

Abstract
Cellular heme is thought to be distributed between a pool of sequestered heme that is tightly bound within hemeproteins and a labile heme pool required for signaling and transfer into proteins. A heme chaperone that can hold and allocate labile heme within cells has long been proposed but never been identified. Here, we show that the glycolytic protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) fulfills this role by acting as an essential repository and allocator of bioavailable heme to downstream protein targets. We identified a conserved histidine in GAPDH that is needed for its robust heme binding both in vitro and in mammalian cells. Substitution of this histidine, and the consequent decreases in GAPDH heme binding, antagonized heme delivery to both cytosolic and nuclear hemeprotein targets, including inducible nitric-oxide synthase (iNOS) in murine macrophages and the nuclear transcription factor Hap1 in yeast, even though this GAPDH variant caused cellular levels of labile heme to rise dramatically. We conclude that by virtue of its heme-binding property, GAPDH binds and chaperones labile heme to create a heme pool that is bioavailable to downstream proteins. Our finding solves a fundamental question in cell biology and provides a new foundation for exploring heme homeostasis in health and disease.
Keywords: heme, hemostasis, kinetics, iron, intracellular trafficking, GAPDH, heme chaperone, heme trafficking, porphyrin, tetrapyrroles, heme toxicity
Introduction
Intracellular heme trafficking enables hemeproteins to mature and function outside the mitochondria and participate in diverse activities such as gas exchange, catalysis, electron transfer, transcription, and the initiation and propagation of signaling cascades (1–3). Details of the intracellular heme transport and delivery pathways and their regulation have remained elusive. In particular, what protein(s) may help traffic heme after it is made and released from the mitochondria has long remained a mystery, with a number of protein candidates proposed over the years (4–7). Our previous work (8, 9) implicated a most unlikely protein, glyceraldehyde-3-phosphate dehydrogenase (GAPDH),4 as having some role in the process. Knockdown of GAPDH in mammalian cells led to a diminished heme content and activity of the soluble hemeprotein-inducible nitric-oxide synthase (iNOS) (9), whereas a yeast knockout strain lacking the GAPDH homolog TDH3 showed increased intracellular “labile” heme levels as determined by a fluorescent heme sensor, yet had low Hap1 transcription factor activity, which requires Hap1 heme binding (8). This resulted in GAPDH joining the list of possible heme transport proteins (10, 11), but it did not lead to an understanding that GAPDH was in fact responsible for heme allocation in cells. Essentially, the previous work fell short because it did not test whether GAPDH heme binding is of central importance to its involvement. In fact, to date there has been no demonstration that GAPDH can even bind mitochondrially-generated heme in intact cells or whether such intracellular heme binding by GAPDH, if it exists, is actually required for downstream heme deliveries.
To address these gaps, we first identified a GAPDH histidine residue that is involved in its in vitro heme binding. This allowed us to create a GAPDH variant that demonstrated a decreased heme-binding affinity that we then used alongside wildtype (WT) GAPDH in mammalian cell assays to measure their intracellular heme-binding capacity and their impact on downstream heme delivery to distinct cytosolic and nuclear protein targets. Our findings provide the first demonstration that GAPDH binds heme that is naturally generated by mitochondria in cells and that its heme-binding function is the essential aspect that enables downstream heme deliveries, rather than any other property of GAPDH itself. Thus, our work reveals that GAPDH acts as a middleman to shuttle labile heme, in effect making it bioavailable to target proteins that reside outside the mitochondria in mammalian cells and yeast.
Results
Identifying a heme-binding residue in GAPDH
Our previous study with rabbit GAPDH (rGAPDH) showed that purified rGAPDH ligates heme through an unidentified histidine (His) residue (6). To identify this residue, we considered three highly-conserved His in human GAPDH (hGAPDH) (Figs. 1 and 2A). Of these, His-53 (His-51 in rGAPDH) is located on a flexible loop at the inter-protomer interface in the GAPDH tetramer. Our computer modeling showed that His-53 could form either a mono-coordinated heme complex or a bis-coordinated His heme complex with a His-53 located in the neighboring protomer (Fig. 2, A and B) or, alternatively, with the nearby nonconserved His-57. Accordingly, we generated H53A and H57A hGAPDH mutants and the corresponding H51A and H55A rGAPDH mutants, and we found that whereas none of the His mutations affected the in vitro glycolytic activity of GAPDH (Fig. 2C), the H51A substitution in rGAPDH did eliminate the characteristic Soret spectral shift to 415 nm that indicates His ligation to heme (Fig. 2D) (6). The failure of the H51A rGAPDH to produce this spectral shift indicates that His-51 is indeed the heme-ligating histidine. Changes to the heme spectrum in the presence of H51A rGAPDH indicate that there is residual binding either in the heme-binding pocket or at a secondary, nonspecific site. However, whatever the nature of this heme binding, it is no longer His-ligated.
Figure 1.

GAPDH protein alignment. The three strictly conserved histidine residues are highlighted in red (human, His-53) and magenta (human, His-111 and His-179). In blue is His-57 (human) found in human and rabbit GAPDH. His-111 and His-179 are found in stable secondary structure motifs, whereas His-53 and His-57 are located on a flexible loop at the interprotomer interface.
Figure 2.

GAPDH heme binding involves a specific His residue. A, human GAPDH tetramer from PDB code 1znq. Conserved histidines are shown as sticks; His-53 is in red; His-57 is in blue, and His-111 and His-179 are in magenta. B, close-up of subunit interface showing a representative structure from molecular dynamics simulations of the modeled human GAPDH–heme complex. C, glycolytic activity of GAPDH WT and mutants measured using conversion of NAD+ to NADH. Values represent mean ± S.E. (n = 5, no significance). D, UV-visible spectra of rabbit GAPDH in the presence of heme. Free hemin displays a Soret peak at 390 nm. Histidine ligation of the heme shifts this peak to 415 nm. WT but not H51A (His-53 equivalent) rGAPDH displays this spectral shift indicative of heme binding through histidine ligation. Changes to the heme absorbance trace in the presence of H51A rGAPDH indicates protein–heme binding without histidine ligation.
Altering a specific histidine residue in GAPDH disables its heme binding
To further explore the effect of the histidine mutations on GAPDH heme binding, we measured in vitro heme binding to purified hGAPDH proteins. Only the H53A mutation significantly diminished hGAPDH heme-binding affinity, as judged by a 4.5-fold increase in heme Kd values (Fig. 3A) and a 12-fold faster heme koff value (Fig. 3, B and C). Using these measured koff values and a kon value that was determined previously for heme binding to rGAPDH (17,800 m−1 s−1 (12)), we estimated Kd values of heme binding to the WT, H57A, and H53A hGAPDH proteins to be 0.15, 0.06, and 1.9 μm, respectively. Thus, our binding studies showed that Ala substitution of His-53 but not His-57 caused an approximate 10-fold loss in heme-binding affinity toward hGAPDH. This matches the spectroscopic results we obtained with the analogous rGAPDH proteins and implies that His-53 is involved in heme binding to hGAPDH.
Figure 3.
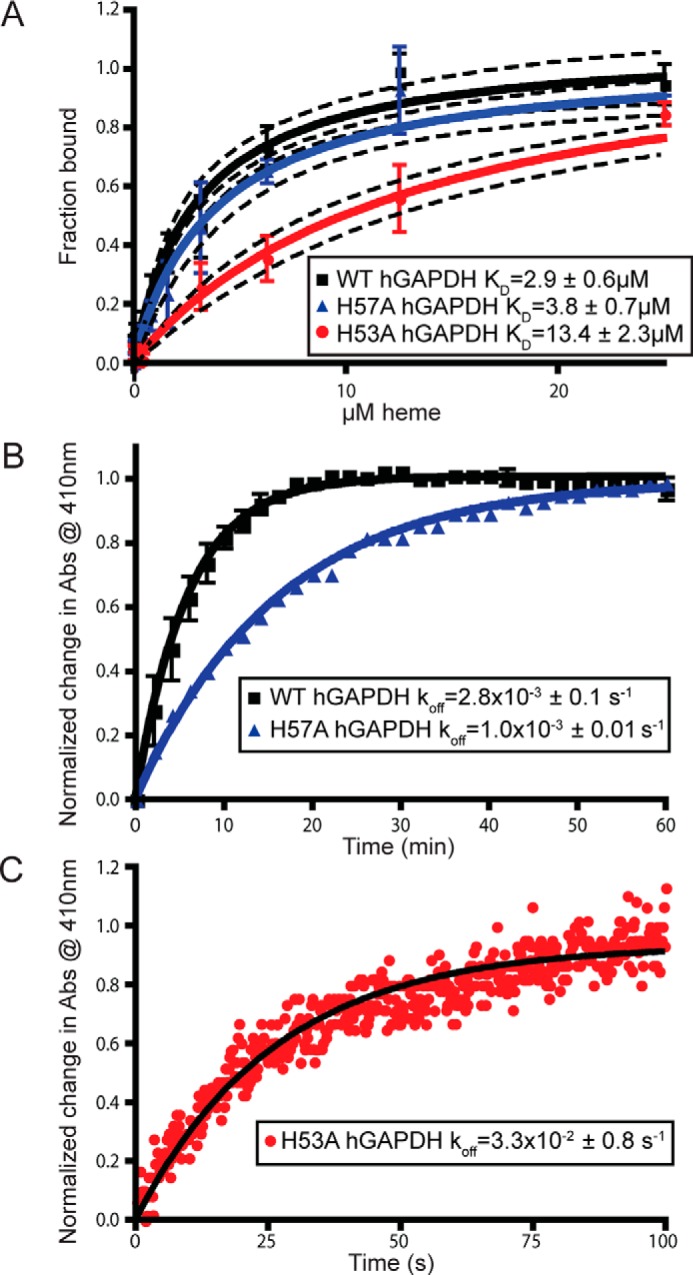
Mutation of histidine 53 antagonizes heme binding in vitro. A, heme binding to human GAPDH WT, H57A, and H53A measured using nanoscale thermophoresis on a label-free Nanotemper device. Values represent mean ± S.E. (H53A and H57A, n = 4, and WT, n = 7). Dashed lines represent 95% confidence interval. B, koff measurement of WT and H57A GAPDH using heme binding to apo-myoglobin as measured at 410 nm. Difference in koff values is not statistically significant. C, koff measurement of H53A GAPDH using heme binding to apomyoglobin as measured at 410 nm using stopped-flow.
Cellular hGAPDH binds heme and its binding is destabilized by the H53A mutation
To examine whether GAPDH binds heme in cells, and whether the His mutations would affect its heme binding under this circumstance, we expressed hGAPDH proteins with a hemagglutinin (HA) tag in mammalian cells that were given the labeled heme biosynthetic precursor δ-5-[14C]aminolevulinic acid (14C-δALA), which can be naturally converted into 14C-labeled heme (13). Pulldowns of the HA-tagged hGAPDH proteins showed that WT HA–hGAPDH displayed robust 14C-heme binding (Fig. 4, A and B), which was severely diminished in the H53A mutant but only slightly diminished in H57A mutant (Fig. 4B). The 14C counts in the HA pulldowns were confirmed to be 14C-heme through HPLC analysis (Fig. 4A). These data reveal that hGAPDH in mammalian cells binds endogenously-generated heme and that its heme binding is diminished by the H53A substitution.
Figure 4.

GAPDH binds intracellular heme in mammalian cells, and this binding is disrupted by the H53A mutation. A, representative HPLC fractions with 14C counts from HA pulldowns. HA–GAPDH WT 14C peak corresponds to heme elution. B, 14C counts from HA pulldowns of HA–GAPDH-transfected GlyA cells. Values represent mean ± S.E. n = 4. Western blots of GlyA cells show expression of HA–GAPDH and β-actin. *, p ≤ 0.02; **, p < 0.005; ***, p < 0.0001, two-tailed t tests. mAU, milli-absorbance units.
GAPDH heme-binding mutant H53A antagonizes heme insertion into iNOS
To determine whether diminished GAPDH heme binding can impact heme delivery in mammalian cells, we assessed the effect of the H53A GAPDH mutant on iNOS heme insertion and activity. After transfecting the various HA–hGAPDH constructs into heme-depleted mouse macrophage (RAW264.7) cells, we induced their apo-iNOS expression for 16 h, added in exogenous 55Fe-heme for the final 2 h, lysed the cells, and performed 2′,5′-ADP resin pulldowns on the cell supernatants to measure the extent of iNOS heme binding (Fig. 5A). Expressing the WT HA–hGAPDH in cells led to robust 55Fe-heme binding by apo-iNOS. However, expressing the H53A and to a lesser extent H57A HA–hGAPDH proteins diminished the extent of 55Fe-heme binding by the apo-iNOS (Fig. 5A). This indicates that the heme-binding–defective hGAPDH mutant antagonized 55Fe-heme insertion into the accumulated apo-iNOS. To further explore this observation, we tested the effect of the HA–hGAPDH proteins on the NO synthesis activity of iNOS, which requires that iNOS obtain a bound heme (Fig. 5, B and C). This was done under two conditions: 1) in heme-depleted cells treated with exogenous heme as above (Fig. 5B), and 2) in cells grown in normal media with endogenous heme levels (Fig. 5C). Under both conditions, expressing the WT HA–hGAPDH as well as the H57A mutant supported development of full NO synthesis activity of iNOS. However, the expression of the H53A mutant suppressed iNOS activity significantly (Fig. 5, B and C). Because the HA–hGAPDH mutants must operate alongside the cell's endogenous pool of WT GAPDH, the inhibitory effect of the H53A mutant is especially striking. The results imply that intracellular heme binding by GAPDH is an essential feature that enabled downstream heme delivery to iNOS.
Figure 5.

Mutation of heme-binding histidine in GAPDH impairs heme binding and activity of iNOS in mammalian cells. A, heme binding to iNOS as measured by 55Fe counts on ADP pulldowns. Values represent mean ± S.E. n = 2. B and C, iNOS in activated RAW264.7 lysates was assayed for activity by measuring the conversion of oxyHb to metHb. The initial rate of oxidation of oxyHb to metHb by NO was measured by increases in absorbance at 401 nm, and activity relative to WT GAPDH was plotted. B, cells were pretreated with SA, and exogenous heme was added. Values represent mean ± S.E. n = 3. C, activity of lysates with endogenous heme levels. Values represent means ± S.E. n = 4. D, Western blots of RAW264.7 cells used for the iNOS activity assays show expression of iNOS, HA–GAPDH, and β-actin. *, p ≤ 0.02; **, p < 0.005; ns, not significant.
Heme binding allows GAPDH to buffer the intracellular labile heme concentration and to deliver heme to nuclear transcription factor Hap1 in yeast
Next, we took advantage of a GAPDH yeast knockout strain to further explore the importance of GAPDH heme binding in heme homeostasis and allocation. Saccharomyces cerevisiae expresses three GAPDH isoforms, and through a gene knockout approach, the expression of one isoform (TDH3) was previously found to be required for controlling the intracellular labile heme level and for enabling heme delivery to the heme-regulated nuclear transcription factor Hap1 (8). We utilized the tdh3 yeast knockout strain (tdh3Δ) that also expressed a ratiometric fluorescent heme sensor (8) that can measure the level of intracellular labile heme. The heme sensor readings confirmed that labile heme levels are dramatically elevated in tdh3Δ compared with the parent yeast strain (Fig. 6A), as reported previously (8). Transforming the tdh3Δ cells to express either their native TDH3 or WT hGAPDH both resulted in the labile heme readings being restored to their normal lower levels. In contrast, expressing the TDH3 H51A mutant (the H53A equivalent in S. cerevisiae) or the hGAPDH H53A mutant did not rescue the tdh3Δ phenotype regarding its higher than normal labile heme level (Fig. 6A). This reveals that the heme-binding ability of GAPDH is what allows it to control the level of intracellular labile heme in yeast, with apparent conservation of the GAPDH heme-binding site and function between yeast and human. Regarding downstream heme delivery to the transcription factor Hap1, the tdh3Δ cells displayed the expected diminished Hap1 activity (Fig. 6B). This defect was rescued by expression of either the TDH3 WT or hGAPDH WT proteins but not by expression of either the TDH3 H51A or the hGAPDH H53A heme-binding mutants (Fig. 6B). Thus, GAPDH heme binding was also required for heme delivery to the nucleus-localized yeast protein, HAP1. The similar functional importance of GAPDH heme binding in our yeast and mammalian cell experiments suggests that these aspects are broadly conserved.
Figure 6.
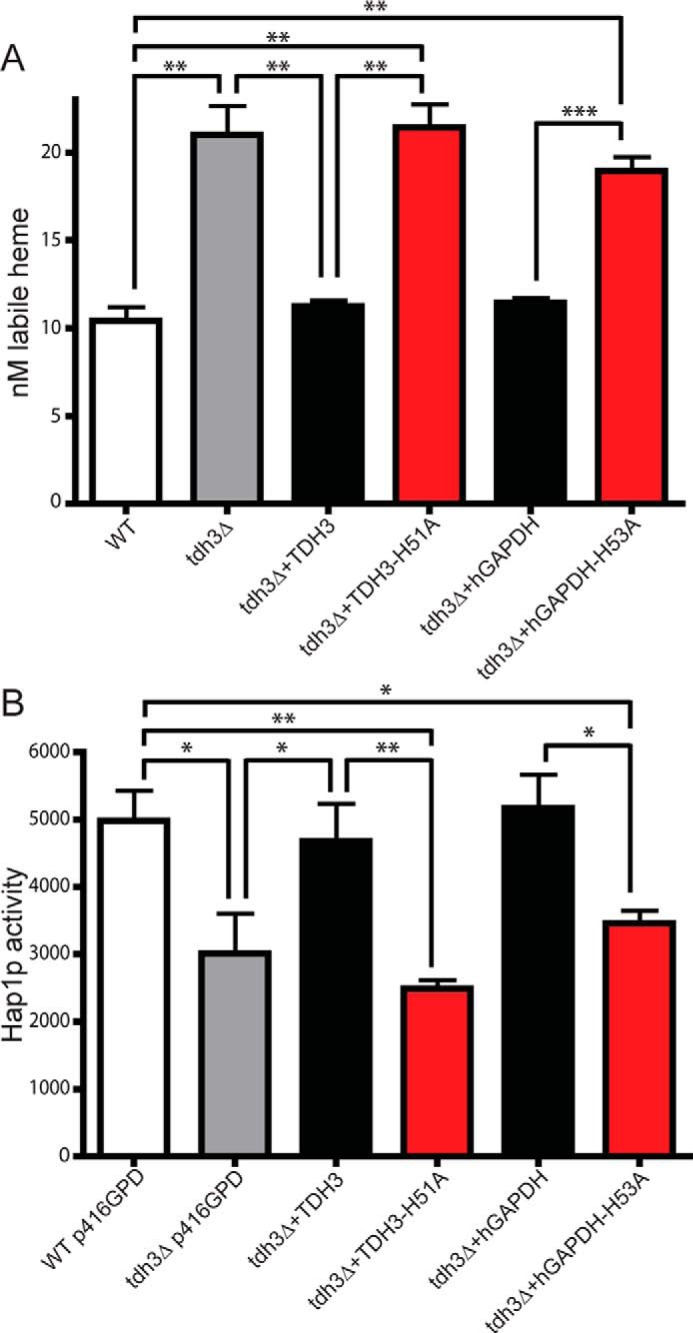
GAPDH heme binding is conserved in yeast and essential for controlling cell heme tone and delivery to nuclear Hap1p. A, sensor-detected labile heme levels in WT and tdh3Δ (GAPDH deletion) yeast strains with or without transfection with yeast and human GAPDH variants. Values represent mean ± S.E. n = 3. B, corresponding Hap1p activities in yeast. Values represent mean ± S.E. n = 3, *, p ≤ 0.02; **, p < 0.005; ***, p < 0.0001, two-tailed t test.
Discussion
Intracellular heme trafficking is a fundamental process in biology, but the transport and delivery pathways and their regulation are only recently being clarified. Besides GAPDH, several other proteins have been proposed to possibly serve in heme transport, including GSH S-transferase (4), heme-binding proteins (5, 6), and fatty acid–binding proteins (7), but in all cases their in-cell heme-binding abilities were not demonstrated nor found linked to heme delivery to downstream hemeprotein targets. We reasoned that if GAPDH is involved in intracellular heme trafficking, then it should be demonstrated to bind heme in cells, and its binding capacity should be directly linked to successful heme delivery to downstream target proteins. Our findings reveal that GAPDH fulfills both tenets: it binds endogenous and exogenous heme in cells, and its heme-binding capacity is directly linked to successful heme delivery to a cytosolic (iNOS) and a nuclear (Hap1) hemeprotein target. This helps to clarify how we think about heme sequestration and allocation within the cell. Rather than acting as a passive heme sink to protect the cell from toxicity (14), GAPDH appears to be a critical middleman in a dynamic network that makes heme bioavailable for directed mobilization to downstream targets.
Fig. 7 depicts a model that highlights a role for GAPDH in heme delivery and is consistent with the results to date. Heme that is generated naturally in cells (i.e. from δ-ALA) is transported out of the mitochondria by membrane transporters such as FLVCR1b (15) to reach the cytosol, where it, either directly or through an unknown intermediary, binds to GAPDH to create a pool of safely-sequestered but bioavailable heme. If exogenous heme arrives at the cell surface, it can be internalized by cell membrane transporters like HRG-4 (16, 17) and also join the GAPDH heme pool. Our study suggests that the GAPDH heme pool is in equilibrium with the more general pool of labile heme that is detected by the fluorescent heme sensor and that GAPDH sequesters a significant portion of the cell-labile heme pool. Once heme is bound to GAPDH, it becomes bioavailable for insertion into cytosolic proteins like iNOS and for delivery to nuclear proteins like Hap1. Whether heme delivery into the nucleus involves membrane transporters like BVR or involves transfer of the GAPDH–heme complex itself, perhaps similar to GAPDH being imported into the nucleus during apoptosis (18, 19), is a fundamental question that can now be investigated.
Figure 7.

Model of GAPDH as a cellular depot and allocator of bioavailable heme. Heme can be synthesized endogenously by the cell or imported through the plasma membrane. Endogenous heme biosynthesis occurs from glycine through the intermediates δ-ALA and coproporphyrinogen III (CPgenIII) catalyzed by enzymes found in the cytoplasm and mitochondria and finally exported into the cytoplasm by the transporter feline leukemia virus subgroup C receptor 1b (FLVCR1b). Exogenous heme can be imported into the cell through transporters such as histidine-rich glycoprotein 4 (HRG-4). GAPDH can bind both endogenous and exogenous heme sources thereby sequestering heme into a bioavailable pool. Heme binding to the heme sensor HS1 causes a decrease in EGFP (green) fluorescence but has no effect on mKATE (red) fluorescence. The labile heme pool detected by the heme sensor is distinct from the GAPDH-bound bioavailable heme pool. The GAPDH-bound heme can be delivered to soluble proteins such as iNOS or to nuclear proteins such as the transcription factor Hap1p. Heme transport into the nucleus may occur through a hand-off to the transporter of BVR or directly carried into the nucleus by GAPDH.
Notably, our work reveals that cellular heme is distributed into at least three groups, instead of two, due to a functional splitting of the labile heme pool. Specifically, our findings distinguish the GAPDH heme pool, whose heme is available for transfer into the two target proteins that we studied, from the cell's more general labile heme pool, whose heme is unavailable for transfer into the two targets in the absence of GAPDH. Given that the fluorescent heme sensor is itself derived from a heme-binding protein (it contains the heme-binding domain of cytochrome b562) (8), our results imply that a subset of cellular hemeproteins rely on GAPDH for heme delivery, whereas another subset may obtain heme independent of GAPDH. It will now be important to confirm and investigate these two modes of heme delivery.
As noted above, the size of the GAPDH–heme pool appears to be physically significant, and GAPDH binds heme with an affinity that allows it to diminish the cell's general pool of labile heme (as detected by the sensor) while at the same time allowing heme delivery to downstream targets. Indeed, deletion of TDH3 in yeast resulted in an increase in labile heme from 10 to 21 nm. This indicates that GAPDH binds ∼50% of the cytosolic labile heme in yeast. Expression of either the H53A hGAPDH or H51A TDH3 proteins, which are both heme-binding defective, in the tdh3Δ yeast was unable to decrease the labile heme level to its normal value. This behavior is consistent with GAPDH functioning as an intermediary, with its heme-binding affinity poised to be in equilibrium with an upstream heme source, while being lower than the heme affinities of the downstream protein delivery targets. In this regard, our estimate of the hGAPDH heme-binding affinity (Kd of 1.5 × 10−7 m) derived from the kon and koff measures is similar to the heme Kd value estimated for rGAPDH (12), and it is poorer than the heme Kd values reported for hemeproteins like hemoglobin, myoglobin, eIF2α, and others (20). The GAPDH heme Kd is also consistent with the recent estimate of the labile heme concentration in cells that was determined using the fluorescent heme sensor that we used here (8). Apparently, a 10-fold loss in the hGAPDH heme-binding affinity that is caused by the H53A mutation disrupted the heme binding continuum within the cell, by preventing the buildup of a sufficient GAPDH–heme complex needed to support downstream heme deliveries. Given that mammalian GAPDH exists as a tetramer, it will be interesting to understand how the H53A mutation impacts the heme-binding affinity within a mixed tetramer that contains both WT and mutant GAPDH subunits, which are likely to form when the mutant is expressed in cells. The dynamics of the GAPDH heme flux (i.e. heme loading onto and transfer from GAPDH) is also an important parameter that needs to be studied. In preliminary experiments, we found that mammalian cells needed to be made heme-deficient in order for the expressed HA–hGAPDH to bind detectable quantities of newly-synthesized 14C-heme. This implies that the existing GAPDH needed to be (or become) depleted of bound heme before it could bind significant amounts of the newly-generated 14C-heme. Investigating the dynamics of GAPDH heme loading and depletion will help in understanding how this critical pool is utilized and regulated.
Exposure to NO causes the cell-labile heme level to temporarily increase (8). An intriguing open question is whether the GAPDH-bound heme is released upon such NO exposure and whether such change is related to NO's ability to enable versus inhibit heme insertion into apo-protein targets such as the β subunit of soluble guanylate cyclase (21) or into NOS enzymes, hemoglobin, cytochrome P450, and catalase (22, 23). We also do not know how GAPDH engages in heme transfer to the downstream targets. In this regard, there are likely to be mechanistic connections between GAPDH and the cellular chaperone hsp90, which participates in the process by binding to the heme-free forms of several proteins to help drive their heme insertion (21, 24). The discovery that GAPDH is a provider of bioavailable heme helps to build a foundation for ultimately understanding how intracellular heme trafficking and homeostasis is related to life, health, and disease.
Experimental procedures
Reagents
Chemicals were from Sigma unless otherwise noted. 14C-δALA was purchased from ChemDepo and 55Fe-heme from RI Consultants. HA-tagged hGAPDH and variants were cloned into pcDNA3.1 expression plasmids.
Cell lines
GlyA CHO were a gift from Dr. P. J. Stover, Cornell University, and were confirmed in-house to be glycine-auxotrophic for growth. RAW264.7 cells were obtained from ATCC.
GAPDH alignment
Sequences for GAPDH from a number of species were downloaded from the NCBI database and aligned using MacVector.
GAPDH–heme model building and molecular dynamics simulation
The structure of the heme group used to build the GAPDH–heme model was taken from the bis-ligated protoporphyrin IX containing iron of the NMR-solved cytochrome b5 (PDB code 1jex). All cysteines (including the catalytic ones) were represented in their reduced form, whereas the protonation state of histidines was determined with the H++ server (25). Using PyMOL version 1.7.6.3 (26), the heme group was manually accommodated in the inter-monomer cleft between the two flexible loops containing His-53 in the hGAPDH (PDB code 1znq). Using the Molefacture plug-in version 1.3 of VMD 1.9.2 (27), the histidines were rotated making both imidazole rings point toward the middle of the cleft, at bonding distance from the iron atom of the heme group. The imidazole group of His-53 was predicted by H++ to be protonated in ϵ and δ. Consequently, the ϵ proton was removed to make possible the binding of the NE2 atom of histidine with the iron center of the heme group. The model was then minimized in vacuo, neutralized (with 16 Cl−), solvated (with explicit waters and 0.15 m NaCl), and minimized in solution with positional restraints on the solute using our well-established multistep protocol (28, 29). To produce the final model, the minimized structure was thermalized to 298 °C at NVT and then simulated during 150 ns by means of Molecular Dynamics simulations at NPT (p = 1 atm). GAPDH was represented by the state-of-the-art ff14SB force field (30), surrounded by a truncated octahedral box of ∼22,000 TIP3P water molecules (31), Dang parameters for ions (32), Walker's parameters for NAD+ (33), and the heme hexacoordinate parameters developed by Estrin and co-workers (34, 35). Ions were initially placed randomly, at a minimum distance of 5 Å from the solute and 3.5 Å from one another. All systems were simulated using the Berendsen algorithm (36) to control the temperature and the pressure, with a coupling constant of 5 ps. Center of mass motion was removed every 10 ps. SHAKE (37) was used to keep all bonds involving hydrogen at their equilibrium values, which allowed us to use a 2-fs step for the integration of Newton equations of motion. Long-range electrostatic interactions were accounted for by using the Particle Mesh Ewald method (38) with standard defaults and a real-space cutoff of 9 Å. All simulations were carried out using the PMEMD CUDA code module (39) of AMBER 16 (40) and analyzed with CPPTRAJ (41).
Protein purification
WT human GAPDH in a pGEX 4T-2 vector and mutants, made using site-directed mutagenesis, were purified from BL21 (DE3) Escherichia coli using standard techniques. Briefly, E. coli cells carrying the GAPDH construct were grown to an optical density of 0.8–1.0 and induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 48 h at room temperature. After cell lysis, purification of WT and mutant GAPDH was carried out using GST affinity chromatography as per the manufacturer's instructions (Sigma). After subsequent cleavage of the GST tag using thrombin, the GST and thrombin were removed using a GST column followed by a benzamidine FF column (GE Healthcare). Before use in heme-binding assays, the proteins were run over a Superdex 200 column (GE Healthcare) and assessed for purity by SDS-PAGE.
WT rabbit GAPDH and H51A mutant, made using site-directed mutagenesis, were purified as described previously (12) using nickel-nitrilotriacetic acid affinity chromatography (Amersham Biosciences) followed by thrombin cleavage of the His6 tag. Before use in heme-binding assays, the proteins were assessed for purity by SDS-PAGE.
UV-visible spectroscopy
UV-visible spectra of hemin, rGAPDH (WT and H51A), and rGAPDH-hemin complexes were measured on a Shimadzu UV-2600 using freshly prepared rGAPDH and hemin stocks. Heme binding via His ligation was determined by the shift in Soret peak from 390 nm (free heme) to 412 nm (WT rGAPDH–heme) (12).
Heme binding using nanoscale thermophoresis
Exact hemin concentration was determined by diluting the stock solution in pure DMSO and measuring absorbance at 406 nm on a Hitachi U-2000 spectrophotometer (extinction coefficient 170 mm−1 cm−1) (42). Heme was diluted from the stock solution to 1.6 mm in PBS. Sixteen serial dilutions of heme were then made from 50 μm to 1.53 nm in 50 mm Tris-HCl, pH 7.4, 10 mm MgCl2, 150 mm NaCl, and 1 mm DTT (TMND) with 0.65% DMSO. Purified hGAPDH WT and mutants were diluted to 1 μm in TMND with 0.1% Pluronic F-127 (Nanotemper). 10 μl of GAPDH protein was added to 10 μl of each heme dilution and after mixing transferred to capillary tubes (Nanotemper). Capillary tubes were loaded onto and analyzed using a Monolith NT.LabelFree Nanotemper device. Each capillary series was analyzed at three different LED:MST settings, and binding curves were obtained using thermophoresis (movement of particles due to temperature gradients, which changes based on binding events) using the intrinsic tryptophan fluorescence of GAPDH. Data from individual Nanotemper runs were combined using Prism and fit with a one-site binding curve.
Measurement of GAPDH dehydrogenase activity
WT and GAPDH variants were assayed for their enzymatic activity by measuring conversion of NAD+ to NADH in 96-well clear plates as described in the literature (43). Briefly, GAPDH was diluted to a concentration of 6.9 nm in 10 mm tetrasodium pyrophosphate, pH 8.5, 20 mm sodium phosphate, 2 mm NAD+, and 0.3 μm DTT. The enzymatic reaction was initiated by addition of 2 mm dl-glyceraldehyde 3-phosphate to the wells. After mixing, the absorbance at 340 nm was measured for 5 min. The initial linear portion of the curve was used to determine the rate of NADH production using the extinction coefficient for NADH of 6.22 mm−1 cm−1.
Heme koff measurements using apo-myoglobin
The kinetics of ferric heme transfer from hGAPDH (10 μm GAPDH, 2 μm heme) to apo-myoglobin (50 μm) were measured as described (12) in either a Shimadzu UV-2600 or a Hi-Tech Scientific stop-flow using Kinetic Studio software for data acquisition and analysis. Normalized change in absorbance at 410 nm was plotted using Prism and fit using a one-phase exponential association.
Cell culture
RAW264.7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and penicillin/streptomycin (100 units/ml and 100 μg/ml, respectively) at 37 °C with 5% CO2 in a humidified incubator. The day before transfection, RAW264.7 cells were seeded out at a density of 3 × 106 cells per 10-cm dish into DMEM with 10% FBS, 3 mm Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME), and no antibiotic. Cells were transfected with 5 μg of HA-tagged GAPDH DNA and 15 μl of Lipofectamine 2000 (ThermoFisher Scientific) per sample. Sixteen hours after transfection, the media were changed to DMEM with 10% FBS, 3 mm l-NAME, and penicillin/streptomycin. Transfection efficiency was assessed by imaging an mVenus control plate. Efficiency was generally 85–95%. Cells were activated to induce iNOS expression 24 h after transfection with 10 units/ml interferon γ (IFNγ, PeproTech) and 25 μg of lipopolysaccharide. After 16 h of activation, the cells were harvested and lysed in lysis buffer (40 mm EPPS, pH 7.6, 150 mm NaCl, 10% glycerol, 1% Nonidet P-40, and protease inhibitors). Crude lysates were cleared of insoluble debris by centrifugation at 13,000 rpm. Lysate protein concentration was assessed using a DC protein assay from Bio-Rad and subsequently used in HA pulldowns, 2′,5′-ADP affinity resin pulldowns, OxyPEGHb assays, and Western blottings. For HA and ADP pulldown experiments, cells were grown in media made with heme-depleted FBS and treated with succinyl acetone (SA) to inhibit heme synthesis for 3 days before transfection. Before cell lysis, activated cells were incubated with 55Fe-heme for 2 h. For iNOS activity assays, cells were heme-depleted as above and then incubated with cold heme for 2 h prior to lysis.
GlyA cells, a CHO cell variant lacking mSHMT activity, were used to minimize native δ-ALA production from glycine (44, 45). Cells were thawed and grown in media containing glycine (Kaighn's modified medium). Cells were transfected with HA–hGAPDH constructs as above using Lipofectamine 2000. After 16–20 h post-transfection, media were changed to glycine-deficient medium (F-12 media with 10% dialyzed FBS) for 3 h. The media were then replaced with F-12 media containing 14C-labeled δ-ALA and incubated for 3 h at 37 °C. After incubation, cells were washed with 0.1% BSA twice and PBS twice. Counts in the last wash were tested to avoid any leftover external contamination. Cells were then transferred on ice and harvested with RIPA buffer (50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 1% Nonidet P-40, 1% sodium -deoxycholate) containing protease inhibitors and phenylmethylsulfonyl fluoride. Crude lysates were cleared of insoluble debris by centrifugation at 13,000 rpm. Lysate protein concentration was assessed using a DC protein assay from Bio-Rad and subsequently used in HA-pulldown experiments.
Measuring radiolabeled 14C and 55Fe counts
Lysates containing equal amounts of protein were incubated with HA monoclonal antibody (GlyA or RAW264.7) overnight at 4 °C with rotation. For HA-pulldowns, protein G-agarose beads (Millipore) were washed twice with PBS and then equilibrated with RIPA buffer twice at 2000 × g for 2 min. The overnight rotating cell lysate and antibody complexes were then incubated with equilibrated beads at 4 °C with rotation for 3 h. For iNOS, pulldowns using ADP-affinity resin (RAW264.7) beads were washed as above and incubated with lysates at 4 °C with rotation for 3 h. After incubation, the beads were washed three times with RIPA buffer. Washed beads (HA pulldowns and ADP pulldowns) were added to scintillation liquid and counted on a Beckman Coulter LS 6500 Multi-Purpose Scintillation Counter for 14C or 55Fe counts. Alternatively, heme was extracted from the beads using acidified acetone, and the extracts were run on an Agilent HPLC. Fractions were then assessed for radioactive counts as above.
Measuring iNOS NO synthesis activity
The oxyHb assay for NO was carried out in a 96-well clear plate in a 200-μl volume. The final reaction conditions included 5 μm tetrahydrobiopterin (Schircks), 5 μm FMN, 5 μm FAD, 0.3 mm DTT, 0.1 mg/ml BSA, protease inhibitor, 60 units/ml superoxide dismutase, 100 units/ml catalase, 10 mm l-arginine, 5 μm Oxy(PEG)Hb, and 80 μg of lysate or 0.1 μm purified iNOS protein as a positive control. Activity of iNOS in the cell lysates was assessed by measuring the conversion of OxyHb to MetHb by monitoring increases in absorbance at 401 nm. Absorbance was read on a SpectraMax M2e (Molecular Devices) plate reader at 401 nm for 10 min, and the initial rate was used to determine Vmax.
Western blot analysis
SDS-PAGE and Western blot analysis were performed using standard procedures with antibodies against HA for HA–GAPDH (Sigma), iNOS (Cell Signaling), and β-actin (Origene). Horseradish peroxidase-linked secondary antibodies (Santa Cruz Biotechnology) were used to visualize the proteins using a Western Lightning Plus-ECL kit from PerkinElmer Life Sciences.
Yeast strains, transformations, and growth conditions
S. cerevisiae strains used in this study were derived from BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0). The tdh3Δ::KanMX4 strain was obtained from the yeast gene deletion collection (ThermoFisher Scientific). Yeast transformations were performed by the lithium acetate procedure (46). Strains were maintained at 30 °C on either enriched yeast extract (1%)-peptone (2%)–based medium supplemented with 2% glucose (YPD) or synthetic complete medium (SC) supplemented with 2% glucose and the appropriate amino acids to maintain selection (8). Cells cultured on solid media plates were done so with YPD or SC media supplemented with 2% agar (8). Selection for yeast strains containing the KanMX4 marker was done with YPD agar plates supplemented with G418 (200 μg/ml) (8). WT cells treated with the heme synthesis inhibitor, SA, and hem1Δ cells were cultured in YPD or SC media supplemented with 50 μg/ml ALA or 15 mg/ml ergosterol and 0.5% Tween 80 (YPDE or SCE, respectively) (8, 47). All liquid cultures were maintained at 30 °C and shaken at 220 rpm.
Yeast plasmids
Yeast plasmids expressing the cytosolic heme sensor, HS1-M7A, which was subcloned into pRS415 and driven by the GPD promoter, and the Hap1 reporter plasmid in which EGFP is driven by the CYC1 promoter were previously described (8). The yeast Tdh3 expression plasmid was generated by amplifying TDH3 from yeast genomic DNA and sub-cloning it into the SpeI/BamHI sites of p416-GPD (48) to generate plasmid pAR1031. The yeast Tdh3-H51A mutation was generated by QuikChange mutagenesis (Agilent Technologies) using pAR1031 as a template, generating pAR1033. The yeast hGAPDH and hGAPDH-H53A expression plasmids were generated by amplifying hGAPDH from pGex4T2-GST-hGAPDH or pGex4T2-GST-hGAPDH-H53A and sub-cloning it into the SpeI/BamHI sites of p416-GPD (48) to generate plasmids pAR1035 and pAR1036, respectively.
Labile heme measurements using a fluorescent heme sensor
WT or tdh3Δ cells co-expressing HS1-M7A and the indicated GAPDH variant or empty vector (p416-GPD) (48) were cultured in SCE/LEU/URA to an A600 nm of 1.0 (2 × 107 cells/ml). Cells were washed in ultrapure water and resuspended in phosphate-buffered saline (PBS) solution to give a concentration of 1 × 108 cells/ml, prior to recording EGFP (excitation 488 nm and emission 510 nm) and mKATE2 (excitation 588 nm and emission 620 nm) fluorescence in a Synergy H1 hybrid multimode microplate reader (BioTek). Labile heme concentration was calculated using Equation 1,
| (Eq. 1) |
where KD is the HS1–M7A–ferrous heme dissociation constant at pH 7.0, 25 nm; Rexpt is the EGFP/mKATE2 fluorescence ratio under any given condition; Rmin is the EGFP/mKATE2 fluorescence ratio when 0% of the sensor is bound to heme; Rmax is the EGFP/mKATE2 fluorescence ratio when 100% of the sensor is bound to heme; FminmKATE2 is the mKATE2 emission intensity when 0% of the sensor is bound to heme, and FmaxmKATE2 is the mKATE2 emission intensity when 100% of the sensor is bound to heme (8).
Determination of Rmax and FmaxmKATE2 involves recording EGFP and mKATE2 fluorescence after digitonin permeabilization of cells and incubation with 50 μm heme (8). Briefly, 1 × 108 cells/ml of cells are resuspended in PBS with 200 μg/ml digitonin, 1 mm ascorbate, and 50 μm hemin chloride. After a 30-min incubation at 30 °C, cells were harvested, washed, and resuspended in PBS buffer prior to recording of fluorescence. Determination of Rmin and FminmKATE2 involves recording EGFP and mKATE2 fluorescence from parallel cultures treated with the heme biosynthesis inhibitor succinylacetone (8, 49).
Hap1 activity
WT or tdh3Δ cells co-expressing p415–CYC1–EGFP or EGFP driven by the Hap1p-regulated CYC1 promoter (8) and the indicated GAPDH variant or empty vector (p416–GPD) (42) were cultured in 10 ml of SCE/LEU/URA medium for 15 h to 1 A600 nm/ml. Cells were resuspended in PBS to a concentration of 1 × 108 cells/ml, and 100 μl was used to measure EGFP fluorescence (excitation 488 nm and emission 510 nm). As a positive and negative control, WT and hem1Δ cells were cultured for each experiment. Background autofluorescence of cells not expressing EGFP was recorded and subtracted from the EGFP-expressing strains.
Author contributions
E. A. S., A. B. S., R. C., L. H., A. R. R., and D. J. S. conceptualization; E. A. S., A. B. S., P. D. D., A. R. R., and D. J. S. data curation; E. A. S., A. B. S., O. M.-G., A. S., M. M. H., G. G., P. D. D., L. H., A. R. R., and D. J. S. formal analysis; E. A. S., A. R. R., and D. J. S. funding acquisition; E. A. S., A. B. S., R. C., O. M.-G., A. S., M. M. H., G. G., P. D. D., and L. H. investigation; E. A. S., R. C., P. D. D., A. R. R., and D. J. S. methodology; E. A. S. and D. J. S. writing-original draft; E. A. S., A. B. S., L. H., A. R. R., and D. J. S. writing-review and editing; A. R. R. and D. J. S. supervision.
Acknowledgments
We thank S. Schlanger and D. Durra for technical assistance and Dr. J. S. Olson (Rice University) for helpful advice.
This work was supported by National Institutes of Health Grants RO1 GM097041 (to D. J. S.), PO1 HL081064 Project 3 and PO1 HL103453 Project 3 (to D. J. S.), and R21 ES025661 (to A. R. R.), National Science Foundation Grant MCB-1552791 (to A. R. R.), an American Heart Association Postdoctoral Fellowship (to E. A. S.), the Blanchard Professorship (to A. R. R.), and Georgia Institute of Technology start-up funds (to A. R. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- iNOS
- inducible nitric-oxide synthase
- EGFP
- enhanced green fluorescent protein
- ALA
- aminolevulinic acid
- hGAPDH
- human GAPDH
- rGAPDH
- rabbit GAPDH
- HA
- hemagglutinin
- EPPS
- 4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- GST
- glutathione S-transferase
- BVR
- biliverdin reductase
- PDB
- Protein Data Bank
- l-NAME
- Nω-nitro-l-arginine methyl ester hydrochloride
- SA
- succinyl acetone.
References
- 1. Ponka P. (1999) Cell biology of heme. Am. J. Med. Sci. 318, 241–256 [DOI] [PubMed] [Google Scholar]
- 2. Mense S. M., and Zhang L. (2006) Heme: a versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 16, 681–692 10.1038/sj.cr.7310086 [DOI] [PubMed] [Google Scholar]
- 3. Adachi Y., Umeda M., Kawazoe A., Sato T., Ohkawa Y., Kitajima S., Izawa S., Sagami I., and Taketani S. (2017) The novel heme-dependent inducible protein, SRRD regulates heme biosynthesis and circadian rhythms. Arch. Biochem. Biophys. 631, 19–29 10.1016/j.abb.2017.08.006 [DOI] [PubMed] [Google Scholar]
- 4. Harvey J. W., and Beutler E. (1982) Binding of heme by glutathione S-transferase: a possible role of the erythrocyte enzyme. Blood 60, 1227–1230 [PubMed] [Google Scholar]
- 5. Iwahara S., Satoh H., Song D. X., Webb J., Burlingame A. L., Nagae Y., and Muller-Eberhard U. (1995) Purification, characterization, and cloning of a heme-binding protein (23 kDa) in rat liver cytosol. Biochemistry 34, 13398–13406 10.1021/bi00041a017 [DOI] [PubMed] [Google Scholar]
- 6. Taketani S., Adachi Y., Kohno H., Ikehara S., Tokunaga R., and Ishii T. (1998) Molecular characterization of a newly identified heme-binding protein induced during differentiation of urine erythroleukemia cells. J. Biol. Chem. 273, 31388–31394 10.1074/jbc.273.47.31388 [DOI] [PubMed] [Google Scholar]
- 7. Vincent S. H., and Muller-Eberhard U. (1985) A protein of the Z class of liver cytosolic proteins in the rat that preferentially binds heme. J. Biol. Chem. 260, 14521–14528 [PubMed] [Google Scholar]
- 8. Hanna D. A., Harvey R. M., Martinez-Guzman O., Yuan X., Chandrasekharan B., Raju G., Outten F. W., Hamza I., and Reddi A. R. (2016) Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. U.S.A. 113, 7539–7544 10.1073/pnas.1523802113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chakravarti R., Aulak K. S., Fox P. L., and Stuehr D. J. (2010) GAPDH regulates cellular heme insertion into inducible nitric-oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 18004–18009 10.1073/pnas.1008133107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Severance S., and Hamza I. (2009) Trafficking of heme and porphyrins in metazoa. Chem. Rev. 109, 4596–4616 10.1021/cr9001116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuan X., Fleming M. D., and Hamza I. (2013) Heme transport and erythropoiesis. Curr. Opin. Chem. Biol. 17, 204–211 10.1016/j.cbpa.2013.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hannibal L., Collins D., Brassard J., Chakravarti R., Vempati R., Dorlet P., Santolini J., Dawson J. H., and Stuehr D. J. (2012) Heme binding properties of glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 51, 8514–8529 10.1021/bi300863a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shedlofsky S. I., Sinclair P. R., Bonkovsky H. L., Healey J. F., Swim A. T., and Robinson J. M. (1987) Haem synthesis from exogenous 5-aminolaevulinate in cultured chick-embryo hepatocytes. Effects of inducers of cytochromes P-450. Biochem. J. 248, 229–236 10.1042/bj2480229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Huang Y., Zhang P., Yang Z., Wang P., Li H., and Gao Z. (2017) Interaction of glyceraldehyde-3-phosphate dehydrogenase and heme: the relevance of its biological function. Arch. Biochem. Biophys. 619, 54–61 10.1016/j.abb.2017.03.005 [DOI] [PubMed] [Google Scholar]
- 15. Chiabrando D., Marro S., Mercurio S., Giorgi C., Petrillo S., Vinchi F., Fiorito V., Fagoonee S., Camporeale A., Turco E., Merlo G. R., Silengo L., Altruda F., Pinton P., and Tolosano E. (2012) The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Invest. 122, 4569–4579 10.1172/JCI62422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun F., Cheng Y., and Chen C. (2015) Regulation of heme biosynthesis and transport in metazoa. Sci. China Life Sci. 58, 757–764 10.1007/s11427-015-4885-5 [DOI] [PubMed] [Google Scholar]
- 17. Khan A. A., and Quigley J. G. (2011) Control of intracellular heme levels: heme transporters and heme oxygenases. Biochim. Biophys. Acta 1813, 668–682 10.1016/j.bbamcr.2011.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., and Sawa A. (2005) S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 10.1038/ncb1268 [DOI] [PubMed] [Google Scholar]
- 19. Sen N., Hara M. R., Kornberg M. D., Cascio M. B., Bae B. I., Shahani N., Thomas B., Dawson T. M., Dawson V. L., Snyder S. H., and Sawa A. (2008) Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 10, 866–873 10.1038/ncb1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hannibal L., and Stuehr D. J. (2013) Handbook of Porphyrin Science, Vol. 30, pp. 55–102, World Scientific Publishing Co., Singapore [Google Scholar]
- 21. Ghosh A., Stasch J. P., Papapetropoulos A., and Stuehr D. J. (2014) Nitric oxide and heat shock protein 90 activate soluble guanylate cyclase by driving rapid change in its subunit interactions and heme content. J. Biol. Chem. 289, 15259–15271 10.1074/jbc.M114.559393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Waheed S. M., Ghosh A., Chakravarti R., Biswas A., Haque M. M., Panda K., and Stuehr D. J. (2010) Nitric oxide blocks cellular heme insertion into a broad range of hemeproteins. Free Radic. Biol. Med. 48, 1548–1558 10.1016/j.freeradbiomed.2010.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chakravarti R., Gupta K., Majors A., Ruple L., Aronica M., and Stuehr D. J. (2015) Novel insights in mammalian catalase heme maturation: effect of NO and thioredoxin-1. Free Radic. Biol. med. 82, 105–113 10.1016/j.freeradbiomed.2015.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ghosh A., Garee G., Sweeny E. A., Nakamura Y., and Stuehr D. J. (2018) Hsp90 chaperones hemoglobin maturation in erythroid and nonerythroid cells. Proc. Natl. Acad. Sci. U.S.A. 115, E1117–E1126 10.1073/pnas.1717993115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Anandakrishnan R., Aguilar B., and Onufriev A. V. (2012) H++ 3.0: automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 40, W537–W541 10.1093/nar/gks375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schrödinger L. (2015) The PyMOL Molecular Graphics System, Version 1.7, Schrödinger, LLC, New York [Google Scholar]
- 27. Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38, 27–28 10.1016/0263-7855(96)00018-5 [DOI] [PubMed] [Google Scholar]
- 28. Pérez A., Luque F. J., and Orozco M. (2007) Dynamics of B-DNA on the microsecond time scale. J. Am. Chem. Soc. 129, 14739–14745 10.1021/ja0753546 [DOI] [PubMed] [Google Scholar]
- 29. Dans P. D., Danilāne L., Ivani I., Dršata T., Lankaš F., Hospital A., Walther J., Pujagut R. I., Battistini F., Gelpí J. L., Lavery R., and Orozco M. (2016) Long-timescale dynamics of the Drew-Dickerson dodecamer. Nucleic Acids Res. 44, 4052–4066 10.1093/nar/gkw264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., and Simmerling C. (2015) ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 10.1021/acs.jctc.5b00255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 32. Smith D. E., and Dang L. X. (1994) Computer-simulations of Nacl association in polarizable water. J. Chem. Phys 100, 3757–3766 10.1063/1.466363 [DOI] [Google Scholar]
- 33. Walker R. C., de Souza M. M., Mercer I. P., Gould I. R., and Klug D. R. (2002) Large and fast relaxations inside a protein: calculation and measurement of reorganization energies in alcohol dehydrogenase. J. Phys. Chem. B 106, 11658–11665 10.1021/jp0261814 [DOI] [Google Scholar]
- 34. Capece L., Marti M. A., Bidon-Chanal A., Nadra A., Luque F. J., and Estrin D. A. (2009) High pressure reveals structural determinants for globin hexacoordination: neuroglobin and myoglobin cases. Proteins 75, 885–894 10.1002/prot.22297 [DOI] [PubMed] [Google Scholar]
- 35. Nadra A. D., Martí M. A., Pesce A., Bolognesi M., and Estrin D. A. (2008) Exploring the molecular basis of heme coordination in human neuroglobin. Proteins 71, 695–705 10.1002/prot.21814 [DOI] [PubMed] [Google Scholar]
- 36. Berendsen H. J. C., Postma J. P. M., Vangunsteren W. F., Dinola A., and Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 37. Ryckaert J. P., Ciccotti G., and Berendsen H. J. C. (1977) Numerical-integration of cartesian equations of motion of a system with constraints- molecular-dynamics of N-alkanes. J. Comput. Phys. 23, 327–341 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 38. Darden T., York D., and Pedersen L. (1993) Particle Mesh Ewald-an N. Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 39. Salomon-Ferrer R., Götz A. W., Poole D., Le Grand S., and Walker R. C. (2013) Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 10.1021/ct400314y [DOI] [PubMed] [Google Scholar]
- 40. Case D. A., Cheatham T. E. III, Darden T. A., Duke R. E., Giese T. J., Gohlke H., Goetz A. W., Greene D., Homeyer N., Izadi S., Kovalenko A., Lee T. S., LeGrand S., Li P., Lin C., et al. (2017) AMBER 16, University of California, San Francisco [Google Scholar]
- 41. Roe D. R., and Cheatham T. E. 3rd. (2013) PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095 10.1021/ct400341p [DOI] [PubMed] [Google Scholar]
- 42. Rish K. R., Swartzlander R., Sadikot T. N., Berridge M. V., and Smith A. (2007) Interaction of heme and heme-hemopexin with an extracellular oxidant system used to measure cell growth-associated plasma membrane electron transport. Biochim. Biophys. Acta 1767, 1107–1117 10.1016/j.bbabio.2007.06.003 [DOI] [PubMed] [Google Scholar]
- 43. Zhang J., and Snyder S. H. (1992) Nitric oxide stimulates auto-ADP-ribosylation of glyceraldehyde-3-phosphate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 89, 9382–9385 10.1073/pnas.89.20.9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Stover P. J., Chen L. H., Suh J. R., Stover D. M., Keyomarsi K., and Shane B. (1997) Molecular cloning, characterization, and regulation of the human mitochondrial serine hydroxymethyltransferase gene. J. Biol. Chem. 272, 1842–1848 10.1074/jbc.272.3.1842 [DOI] [PubMed] [Google Scholar]
- 45. Narkewicz M. R., Sauls S. D., Tjoa S. S., Teng C., and Fennessey P. V. (1996) Evidence for intracellular partitioning of serine and glycine metabolism in Chinese hamster ovary cells. Biochem. J. 313, 991–996 10.1042/bj3130991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gietz R. D., and Schiestl R. H. (1991) Applications of high efficiency lithium acetate transformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast 7, 253–263 10.1002/yea.320070307 [DOI] [PubMed] [Google Scholar]
- 47. Ness F., Achstetter T., Duport C., Karst F., Spagnoli R., and Degryse E. (1998) Sterol uptake in Saccharomyces cerevisiae heme auxotrophic mutants is affected by ergosterol and oleate but not by palmitoleate or by sterol esterification. J. Bacteriol. 180, 1913–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mumberg D., Müller R., and Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 10.1016/0378-1119(95)00037-7 [DOI] [PubMed] [Google Scholar]
- 49. Ebert P. S., Hess R. A., Frykholm B. C., and Tschudy D. P. (1979) Succinylacetone, a potent inhibitor of heme biosynthesis: effect on cell growth, heme content and δ-aminolevulinic acid dehydratase activity of malignant murine erythroleukemia cells. Biochem. Biophys. Res. Commun. 88, 1382–1390 10.1016/0006-291X(79)91133-1 [DOI] [PubMed] [Google Scholar]