934, 2012. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheng CJ, Baum M, Huang CL. Kidney-specific WNK1 regulates sodium reabsorption and potassium secretion in mouse cortical collecting duct. Am J Physiol Renal Physiol 304: F397–F402, 2013. doi: 10.1152/ajprenal.00589.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cope G, Murthy M, Golbang AP, Hamad A, Liu CH, Cuthbert AW, O’Shaughnessy KM. WNK1 affects surface expression of the ROMK potassium channel independent of WNK4. J Am Soc Nephrol 17: 1867–1874, 2006. doi: 10.1681/ASN.2005111224. [DOI] [PubMed] [Google Scholar]
- 4.Fang L, Garuti R, Kim BY, Wade JB, Welling PA. The ARH adaptor protein regulates endocytosis of the ROMK potassium secretory channel in mouse kidney. J Clin Invest 119: 3278–3289, 2009. doi: 10.1172/JCI37950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giebisch G, Hebert SC, Wang WH. New aspects of renal potassium transport. Pflügers Arch 446: 289–297, 2003. doi: 10.1007/s00424-003-1029-8. [DOI] [PubMed] [Google Scholar]
- 6.Hadchouel J, Soukaseum C, Büsst C, Zhou XO, Baudrie V, Zürrer T, Cambillau M, Elghozi JL, Lifton RP, Loffing J, Jeunemaitre X. Decreased ENaC expression compensates the increased NCC activity following inactivation of the kidney-specific isoform of WNK1 and prevents hypertension. Proc Natl Acad Sci USA 107: 18109–18114, 2010. doi: 10.1073/pnas.1006128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He G, Wang HR, Huang SK, Huang C-L. Intersectin links WNK kinases to endocytosis of ROMK1. J Clin Invest 117: 1078–1087, 2007. doi: 10.1172/JCI30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev 85: 319–371, 2005. doi: 10.1152/physrev.00051.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet 35: 372–376, 2003. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 10.Kahle KT, Gimenez I, Hassan H, Wilson FH, Wong RD, Forbush B, Aronson PS, Lifton RP. WNK4 regulates apical and basolateral Cl- flux in extrarenal epithelia. Proc Natl Acad Sci USA 101: 2064–2069, 2004. doi: 10.1073/pnas.0308434100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahle KT, Ring AM, Lifton RP. Molecular physiology of the WNK kinases. Annu Rev Physiol 70: 329–355, 2008. doi: 10.1146/annurev.physiol.70.113006.100651. [DOI] [PubMed] [Google Scholar]
- 12.Lazrak A, Liu Z, Huang CL. Antagonistic regulation of ROMK by long and kidney-specific WNK1 isoforms. Proc Natl Acad Sci USA 103: 1615–1620, 2006. doi: 10.1073/pnas.0510609103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leng Q, Kahle KT, Rinehart J, MacGregor GG, Wilson FH, Canessa CM, Lifton RP, Hebert SC. WNK3, a kinase related to genes mutated in hereditary hypertension with hyperkalaemia, regulates the K+ channel ROMK1 (Kir1.1). J Physiol 571: 275–286, 2006. doi: 10.1113/jphysiol.2005.102202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin DH, Sterling H, Lerea KM, Welling P, Jin L, Giebisch G, Wang WH. K depletion increases protein tyrosine kinase-mediated phosphorylation of ROMK. Am J Physiol Renal Physiol 283: F671–F677, 2002. doi: 10.1152/ajprenal.00160.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin DH, Yue P, Yarborough O III, Scholl UI, Giebisch G, Lifton RP, Rinehart J, Wang WH. Src-family protein tyrosine kinase phosphorylates WNK4 and modulates its inhibitory effect on KCNJ1 (ROMK). Proc Natl Acad Sci USA 112: 4495–4500, 2015. doi: 10.1073/pnas.1503437112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Xu S, Woda C, Kim P, Weinbaum S, Satlin LM. Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol 285: F998–F1012, 2003. doi: 10.1152/ajprenal.00067.2003. [DOI] [PubMed] [Google Scholar]
- 17.Liu Z, Xie J, Wu T, Truong T, Auchus RJ, Huang CL. Downregulation of NCC and NKCC2 cotransporters by kidney-specific WNK1 revealed by gene disruption and transgenic mouse models. Hum Mol Genet 20: 855–866, 2011. doi: 10.1093/hmg/ddq525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Najjar F, Zhou H, Morimoto T, Bruns JB, Li HS, Liu W, Kleyman TR, Satlin LM. Dietary K+ regulates apical membrane expression of maxi-K channels in rabbit cortical collecting duct. Am J Physiol Renal Physiol 289: F922–F932, 2005. doi: 10.1152/ajprenal.00057.2005. [DOI] [PubMed] [Google Scholar]
- 19.Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J, Korbmacher C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303: F1289–F1299, 2012. doi: 10.1152/ajprenal.00247.2012. [DOI] [PubMed] [Google Scholar]
- 20.O’Reilly M, Marshall E, Macgillivray T, Mittal M, Xue W, Kenyon CJ, Brown RW. Dietary electrolyte-driven responses in the renal WNK kinase pathway in vivo. J Am Soc Nephrol 17: 2402–2413, 2006. doi: 10.1681/ASN.2005111197. [DOI] [PubMed] [Google Scholar]
- 21.Palmer LG. Potassium secretion and the regulation of distal nephron K channels. Am J Physiol 277: F821–F825, 1999. doi: 10.1152/ajprenal.1999.277.6.F821. [DOI] [PubMed] [Google Scholar]
- 22.Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci USA 104: 4025–4029, 2007. doi: 10.1073/pnas.0611728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Terker AS, Zhang C, Erspamer KJ, Gamba G, Yang CL, Ellison DH. Unique chloride-sensing properties of WNK4 permit the distal nephron to modulate potassium homeostasis. Kidney Int 89: 127–134, 2016. doi: 10.1038/ki.2015.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vidal-Petiot E, Cheval L, Faugeroux J, Malard T, Doucet A, Jeunemaitre X, Hadchouel J. A new methodology for quantification of alternatively spliced exons reveals a highly tissue-specific expression pattern of WNK1 isoforms. PLoS One 7: e37751, 2012. doi: 10.1371/journal.pone.0037751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300: F1385–F1393, 2011. doi: 10.1152/ajprenal.00592.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wade JB, Fang L, Liu J, Li D, Yang CL, Subramanya AR, Maouyo D, Mason A, Ellison DH, Welling PA. WNK1 kinase isoform switch regulates renal potassium excretion. Proc Natl Acad Sci USA 103: 8558–8563, 2006. doi: 10.1073/pnas.0603109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang MX, Cuevas-Gallardo C, Su XT, Wu P, Gao Z-X, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium (K+) intake modulates NCC activity via the K+ channel. Kir4.1. Kidney Int 93: 893–902, 2018. doi: 10.1016/j.kint.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang C, Wang L, Su XT, Zhang J, Lin DH, Wang WH. ENaC and ROMK activity are inhibited in the DCT2/CNT of TgWnk4PHAII mice. Am J Physiol Renal Physiol 312: F682–F688, 2017. doi: 10.1152/ajprenal.00420.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang C, Wang L, Thomas S, Wang K, Lin DH, Rinehart J, Wang WH. Src family protein tyrosine kinase regulates the basolateral K channel in the distal convoluted tubule (DCT) by phosphorylation of KCNJ10 protein. J Biol Chem 288: 26135–26146, 2013. doi: 10.1074/jbc.M113.478453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang C, Wang L, Zhang J, Su X-T, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111: 11864–11869, 2014. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]