

Abstract
Cells organize actin filaments into contractile bundles known as stress fibers that resist mechanical stress, increase cell adhesion, remodel the extracellular matrix, and maintain tissue integrity. α-actinin is an actin filament bundling protein that is thought to be essential for stress fiber formation and stability. However, previous studies have also suggested that α-actinin might disrupt fibers, making the true function of this biomolecule unclear. Here we use fluorescence imaging to show that kidney epithelial cells depleted of α-actinin-4 via shRNA or CRISPR/Cas9, or expressing a disruptive mutant make more massive stress fibers that are less dynamic than those in WT cells, leading to defects in cell motility and wound healing. The increase in stress fiber mass and stability can be explained, in part, by increased loading of the filament component tropomyosin onto stress fibers in the absence of α-actinin, as monitored via immunofluorescence. We show using imaging and cosedimentation that α-actinin and tropomyosin compete for binding to F-actin and that tropomyosin shields actin filaments from cofilin-mediated disassembly in vitro and in cells. Perturbing tropomyosin in cells lacking α-actinin-4 results in a complete loss of stress fibers. Our results with α-actinin-4 on stress fiber organization are the opposite of what might have been predicted from previous in vitro biochemistry and further highlight how the complex interactions of multiple proteins competing for filament binding lead to unexpected functions for actin-binding proteins in cells.
Keywords: actin, cofilin, tropomyosin, epithelial cell, migration, kidney, focal adhesions, α-actinin, contractility, motility, paxillin, stress fibers
Introduction
Stress fibers are conspicuous bundles of actin filaments composed primarily of actin, myosin II, α-actinin, and tropomyosin (1, 2). The fibers span nearly the entire length of the cell and are anchored at both of their ends in focal adhesions. Although stress fibers are contractile (3–5), their presence is anticorrelated with cell motility (6–8), but they are especially prominent in nonmotile cells that are strongly attached to rigid substrates (9, 10) or in vivo in cells that are exposed to mechanical stress (11, 12), which gives them their name. They provide mechanical stability to cells and tissues by promoting strong adhesion and remodeling the extracellular matrix (2).
α-Actinin is a conserved, abundant, and ubiquitous F-actin–binding protein that bundles actin filaments in vitro (13, 14) and localizes to actin stress fibers in vivo (15). Its role in stabilizing F-actin bundles in stress fibers seems axiomatic and has experimental support. Pavalko and Burridge (16) used a dominant-negative approach, which was the method available at the time, to test the role of α-actinin in stress fiber organization. They showed that proteolytic fragments of α-actinin injected into cells disrupted stress fiber organization (16). Since then, five separate studies have used RNAi to show that α-actinin plays some role in assembling or maintaining actin stress fibers (17–21). However, in addition to these studies, two other laboratories observed an increase in actin stress fiber mass in cells depleted of α-actinin (22, 23). Although these authors did not investigate or discuss an underlying mechanism or examine the physiological significance of α-actinin depletion, discrepancies between their results and previous studies clearly show that the function of α-actinin in cells cannot be predicted easily from in vitro biochemistry.
To further understand the role of α-actinin in cellular actin organization, we reinvestigated the function of α-actinin in stress fiber organization and dynamics in kidney epithelial cells (MDCK).3 MDCK cells make basal actin stress fibers, like many cell types throughout the kidney in vivo (24–27). In addition, the kidney expresses α-actinin-4, which is of particular interest in human health because mutations in α-actinin-4 cause an inherited kidney disease known as focal segmental glomerulosclerosis (28).
Results
We examined the role of α-actinin in MDCK cells, which, like kidney, expresses α-actinin-4 (28, 29). Fluorescence imaging showed actin stress fibers at the basal surface of polarized MDCK cells that were decorated with α-actinin-4 (Fig. 1, A and A′). Stress fibers inserted end-on into focal adhesions marked by paxillin at either end of the bundle (Fig. 1, A and A′).
Figure 1.

α-Actinin-4 depletion in MDCK cells leads to an increase in stress fiber mass. A, basal surface of WT MDCK cells stained for α-actinin-4, paxillin, and actin showing stress fibers. Micrographs have been adjusted the same as in B for comparison. A′, micrographs are same as above, but the contrast was adjusted to better show the stress fibers. Scale bars are 20 μm. B, basal surface of α-actinin-4–depleted cells stained for α-actinin-4, paxillin, and actin. C, graph of the mean average intensity ±95% confidence interval of actin along filaments. The mean intensity is significantly greater in α-actinin-4–depleted cells. D, graph of the mean fiber length ±95% confidence interval in WT and AKD cells. The mean length is not significantly different in depleted cells. E, Western blotting, with molecular mass markers, of WT and α-actinin-4–depleted cell lines demonstrating α-actinin-4 is depleted. F, region of interest from A′ and B showing examples of actin stress fibers that were counted in WT cells and actinin–depleted cells. Stress fibers are marked with arrows. Scale bars are 20 μm. G, Western blotting, with molecular mass markers, of WT and α-actinin-4–depleted cell lines demonstrating α-actinin-1 is unchanged in actinin-4–depleted cells.
To test the role of α-actinin-4 in stress fiber organization, we depleted it from cells using shRNA. Surprisingly, cells depleted of α-actinin-4 had more prominent actin stress fibers that were still anchored to paxillin-positive focal adhesions (Fig. 1B). Note that the images of α-actinin-4–depleted cells in Fig. 1B are to be compared with Fig. 1A of WT cells that were acquired with the same settings (the contrast of images in Fig. 1A′ of WT cells was increased to better show the stress fibers). Quantification of the micrographs showed that more actin polymer is incorporated into stress fibers in the absence of α-actinin-4 than in its presence (Fig. 1C), but stress fiber length remained the same (Fig. 1D). Fig. 1F zooms in on examples of stress fibers, demarcated with arrows, that were used to quantify the images. Western blotting confirmed that α-actinin-4 protein was substantially depleted from the cells, and there was no detectable, compensatory increase in the expression of α-actinin-1 (Fig. 1, E and G). Detecting the low amount of α-actinin-1 in these cells requires loading three times as much cell extract (60 μg of total protein) and exposing the film for a long time (longer than 10 min compared with less than 30 s) when compared with probing for α-actinin-4 with either monoclonal or polyclonal antibodies for α-actinin-4.
To further test whether the brighter fibers were a consequence of a loss of F-actin–α-actinin interactions, we overexpressed a mutant version of α-actinin-4 that lacked the actin-binding domain fused to GFP (Fig. 2A). We refer to this truncated version of α-actinin-4 as SREF because it contains the spectrin repeats and EF hand domain but lacks the actin-binding domain. Our expectation was that this mutant protein would form heterodimers with endogenous α-actinin, disrupt its ability to bundle actin, and decrease its binding to F-actin as the new protein will only have one actin-binding domain, which has a low affinity for actin (30). To test whether SREF–GFP formed heterodimers with wildtype α-actinin-4, we immunoprecipitated the mutant protein with an antibody against EGFP and Western blotted with an antibody to the actin-binding domain of α-actinin-4. This experiment showed that the mutant protein was able to form heterodimers with the endogenous protein (Fig. 2C). Fluorescence microscopy showed that cells expressing the mutant protein have more conspicuous stress fibers relative to cells not expressing the dominant-negative version of α-actinin-4 (EGFP-positive versus -negative cells in Fig. 2A). Quantification of the mean actin fluorescence showed that actin fibers in cells expressing the dominant-negative version were brighter than nonexpressing cells (Fig. 2B). We interpret these results to mean that the mutant protein acts as a dominant-negative blocking α-actinin's interaction with actin in overexpressing cells. The consequence of inhibiting α-actinin's ability to bundle F-actin in cells is an increase in stress fibers.
Figure 2.
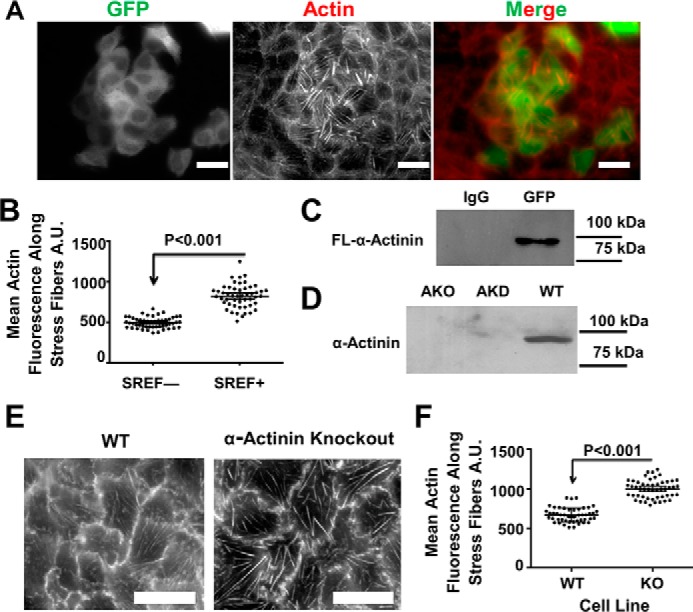
Disrupting α-actinin function via a dominant-negative or knockout approach also increases stress fiber mass. A, basal surface of MDCK cells overexpressing an actinin mutant lacking the actin-binding domain with a GFP tag and stained with phalloidin showing stress fibers. Scale bars are 20 μm. B, graph of the mean average intensity ± 95% confidence interval of actin along stress fibers. A.U., arbitrary units. C, Western blotting probing endogenous actinin from an immunoprecipitation of GFP from the overexpressing cells in A. D, Western blotting of WT, actinin knockdown, and knockout cell lines demonstrating that the knockout cell line lacks actinin-4 expression. E, micrographs of WT and knockout cell lines stained with phalloidin showing the stress fibers. F, graph of the mean average intensity ±95% confidence interval of actin along stress fibers from WT and KO cell lines. The mean intensity is significantly greater in the knockout cell line just as the knockdowns from Fig. 1.
We also knocked out α-actinin-4 in MDCK cells using CRISPR/Cas9 and selected for clonal cells that lacked expression of α-actinin-4. Western blotting demonstrated that there was no detectable expression of α-actinin-4 in these cells (Fig. 2D). Knocking out α-actinin-4 resulted in brighter and more pronounced stress fibers at the basal surface (Fig. 2, E and F). Thus, perturbing α-actinin function in MDCK cells using three different methods all result in more prominent stress fibers, not less.
The α-actinin-4 knockout cell line allowed us to test the ability of various α-actinin constructs to rescue the stress fiber phenotype. Expressing EGFP-tagged α-actinin-4 in the knockout cells rescued the knockout phenotype by reducing actin polymer mass within stress fibers back toward that seen in WT cells (Fig. 3, A, top panels, and B, 1st graph). α-Actinin-1 is another nonmuscle isoform highly homologous to α-actinin-4 (31). We wanted to know whether α-actinin-1 could also rescue the knockout phenotype if we overexpressed it in the knockout cells. Indeed, overexpressing α-actinin-1 rescued the phenotype (Fig. 3, A, middle panels, and B, middle graph). Therefore, both α-actinin-4 and α-actinin-1 suppress stress fiber formation in MDCK cells.
Figure 3.
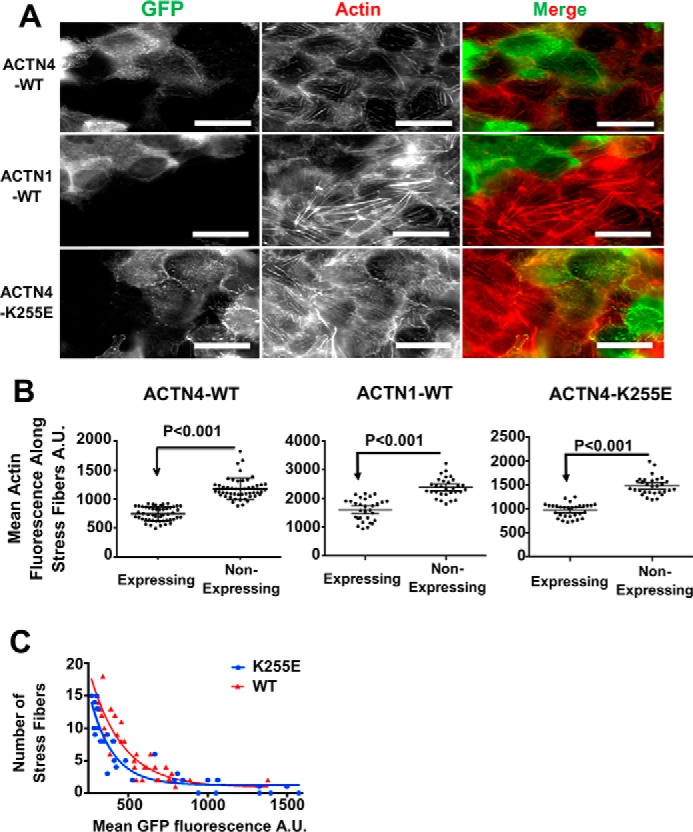
α-Actinin-4 knockout is rescued by either expression of α-actinin-4–GFP or α-actinin-1–GFP. The numbers of stress fibers in rescue cells are negatively correlated to the amount of expression and the affinity of actinin for actin. A, micrographs α-actinin-4 knockout cells rescued with either actinin-4, actinin-1, or mutant K255E–actinin-4 tagged with GFP and stained for actin. A.U., arbitrary units. Scale bars are 20 μm. B, graphs showing the mean fluorescent intensity of actin along stress fibers as a function of different α-actinin's expression in the knockout cell line ±95% confidence interval. C, graph showing the number of stress fibers as a function of the mean GFP fluorescence in that cell in WT and K255E rescued cells.
Having established that loss of α-actinin expression or perturbing α-actinin's ability to bind F-actin produced more prominent stress fibers, we wanted to test whether we could decrease stress fiber mass by increasing the amount of α-actinin binding to actin. We used both WT α-actinin-4 and a point mutant (K255E) of α-actinin-4, which causes a large increase in the affinity of the protein for F-actin (28, 30). If α-actinin was preventing formation of robust stress fibers through its actin-binding activity, then increasing expression in an α-actinin-4 knockout background should result in fewer and less prominent stress fibers. Also, it should take less K255E expression than WT expression to suppress stress fibers. Fluorescence imaging showed that K255E, like WT α-actinin-4, could rescue the phenotype of brighter stress fibers (Fig. 3, A, bottom panels, and B, last graph). Plotting the number of stress fibers in the cell as a function of α-actinin-4 expression, as determined from the mean EGFP fluorescence, showed that increasing expression of either WT or K255E α-actinin correlated with the reduced numbers of stress fibers (Fig. 3C). The quantification also showed that it required less K255E expression to suppress the number of stress fibers than it did for WT expression (Fig. 3C). This is consistent with our hypothesis that α-actinin binding to F-actin is suppressing stress fibers in MDCK cells.
Stress fibers are anticorrelated with cell movement (6–8). Rather than drive cell motility, stress fibers help cells generate strong adhesive contacts to the extracellular matrix, and adhesion can become so strong that it impedes movement (32–34). MDCK cells do not move once they reach confluence, but they do move in response to wounding (35). Cells might need to disassemble their stress fibers to allow them to move and repair the wound. We examined the fate of actin stress fibers following wounding of epithelial sheets in WT versus α-actinin–depleted cells. Stress fibers in WT cells quickly dissolved in a gradient from the wound edge (Fig. 4A). In contrast, stress fibers did not disassemble in α-actinin-4–depleted cells following wounding (Fig. 4B). Quantification of the results showed that the number of actin stress fibers proximal to the wound dropped precipitously in WT cells but not in α-actinin-4–depleted cells demonstrating that actin stress fibers are less stable in the presence of α-actinin-4 than in its absence (Fig. 4C). Consistent with earlier results that stress fibers are anticorrelated with movement, α-actinin-4–depleted cells did not advance as far into the wound as WT cells (Fig. 4, D versus E). To better understand the cellular basis of the wound-healing defect, we tracked cells during the 1st h of wound healing (Fig. 4, D and E, and Videos S1 and S2). WT cells moved in a coordinated fashion into the wound (Fig. 4, D and F). In contrast, the α-actinin–depleted cells either moved very little or moved in random directions and often away from the wound edge (Fig. 4, E and G). To fully appreciate the movements we observed, we suggest watching Videos S1 and S2. To visualize the data in another way, we plotted the net distance traveled by the cells in Fig. 4, D and E, where a positive movement is toward the wound and negative is away from the wound edge (Fig. 4H). WT cells made a 10-μm net, average advancement into the wound over 1 h compared with a 2-μm net advancement by depleted cells. However, cells in the depleted monolayer also moved away from the wound edge as seen in the cell trajectories (Fig. 4, G and H). These results imply that actin stress fibers in α-actinin–depleted cells are more stable than those in WT cells and demonstrate that loss of α-actinin in MDCK cells disrupts cell motility and wound healing.
Figure 4.

Stress fibers in α-actinin-4–depleted cells are more stable than their WT counterparts, and depleted cells show less directed movement into the wound. A, stitched micrograph of a WT wounded monolayer stained with phalloidin. Regions of interest show actin fibers proximal and distal to the wound edge. Scale bars are 20 μm. B, stitched micrograph of an α-actinin-4–depleted wounded monolayer stained with phalloidin. Regions of interest show actin fibers proximal and distal to the wound edge. Scale bars are 20 μm. C, graph of the mean number of filaments per cell, ±95% confidence interval, proximal and distal to the wound edge in WT and depleted monolayers. D, representative image at the beginning of the wound healing experiment of WT cells with the green line outlining the wound edge at time 0 and the magenta line outlining the wound edge at the end of the experiment. Scale bar is 50 μm. E, representative image at the beginning of wound healing experiment of α-actinin–depleted cells with the green line outlining the wound edge at time 0 and the magenta line outlining the wound edge at the end of the experiment. Scale bar is 50 μm. F, image from D with arrows marking the direction of movement of cells during the experiment. Scale bar is 50 μm. G, image from E with arrows marking the direction of movement of cells during the experiment. Scale bar is 50 μm. H, plot of the distances traveled by cells from wound healing experiments. Positive is toward the wound and negative is away from the wound edge.
To further test whether stress fibers are less dynamic in α-actinin–depleted cells, we imaged actin flow in live monolayers. We expressed EGFP–actin in both WT and knockout cell lines and viewed the basal surfaces of these cells over a period of 10 min. Actin on the basal surface of WT cells was very dynamic with an overall inward flow of actin from the cell edge (Fig. 5A and Video S3). Fig. 5, B and C, shows representative kymographs generated from the two lines shown in Fig. 5A. The kymograph in Fig. 5B corresponds to line 1, which is drawn along the length of a stress fiber. It shows actin flux through the fiber toward the center of the cell. Fig. 5C is a kymograph corresponding to line 2 in Fig. 5A, which is drawn orthogonally across a few stress fibers. The kymograph shows the fibers being pinched toward a midline. All WT cells that were imaged showed these dynamics and flow (n = 30). Because there is no accretion of actin polymer in the middle of the cell, the fast actin assembly and centripetal flux must be balanced by fast actin disassembly. In contrast, live imaging of EGFP–actin showed that actin stress fibers in α-actinin-4 knockout cells were far less dynamic than those in WT cells and showed little to no centripetal actin flow (Fig. 5, D–F, and Video S4). From the kymographs, we estimated the actin flow rate to be ∼17 nm/s in WT cells in agreement with previous results (32), but there was no detectable actin flow in knockout cells (Fig. 5G). Next, we wanted to test whether this was because of actin turnover dynamics. To test this, we imaged EGFP–actin-expressing cells after photobleaching areas along stress fibers (Fig. 5H and Videos S5 and S6). We watched recovery of the bleached spots over time and visualized them in kymographs (Fig. 5I). When we quantified the recovery of the photobleached area for nine cells from each WT and depleted cells, we can see that the depleted cells are unable to recover as quickly as the WT cells (Fig. 5J). This suggests that there is a more stable pool of actin in the stress fibers of depleted cells. The live imaging further supports our hypothesis that α-actinin is maintaining stress fiber dynamics in MDCK cells.
Figure 5.
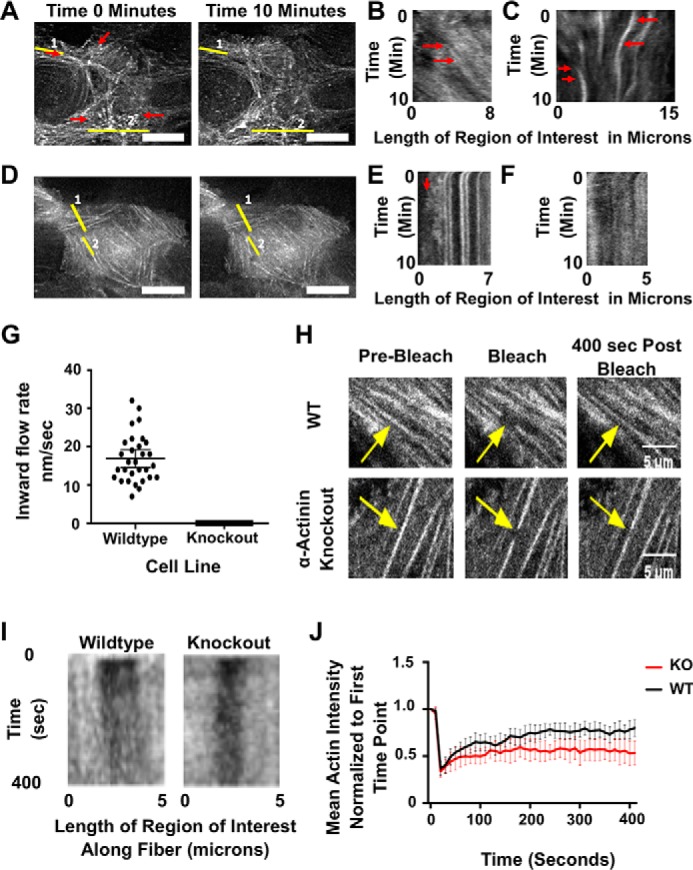
α-Actinin-4 is necessary for actin flow in nonmotile MDCK cells. A, micrographs from start and end of video of GFP-actin in a WT cell. Scale bar is 10 μm. Red arrows show direction of actin flow. B, kymograph from area of interest 1 from A, which is a line along a stress fiber. Notice the flow of actin through the stress fiber toward the center of the cell, which is marked with a red arrow. C, kymograph from area of interest 2 from A, which cuts across stress fibers. This kymograph shows the convergence of the actin into the middle over time as marked out with the red arrows. D, micrographs from start and end of video of GFP-actin in an α-actinin knockout cell. Scale bar is 10 μm. E, kymograph of area of interest 1 from D. Notice there is no flow of actin inward and only protrusive activity at the edge of the cell as pointed out with a red arrow. F, kymograph of area of interest 2 along a stress fiber showing no flow along the fiber. G, graph showing inward flow rates of actin from 30 cells. Error bars are 95% confidence interval. H, micrographs of GFP-actin in WT and α-actinin knockout cells in a FRAP experiment. Arrows point to area of photobleach along stress fibers in each cell. Time points are pre-bleach, immediately post-bleach, and 400 s post-bleach. Scale bar is 50 μm. I, kymographs of stress fibers from H with bleach site in the middle along the x axis and time along the y axis. J, graph of the mean actin intensity at the photobleach site from nine different stress fibers in individual cells normalized to the intensity to the bleach area at the first time point.
Because stress fibers did not disassemble in α-actinin-4–depleted cells following wounding, and because stress fibers appeared to be less dynamic in the live imaging, we hypothesized that α-actinin is somehow permitting actin stress fiber disassembly in the cell. Cofilin is the primary actin-binding protein driving actin filament disassembly (36), and inhibition of cofilin-mediated actin filament disassembly is thought to promote stress fiber formation (37). Cofilin activity is regulated through phosphorylation by LIM kinase, which inactivates cofilin preventing it from binding and severing actin filaments (38, 39). To test whether actin stress fibers in α-actinin-4–depleted cells were less susceptible to cofilin-mediated actin disassembly, we treated cells with the LIM kinase inhibitor, LIMKi 3 (40, 41). In WT cells, stress fibers quickly disassembled within minutes of up-regulating cofilin activity by applying the inhibitor (Fig. 6, A, upper panels, and B). In contrast, actin stress fibers in cells depleted of α-actinin-4 resisted disassembly upon deactivation of LIM kinase (Fig. 6, A, lower panels, and B). The increased stability of stress fibers in α-actinin–depleted cells could not be explained by changes in cofilin expression, and, if anything, cofilin levels were slightly increased in α-actinin–depleted cells (Fig. 6C) Western blotting showed that phospho-cofilin levels were the same in both WT and α-actinin knockdown cells and confirmed that the LIM kinase inhibitor had the same effect on phospho-cofilin levels in both WT and α-actinin–depleted cells (Fig. 6D). Thus, actin stress fibers in α-actinin-4–depleted cells are resistant to cofilin-mediated actin disassembly.
Figure 6.
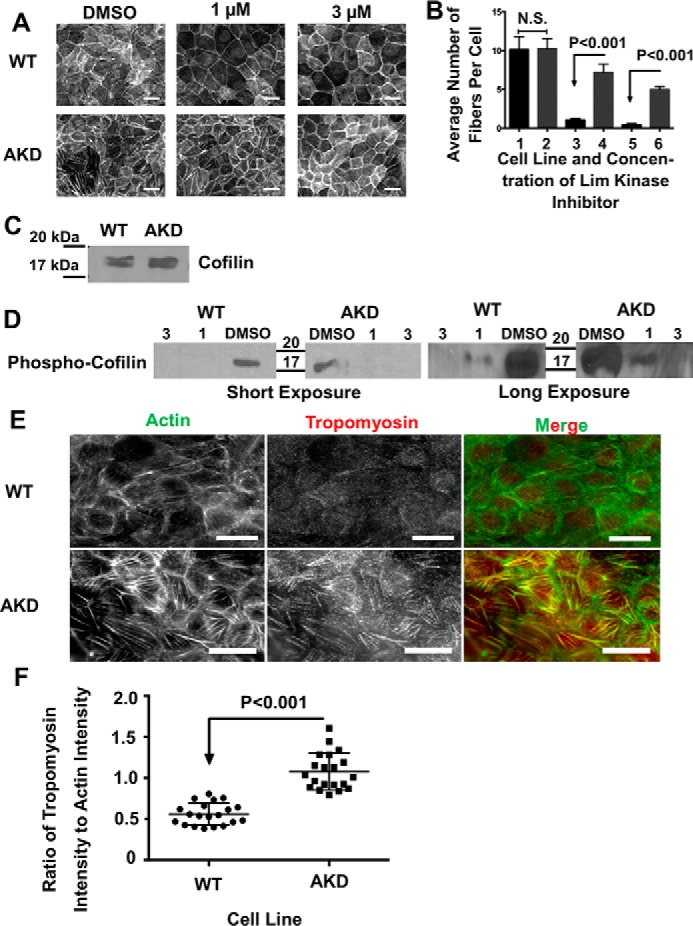
Stress fibers in α-actinin–depleted cells are less susceptible to cofilin-mediated disassembly and contain more tropomyosin. A, micrographs of the basal surface of WT and AKD cells treated with DMSO and 1 or 3 μm Lim kinase inhibitor. Scale bars are 20 μm. B, graph of the average number of filaments per cell, ±95% confidence interval, in WT and AKD cells as a function of the amount of Lim kinase inhibitor. Bar 1 is WT control cells; bar 2 is AKD control-treated cells; bar 3 is WT cells treated with 1 μm inhibitor; bar 4 is AKD cells treated with 1 μm inhibitor; bar 5 is WT cells treated with 3 μm inhibitor; and bar 6 is AKD cells treated with 3 μm inhibitor. C, developed Western blotting probing cofilin levels in WT and AKD cells demonstrating no significant change in protein levels. D, developed Western blots of cells treated with DMSO or Lim kinase inhibitor and probed for phospho-cofilin. Both a short and long exposure are given. and 20 μg of protein was added to each lane. E, micrographs of WT or actinin–depleted cells stained for actin and tropomyosin. Scale bars are 20 μm. F, graph of the ratio of tropomyosin to actin along stress fibers in WT and depleted cells. The mean ratio was significantly greater in the depleted cells.
We hypothesized that another actin-binding protein might substitute for α-actinin for binding to stress fibers and confer resistance to cofilin-mediated disassembly. Classic experiments have suggested that tropomyosin might compete with α-actinin for binding to F-actin (42, 43), and tropomyosin stabilizes F-actin (44). Immunofluorescence of tropomyosin 1 showed more tropomyosin-decorating actin stress fibers in the absence of α-actinin-4 than in its presence (Fig. 6, E and F) providing a possible explanation for the increased stability of stress fibers in the absence of α-actinin. Increased loading of tropomyosin onto stress fibers in the absence of α-actinin was not due to an up-regulation of tropomyosin expression in MDCK cells (Fig. 7G, left lanes).
Figure 7.

Tropomyosin provides stability to actin fibers both in vitro and in vivo, and α-actinin competes for access to actin filaments in vitro. A, consecutive micrographs from an in vitro severing assay of actin filaments either in the presence or absence of tropomyosin. The arrows point to a filament that severs in the presence of cofilin. Scale bars are 5 μm. B, graph of the severing rate of actin filaments via cofilin as a function of tropomyosin concentration. C, Coomassie Blue-stained gel of the pellets from spin-down assays where tropomyosin concentration was maintained and α-actinin concentration was increased from left to right. D, graph of the amount of tropomyosin in the pellet as a function of actinin concentration from repeated experiments. E, Coomassie Blue-stained gel of the pellets from spin-down assays where actinin concentration was maintained and tropomyosin concentration was increased from left to right. F, graph of the amount of actinin in the pellet as a function of tropomyosin concentration from repeated experiments. G, Western blotting probing tropomyosin in WT, actinin knockout, and actinin knockout transfected with tropomyosin ShRNA1 or ShRNA2. H, micrographs of actinin knockout cell line or actinin knockout transfected with SH1 or SH2 and stained for actin. I, graph quantifying the percentage of cells with stress fibers as a function of transfection with SH1 or SH2.
One mechanism through which tropomyosin is thought to stabilize F-actin is by competing with cofilin for binding to F-actin (45, 46). However, cofilin-mediated actin filament fragmentation is complicated and is highest when the filaments are partially decorated with cofilin (47, 48). This opens the possibility that factors like tropomyosin that compete with cofilin for binding to the sides of actin filaments might promote filament severing as opposed to suppressing it, as has been shown for phalloidin, which also competes with cofilin for binding to F-actin (48). To distinguish whether tropomyosin promotes or inhibits cofilin-mediated actin severing, we imaged single actin filaments in the presence of cofilin and increasing concentrations of tropomyosin. Tropomyosin suppressed cofilin-mediated actin severing (Fig. 7, A and B), which is consistent with previous results showing that tropomyosin stabilizes F-actin (45, 46, 49, 50). Using cosedimentation, we found that α-actinin competes with tropomyosin for binding to F-actin (Fig. 7, C and D). Similarly, increasing concentrations of tropomyosin could compete α-actinin off F-actin (Fig. 7, E and F). These in vitro experiments confirm that α-actinin and tropomyosin compete for access to the filament and provide an explanation for why stress fibers in cells lacking α-actinin-4 contain more tropomyosin.
If α-actinin maintains actin dynamics by preventing tropomyosin from loading onto actin stress fibers, then depleting tropomyosin in the α-actinin knockout cell line should result in diminished stress fibers as they would be susceptible to cofilin-mediated disassembly. Tropomyosin 1 has been shown to stabilize actin filaments in vitro and is associated with stress fibers in cells (49). We designed shRNAs against exons 7 or 8 of tropomyosin 1 because our analysis of the NCBI genome data shows that these exons are present in every known isoform of tropomyosin 1. We used a commercial antibody (see under “Materials and methods”) raised against amino acids 1–284 of tropomyosin 1 to test whether the shRNAs could knock down expression of tropomyosin 1. According to the manufacturer, the antibody recognizes all known isoforms of tropomyosin 1 as well as tropomyosins β, γ, and tropomyosin 4. Western blottings using this antibody revealed one band in cell extracts from WT cells (Fig. 7G, 1st lane). Tropomyosin levels were not affected by knocking out α-actinin-4 (Fig. 7G, 2nd lane). Western blotting confirmed that we could deplete tropomyosin from the knockout cell line using the two different shRNAs directed against tropomyosin 1 (Fig. 7G, 3rd and 4th lanes). Based upon these results, we conclude that tropomyosin 1 is the main tropomyosin family member expressed in MDCK cells. Fluorescence imaging of actin showed that depleting tropomyosin from cells lacking α-actinin-4 resulted in a dramatic reduction in stress fibers (Fig. 7H). Greater than 95% of the α-actinin-4 knockout cells had prominent stress fibers, but only 10% of the tropomyosin-depleted cells had stress fibers (Fig. 7I). From this, we believe that the prominent stress fibers in α-actinin–depleted cells are a result of increased tropomyosin on those fibers, which in turn shields them from cofilin-mediated disassembly.
The results in cells suggest that cofilin can sever actin filaments in the presence of α-actinin-4 better than in the presence of tropomyosin. The bundling activity of α-actinin-4 made it too difficult to quantify severing by single-filament imaging. Therefore, we used a pyrene-based spectroscopy assay to monitor actin polymerization in the presence of cofilin and α-actinin-4 or tropomyosin. Actin polymerization is usually preceded by a long lag phase reflecting the kinetic barrier to the formation of new actin seeds capable of polymerizing (Fig. 8A, brown circles). α-Actinin-4 had little effect on the assembly kinetics (Fig. 8A, blue triangles). In the presence of cofilin, assembly was preceded by a long lag phase but then quickly accelerated because cofilin severing created more actin filament (+) ends that could quickly grow (Fig. 8A, orange circles). We can use this assay to test inhibition of cofilin-mediated severing. Fig. 8B shows the effect of increasing concentrations of α-actinin-4 on cofilin's ability to accelerate actin assembly. Interestingly, low concentrations of α-actinin-4 promoted cofilin-mediated severing, whereas higher concentrations either had no effect or slightly inhibited cofilin severing (Fig. 8B). In contrast, tropomyosin only inhibited cofilin-mediated filament severing (Fig. 8C).
Figure 8.
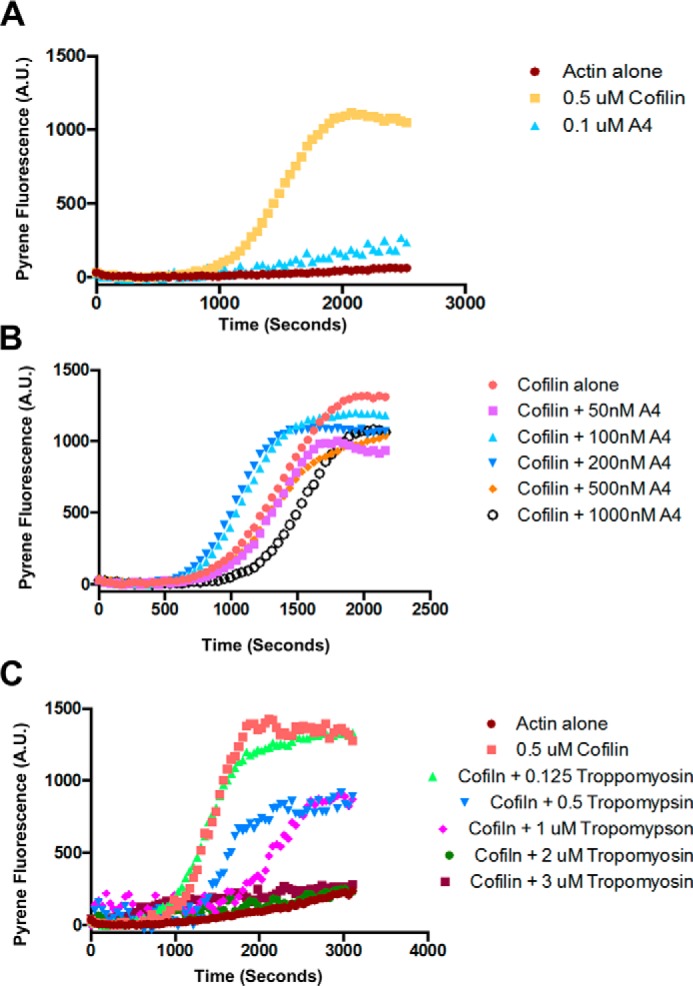
α-Actinin enhances cofilin-mediated severing at low concentrations and blocks or has no effect at higher concentrations. However, tropomyosin blocks cofilin-mediated severing of filaments in vitro. A, graph of a pyrene assay demonstrating how cofilin enhances actin polymerization, and α-actinin has no effect. B, graph of a pyrene assay demonstrating that low concentrations of α-actinin enhance cofilin-mediated severing and high concentrations either have no effect or inhibit cofilin severing. C, graph of a pyrene assay demonstrating that tropomyosin inhibits cofilin-mediated severing of actin filaments in a concentration-dependent manner.
Discussion
We have shown that α-actinin-4 suppresses actin stress fibers, limits the size of focal adhesions, and promotes (or permits) fast actin turnover dynamics. Physiologically, α-actinin-4 is important for wound healing in MDCK cells. Stress fibers normally dissolve rapidly in MDCK cells in response to wounding, but this is blocked in α-actinin-4–depleted cells because the fibers are too stable. The normally coordinated advance of interconnected cells into the wound is disrupted in α-actinin-4–depleted cells, and some cells even moved away from the wound thus revealing an important role for this actin-bundling protein in collective cell migration.
Suppression of stress fibers and increased actin turnover dynamics are the opposite results of what we predicted for α-actinin. A number of previous studies showed that α-actinin is necessary for the formation of stress fibers and focal adhesions (16–21), which seemed consistent with its actin filament–bundling activity and its early identification as an integrin-binding protein that could link integrins to the actin cytoskeleton (51). However, two other studies found an antagonistic relationship between α-actinin and stress fiber formation (22, 23). Although those studies did not explore any potential mechanisms or physiological consequences, it is now clear that α-actinin's function in organizing the cytoskeleton depends on cell type or cell context.
The increase in actin polymer mass in stress fibers in α-actinin-4–depleted cells might be due to a simple competition between α-actinin, tropomyosin, and cofilin for sides of actin filaments. Cofilin is the primary actin filament disassembly factor necessary for fast actin turnover dynamics (36, 52, 53). Tropomyosin, however, stabilizes actin filaments and shields them from cofilin action (45, 46, 54). We found that although α-actinin can also compete with cofilin for binding to F-actin, α-actinin promotes cofilin-mediated severing at low concentrations and only starts to inhibit severing at high concentrations. α-Actinin-4 therefore helps maintain fast turnover dynamics by competing with tropomyosin while largely permitting or even promoting cofilin-mediated severing. In this model, increased actin polymer in stress fibers reflects slower actin turnover dynamics. Increased tropomyosin loading in α-actinin-4 knockdown cells could also account for the inability of these cells to dissolve actin stress fibers in response to wounding.
Alternatively, α-actinin's complex role in stress fiber formation might be explained, in part, by recent work that used biomimetic systems and simulations to understand how disorganized actomyosin networks generate contractile force (55–58). Unlike sarcomeres, which produce tensile force through myosin-dependent filament transport, disordered actin networks contract through myosin-mediated actin filament buckling (55, 58, 59). Buckling dependent contractility requires the filaments to be highly connected. Therefore, buckling-mediated contractility is promoted by actin filament cross-linkers like α-actinin (55) that increase connectivity, but it is suppressed by factors that increase filament rigidity, like tropomyosin (60). Transport-mediated contractility, however, does not require cross-linkers and is not affected by filament rigidity. α-Actinin-4 promotes buckling-mediated contractility by increasing the connectivity of the basal actin network and by preventing tropomyosin from binding to F-actin, which would stiffen filaments (61). In addition, low concentrations of α-actinin permit cofilin binding making actin filaments more flexible (62, 63) and easier to buckle. Cofilin-mediated alteration of filament twist (64) can also create more α-actinin–binding sites (65) to increase network connectivity (65, 66). Finally, experimentation and computation show that a degree of filament turnover, which cofilin provides, is necessary for contractility of disordered actin networks (67, 68). In all events, loss of α-actinin decreases the connectivity of the actin network while freeing space for tropomyosin to bind to the sides of actin filaments to increase their rigidity. These changes cause the basal actin network to convert from a buckling dominated regime that produces biaxial contractility and centripetal and cortical actin flow to a sliding regime that produces uniaxial stress fibers (Fig. 9).
Figure 9.
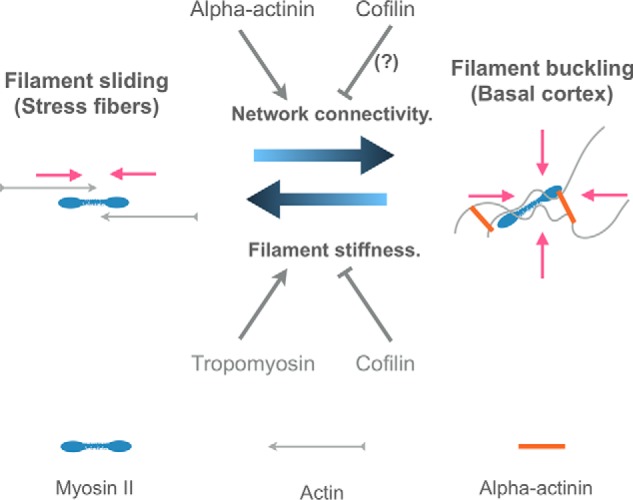
Model depicting one way in which α-actinin-4 might suppress stress fiber formation. In the presence of α-actinin, a highly interconnected network of disordered actin filaments forms at the basal surface of cells that undergoes biaxial contraction through filament buckling. In the absence of α-actinin, network connectivity is lost, favoring stress fiber formation. Loss of α-actinin also allows tropomyosin to bind to F-actin, which stiffens the filaments to further inhibit buckling and favor stress fibers. Pink arrows show the directions of contractile forces. See under “Discussion” for further details.
Buckling-dependent biaxial contractility in vitro using a minimal system of actin, myosin, and cross-linker produces stable actin filament asters (55), but stable asters do not accumulate in MDCK cells. The most likely reason is actin turnover dynamics. Even though α-actinin can suppress cofilin-mediated severing, it is not as effective as tropomyosin. In fact, we find that lower concentrations of α-actinin-4 promote cofilin-mediated severing. Furthermore, cofilin is thought to preferentially sever bent filaments (66), and α-actinin cross-linking promotes myosin-dependent filament bending. Finally, α-actinin itself can promote myosin-dependent filament fragmentation that occurs during buckling (55). All of these mechanisms help explain how α-actinin contributes to fast actin turnover dynamics to prevent the formation of stable asters and maintain a constant, centripetal actin flux on the basal surface. How disordered actin networks remain sufficiently interconnected to contract in the face of high cofilin severing rates is a challenge for the future with computational approaches providing a framework for future studies (67). Perhaps a highly cooperative mode of actin filament disassembly (69, 70) can replenish actin monomer pools necessary for polymerization without having to sever many actin filaments.
α-Actinin is also involved in focal adhesion assembly, and α-actinin-4's effect on stress fibers could be a downstream consequence of its effects on focal adhesions. In most systems examined thus far, α-actinin promotes focal adhesion growth and stabilizes the linkage between focal adhesions and actin stress fibers (17, 23, 71–74). The opposite is true in MDCK cells where loss of α-actinin-4 produces bigger focal adhesions. Whether the primary function of α-actinin in stress fiber formation is to control actin dynamics or focal adhesion dynamics will probably depend on cell type, cell context, and the mode of stress fiber formation operating in them.
Materials and methods
Cell culture and constructs
MDCK II cells were maintained in and all experiments were performed in minimal essential medium with l-glutamine and 5% bovine calf serum. Cells were grown at 37 °C and 5% CO2. Iron-supplemented calf serum was from VWR product no. 2100-500, lot 141A15. Media were changed every 3 days. To make the α-actinin knockdown cell line, we used the shRNA cloning plasmid pLKO.1-TRC cloning vector. This was a gift from David Root (Addgene plasmid no. 10878) (58). The target sequence was designed using the broad institute GPP web portal, and we followed the Addgene protocol as described on line. The target sequences used were shRNA#1 GCTCAATGAGCTGGACTATTA and shRNA#2 GGCCACCCTGTCGGATATTAA; the cell line was made from a transfection of both. After 1 week of selection, all positive colonies were kept and used as the cell line. The AKD cell line, as well as WT cells, were frozen in aliquots at this time point and stored for future use.
For making the α-actinin-4 knockout cell line, WT MDCK cells were transfected with CRISPR/Cas9 plasmid px459. pSpCas9(BB)-2A-Puro (PX459) V2.0 was a gift from Feng Zhang (Addgene plasmid no. 62988) (59). The sequences used were designed using the Broad Institute webpage (http://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design) and the RGen website to check for off-site targets (http://www.rgenome.net/cas-offinder/) (60).4 Target sequences used were as follows: 1) CGGAATGGTGGACTACCACG; 2) GGTACGACTGGTTCGCCGCG; and 3) ACAGATAGAGAACATCGACG. Cells were transfected as before; however, 24 h after transfecting, they were trypsinized and placed into fresh media with 4 μg/ml puromycin and left for 24 h of selection. Then the media were replaced with fresh media lacking puromycin. 48 h after selection and 4 days after transfection, the cells were trypsinized again and diluted to single cells in multiwell plates. After 1 week, the plates were split in two, and cells were checked for expression of actinin using an antibody and immunofluorescence. Wells lacking fluorescence were selected, and those cells were checked again using Western blottings, and negative clones were saved. One clone was used for further experiments and that came from using target sequence 1. Also target sequence 2 failed to give any knockouts.
α-Actinin constructs were all cloned into the plasmid pLenti-III-HA from abmGood. First, we cloned EGFP into pLenti-III-HA either in between NheI-ClaI or XbaI-XhoI to make plasmids pLGN or pLGC, respectively. α-Actinin that lacked the ABD was cloned into pLGN from EcoRI to XhoI. α-Actinin-4 WT and K255E were cloned into pLGC from KpnI to EcoRI. pEGFP-N1 α-actinin 1 was a gift from Carol Otey (Addgene plasmid no. 11908) (61). Rescue lines were made by transfecting the knockout cell line with the constructs mentioned above and selecting with drugs 48 h after transfection by plating into fresh media with selection. The cells surviving were collected and maintained as a heterogeneous mixture. EGFP–actin was cloned into pLenti-III-HA into sites NheI and XbaI. EGFP–actin cell lines were made as above where the parent cell line was transfected and then positive cells were selected 48 h after transfection for 1 week, and all positive cells were collected into a new cell line. For knocking down tropomyosin in the α-actinin-4 knockout cell line, we transiently transfected the knockout cell line with tropomyosin shRNA, and images and Western blottings were performed on cells 48 h after transfection. Tropomyosin shRNAs were designed as described above, except that we chose to target dog exons 7 and 8 of dog TPM1 to have the highest probability of knocking down any tropomyosin 1 isoforms. The target sequences were sh1, TCGCAGAAGGAAGACAAATAT, and sh2, GGAGAGGTCAGTAACTAAATT.
Antibodies and reagents
Secondary antibodies were all purchased from Life Technologies, Inc. Primary antibodies used were mouse anti-α-actinin-4 (1:100 immunofluorescence; 1:1000 WB), rabbit anti-tropomyosin (1:100 immunofluorescence; 1:500 WB), and rabbit anti-myosin IIB (1:100 immunofluorescence), from Santa Cruz Biotechnology SC393495, SC28543, and SC47204, respectively. Mouse anti-paxillin (1:100 immunofluorescence) was purchased from BD Transduction Laboratories catalog no. 612405. Rabbit anti-α-actinin-4 was produced in-house and described before (25). Mouse anti-α-actinin 1 (1:200 WB) was purchased from Abnova H00000087-M01A. All α-actinin-4 antibodies used detect the N terminus of α-actinin-4 and do not cross-react with other actinins. Mouse anti-GFP 12A6 used in the immunoprecipitation experiments was obtained from Developmental Studies Hybridoma Bank (DSHB). FITC-phalloidin was purchased from American Peptide product no. 92-0-11A and diluted in DMSO to 1 mg/ml as a stock solution and frozen in small aliquots at −20 °C. Working aliquots were kept at 4 °C to prevent freeze–thaws and used within a couple of months. Working concentration of phalloidin was 1 μg/ml. Formaldehyde was purchased from Polysciences Inc., catalog no. 18814, and aliquots were made and kept at −20 °C until used. (−)-Blebbistatin item 13013 was purchased from Cayman Chemical Co., and a stock solution was made at 100 mm in DMSO. Lim kinase inhibitor, LIMKi 3, was purchased from Tocris Bioscience 4745, and stock solution was 20 mm in DMSO. All stocks were kept at −20 °C. Working concentrations were 50 μm blebbistatin and 1 or 3 μm Lim kinase inhibitor. ProLong Gold was purchased from Life Technologies, Inc. Collagen type 1 was purchased from BD Biosciences, and a stock solution was made at 50 μg/ml in 0.02 n acetic acid and kept at 4 °C. Working solution was made fresh the day of use and was diluted from the stock solution to 0.5 μg/ml into 0.02 n acetic acid.
Cell experiments
Cells were plated on glass coverslips coated with 0.5 μg/ml collagen. This was accomplished by placing 300 μl of the solution onto coverslips and incubating for 30 min in the cell incubator. Collagen was then removed, and medium was added to the dish. Cells were plated on coverslips such that they reached confluence within 48 h. All experiments took place 24 h after cells reached confluency, which was 48–72 h after being plated on the coverslips. For the wounded monolayer experiment, a monolayer of cells was poked with an 18-gauge needle that was grounded flat and sharpened to resemble a hole punch. The monolayer was then poked with the needle. In our hands this allowed for monolayers to be wounded without cells being ripped off the coverslip during wounding. Cells were then processed 30 min after wounding. Live cell wound-healing experiments were performed by first incubating monolayers, created as before, with PBS with Mg2+ but not Ca2+ for 10 min. The monolayers were then scratched with a 18-gauge needle, and fresh medium was added. Monolayers were allowed to recover for 10–30 min until the first lamellipodia was seen. Then, the coverslip was placed on top of two layers of silicone tape attached to a glass slide. We cut out a square in the tape just smaller than the coverslip, and the sides of the silicone tape were sealed to the coverslip with lanolin. Medium was added to the chamber, and the coverslip was placed upside down into the chamber sealing the cells into the chamber with medium. The bottom of the coverslip, now facing up, was washed, and then the slide was placed onto the microscope and imaged via phase contrast for 60 min with micrographs taken every minute. For drug experiments, the medium was removed, and the drug or DMSO control was added, and then the medium was immediately replaced to the dish. Cells were then processed for staining and imaging. For the Lim kinase inhibition where we blotted for phospho-cofilin, 35-mm dishes of cells were processed as the fixed samples, but after 30 min of drug treatment, the medium was replaced with 300 μl of 1% SDS in fixation buffer. The cells were removed and placed through a needle to shear the DNA. Protein concentration was calculated, and 20 μg was added per well for each condition and then processed for blotting using anti-phospho-cofilin antibody from Santa Cruz Biotechnology (sc-12912-R). Cells used for immunofluorescence were fixed in 0.5% formaldehyde in fixation buffer (10 mm Hepes, pH 6.8, and 150 mm NaCl) for 15 min at room temperature. Then the buffer was removed and replaced with fixation buffer with 0.5% Triton X-100 and 100 mm glycine and incubated for 60 min at room temperature. Then primary antibodies were added in fixation buffer and incubated with the cells for 60 min at room temperature. Cells were then washed two times in fixation buffer. Secondary antibodies were then added in fixation buffer for 30 min at room temperature, and cells were washed twice. Coverslips were then mounted onto glass slides with ProLong Gold. Micrographs were taken using a ×20 objective lens for fixed wound-healing experiments, with a ×10 objective for live cell experiments, and with a ×63 objective lens (NA 1.4) for all other experiments attached to a 1000 × 1000 charge-coupled device camera (ORCA-ER; Hamamatsu Photonics) on a Zeiss AxioImager with the Colibri illumination system using Axiovision Zeiss acquisition software (Carl Zeiss). Live cell experiments where we imaged EGFP–actin were performed using 35-mm Petri dishes from Matek Corp., part no. P35G-1.5-14-C. Images were taken every 5 s for 10–20 min. Only the first 10 min were analyzed for quantifications because after 10 min of imaging the cells started showing signs of phototoxicity. Data were collected on a DeltaVision OMX V4 microscope (GE Healthcare) with solid-state illumination using a ×100 (N.A. 1.4) oil immersion objective (Olympus) and an EM-CCD camera (Photometrics Evolve).
For FRAP experiments, cells were grown as above and imaged on a Zeiss LSM710. For the immunoprecipitation experiment to test whether mutant α-actinin lacking the actin-binding domain could form heterodimers with endogenous full-length protein, we used the monoclonal antibody DSHB-GFP-12A6, which was deposited to the DSHB (DSHB Hybridoma Product DSHB-GFP-12A6). We used 10 μg of either the GFP or a control IGG antibody to demonstrate that the overexpressed mutant protein was able to form heterodimers with the WT protein as expected. Mouse antibodies were bound to protein G beads. Then a cytoplasmic extract from two 10-cm confluent dishes of overexpressing cells was created for each the control and anti-GFP beads. The confluent dishes were first washed in PBS and then placed in a total of 2 ml of RIPA buffer. The cells were scraped off the dishes and then lysed through a 30-gauge needle. The extracts were spun for 10 min at 10,000 × g to remove insoluble material. The supernatant was considered the cytoplasmic extract, and this was placed over columns with control or anti-GFP beads. After binding, the columns were washed with 10 column volumes of RIPA and RIPA with 500 mm NaCl. Then the columns were eluted with 200 mm glycine, pH 2.3. 10% of the eluate was run on a 10% SDS-polyacrylamide gel and transferred to nitrocellulose. Then we probed for full-length α-actinin.
Quantification of cell experiments
Images were processed using Fiji software (62). All graphs were made using GraphPad software, and p values were calculated using the Student's t test in the software program. All error bars are 95% confidence interval for the data, except for the severing graph where we used standard deviation. For filament length and intensity in Fig. 1, 50 filaments were measured using the line scan in Fiji, and lengths were measured along filaments between paxillin puncta. For those same filaments, the mean intensity was measured and that was the intensity used. For the wounded monolayers, three different monolayers of cells were wounded, and images were taken of the wound edge and continuing distally. Fibers were counted manually in the image. The total number of fibers counted was divided by the total number of cells in the image to give us the number of fibers per cell. This number was used, and Fig. 4C shows the bar graphs of the averages from the three experiments. The total number of cells used were as follows: AKD 73 cells proximally and 181 distally; WT 88 proximally and 172 distally. Proximal cells were considered as within the first three cell layers, and distal were more than 30 cell layers away from wound edge. For Fig. 6B, the fibers were counted manually in Fiji, and total fibers counted in a given image were divided by the total number of cells in the image. Six images were used from three different replicates. A total of 200 or more cells were counted for each experiment. For Fig. 6F, the mean actin and tropomyosin intensities were measured along 20 different stress fibers in both WT and AKD cells. These numbers were used to make the ratios used in the graphs. For Fig. 4, D–G, the Cell Tracker program was used (Piccinini et al. (76)). 20 cells were chosen from either Video S1 (wildtype) or Video S2 α-actinin-4–depleted cells), and their movements were calculated using the semi-automated function and only adjusted if the program was unable to maintain the cell position over time. In Fig. 4H, net distances traveled toward (positive) or away (negative) from the wound edge were calculated and plotted using PRISM. In Fig. 5, line scans were drawn, and kymographs built using ImageJ. Inward flow rates were calculated from kymographs where line scans were drawn from the edge of cells, and EGFP–actin pixels were tracked for at least 10 frames, and their rates of movement were calculated. Rates of flow were calculated from 30 cells for each cell line.
In vitro severing reactions
Actin filament severing assays in the presence of cofilin were performed by imaging single actin filaments in perfusion chambers as described (63). Briefly, a 2 μm solution of actin containing 20% Cy5-labeled actin was polymerized in 1× KMEI buffer for 4 min and then introduced into perfusion chambers that were previously blocked with saturated casein solution contain 0.5% Tween 20 and 0.2% Pluronic F-127. After introducing the filaments into the chamber, the chamber was washed once with 1× KMEI containing glucose, glucose oxidase, catalase, and Trolox to limit photobleaching and 0.5% methylcellulose to hold the filaments in the plane of the microscope. This solution was then replaced with solutions containing 4 μm human cofilin-1 and increasing concentrations of tropomyosin that had been purified from chicken gizzards as described previously (64). A time-lapse movie was then acquired using a ×63 1.4 N.A objective on a Zeiss Axioscope M1 with Colibri LED illumination and a Hamamatsu Orca digital camera. Images were acquired using Zeiss software and severing events quantified using FIJI software.
Pyrene actin assembly assays
Pyrene actin assembly assays were performed as described previously (75). To prepare pyrene actin, a 50 μm solution of G-actin in G-buffer (5 mm Tris, pH 8.0, 0.2 mm CaCl2, 0.2 mm ATP, 0.5 mm DTT) was dialyzed into the same buffer lacking DTT to remove free sulfhydryls. A 2 m solution of pyrene iodoacetamide was prepared in DMSO and added dropwise to the dialyzed solution of G-actin to a final concentration of 55 μm pyrene while vortexing. After the pyrene was added, 100 mm KCl, 2 mm MgCl2, and 1 mm ATP were added. The tube was covered in foil and allowed to react overnight with gentle tumbling at room temperature. After 16 h, the actin filaments were recovered by centrifugation for 20 min at 150,000 × g in a Beckman TLA100.3 centrifuge rotor. The supernatant was discarded, and the labeled F-actin pellet was suspended in G-buffer and dialyzed against the same in the dark. Following dialysis, the labeled G-actin was centrifuged for 20 min at 200,000 × g in a TLA100.3 rotor to remove insoluble material. The protein concentration and percent labeling were determined using 344 nm to measure the optical density of pyrene and 290 nm to measure the optical density of actin. Total actin concentration was determined using Equation 1,
| (Eq. 1) |
Using an extinction coefficient of 22,000 m−1 cm−1 for pyrene, the pyrene/actin ratio was calculated using pyrene/actin = (OD344/22.0)/mm actin.
Labeling ratios were always between 0.8 and 0.9
To measure actin assembly kinetics, pyrene-labeled G-actin was mixed with unlabeled G-actin to 20% pyrene label, final. This solution was then diluted down to 20 μm with G-buffer. Polymerization reactions were conducted in a final volume of 200 μl containing a final concentration of 1 μm pyrene actin and varying concentrations of cofilin, tropomyosin, or α-actinin. For each reaction, 10 μl of actin was mixed with water and the other proteins to a final volume of 180 μl. Polymerization was induced by adding 20 μl of 10× polymerization buffer (200 mm imidazole, pH 7.4, 500 mm KCl, 10 mm MgCl2, 10 mm ATP, 10 mm EGTA). Polymerization was monitored on a fluorescent plate reader (Spectramax M2, Molecular Devices) using λEM = 365 nm and λEX = 410 nm.
In vitro actin binding reactions
α-Actinin and tropomyosin competition assays were performed in 50-μl reactions. 5 μm actin was used for all reactions in KMEI buffer. For Fig. 8C, tropomyosin was held constant at 1 μm, and actinin concentration was increased from 0 to 3 μm. For Fig. 8E, actinin was held constant at 1 μm, and tropomyosin was increased from 0 to 3 μm. Proteins were added together, and actin was polymerized for 15 min and centrifuged at 65,000 rpm in a Beckman TLA-100 rotor. The supernatants were removed, and SDS loading buffer was added. SDS loading buffer was also added to the pellets and allowed to solubilize the pellets overnight, and then 1× KMEI was added to the pellets. The samples from the pellets were then run out on a 10% SDS-polyacrylamide gel and stained with Coomassie Blue. After destaining, the gels were scanned, and ImageJ was used to quantify the intensity of the bands for the plots in Fig. 7, D and F. The spin downs were repeated three times, and representative gels and quantification are shown.
Author contributions
J. P. K. and W. M. B. conceptualization; J. P. K. and W. M. B. resources; J. P. K. and W. M. B. data curation; J. P. K. and W. M. B. formal analysis; J. P. K. and W. M. B. validation; J. P. K. and W. M. B. investigation; J. P. K. visualization; J. P. K. and W. M. B. methodology; J. P. K. writing-original draft; W. M. B. supervision; W. M. B. funding acquisition; W. M. B. project administration; W. M. B. writing-review and editing.
Supplementary Material
This work was supported by National Institutes of Health Grant R01-GM106106 (to W. M. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Videos S1–S6.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MDCK
- Madin-Darby canine kidney cell
- EGFP
- enhanced green fluorescent protein
- WB
- Western blotting
- FRAP
- fluorescence recovery after photobleaching
- shRNA
- short hairpin RNA.
References
- 1. Burridge K., and Guilluy C. (2016) Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 343, 14–20 10.1016/j.yexcr.2015.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pellegrin S., and Mellor H. (2007) Actin stress fibres. J. Cell Sci. 120, 3491–3499 10.1242/jcs.018473 [DOI] [PubMed] [Google Scholar]
- 3. Isenberg G., Rathke P. C., Hülsmann N., Franke W. W., and Wohlfarth-Bottermann K. E. (1976) Cytoplasmic actomyosin fibrils in tissue culture cells: direct proof of contractility by visualization of ATP-induced contraction in fibrils isolated by laser micro-beam dissection. Cell Tissue Res. 166, 427–443 [DOI] [PubMed] [Google Scholar]
- 4. Katoh K., Kano Y., Masuda M., Onishi H., and Fujiwara K. (1998) Isolation and contraction of the stress fiber. Mol. Biol. Cell 9, 1919–1938 10.1091/mbc.9.7.1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kreis T. E., and Birchmeier W. (1980) Stress fiber sarcomeres of fibroblasts are contractile. Cell 22, 555–561 10.1016/0092-8674(80)90365-7 [DOI] [PubMed] [Google Scholar]
- 6. Badley R. A., Couchman J. R., and Rees D. A. (1980) Comparison of the cell cytoskeleton in migratory and stationary chick fibroblasts. J. Muscle Res. Cell Motil. 1, 5–14 10.1007/BF00711922 [DOI] [PubMed] [Google Scholar]
- 7. Cramer L. P., Siebert M., and Mitchison T. J. (1997) Identification of novel graded polarity actin filament bundles in locomoting heart fibroblasts: implications for the generation of motile force. J. Cell Biol. 136, 1287–1305 10.1083/jcb.136.6.1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Herman I. M., Crisona N. J., and Pollard T. D. (1981) Relation between cell activity and the distribution of cytoplasmic actin and myosin. J. Cell Biol. 90, 84–91 10.1083/jcb.90.1.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Willingham M. C., Yamada K. M., Yamada S. S., Pouysségur J., and Pastan I. (1977) Microfilament bundles and cell shape are related to adhesiveness to substratum and are dissociable from growth control in cultured fibroblasts. Cell 10, 375–380 10.1016/0092-8674(77)90024-1 [DOI] [PubMed] [Google Scholar]
- 10. Yeung T., Georges P. C., Flanagan L. A., Marg B., Ortiz M., Funaki M., Zahir N., Ming W., Weaver V., and Janmey P. A. (2005) Effects of substrate stiffness on cell morphology, cytoskeletal structure, and adhesion. Cell Motil. Cytoskeleton 60, 24–34 10.1002/cm.20041 [DOI] [PubMed] [Google Scholar]
- 11. Gabbiani G., Badonnel M. C., and Rona G. (1975) Cytoplasmic contractile apparatus in aortic endothelial cells of hypertensive rats. Lab. Invest. 32, 227–234 [PubMed] [Google Scholar]
- 12. Wong A. J., Pollard T. D., and Herman I. M. (1983) Actin filament stress fibers in vascular endothelial cells in vivo. Science 219, 867–869 10.1126/science.6681677 [DOI] [PubMed] [Google Scholar]
- 13. Kawamura M., Masaki T., Nonomura Y., and Maruyama K. (1970) An electron microscopic study of the action of the 6S component of α-actinin on F-actin. J. Biochem. 68, 577–580 10.1093/oxfordjournals.jbchem.a129388 [DOI] [PubMed] [Google Scholar]
- 14. Maruyama K., and Ebashi S. (1965) α-Actinin, a new structural protein from striated muscle. II. Action on actin. J. Biochem. 58, 13–19 10.1093/oxfordjournals.jbchem.a128158 [DOI] [PubMed] [Google Scholar]
- 15. Lazarides E., and Burridge K. (1975) α-Actinin: immunofluorescent localization of a muscle structural protein in nonmuscle cells. Cell 6, 289–298 10.1016/0092-8674(75)90180-4 [DOI] [PubMed] [Google Scholar]
- 16. Pavalko F. M., and Burridge K. (1991) Disruption of the actin cytoskeleton after microinjection of proteolytic fragments of α-actinin. J. Cell Biol. 114, 481–491 10.1083/jcb.114.3.481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choi C. K., Vicente-Manzanares M., Zareno J., Whitmore L. A., Mogilner A., and Horwitz A. R. (2008) Actin and α-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat. Cell Biol. 10, 1039–1050 10.1038/ncb1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Feng Y., Ngu H., Alford S. K., Ward M., Yin F., and Longmore G. D. (2013) α-Actinin1 and 4 tyrosine phosphorylation is critical for stress fiber establishment, maintenance and focal adhesion maturation. Exp. Cell Res. 319, 1124–1135 10.1016/j.yexcr.2013.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kovac B., Teo J. L., Mäkelä T. P., and Vallenius T. (2013) Assembly of non-contractile dorsal stress fibers requires α-actinin-1 and Rac1 in migrating and spreading cells. J. Cell Sci. 126, 263–273 10.1242/jcs.115063 [DOI] [PubMed] [Google Scholar]
- 20. Meacci G., Wolfenson H., Liu S., Stachowiak M. R., Iskratsch T., Mathur A., Ghassemi S., Gauthier N., Tabdanov E., Lohner J., Gondarenko A., Chander A. C., Roca-Cusachs P., O'Shaughnessy B., Hone J., and Sheetz M. P. (2016) α-Actinin links extracellular matrix rigidity-sensing contractile units with periodic cell-edge retractions. Mol. Biol. Cell 27, 3471–3479 10.1091/mbc.e16-02-0107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oakes P. W., Beckham Y., Stricker J., and Gardel M. L. (2012) Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 196, 363–374 10.1083/jcb.201107042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fukumoto M., Kurisu S., Yamada T., and Takenawa T. (2015) α-Actinin-4 enhances colorectal cancer cell invasion by suppressing focal adhesion maturation. PLoS ONE 10, e0120616 10.1371/journal.pone.0120616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu S., Dasbiswas K., Guo Z., Tee Y. H., Thiagarajan V., Hersen P., Chew T. L., Safran S. A., Zaidel-Bar R., and Bershadsky A. D. (2017) Long-range self-organization of cytoskeletal myosin II filament stacks. Nat. Cell Biol. 19, 133–141 10.1038/ncb3466 [DOI] [PubMed] [Google Scholar]
- 24. Goto K., and Ishikawa H. (1998) Differential distribution of actin and cytokeratin in isolated full-length rabbit renal tubules. Cell Struct. Funct. 23, 73–84 10.1247/csf.23.73 [DOI] [PubMed] [Google Scholar]
- 25. Mulholland D. J., Wilson J., Vogl W., and Webber W. (1999) Distribution of actin bundles in Bowman's capsule of rat kidney. Tissue Cell 31, 610–616 10.1054/tice.1999.0075 [DOI] [PubMed] [Google Scholar]
- 26. Murakami T., and Ishikawa H. (1991) Stress fibers in situ in proximal tubules of the rat kidney. Cell Struct. Funct. 16, 231–240 10.1247/csf.16.231 [DOI] [PubMed] [Google Scholar]
- 27. Tanaka K., Shibata N., and Tatsumi N. (1977) Electron microscopic studies on the myofibrils in the epithelial cells of the Bowman's capsule and of proximal tubules in rat renal cortex (first report). Jpn. Circ. J. 41, 1329–1336 [DOI] [PubMed] [Google Scholar]
- 28. Kaplan J. M., Kim S. H., North K. N., Rennke H., Correia L. A., Tong H. Q., Mathis B. J., Rodríguez-Pérez J. C., Allen P. G., Beggs A. H., and Pollak M. R. (2000) Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat. Genet. 24, 251–256 10.1038/73456 [DOI] [PubMed] [Google Scholar]
- 29. Tang V. W., and Brieher W. M. (2012) α-Actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J. Cell Biol. 196, 115–130 10.1083/jcb.201103116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weins A., Schlondorff J. S., Nakamura F., Denker B. M., Hartwig J. H., Stossel T. P., and Pollak M. R. (2007) Disease-associated mutant α-actinin-4 reveals a mechanism for regulating its F-actin-binding affinity. Proc. Natl. Acad. Sci. U.S.A. 104, 16080–16085 10.1073/pnas.0702451104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Otey C. A., and Carpen O. (2004) α-Actinin revisited: a fresh look at an old player. Cell Motil. Cytoskeleton 58, 104–111 10.1002/cm.20007 [DOI] [PubMed] [Google Scholar]
- 32. Calof A. L., and Lander A. D. (1991) Relationship between neuronal migration and cell-substratum adhesion: laminin and merosin promote olfactory neuronal migration but are anti-adhesive. J. Cell Biol. 115, 779–794 10.1083/jcb.115.3.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Huttenlocher A., Ginsberg M. H., and Horwitz A. F. (1996) Modulation of cell migration by integrin-mediated cytoskeletal linkages and ligand-binding affinity. J. Cell Biol. 134, 1551–1562 10.1083/jcb.134.6.1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ilić D., Furuta Y., Kanazawa S., Takeda N., Sobue K., Nakatsuji N., Nomura S., Fujimoto J., Okada M., and Yamamoto T. (1995) Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 377, 539–544 10.1038/377539a0 [DOI] [PubMed] [Google Scholar]
- 35. Tang V. W., and Brieher W. M. (2013) FSGS3/CD2AP is a barbed-end capping protein that stabilizes actin and strengthens adherens junctions. J. Cell Biol. 203, 815–833 10.1083/jcb.201304143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lappalainen P., and Drubin D. G. (1997) Cofilin promotes rapid actin filament turnover in vivo. Nature 388, 78–82 10.1038/40418 [DOI] [PubMed] [Google Scholar]
- 37. Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., and Narumiya S. (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895–898 10.1126/science.285.5429.895 [DOI] [PubMed] [Google Scholar]
- 38. Arber S., Barbayannis F. A., Hanser H., Schneider C., Stanyon C. A., Bernard O., and Caroni P. (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393, 805–809 10.1038/31729 [DOI] [PubMed] [Google Scholar]
- 39. Ressad F., Didry D., Xia G. X., Hong Y., Chua N. H., Pantaloni D., and Carlier M. F. (1998) Kinetic analysis of the interaction of actin-depolymerizing factor (ADF)/cofilin with G- and F-actins. Comparison of plant and human ADFs and effect of phosphorylation. J. Biol. Chem. 273, 20894–20902 10.1074/jbc.273.33.20894 [DOI] [PubMed] [Google Scholar]
- 40. Ross-Macdonald P., de Silva H., Guo Q., Xiao H., Hung C. Y., Penhallow B., Markwalder J., He L., Attar R. M., Lin T. A., Seitz S., Tilford C., Wardwell-Swanson J., and Jackson D. (2008) Identification of a nonkinase target mediating cytotoxicity of novel kinase inhibitors. Mol. Cancer Ther. 7, 3490–3498 10.1158/1535-7163.MCT-08-0826 [DOI] [PubMed] [Google Scholar]
- 41. Scott R. W., Hooper S., Crighton D., Li A., König I., Munro J., Trivier E., Wickman G., Morin P., Croft D. R., Dawson J., Machesky L., Anderson K. I., Sahai E. A., and Olson M. F. (2010) LIM kinases are required for invasive path generation by tumor and tumor-associated stromal cells. J. Cell Biol. 191, 169–185 10.1083/jcb.201002041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Drabikowski W., and Nowak E. (1968) The interaction of α-actinin with F-actin and its abolition by tropomyosin. Eur. J. Biochem. 5, 209–214 10.1111/j.1432-1033.1968.tb00359.x [DOI] [PubMed] [Google Scholar]
- 43. Goli D. E., Suzuki A., Temple J., and Holmes G. R. (1972) Studies on purified α-actinin. I. Effect of temperature and tropomyosin on the α-actinin-F-actin interaction. J. Mol. Biol. 67, 469–488 10.1016/0022-2836(72)90464-0 [DOI] [PubMed] [Google Scholar]
- 44. Gunning P. W., Hardeman E. C., Lappalainen P., and Mulvihill D. P. (2015) Tropomyosin–master regulator of actin filament function in the cytoskeleton. J. Cell Sci. 128, 2965–2974 10.1242/jcs.172502 [DOI] [PubMed] [Google Scholar]
- 45. Bernstein B. W., and Bamburg J. R. (1982) Tropomyosin binding to F-actin protects the F-actin from disassembly by brain actin-depolymerizing factor (ADF). Cell Motil. 2, 1–8 10.1002/cm.970020102 [DOI] [PubMed] [Google Scholar]
- 46. Ono S., and Ono K. (2002) Tropomyosin inhibits ADF/cofilin-dependent actin filament dynamics. J. Cell Biol. 156, 1065–1076 10.1083/jcb.200110013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Andrianantoandro E., and Pollard T. D. (2006) Mechanism of actin filament turnover by severing and nucleation at different concentrations of ADF/cofilin. Mol. Cell 24, 13–23 10.1016/j.molcel.2006.08.006 [DOI] [PubMed] [Google Scholar]
- 48. Elam W. A., Kang H., and De La Cruz E. M. (2013) Competitive displacement of cofilin can promote actin filament severing. Biochem. Biophys. Res. Commun. 438, 728–731 10.1016/j.bbrc.2013.07.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gateva G., Kremneva E., Reindl T., Kotila T., Kogan K., Gressin L., Gunning P. W., Manstein D. J., Michelot A., and Lappalainen P. (2017) Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr. Biol. 27, 705–713 10.1016/j.cub.2017.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gupton S. L., Anderson K. L., Kole T. P., Fischer R. S., Ponti A., Hitchcock-DeGregori S. E., Danuser G., Fowler V. M., Wirtz D., Hanein D., and Waterman-Storer C. M. (2005) Cell migration without a lamellipodium: translation of actin dynamics into cell movement mediated by tropomyosin. J. Cell Biol. 168, 619–631 10.1083/jcb.200406063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Otey C. A., Pavalko F. M., and Burridge K. (1990) An interaction between α-actinin and the β1 integrin subunit in vitro. J. Cell Biol. 111, 721–729 10.1083/jcb.111.2.721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Carlier M. F., Laurent V., Santolini J., Melki R., Didry D., Xia G. X., Hong Y., Chua N. H., and Pantaloni D. (1997) Actin depolymerizing factor (ADF/cofilin) enhances the rate of filament turnover: implication in actin-based motility. J. Cell Biol. 136, 1307–1322 10.1083/jcb.136.6.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rosenblatt J., Agnew B. J., Abe H., Bamburg J. R., and Mitchison T. J. (1997) Xenopus actin depolymerizing factor/cofilin (XAC) is responsible for the turnover of actin filaments in Listeria monocytogenes tails. J. Cell Biol. 136, 1323–1332 10.1083/jcb.136.6.1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nishida E., Maekawa S., and Sakai H. (1984) Cofilin, a protein in porcine brain that binds to actin filaments and inhibits their interactions with myosin and tropomyosin. Biochemistry 23, 5307–5313 10.1021/bi00317a032 [DOI] [PubMed] [Google Scholar]
- 55. Murrell M. P., and Gardel M. L. (2012) F-actin buckling coordinates contractility and severing in a biomimetic actomyosin cortex. Proc. Natl. Acad. Sci. U.S.A. 109, 20820–20825 10.1073/pnas.1214753109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ennomani H., Letort G., Guérin C., Martiel J. L., Cao W., Nédélec F., De La Cruz E. M., Théry M., and Blanchoin L. (2016) Architecture and connectivity govern actin network contractility. Curr. Biol. 26, 616–626 10.1016/j.cub.2015.12.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bendix P. M., Koenderink G. H., Cuvelier D., Dogic Z., Koeleman B. N., Brieher W. M., Field C. M., Mahadevan L., and Weitz D. A. (2008) A quantitative analysis of contractility in active cytoskeletal protein networks. Biophys. J. 94, 3126–3136 10.1529/biophysj.107.117960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Murrell M., Oakes P. W., Lenz M., and Gardel M. L. (2015) Forcing cells into shape: the mechanics of actomyosin contractility. Nat. Rev. Mol. Cell Biol. 16, 486–498 10.1038/nrm4012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lenz M., Thoresen T., Gardel M. L., and Dinner A. R. (2012) Contractile units in disordered actomyosin bundles arise from F-actin buckling. Phys. Rev. Lett. 108, 238107 10.1103/PhysRevLett.108.238107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stam S., Freedman S. L., Banerjee S., Weirich K. L., Dinner A. R., and Gardel M. L. (2017) Filament rigidity and connectivity tune the deformation modes of active biopolymer networks. Proc. Natl. Acad. Sci. U.S.A. 114, E10037–E10045 10.1073/pnas.1708625114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kojima H., Ishijima A., and Yanagida T. (1994) Direct measurement of stiffness of single actin filaments with and without tropomyosin by in vitro nanomanipulation. Proc. Natl. Acad. Sci. U.S.A. 91, 12962–12966 10.1073/pnas.91.26.12962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. McCullough B. R., Blanchoin L., Martiel J. L., and De la Cruz E. M. (2008) Cofilin increases the bending flexibility of actin filaments: implications for severing and cell mechanics. J. Mol. Biol. 381, 550–558 10.1016/j.jmb.2008.05.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. McCullough B. R., Grintsevich E. E., Chen C. K., Kang H., Hutchison A. L., Henn A., Cao W., Suarez C., Martiel J. L., Blanchoin L., Reisler E., and De La Cruz E. M. (2011) Cofilin-linked changes in actin filament flexibility promote severing. Biophys. J. 101, 151–159 10.1016/j.bpj.2011.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. McGough A., Pope B., Chiu W., and Weeds A. (1997) Cofilin changes the twist of F-actin: implications for actin filament dynamics and cellular function. J. Cell Biol. 138, 771–781 10.1083/jcb.138.4.771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bonet C., Maciver S. K., and Mozo-Villarías A. (2010) The regulatory action of α-actinin on actin filaments is enhanced by cofilin. Eur. Biophys. J. 39, 1143–1153 10.1007/s00249-009-0566-2 [DOI] [PubMed] [Google Scholar]
- 66. Maciver S. K., Zot H. G., and Pollard T. D. (1991) Characterization of actin filament severing by actophorin from Acanthamoeba castellanii. J. Cell Biol. 115, 1611–1620 10.1083/jcb.115.6.1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McFadden W. M., McCall P. M., Gardel M. L., and Munro E. M. (2017) Filament turnover tunes both force generation and dissipation to control long-range flows in a model actomyosin cortex. PLoS Comput. Biol. 13, e1005811 10.1371/journal.pcbi.1005811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hiraiwa T., and Salbreux G. (2016) Role of turnover in active stress generation in a filament network. Phys. Rev. Lett. 116, 188101 10.1103/PhysRevLett.116.188101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kueh H. Y., Charras G. T., Mitchison T. J., and Brieher W. M. (2008) Actin disassembly by cofilin, coronin, and Aip1 occurs in bursts and is inhibited by barbed-end cappers. J. Cell Biol. 182, 341–353 10.1083/jcb.200801027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nadkarni A. V., and Brieher W. M. (2014) Aip1 destabilizes cofilin-saturated actin filaments by severing and accelerating monomer dissociation from ends. Curr. Biol. 24, 2749–2757 10.1016/j.cub.2014.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rajfur Z., Roy P., Otey C., Romer L., and Jacobson K. (2002) Dissecting the link between stress fibres and focal adhesions by CALI with EGFP fusion proteins. Nat. Cell Biol. 4, 286–293 10.1038/ncb772 [DOI] [PubMed] [Google Scholar]
- 72. Roca-Cusachs P., del Rio A., Puklin-Faucher E., Gauthier N. C., Biais N., and Sheetz M. P. (2013) Integrin-dependent force transmission to the extracellular matrix by α-actinin triggers adhesion maturation. Proc. Natl. Acad. Sci. U.S.A. 110, E1361–E1370 10.1073/pnas.1220723110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shao H., Wang J. H., Pollak M. R., and Wells A. (2010) α-Actinin-4 is essential for maintaining the spreading, motility and contractility of fibroblasts. PLoS One 5, e13921 10.1371/journal.pone.0013921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ciobanasu C., Faivre B., and Le Clainche C. (2013) Integrating actin dynamics, mechanotransduction and integrin activation: the multiple functions of actin-binding proteins in focal adhesions. Eur. J. Cell Biol. 92, 339–348 10.1016/j.ejcb.2013.10.009 [DOI] [PubMed] [Google Scholar]
- 75. Yu-Kemp H. C., and Brieher W. M. (2016) Collapsin response mediator protein-1 regulates Arp2/3-dependent actin assembly. J. Biol. Chem. 291, 658–664 10.1074/jbc.C115.689265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Piccinini F., Kiss A., and Horvath P. (2016) CellTracker (not only) for dummies. Bioinformatics 32, 955–957 10.1093/bioinformatics/btv686 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.