

Abstract
Human islet amyloid polypeptide (hIAPP) is the principal constituent of amyloid deposits and toxic oligomers in the pancreatic islets. Together with hyperglycemia, hIAPP-derived oligomers and aggregates are important culprits in type 2 diabetes mellitus (T2DM). Here, we explored the role of the cell's main proteolytic complex, the proteasome, in hIAPP turnover in normal and stressed β-cells evoked by chronic hyperglycemia. Moderate inhibition (10–35%) of proteasome activity/function in cultured human islets by the proteasome inhibitor lactacystin enhanced intracellular accumulation of hIAPP. Unexpectedly, prolonged (>1 h) and marked (>50%) impairment of proteasome activity/function had a strong inhibitory effect on hIAPP transcription and secretion from normal and stressed β-cells. This negative compensatory feedback mechanism for controlling IAPP turnover was also observed in the lactacystin-treated rat insulinoma β-cell line (INS 832/13), demonstrating the presence of an evolutionarily conserved mechanism for IAPP production. In line with these in situ studies, our current ex vivo data showed that proteasome activity and hIAPP expression are also down-regulated in islets isolated from T2DM subjects. Gene expression and promoter activity studies demonstrated that the functional proteasome complex is required for efficient activation of the hIAPP promoter and for full expression of IAPP's essential transcription factor, FOXA2. ChIP studies revealed that promoter occupancy of FoxA2 at the rat IAPP promoter region is an important and limiting factor for amylin expression in proteasome-impaired murine cells. This study suggests a novel regulatory pathway in β-cells involving proteasome, FOXA2, and IAPP, which can be possibly targeted to regulate hIAPP levels and islet amyloidosis in T2DM.
Keywords: amyloid, insulin, secretion, proteasome, transcription, type 2 diabetes, FoxA2, hyperglycemia, Islet amyloid polypeptide
Introduction
Type 2 diabetes mellitus (T2DM)2 is a complex, multifactorial metabolic disorder characterized by progressive impairment of pancreatic β-cell secretory functions, β-cell death, peripheral insulin resistance, and resulting hyperglycemia (1). Although the mechanisms and cellular factors implicated in β-cell loss or their dysfunction during T2DM are diverse and still emerging, the accumulated data suggest that human islet amyloid polypeptide (hIAPP)–derived toxic oligomers and aggregates contribute significantly toward the progressive loss of β-cells during T2DM (1–5). hIAPP or amylin is a 37-amino acid pancreatic peptide hormone encoded by a single copy gene on chromosome 12 in humans (6). It is an important signaling molecule and a hormone that regulates food intake, energy homeostasis, and insulin secretion from pancreatic β-cells (7–11). IAPP is produced and co-released with insulin from islet β-cells in a glucose-dependent manner (5, 12). Likewise, IAPP and insulin promoters share common glucose-responsive regulatory elements and transcription factors such as PDX1 and ISL1 (13). In addition to these two transcription factors, forkhead box protein A2 (FOXA2) has been also implicated in the regulation of IAPP expression in β-cells (14, 15). IAPP is first synthesized as an 89-amino acid prepropeptide (16, 17). The prepro-IAPP form, together with nascent insulin, undergoes a series of post-translational and proteolytic processing in the endoplasmic reticulum (ER), Golgi, and secretory vesicles by prohormone convertase 2 (PC2) and 1/3 (PC1/3) and carboxypeptidase E (5). Fully processed IAPP and insulin are then stored in the same secretory granules of pancreatic islet β-cells. In normal subjects, upon physiological stimulation (such as elevated glucose/or nutrients), insulin and IAPP are co-secreted to regulate glucose homeostasis in the body (5, 18, 19). However, under conditions that favor the development of T2DM, hIAPP misfolds and forms toxic amyloid oligomers and aggregates (5). At present, it is not clear which cellular processes and factors regulate hIAPP-mediated cytotoxicity, but it has been suggested that impaired turnover and cellular processing of hIAPP contribute significantly toward the progressive β-cell failure during T2DM (4, 5).
Several independent studies have linked impaired ubiquitin–proteasome system (UPS) as a risk factor for age-related diseases such as T2DM (3, 20). The primary component of UPS is the 26S proteasome complex, nonlysosomal protein degradation machinery in eukaryotes. The 26S proteasome is composed of a 20S proteolytic core and 19S regulatory components. The 20S core is a cylinder-like structure, consisting of α (α1-α7) and β (β1-β7) subunits. Within the seven β-subunits, β5, β2, and β1 are catalytically active and responsible for chymotrypsin-like, trypsin-like, and post-acidic or caspase-like proteolytic activities, respectively (21). The 26S proteasome complex is responsible for degradation of polyubiquitinated proteins in an ATP-dependent manner. However, recent reports also provide evidence for nonubiquitin and non-ATP–dependent degradation mechanisms of the 20S proteasome (22, 23). Intriguingly, the UPS has also been implicated in transcriptional regulation of several eukaryotic genes. Studies showed that proteolytic and nonproteolytic activities of the 26S proteasome complex regulate the availability, localization, and promoter recruitment of various transcription factors. In this way, UPS controls the key stages of eukaryotic gene expression; transcription initiation, elongation, maturation, and nuclear export of mRNA (24).
Although the exact role of UPS in the pathology of T2DM is still emerging, microarray analyses of human pancreatic islets revealed down-regulation of several proteasome subunits in T2DM patients, indicating its possible role in disease onset and progression (25). Studies using pancreatectomy-induced diabetic rat models showed an initial increase followed by gradual down-regulation of rodent IAPP mRNA levels, together with ensuing hyperglycemia (26). Previous studies in our laboratory demonstrated the crucial role of the proteasome in the degradation of internalized hIAPP, thereby preventing hIAPP-induced β-cell toxicity (27). However, the role of the proteasome in the production, degradation, and secretion, hereafter collectively referred as “turnover,” of endogenous hIAPP in normal and disease states has yet to be determined. Given the emerging role of the 26S proteasome complex in the regulation of eukaryotic gene transcription and the important pathophysiological roles of hIAPP, in this study, we explored the role of the proteasome in IAPP turnover in rodent and human pancreatic β-cells. This study points to the essential and novel role of proteasome complex in IAPP synthesis, secretion, and degradation in β-cells. This proteasome-regulated pathway may have important ramifications for amylin-induced amyloid formation in human islets and its pathological role in T2DM.
Results
Intracellular hIAPP levels and proteasome activity are down-regulated in diabetic human islet cells
Previous studies revealed that chronic hyperglycemia may impair proteasome activity and alter insulin and IAPP biosynthesis in human and rodent pancreatic β-cells (15, 28, 29). However, causal connection between these two confounding factors in T2DM is unclear. It is also not known whether the hIAPP production and proteasome function are co-impaired in T2DM. Here, we analyzed the intracellular hIAPP content with respect to proteasome activity and function in freshly isolated intact human pancreatic islets procured from adult T2DM and control nondiabetic donors. Protein and gene expression analysis revealed that the intracellular hIAPP mRNA and protein levels were significantly decreased in the T2DM human islet cells as compared with age-matched nondiabetic control islets (Fig. 1, A–C). Moreover, Western blot analysis showed that levels of polyubiquitinated proteins were markedly elevated (>3-fold) in the islets of T2DM donors, as compared with control nondiabetic islets indicating severe protein stress (Fig. 1, A and B). Consistent with this finding, we found a similar ∼3-fold decline in proteasome proteolytic activity in T2DM islets as compared with nondiabetic donor islets (Fig. 1D). This decline was comparable with that observed with the selective and potent proteasome inhibitor, lactacystin (LC), indicating almost complete loss of proteasome function in those diabetic patients. Furthermore, gene expression analysis demonstrated a significant and substantial (∼1.7-fold) drop in mRNA levels of IAPP's crucial transcription factor, FOXA2, in the islets procured from T2DM donors (Fig. 1E). These results suggested a causal link between proteasomes and hIAPP transcription/translation in human islets.
Figure 1.

Diabetic islets exhibit altered hIAPP gene expression and proteasome activity. A, Western blot analysis of hIAPP and polyubiquitinated protein levels in the pancreatic islet cells of nondiabetic and T2DM donors. Glycosylated hemoglobin level (HbA1C) and body mass index (BMI) are provided for each sample. Nondiabetic and T2DM samples are grouped by one empty lane in the Western blotting. B, band intensity analysis of hIAPP (left panel) and ubiquitinated (right panel) protein levels. Significance was established at *, p < 0.05, nondiabetic n = 3 versus diabetic n = 4, Student's t test. C, hIAPP gene expression analysis by qPCR in the pancreatic islet cells of nondiabetic and T2DM donors. Significance was established at **, p < 0.01, n = 6, Student's t test. D, quantification of 20S proteasome activity in nondiabetic, diabetic, and LC-treated human islet cells. Significance was established at *, p < 0.05; **, p < 0.01, n = 4, ANOVA followed by Tukey's post hoc comparison test. E, FOXA2 gene expression analysis by qPCR in the pancreatic islet cells of nondiabetic and T2DM donors. Significance was established at *, p < 0.05, n = 6, Student's t test.
Functional proteasome complex is required for the hIAPP expression in human islet cells
To verify ex vivo expression studies described above and to clarify the causal relationship between proteasome activity/function and hIAPP turnover, we performed inhibitory studies in human islets cultured under normal (5 mm) or high glucose (20 mm) conditions, either in the absence (control) or presence of LC. Thereafter, the temporal changes in hIAPP protein levels were determined by Western blotting and ELISA (Fig. 2, A–C). Consistent with the fact that hIAPP is a proteasome client (27), a moderate (∼30%) and transient (1 h) decrease in proteasome activity induced by 10 μm LC (5 mm Glc + LC, 1 h, Fig. 2E) raised hIAPP intracellular protein levels in these islet cells by ∼2-fold compared with control cells maintained under normal glucose conditions (Fig. 2, A–C). In support of the Western blotting results, IAPP-specific ELISA confirmed progressive accumulation of hIAPP in human islets during early time points of proteasome inhibition (5 mm Glc + 10 μm LC, 0–60 min) (Fig. 2B, right panel). A similar stimulatory effect of LC on intracellular hIAPP accumulation was also observed at 12 h using a lower (1 μm) LC concentration (5 mm Glc + 1 μm LC, Fig. 3, A–C). This treatment also induced a moderate (∼35%) decrease in proteasome activity (5 mm Glc + 1 μm LC, Fig. 3E). Interestingly, under hyperglycemia-mimicking conditions (20 mm glucose), a short-term (1 h) LC treatment had the opposite (inhibitory) effect on hIAPP expression (20 mm Glc + 10 μm LC, 1 h, Fig. 2, A and C). This is likely due to a sensitizing effect of high glucose on the LC-evoked proteasome dysfunction observed during an early stage of proteasome inhibition (5 mm Glc + 10 μm LC versus 20 mm Glc + 10 μm LC, 1 h, Fig. 2E). Similarly, under conditions of a sustained (12 h) and marked (>50%) impairment of proteasome activity/function by LC, there was a significant decline in hIAPP production in human islet β-cells (Fig. 3, A–E). In agreement with these biochemical results (Figs. 2 and 3, A–E), immunoconfocal microscopy analysis revealed a similar dose-dependent decrease of intracellular hIAPP levels by LC (Fig. 3F). In contrast to hIAPP, neither transient (1 h) nor prolonged (12 h) inhibition of proteasome activity by LC significantly changed the expression levels of insulin in pancreatic β-cells (Figs. 2, A and D, and 3, A and D). In addition, LC did not interrupt insulin trafficking, as numerous insulin-positive secretory granules were observed in the cytosol and near the plasma membrane of control and LC-treated human islet β-cells (Fig. 3F). These results indicate a rather selective regulation of hIAPP turnover in β-cells by active proteasome complex.
Figure 2.

Effect of proteasome inhibition on hIAPP and insulin expression in human islets. Human islet cells were cultured in 5 or 20 mm glucose containing media for 6 days, in the absence or presence of LC, following which hIAPP and insulin levels were determined by Western blotting or ELISA. Samples of 5 and 20 mm glucose (Glc)-containing media are separated by an empty lane in the Western blotting. A, Western blot analysis of hIAPP and insulin protein levels following temporal (0–6 h, 10 μm LC) inhibition of proteasome activity with LC (10 μm). The same actin blot was used in A and Fig. 4B. B, Western blotting (left panel) and ELISA (right panel) analysis of the hIAPP intra-islet protein levels cultured short term (60 min) in 5 mm glucose (Glc)-containing media in the absence or presence of LC (10 μm). C and D, quantifications of band intensities reflecting relative protein expression in the Western blottings are depicted by histograms. Significance was established at *, p < 0.05 (control versus LC + 5 mm Glc) and #, p < 0.05; ##, p < 0.01; ###, p < 0.001 (control versus LC + 20 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test. E, quantification of 20S proteasome activity in human islet cells following temporal (0–6 h) inhibition by LC (10 μm). Significance was established at **, p < 0.01; ****, p < 0.0001 (control versus LC + 5 mm Glc); ####, p < 0.0001 (control versus LC + 20 mm Glc); @@, p < 0.01 (LC + 5 mm Glc versus LC + 20 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test.
Figure 3.

Proteasome activity is required for hIAPP but not insulin turnover in human islet β-cells. Human islet cells were cultured in 5 or 20 mm glucose-containing media for 6 days, in the absence or presence of increasing concentrations of LC, following which hIAPP and insulin levels were determined by Western blotting method. A and B, Western blot analysis of hIAPP and insulin protein levels following prolonged (12 h) and dose-dependent (0–10 μm LC) inhibition of proteasome activity with LC. By extending blot exposure time, the changes in IAPP signal in euglycemic samples (left panel of Western blotting) are more apparent due to improved S/N (B). C and D, quantifications of band intensities reflecting relative protein expression in the Western blottings are depicted by histograms. Significance was established at *, p < 0.05; **, p < 0.01 (control versus LC + 5 mm Glc) and ###, p < 0.001 (control versus LC + 20 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test. E, 20S proteasome activity in human islet cells following dose-dependent (0–10 μm LC, 12 h) inhibition of proteasome activity with LC. Significance was established at *, p < 0.05; **, p < 0.01 (control versus LC + 5 mm Glc); ##, p < 0.01 (control versus LC + 20 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test. F, human islet cells were cultured in 20 mm glucose containing media for 6 days in the absence or presence of LC. Immunoconfocal microscopy analysis of hIAPP and insulin levels and trafficking following inhibition of proteasome activity with LC (12 h) was then performed. Bars, 2 μm.
We also investigated the effect of LC on levels of the proteins known to be affected by stress such as heat-shock proteins. HSP70 and HSP90 are chaperone proteins that play central roles in protein refolding and are up-regulated during cellular stress (30). In agreement with their well-known chaperone functions, a dose-dependent inhibition of proteasome activity by LC resulted in a stepwise increase in the intracellular levels of HSP70 and HSP90 (Fig. 4A). However, inhibition of proteasome activity by LC did not significantly change the expression levels of proteasome catalytic subunits (β1 and α4) in human islet cells (Fig. 4A). Additionally, inhibition of proteasome activity by LC promptly raised the levels of polyubiquitinated proteins, indicating protein stress in these cells (Fig. 4B). In contrast, levels of cleaved caspase-3, a cell apoptotic marker protein, were similar in proteasome-impaired and control islets (Fig. 4B).
Figure 4.

Effect of proteasome inhibition on expression levels of heat-shock proteins, proteasome complex subunits, and the viability of human islet cells. A, Western blot analysis of whole-cell extracts of human islet cells cultured in 20 mm glucose (Glc)-containing media for 6 days followed by dose-dependent inhibition of proteasome activity (0–10 μm) with LC for 12 h. Blots were probed with anti-HSP90, anti-HSP-70, 20 α1, 20S β1, and anti-actin antibodies. B, Western blot analysis of whole-cell extracts of human islet cells following temporal (0–6 h) inhibition of proteasome activity with LC (10 μm). Blots were probed with anti-ubiquitin (protein stress marker) and anti-caspase 3 (apoptotic marker) and anti-actin (loading control) antibodies. The actin blot was reused in both this panel and Fig. 2A because proteins analyzed in these Western blottings were resolved on the same gel.
We used two other proteasome-selective pharmacological inhibitors, MG132 and epoxomicin, to confirm the involvement of proteasome in hIAPP turnover in β-cells. Consistent with the LC study, we observed a marked (∼80%) decrease in intracellular hIAPP levels (Fig. 5A) following equipotent inhibition of proteasome activity with MG132 and epoxomicin (5 μm, 6 h) (Fig. 5A). Similar to LC, the decline in the proteasome activity/function by MG132 and epoxomicin induced protein stress, which was evident by the accumulation of polyubiquitinated proteins (Fig. 5A). However, levels of the apoptotic marker, cleaved caspase-3, again remained low in control and proteasome-impaired islets, indicating viable cells (Fig. 5A). Because some of these proteasome inhibitors can also inhibit other proteases, particularly at higher concentrations (>10 μm) (31), we treated the cells with lower (nanomolar) concentrations of MG132 and epoxomicin to reduce their possible off-target effects. We observed a marked decrease of intracellular hIAPP levels in human islets following treatment with sub-micromolar concentrations of MG132 and epoxomicin (0.25–1 μm) (Fig. 5, B and C), indicating a link between hIAPP turnover and proteasome activity/function in islet β-cells.
Figure 5.

MG132 and epoxomicin modulate hIAPP expression and viability in pancreatic islet cells. Human islet cells were cultured in 20 mm glucose-containing media for 6 days in the absence or presence of MG132 and epoxomicin, following which intracellular hormone content was determined. A, left and top right panel, quantitative Western blot analysis of hIAPP, protein stress, and apoptotic marker expression levels following inhibition of proteasome activity with MG132 (5 μm) and epoxomicin (5 μm) for 8 h. Bottom right panel, effect of MG132 and epoxomicin on 20S proteasome activity in human islet cells. B and C, top panel, Western blot analysis reveals changes in hIAPP protein levels following inhibition of proteasome activity with sub-micromolar concentrations of MG132 (B) and epoxomicin (C) (0–1 μm) for 8 h. Bottom panel, effects of sub-micromolar concentrations (0–1 μm) of MG132 and epoxomicin on the 20S proteasome activity in human islet cells. Significance was established at **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 (control versus treatments), n = 3, ANOVA followed by Tukey's post hoc comparison test.
These results suggest that altered proteasome activity/function and associated protein stress, but not compromised cellular viability, account for the regulatory effects of LC and other proteasome inhibitors on hIAPP turnover in β-cells (Figs. 2–5). This notion is further supported by confocal microscopy, which revealed single and clustered islet cells with well-defined nuclei and no visible signs of cell shrinkage, nuclear fragmentation, or nuclear condensation in control and LC-treated cells (Fig. 3F), indicating viable islet cells.
Proteasomes modulate hIAPP but not insulin secretion in human pancreatic β-cells
Protein homeostasis in cells is maintained by a fine balance between biosynthesis, degradation, and secretion of proteins. To this end, we tested whether the increased secretion of hIAPP in proteasome-impaired cells may account for the inhibitory effect of LC on the intracellular accumulation of hIAPP (Fig. 6A). This detoxification mechanism is possible, especially in cells with impaired proteolytic functions and harboring aggregation-prone amyloid proteins (32–34). Contrary to this idea, Western blot analysis of the conditioned media from human islet cells cultured in 5 or 20 mm glucose for 6 days actually showed a decrease in extracellular levels of secreted hIAPP following short-term (8 h) treatment with LC as compared with control cells (Fig. 6, A and B). In contrast to hIAPP, LC did not significantly alter the secretion of insulin (Fig. 6, A and B) suggesting that β-cells' secretory defects, if any, did not cause a decrease in hIAPP extracellular levels in proteasome-impaired cells. Importantly, human islets remained secretory (glucose)-responsive even after 6 days in culture, as evident from the release of these two pancreatic hormones following glucose stimulation (Fig. 6C). These results suggest that secretory mechanisms did not significantly contribute to the hIAPP decrease in cells with compromised proteasome function.
Figure 6.

Proteasome activity and hormone secretion in human islet cells. Human islets were cultured for 6 days in normal (5 mm Glc) or high-glucose (20 mm Glc) media. Old culturing media were replaced with the fresh media containing an increasing concentration of LC (0–10 μm) during the final 8 h of the experiment. A, Western blot analysis of extracellular hIAPP and insulin accumulation following dose-dependent proteasome inhibition with LC (0–10 μm, 12 h). A re-imaged region (5 mm Glc) with improved signal is presented below the original blot. Samples collected from 5 and 20 mm glucose-containing media are separated by two empty lanes in the Western blotting. B, histograms depict fold change in secreted hIAPP and insulin levels relative to control. Significance was established at *, p < 0.05, **, p < 0.01, n = 3, ANOVA followed by Tukey's post hoc comparison test. C, Western blot analysis of extracellular hIAPP and insulin levels in human islet cells cultured in 5 and 20 mm glucose-containing media up to 6 days. Blots showed a significant release in IAPP and insulin release after 48 and 96 h incubation of islets in 20 mm glucose media, respectively.
Proteasomes and lysosomes regulate hIAPP levels in β-cells in a mutually independent manner
To relieve cellular stress inflicted by the accumulation of misfolded and potentially damaging proteins (35), cells with compromised proteasome function may up-regulate the alternative routes of cellular degradation such as the autophagy–lysosome system. Previous studies demonstrated that the autophagy–lysosome system plays an important role in the clearance of toxic hIAPP species in pancreatic β-cells, dysregulation of which leads to significant β-cell stress and glucose intolerance. In view of this, we tested the idea that enhanced degradation of hIAPP by lysosomes may account for the decrease in intra- and extracellular hIAPP content in proteasome-impaired β-cells (Figs. 2–6). To address this possibility, we inhibited the lysosomal lytic function using the proteolytic inhibitor pepstatin (27). In agreement with previous studies (36–38), Western blot analysis showed a significant increase in the hIAPP intracellular levels in pepstatin-treated human islet cells as compared with controls (Fig. 7), demonstrating an important role of lysosome in hIAPP degradation. By contrast, inhibition of proteasome activity by LC depleted the hIAPP intracellular levels (Fig. 7). Interestingly, pepstatin did not reverse (rescue) the inhibitory effect of LC on hIAPP expression in the human islet cells (Fig. 7). This could be expected if the LC-induced hIAPP loss occurred through increased lysosomal degradation of this hormone (Fig. 7). Taken together, these results suggest that proteasomes and lysosomes independently regulate hIAPP turnover in β-cells.
Figure 7.
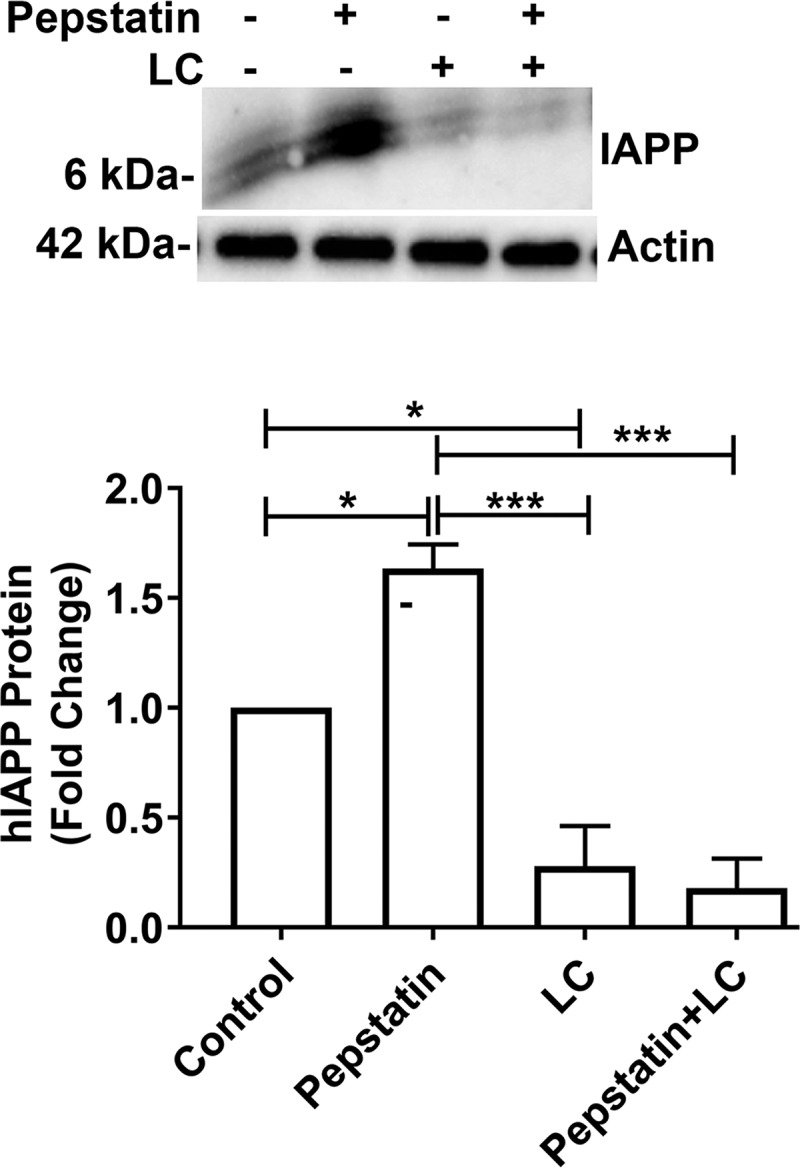
Proteasome activity regulates hIAPP turnover in a lysosome-independent manner. Western blot analysis of intracellular hIAPP turnover in proteasome and lysosome impaired cells was performed. Human islet cells were cultured in 20 mm glucose containing media for 5 days, followed by additions of LC (5 μm), pepstatin (5 μm), or both for additional 12 h. Histogram depicts mean ± S.E. changes in hIAPP protein levels for three independent experiments. Significance was established at *, p < 0.05; ***, p < 0.001, n = 3, ANOVA followed by Tukey's post hoc comparison test.
Proteasomes regulate the turnover of human and rodent IAPP in a transcription-dependent manner
Several reports have linked proteasome activity with the regulation of gene expression, including initiation of transcription, elongation, maturation, export of mRNA from the nucleus, as well as mRNA stability (39–41). These important biosynthetic steps were related to its proteolytic and nonproteolytic activities (39–41). In view of this, we tested if and the extent to which changes in proteasome's proteolytic activity affects the hIAPP gene transcription by analyzing the mRNA levels of hIAPP and its transcription factors FOXA2 and PDX1 in human islet cells (Fig. 8). qPCR analysis showed a robust (4-fold) and prompt (∼2 h) decrease in hIAPP transcript levels following proteasome inhibition, under both physiological (5 mm) and elevated (20 mm) glucose concentrations (Fig. 8, A and B). In the absence of LC, high glucose conditions induced a significant (>4-fold) increase in hIAPP transcript levels (Fig. 8, A and B), in accordance with previous studies (29, 42, 43). Also, in agreement with our protein studies (Figs. 2 and 3), the steady-state insulin mRNA levels did not significantly change following extended (overnight) proteasome inhibition by LC (Fig. 8A).
Figure 8.

Proteasomes regulate hIAPP promoter activity and transcription in human islets. A, histogram depicts changes in hIAPP and insulin gene expression (mRNA) levels following proteasome inhibition by LC. Human islet cells were cultured in 5 and 20 mm glucose (Glc) containing media for 5 days, followed by proteasome inhibition with LC for additional 12 h. Significance was established at *, p < 0.05; ***, p < 0.001; ****, p < 0.0001, n = 3, Student's t test and ANOVA followed by Tukey's post hoc comparison test. B, dynamic analysis of hIAPP gene expression in normal and proteasome-impaired cells. Significance was established at **, p < 0.01 (control versus LC + 5 mm Glc); #, p < 0.05; ##, p < 0.01; ####, p < 0.0001, n = 3, ANOVA followed by Tukey's post hoc comparison test. C, hIAPP promoter activity in normal and proteasome impaired islets. Normalized hIAPP promoter activities are presented as fold changes from control (empty vector). Significance was established at *, p < 0.05; **, p < 0.01; ***, p < 0.001, n = 3, ANOVA followed by Tukey's post hoc comparison test. D–F, time-lapse qPCR analyses of FOXA2, PDX1, and MAFA mRNA following temporal proteasome inhibition. Significance was established at *, p < 0.05; **, p < 0.01 (control versus LC + 5 mm Glc).
To further elucidate the transcriptional mechanism of proteasome-mediated hIAPP biosynthesis, we generated a construct where the expression of firefly luciferase gene is driven by the full-length hIAPP promoter. Luciferase expression (luminescence assay) revealed a comparable hIAPP promoter activity in control and short-term (0–6 h) LC-treated islets cultured under normal or high glucose conditions (Fig. 8C). However, a significant decline of ∼60% hIAPP promoter activity was observed during an extended (12 h) period of proteasome inhibition with LC respective to their controls under both normal (5 mm) and elevated (20 mm) glucose conditions (Fig. 8C). In accordance with hIAPP gene expression data, time-lapse qPCR analysis revealed a rapid (within the first 3 h) down-regulation of mRNA levels of hIAPP's crucial transcription factor, FOXA2, following LC treatment (Fig. 8D). This is within the time frame of LC-mediated IAPP decline in the same cells (Figs. 2, A and C, and 8B). Gene expression levels of the other important hIAPP transcription factor, PDX1, was also significantly down-regulated in LC-treated islets only under normal glucose conditions (Fig. 8E). Interestingly, our studies revealed that the insulin-specific transcription factor, MAFA, was also significantly down-regulated following LC treatment (Fig. 8F). These results suggest that the availability of FOXA2 and/or its promoter occupancy could be major limiting factors for hIAPP transcription in proteasome activity–impaired human islets. After an initial delay (∼6 h), FOXA2 protein levels also start to decline in LC-treated but not in control islets cultured under normal or elevated glucose levels (Fig. 9). By contrast, LC induced a decline in hIAPP protein levels in human islets at least 6 h before the disappearance of FOXA2 protein (Fig. 9).
Figure 9.

Effect of prolonged proteasome inhibition on FOXA2 protein expression in cultured human islet cells. Western blot analysis of FOXA2 and hIAPP protein levels in the whole-cell extracts of human islet cells following progressive (0–12 h) inhibition of proteasome activity by LC was performed. Human islet cells were cultured in 5 and 20 mm glucose (Glc)-containing media for 5 days, followed by proteasome inhibition with LC for additional 0–12 h. A separately imaged region with enhanced signal for hIAPP under the normal (5 mm) Glc levels is presented below the original Western blotting. Samples of 5 or 20 mm glucose-containing media are separated by one empty lane in the Western blotting. Significance was established at *, p < 0.05; **, p < 0.01 (control versus LC + 5 mm Glc); and ##, p < 0.01 (control versus LC + 20 mm Glc); @@, p < 0.01 (1 h LC + 20 mm Glc versus LC (6 and 12 h) + 20 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test.
We also investigated the possible role of the proteasome in the regulation of nonamyloidogenic rat IAPP (rIAPP) expression in rat pancreatic β-cells. To this end, we used the glucose-responsive rat insulinoma cell line (INS 832/13), which shows comparable biosynthetic and secretory characteristics to the primary β-cell (44). We performed inhibitory studies in INS 832/13 cells cultured under basal (11 mm) or high glucose (25 mm) conditions, either in the absence (control) or presence of LC. LC-induced a dose-dependent decrease in proteasome activity (IC50 ∼400 nm) in these cells cultured under both basal and high-glucose conditions (Fig. 10A). qPCR analysis showed a significant decline in rIAPP transcript levels only after pronounced (≥30%) inhibition of proteasome activity was reached (Fig. 10, A and B). Analogous to rIAPP, rat FoxA2 protein levels significantly declined following a marked (≥40%) impairment of proteasome activity (Fig. 10, A and C).
Figure 10.

Proteasomes regulate the expression and interactions of rIAPP and FoxA2 in insulinoma β-cells. INS 832/13 cells were cultured in 11 or 25 mm glucose-containing media for 48 h, followed by proteasome inhibition with various concentrations of LC for 12 h. A, 20S proteasome activity assay in INS 832/13 cells following dose-dependent (0–1 μm) inhibition of proteasome activity by LC. Significance was established at **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 (control versus 11 mm Glc) and ##, p < 0.01; ####, p < 0.0001 (control versus 25 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test. B, qPCR analysis of rIAPP gene expression in INS 832/13 cells in the absence and presence of proteasome inhibitor LC (0–1 μm) for 12 h. Significance was established at **, p < 0.01; ****, p < 0.0001 (control versus 11 mm Glc) and ##, p < 0.01; ####, p < 0.0001 (control versus 25 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test. C, Western blot analysis of FoxA2 protein expression in control and stressed INS 832/13 cells evoked by increasing concentrations of LC (0–1 μm) for 12 h. Significance was established at *, p < 0.05; **, p < 0.01; ***, p < 0.001 (control versus 11 mm Glc), and #, p < 0.05; ###, p < 0.001 (control versus 25 mm Glc), n = 3, ANOVA followed by Tukey's post hoc comparison test. D, ChIP analysis of FoxA2 and histone H3 protein interactions with the rIAPP promoter in control and proteasome-impaired INS 832/13 cells. The amount (enrichment) of FoxA2 and histones (H3 isoform) proteins at the proximal site of the rIAPP promoter were normalized and expressed relative to input. Significance was established at **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, n = 3, ANOVA followed by Tukey's post hoc comparison test.
Previous studies demonstrated that FoxA2 binds to the conserved proximal FoxA2 (606 bp)-binding site of the IAPP promoter (15). Here, we used the ChIP approach and FoxA2-validated antibody to test the possible regulatory role of proteasomes in FoxA2 binding to the rIAPP promoter. Specific detection and quantification of FoxA2 binding to IAPP's promoter were achieved using qPCR and primers flanking this proximal (606 bp) FoxA2-binding region in the IAPP promoter. ChIP analysis revealed a significant and substantial (>90%) decrease in relative FoxA2 protein binding to the proximal region of rIAPP promoter following LC treatment. This inhibitory effect of LC was observed in basal (11 mm Glc) and elevated glucose (25 mm Glc) conditions (Fig. 10D). ChIP analysis revealed negligible levels of the rIAPP promoter DNA in immunoprecipitated samples in which irrelevant IgG antibody was used as a bait (Fig. 10D). We also found significant levels of histones (H3 isoform) bound to the rIAPP promoter in H3Ab immunoisolates demonstrating preservation and isolation of an intact rIAPP promoter region in nucleosomal/immunoprecipitation fractions following enzymatic digestion of chromatin (Fig. 10D). Collectively, the current data suggest the evolutionarily conserved role of proteasomes in FOXA2-mediated IAPP transcription in β-cells.
Discussion
Previous studies by our group revealed a pivotal role of the 20S proteasome complex in the degradation of internalized hIAPP, which prevented its intracellular accumulation and toxicity in pancreatic β-cells (27). However, the precise role of the proteasome in turnover of endogenous hIAPP in normal and stressed (high glucose) conditions is not known and hence was investigated in this study. In human T2DM islets, we found a marked decrease in proteasome activity and the consequent increase of polyubiquitinated proteins indicating a functionally impaired proteasome complex. Moreover, our protein and gene expression studies independently revealed a stark decrease in the hIAPP protein and mRNA levels only in the islets from T2DM donors, indicating a possible link between proteasome malfunction and IAPP imbalance. In support of these findings, the gene expression (mRNA) levels of IAPP's main transcription factor FOXA2 were also down-regulated in the islets procured from T2DM donors. Along with this idea and published studies (27), moderate inhibition (up to 35%) of proteasome activity increased steady-state hIAPP levels in human islets indicating its degradation by the proteasome complex. By contrast, a more severe inhibition of proteasome activity (>50%) was required to deplete intracellular hIAPP protein and mRNA levels. Our studies further revealed that hyperglycemia-mimicking conditions sensitized β-cells to proteasome dysfunction and hIAPP loss. In contrast to hIAPP, total insulin intracellular content or its release was not significantly affected by LC treatment. Despite this, LC reduced the expression of insulin-specific transcription factor MAFA. This finding is in agreement with the study by Kitiphongspattana et al. (45), who showed that proteasome dysfunction affects the production of only newly synthesized (pro)insulin, whereas total intracellular (pro)insulin content remains unaltered (45). It is worth mentioning here that our experimental approach only detects steady-state changes in hIAPP and insulin content. Hence, an increase or decrease in insulin synthesis may not be immediately apparent particularly in the case of long-lived and abundant molecules such as insulin (46).
It is not uncommon, particularly in amyloid-laden diseases such as Alzheimer's and Parkinson's, that cells secrete misfolded proteins to alleviate the burden of toxic intracellular protein aggregates (32–34). Hence, we explored whether hIAPP follows a similar detoxification mechanism in pancreatic β-cells. However, this study revealed that proteasome inhibitors reduced both the basal and glucose-stimulated hIAPP release, thus eliminating hIAPP secretion as a cause for the LC-induced decline in hIAPP intracellular levels. We also investigated hIAPP degradation by autophagy as a possible cause for hIAPP disappearance following proteasome dysfunction. UPS and the autophagy–lysosome systems are the two primary players in cellular catabolism and are critical in maintaining protein homeostasis. Several recent studies indicated a tight cross-talk between these two otherwise independent cellular systems (35). Notably, studies showed that impairment of UPS results in up-regulation of autophagy–lysosome-mediated proteolysis as a compensatory mechanism to reduce the load of accumulated UPS clients (36–38). Nevertheless, inhibition of proteasome activity and function still reduced hIAPP levels in lysosome-impaired cells, thus making a cross-talk between UPS and autophagy–lysosome systems in hIAPP turnover in β-cells unlikely.
In addition to proteolytic degradation and secretion, inhibition of transcription and/or translation may reduce protein levels in cells. Relevant to this study, it was reported that the 26S proteasome complex plays an important role in the regulation of transcription in eukaryotes (24). A study by Bhat et al. (39) demonstrated that transcription of the key adaptive immune response gene, MHC class II, requires the proteolytically active 26S proteasome complex. Our time-lapse gene expression analysis revealed a prompt and robust decline in hIAPP transcript levels following blockage of proteasome function, indicating the crucial regulatory role of proteasomes in hIAPP gene transcription in human pancreatic β-cells. We also observed a significant decrease in the hIAPP promoter activity following extended (≥12 h) proteasome inhibition in human islets. Transcription of the IAPP gene is known to be regulated by a compound promoter region (spanning from −2798 to +450 region with respect to the transcription start site) (47) along with several transcription factors. Although the homeodomain factor PDX1 is primarily responsible for glucose-responsive promoter activity of IAPP (47), binding sites for other transcription factors such as FOXA2 and ISL1 are present in the IAPP promoter region. FOXA2 belongs to the hepatocyte nuclear factor 3 or FoxA family of proteins and binds to DNA using its centrally located DNA-binding forkhead box domain (48). The IAPP promoter includes three FOXA2-binding sites, and FOXA2 has a regulatory role in the glucose-induced expression of IAPP (15). In line with its inhibitory effect on IAPP biosynthesis, LC also down-regulated transcript levels of IAPP key transcription factors, FOXA2 and PDX1, which are required for IAPP biosynthesis. However, despite a marked and prompt (∼2 h) decrease in hIAPP mRNA and protein levels, FOXA2 protein levels start to decline with a delay of ∼6 h. Consistent with this finding, proteasome inhibitor LC was ineffective in inhibiting amylin promoter activity during the early phase (within the first 6 h) of proteasome inhibition, whereas LC effectively blocked amylin promoter activity at later time points. These results suggest the FOXA2- and promoter-independent mechanisms of IAPP turnover in this initial phase of proteasome dysfunction. They also suggest that some post-promoter regulatory event, such as mRNA elongation, processing, or degradation, likely contributed to the onset of LC-induced decline in IAPP mRNA levels in human islets. Eventually, FOXA2 mRNA and protein levels progressively decreased following an extended period of proteasome dysfunction leading to a long-term inhibition of hIAPP promoter activity and expression. Thus, our results suggest that FOXA2-dependent and -independent transcriptional mechanisms operate in parallel to regulate IAPP synthesis in proteasome-dysfunctional islet β-cells. The expression studies involving nonamyloidogenic rIAPP and FoxA2 in rat insulinoma β-cells also implicated FoxA2-dependent and -independent mechanisms in amylin transcription in the proteasome-impaired β-cells. The importance of FoxA2 is further suggested in our ChIP studies that showed reduced binding of FoxA2 protein to the rIAPP's promoter region following inhibition of proteasome activity.
Interestingly, proteasome inhibition down-regulated expression of MAFA (49), a transcription factor required for insulin gene synthesis. These findings imply the possible negative impact of LC treatment on insulin biosynthesis. However, this study revealed no change in steady-state insulin gene and protein levels following LC treatment. It is likely that high stability of insulin transcripts (half-life ∼29 to 77 h depending on the glucose availability), as well as an abundance of insulin protein (>30% of total β-cell protein pool), mitigated any negative effects of proteasome inhibition on insulin synthesis (46). In contrast, inducible and sparse proteins such as IAPP may be quickly and efficiently up-regulated (by a rise in extracellular Glc levels) or down-regulated (by proteasome impairment) as suggested by our study.
In summary, this study revealed a novel, regulatory role of proteasomes in IAPP turnover in pancreatic β-cells. Functional proteasome complex was required for the bulk of produced and secreted IAPP but not for insulin turnover. Proteasomes regulated IAPP synthesis by modulating IAPP promoter activity as well as the availability and promoter occupancy of IAPP's key transcription factor, FOXA2. At least 50% of regular proteasome activity was required for maintaining the expression and secretion of hIAPP. Because hIAPP is a risk factor for T2DM (2, 5), regulation of proteasome activity/function could be beneficial against hyper- and/or hypo-amylinemia in failing β-cells.
Experimental procedures
Cell culture and treatments
Human islets from diabetic and nondiabetic cadavers with >80% purity and viability were obtained through the integrated islet distribution program for human islet research. Upon arrival, islets were handpicked under a dissecting microscope to yield a purity of ≥95% (based on dithizone staining). Intact islets were suspended in CMRL media (catalog no. 11530-037, Gibco) containing 2 mm glutamine (catalog no. 25030-081, Gibco), 1% (v/v) fetal BSA (catalog no. 10437-028, Gibco), and 1% penicillin/streptomycin (catalog no. 30-2300, ATCC) and were plated on 48-well nonadherent cell culture plates (catalog no. 10861-560, VWR) at 50–80 islets per well. Islets were cultured at 37 °C in a humidified incubator with 5% CO2 for a designated period of time. For confocal studies, 20–30 islets per well were gently dissociated in TrypLe cell dissociation medium (catalog no. A12177-01, catalog no. 10861-560), plated on poly-d-lysine–coated 96-well glass bottom black microplates (catalog no. 655892, VWR), and allowed to adhere for 24 h prior to treatments. Human islet cells were treated with the designated concentration of pharmacological inhibitors: lactacystin (catalog no. 426100), MG132 (catalog no. 474790), epoxomicin (catalog no. 324800), and pepstatin (catalog no. 516481) (all from EMD Millipore) for designated periods. For glucose-responsive studies, human islets were cultured in 5 (normal glucose) or 20 mm glucose (high glucose) containing CMRL media for 6 days. Rat insulinoma INS-832/13 cells (a generous gift from Christopher Newgard, Duke University), were cultured in RPMI 1640 medium (catalog no. 11875-093, Gibco) supplemented with 10% (v/v) fetal bovine serum (catalog no. 10437-028, Gibco), 1% penicillin/streptomycin, 10 mm HEPES (catalog no. 15630-080, Gibco), 2 mm l-glutamine (catalog no. 25030-081, Gibco), 1 mm sodium pyruvate (catalog no. 11360-07, Gibco), and 0.05 mm 2-mercaptoethanol. This cell line was cultured at 37 °C in a humidified incubator with 5% CO2 and passaged bi-weekly. Cells of passages 10–20 were used for all of the experiments. For glucose-responsive studies, INS 832/13 cells were first equilibrated with 5 mm glucose containing media for 8–12 h, followed by culturing in high-glucose (25 mm Glc) media for an additional 48 h.
20S proteasome assay
Proteasome activity was determined using a 20S proteasome activity assay kit (catalog no. 10008041, Cayman Chemicals), following the manufacturer's protocol. Briefly, the detection is based on a specific 20S proteasome substrate, N-succinyl-LLVY-7-amino-4-methylcoumarin, which, upon cleavage by the active enzyme, generates a highly fluorescent product. The increase in fluorescence intensity, reflecting proteasome activity in the sample, was measured using M5 Spectromax fluorometer (Molecular Devices) with excitation/emission set at 360/480 nm. Data (relative fluorescence unit) were collected and plotted using GraphPad Prism 7 graphic program.
Insulin and hIAPP release assay
The secretory potential of cultured β-cells was assessed by measuring the release or accumulation of insulin and hIAPP in the conditioned media following treatments. In these secretory studies, human islets were cultured in normal (5 mm) and high (20 mm) glucose containing CMRL medium for 6 days. Old culturing media were then replaced with the fresh media containing a designated concentration of LC during the final 8 h of the experiment. Conditioned media (400 μl) were collected from each experimental group (wells), and residual cells were removed by centrifugation and used for downstream Western blot analysis.
Western blotting
The regulatory roles of proteasome activity on the levels of hIAPP, insulin, and other proteins were analyzed by Western blotting using our published procedure (27). Briefly, whole-cell extracts of human islet cells were collected in RIPA buffer (catalog no. 20-188 EMD Millipore) (supplemented with protease (catalog no. 1862209) and phosphatase inhibitor (catalog no. 1862495) cocktails, Life Technologies, Inc.). An equal amount (4 μg of the cell extracts) or volumes (15 μl of the conditioned media) were collected from each sample/treatment, and proteins were separated using 4–15% Tris-glycine SDS-PAGE (catalog no. 4561086, Bio-Rad). Resolved proteins were electrophoretically transferred to nitrocellulose membranes (catalog no. 1704270, Bio-Rad), and nonspecific IgG-binding sites were blocked by incubation with 5% nonfat dry milk (VWR). Membranes were probed with primary antibodies at 4 °C overnight, followed by addition of the corresponding horseradish peroxidase-conjugated secondary antibodies (1:2000) (Santa Cruz Biotechnology) for 1 h at room temperature. The following primary antibodies were used: mouse monoclonal anti-amylin (1:200, catalog no. sc-377530, Santa Cruz Biotechnology); rabbit polyclonal anti-insulin (1:1000, catalog no. ab181547, Abcam); mouse monoclonal anti-ubiquitin (1:200, catalog no. sc-8017, Santa Cruz Biotechnology); goat polyclonal anti-HSP70 (1:200, catalog no. sc-1060, Santa Cruz Biotechnology); mouse monoclonal anti-HSP90 (1:200, catalog no. sc-13119, Santa Cruz Biotechnology); rabbit polyclonal anti-caspase-3 (1:1000, catalog no. ab184787, Abcam); rabbit polyclonal 20S proteasome β1 (1:200, catalog no. sc-67345, Santa Cruz Biotechnology); mouse monoclonal 20S proteasome α4 (1:200, catalog no. sc-271297, Santa Cruz Biotechnology); rabbit monoclonal FOXA2 (1:1000, catalog no. 108422, Abcam); and goat polyclonal anti-actin (1:200, catalog no. sc-1615, Santa Cruz Biotechnology). Blots were developed using pico- (catalog no. 34580) or femto (catalog no. 34095)-ECL substrate kit (ThermoFisher Scientific) and documented using the Chemi DOC touch gel imaging system (Bio-Rad). To improve the S/N of some weakly expressed proteins such as hIAPP, in some experiments the acquisition time was extended for portions of Western blots containing such samples. Additionally, Bio-Rad Image transform software was used to detect oversaturated pixels in our samples and to maintain the luminescence signal within the linear range during image acquisition. Band intensities were quantified using Bio-Rad Image-Lab software using actin as a loading control.
ELISA
The regulatory roles of proteasome activity on the intracellular levels of hIAPP under normal (5 mm glucose) conditions were quantified by an IAPP ELISA kit (catalog number EZHA-52K, EMD Millipore) according to manufacturer's protocol. Following proteasome inhibition, samples were collected and diluted 100× to maintain the linear range. hIAPP concentrations in the samples were determined by running the standards provided in the kit. The concentrations obtained were normalized to the protein content measured in each sample.
Immunocytochemistry
Relative expression and localization of hIAPP and insulin in human islets were determined using our published immunocytochemistry approach (50). Briefly, fresh and partially dissociated human islets from nondiabetic individuals were incubated with proteasome inhibitors, in normal (5 mm glucose) and high (20 mm glucose) media for an indicated time. Cells were washed in PBS (three times) and fixed with 4% paraformaldehyde for 20 min. hIAPP and insulin levels and their localization were determined by incubating cells with mouse monoclonal anti-amylin (1:200, catalog no. sc-377530, Santa Cruz Biotechnology) and rabbit polyclonal anti-insulin (1:500, catalog no. ab181547, Abcam) antibodies at 4 °C overnight, followed by goat anti-mouse Alexa 647-conjugated (1:500, catalog no. A21236) and anti-rabbit Alexa 488-conjugated (1:1000, catalog no. A21441) secondary antibodies (Invitrogen) for 60 min at room temperature. Three random fields in each well were imaged by confocal microscopy, and single optical sections (1 μm z axis) through the middle of the cells were acquired for each field using a Zeiss LSCM-800 confocal microscope (Carl Zeiss, Thornwood, NY) and ×63 (1.3 N.A.) oil objective. The pinhole was adjusted to keep the same size of z-optical sections for all channels and objectives used. Multitrack imaging was performed to ensure that there was no cross-talk between the channels. Intensity thresholds of the channels were set using the Zeiss cross-hair function to avoid an arbitrary background threshold being set. Images were assembled using Adobe Photoshop software (Adobe Systems Co.).
Quantitative real-time PCR
The regulatory roles of proteasome activity on the gene expression of hIAPP, insulin, and their transcription factors were investigated using quantitative real-time PCR (qPCR) analysis. Total RNA was extracted from INS 832/13 and human pancreatic islets cells using TRIzol reagent (catalog no. 15596026, Life Technologies, Inc.) and the RNeasy mini kit (catalog no. 74134, Qiagen) according to the manufacturers' instructions. Total RNA was further purified from contaminating DNA using in-column DNA digestion with RNase-free DNase, following the manufacturer's instructions (catalog no. 79254, Qiagen). cDNA was synthesized from 500 ng of RNA using multiScribeTM reverse transcriptase and random primers (High Capacity cDNA Reverse Transcription kit, catalog no. 4368814, Applied Biosystem) according to the manufacturers' protocol. qPCR was performed using Bio-Rad CFX 96 real-time system (C1000 Touch thermal cycler). Briefly, IAPP and insulin levels were detected in the reverse-transcribed cDNA using iTaqTM Universal SYBR Green supermix (catalog no. 1725122, Bio-Rad) using the following thermal cycling protocol: polymerase activation 95 °C, 2 min; DNA denaturation 95 °C, 5 s; annealing and extension 60 °C, 30 s; repeat 40 cycles. All qRT-PCR primer sets are presented in Table 1. Gene expression levels of IAPP and insulin were normalized to β-actin expression. Relative levels of IAPP and insulin mRNA expression were calculated using the 2−ΔCT method (51).
Table 1.
Gene-specific primers used for qPCR analysis and cloning
F is forward, and R is reverse.
| Name | Primer sequences |
|---|---|
| hIAPP | F: 5′-AGCTACACCCATTGAAAGTCATC-3′ |
| R: 5′-GATGAGAGAATGGCACCAAAGT-3′ | |
| rIAPP | F: 5′-AGCTGTTCTCCTCATCCTCT-3′ |
| R: 5′-CTGTGTTGCACTTCCGTTTG-3′ | |
| Insulin | F: 5′-TGTCCTTCTGCCATGGCCCT-3′ |
| R: 5′-TTCACAAAGGCTGCGGCTGG-3′ | |
| FoxA2 | F: 5′-TCTCCATCAACAACCTCATGTC-3′ |
| R: 5′-GTAGTGCATCACCTGTTCGTAG-3′ | |
| PDX1 | F: 5′-CCTTGTGCTCGGGTTATGTT-3′ |
| R: 5′-ATCATCCCACTGCCAGAAAG-3′ | |
| MAFA | F: 5′-GCGGAGAACGGTGATTTCTA-3′ |
| R: 5′-AGGAAAGGGAGGCTGAGAAG-3′ | |
| β-Actin (human) | F: 5′-AGGCACCAGGGCGTGAT-3′ |
| R: 5′-GCCCACATAGGAATCCTTCTGAC-3′ | |
| β-Actin (rat) | F: 5′-GCTACAGCTTCACCACCACA-3′ |
| R: 5′-ATCGTACTCCTGCTTGCTGA-3′ | |
| Human IAPP promoter-cloning primer | F: 5′-AAAAAGATCTACAGCTCTGGCATTTATAAC-3′ |
| R: 5′-AAAACCATGGCTTTTAATGTTTCAATGTCAGC-3′ | |
| FoxA2-binding region in rIAPP promoter-ChIP primer set | F: 5′-GCACAAAATGGAAACTCCAAA-3′ |
| R: 5′-CCCTCAACTTGCTTAGCTCTG-3′ |
Promoter activity assay
The total genomic DNA of HEK293T cell was purified using the Qiagen core kit (catalog no. 201223, Qiagen). The full hIAPP promoter region was amplified (from 2000 bp upstream of the transcription start site to the first exon) using hIAPP gene-specific primers (Table 1) and the KOD hot-start PCR kit, following the manufacturer's instruction (catalog no. 71842, EMD Millipore). The isolated promoter was purified using QIAquick gel extraction kit (catalog no. 28704, Qiagen) and ligated into the PCR-Blunt II-TOPO vector (catalog no. K280020, Life Technologies, Inc.). Isolated clones were confirmed by Sanger sequencing (GENEWIZ). Selected clones were then subcloned into a firefly luciferase encoding pGL3 basic vector (a kind gift from Richard Day, Indiana University) using BgIII and NcoI restriction sites, thus generating the hIAPP promoter-firefly luciferase construct. For transfection, dispersed human islet cells were plated on 96-well tissue culture plates using either 5 or 20 mm glucose-containing CMRL media (1% FBS, no antibiotic). At 24 h following plating, islet cells were co-transfected with thymidine kinase Renilla promoter vector (Richard Day, Indiana University) with PGL3 basic vector or hIAPP promoter vector using the FuGENE HD (catalog no. E2311, Promega) transfection reagent. Following transfection, cells were cultured for an additional 24 h. Transfected cells were then treated with LC (5 μm) for the final 8 h. Firefly and Renilla luciferase activities were detected using the Dual-Glo luciferase assay system (catalog no. E2920, Promega) following the manufacturer's instructions. Firefly luminescence was normalized with Renilla luminescence for each sample well to avoid differential transfection efficiency.
Chromatin immunoprecipitation
The regulatory role of proteasomes on FoxA2 binding at the rIAPP promoter site was determined using SimpleChip kit (catalog no. 9003, Cell Signaling Technology) following the manufacturer's protocol. Briefly, cells were fixed with formaldehyde to cross-linked DNA with its binding proteins, and chromatin was digested with micrococcal nuclease. DNA–protein complexes were immunoprecipitated with either 2 μg of the FoxA2 antibody (catalog no. 108422, Abcam), histone H3 (positive control, provided with the kit), or rabbit IgG (negative control, provided with the kit), and the complexes were captured by protein G magnetic beads. Following immunoprecipitation, the DNA–protein cross-links were reversed, and the purified DNA was analyzed by qPCR and the ChIP primers set listed in Table 1. The amount of immunoprecipitated and amplified rIAPP promoter DNA in each sample was normalized and expressed as percent (%) of input using Equation 1,
| (Eq. 1) |
where CT is the threshold cycle of PCR.
Statistical analysis
The GraphPad Prism 7 program was used for plotting and statistical analysis. The histograms depict mean ± S.E. The unpaired Student's t test or one-way ANOVA followed by the Tukey post hoc test were used for pairwise comparisons among groups when appropriate with significance established at p < 0.05.
Author contributions
D. C. B. and A. J. conceptualization; D. C. B. data curation; D. C. B. formal analysis; D. C. B. investigation; D. C. B. methodology; D. C. B. writing-original draft; A. J. supervision; A. J. funding acquisition; A. J. project administration; A. J. writing-review and editing.
Acknowledgments
We thank Christopher Day and Meghana Keswani for preparation of hIAPP promoter plasmid. We thank our colleagues Rob Donaldson and Anne Chiaramello for critical reading of the manuscript and stimulating discussions on the subject. We thank NIDDK-supported Integrated Islet Distribution Program at City of Hope for making human islets available for this study.
This work was supported by National Institute of Health Grant RO1 DK091845 (to A. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- T2DM
- type 2 diabetes mellitus
- IAPP
- islet amyloid polypeptide
- hIAPP
- human islet amyloid polypeptide
- rIAPP
- rat islet amyloid polypeptide
- ANOVA
- analysis of variance
- FoxA2
- forkhead box protein A2
- LC
- lactacystin
- Glc
- glucose
- qPCR
- quantitative PCR
- UPS
- ubiquitin–proteasome system
- S/N
- signal/noise.
References
- 1. Butler A. E., Janson J., Bonner-Weir S., Ritzel R., Rizza R. A., and Butler P. C. (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 10.2337/diabetes.52.1.102 [DOI] [PubMed] [Google Scholar]
- 2. Westermark P., Wernstedt C., Wilander E., Hayden D. W., O'Brien T. D., and Johnson K. H. (1987) Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc. Natl. Acad. Sci. U.S.A. 84, 3881–3885 10.1073/pnas.84.11.3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jaisson S., and Gillery P. (2014) Impaired proteostasis: role in the pathogenesis of diabetes mellitus. Diabetologia 57, 1517–1527 10.1007/s00125-014-3257-1 [DOI] [PubMed] [Google Scholar]
- 4. Mukherjee A., Morales-Scheihing D., Butler P. C., and Soto C. (2015) Type 2 diabetes as a protein misfolding disease. Trends Mol. Med. 21, 439–449 10.1016/j.molmed.2015.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Westermark P., Andersson A., and Westermark G. T. (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol. Rev. 91, 795–826 10.1152/physrev.00042.2009 [DOI] [PubMed] [Google Scholar]
- 6. Mosselman S., Höppener J. W., Zandberg J., van Mansfeld A. D., Geurts van Kessel A. H., Lips C. J., and Jansz H. S. (1988) Islet amyloid polypeptide: identification and chromosomal localization of the human gene. FEBS Lett. 239, 227–232 10.1016/0014-5793(88)80922-0 [DOI] [PubMed] [Google Scholar]
- 7. Hwang J. J., Chan J. L., Ntali G., Malkova D., and Mantzoros C. S. (2008) Leptin does not directly regulate the pancreatic hormones amylin and pancreatic polypeptide: interventional studies in humans. Diabetes Care 31, 945–951 10.2337/dc07-2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lutz T. A. (2006) Amylinergic control of food intake. Physiol. Behav. 89, 465–471 10.1016/j.physbeh.2006.04.001 [DOI] [PubMed] [Google Scholar]
- 9. Lutz T. A. (2010) The role of amylin in the control of energy homeostasis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 298, R1475–R1484 10.1152/ajpregu.00703.2009 [DOI] [PubMed] [Google Scholar]
- 10. Mather K. J., Paradisi G., Leaming R., Hook G., Steinberg H. O., Fineberg N., Hanley R., and Baron A. D. (2002) Role of amylin in insulin secretion and action in humans: antagonist studies across the spectrum of insulin sensitivity. Diabetes Metab. Res. Rev. 18, 118–126 10.1002/dmrr.263 [DOI] [PubMed] [Google Scholar]
- 11. Wagoner P. K., Chen C., Worley J. F., Dukes I. D., and Oxford G. S. (1993) Amylin modulates beta-cell glucose sensing via effects on stimulus-secretion coupling. Proc. Natl. Acad. Sci. U.S.A. 90, 9145–9149 10.1073/pnas.90.19.9145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Scherbaum W. A. (1998) The role of amylin in the physiology of glycemic control. Exp. Clin. Endocrinol. Diabetes 106, 97–102 [DOI] [PubMed] [Google Scholar]
- 13. German M. S., Moss L. G., Wang J., and Rutter W. J. (1992) The insulin and islet amyloid polypeptide genes contain similar cell-specific promoter elements that bind identical beta-cell nuclear complexes. Mol. Cell. Biol. 12, 1777–1788 10.1128/MCB.12.4.1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang M., and Drucker D. J. (1996) Activation of amylin gene transcription by LIM domain homeobox gene isl-1. Mol. Endocrinol. 10, 243–251 10.1210/mend.10.3.8833653 [DOI] [PubMed] [Google Scholar]
- 15. Jing G., Westwell-Roper C., Chen J., Xu G., Verchere C. B., and Shalev A. (2014) Thioredoxin-interacting protein promotes islet amyloid polypeptide expression through miR-124a and FoxA2. J. Biol. Chem. 289, 11807–11815 10.1074/jbc.M113.525022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakazato M., Asai J., Miyazato M., Matsukura S., Kangawa K., and Matsuo H. (1990) Isolation and identification of islet amyloid polypeptide in normal human pancreas. Regul. Pept. 31, 179–186 10.1016/0167-0115(90)90004-G [DOI] [PubMed] [Google Scholar]
- 17. Nishi M., Sanke T., Seino S., Eddy R. L., Fan Y. S., Byers M. G., Shows T. B., Bell G. I., and Steiner D. F. (1989) Human islet amyloid polypeptide gene: complete nucleotide sequence, chromosomal localization, and evolutionary history. Mol. Endocrinol. 3, 1775–1781 10.1210/mend-3-11-1775 [DOI] [PubMed] [Google Scholar]
- 18. Martin C. (2006) The physiology of amylin and insulin: maintaining the balance between glucose secretion and glucose uptake. Diabetes Educ. 32, Suppl 3, 101S–104S 10.1177/0145721706288237 [DOI] [PubMed] [Google Scholar]
- 19. Pillay K., and Govender P. (2013) Amylin uncovered: a review on the polypeptide responsible for type II diabetes. Biomed. Res. Int. 2013, 826706 10.1155/2013/826706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chondrogianni N., Petropoulos I., Grimm S., Georgila K., Catalgol B., Friguet B., Grune T., and Gonos E. S. (2014) Protein damage, repair and proteolysis. Mol. Aspects Med. 35, 1–71 10.1016/j.mam.2012.09.001 [DOI] [PubMed] [Google Scholar]
- 21. Bhaumik S. R., and Malik S. (2008) Diverse regulatory mechanisms of eukaryotic transcriptional activation by the proteasome complex. Crit. Rev. Biochem. Mol. Biol. 43, 419–433 10.1080/10409230802605914 [DOI] [PubMed] [Google Scholar]
- 22. Huang C. J., Lin C. Y., Haataja L., Gurlo T., Butler A. E., Rizza R. A., and Butler P. C. (2007) High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress mediated beta-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 56, 2016–2027 10.2337/db07-0197 [DOI] [PubMed] [Google Scholar]
- 23. Tarcsa E., Szymanska G., Lecker S., O'Connor C. M., and Goldberg A. L. (2000) Ca2+-free calmodulin and calmodulin damaged by in vitro aging are selectively degraded by 26 S proteasomes without ubiquitination. J. Biol. Chem. 275, 20295–20301 10.1074/jbc.M001555200 [DOI] [PubMed] [Google Scholar]
- 24. Kwak J., Workman J. L., and Lee D. (2011) The proteasome and its regulatory roles in gene expression. Biochim. Biophys. Acta 1809, 88–96 10.1016/j.bbagrm.2010.08.001 [DOI] [PubMed] [Google Scholar]
- 25. Bugliani M., Liechti R., Cheon H., Suleiman M., Marselli L., Kirkpatrick C., Filipponi F., Boggi U., Xenarios I., Syed F., Ladriere L., Wollheim C., Lee M. S., and Marchetti P. (2013) Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin-proteasome system in pancreatic beta cell dysfunction. Mol. Cell. Endocrinol. 367, 1–10 10.1016/j.mce.2012.12.001 [DOI] [PubMed] [Google Scholar]
- 26. Jonas J. C., Sharma A., Hasenkamp W., Ilkova H., Patanè G., Laybutt R., Bonner-Weir S., and Weir G. C. (1999) Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem. 274, 14112–14121 10.1074/jbc.274.20.14112 [DOI] [PubMed] [Google Scholar]
- 27. Singh S., Trikha S., Sarkar A., and Jeremic A. M. (2016) Proteasome regulates turnover of toxic human amylin in pancreatic cells. Biochem. J. 473, 2655–2670 10.1042/BCJ20160026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Broca C., Varin E., Armanet M., Tourrel-Cuzin C., Bosco D., Dalle S., and Wojtusciszyn A. (2014) Proteasome dysfunction mediates high glucose-induced apoptosis in rodent beta cells and human islets. PLoS ONE 9, e92066 10.1371/journal.pone.0092066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mulder H., Ahrén B., and Sundler F. (1996) Islet amyloid polypeptide and insulin gene expression are regulated in parallel by glucose in vivo in rats. Am. J. Physiol. 271, E1008–E1014 [DOI] [PubMed] [Google Scholar]
- 30. Back S. H., Kang S. W., Han J., and Chung H. T. (2012) Endoplasmic reticulum stress in the beta-cell pathogenesis of type 2 diabetes. Exp. Diabetes Res. 2012, 618396 10.1155/2012/618396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwarz K., de Giuli R., Schmidtke G., Kostka S., van den Broek M., Kim K. B., Crews C. M., Kraft R., and Groettrup M. (2000) The selective proteasome inhibitors lactacystin and epoxomicin can be used to either up- or down-regulate antigen presentation at nontoxic doses. J. Immunol. 164, 6147–6157 10.4049/jimmunol.164.12.6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marques O., and Outeiro T. F. (2012) α-Synuclein: from secretion to dysfunction and death. Cell Death Dis. 3, e350 10.1038/cddis.2012.94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perez-Gonzalez R., Gauthier S. A., Kumar A., and Levy E. (2012) The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 287, 43108–43115 10.1074/jbc.M112.404467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baranello R. J., Bharani K. L., Padmaraju V., Chopra N., Lahiri D. K., Greig N. H., Pappolla M. A., and Sambamurti K. (2015) Amyloid-β protein clearance and degradation (ABCD) pathways and their role in Alzheimer's disease. Curr. Alzheimer Res. 12, 32–46 10.2174/1567205012666141218140953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Korolchuk V. I., Menzies F. M., and Rubinsztein D. C. (2010) Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy–lysosome systems. FEBS Lett. 584, 1393–1398 10.1016/j.febslet.2009.12.047 [DOI] [PubMed] [Google Scholar]
- 36. Ding W. X., Ni H. M., Gao W., Yoshimori T., Stolz D. B., Ron D., and Yin X. M. (2007) Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 171, 513–524 10.2353/ajpath.2007.070188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Iwata A., Riley B. E., Johnston J. A., and Kopito R. R. (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 280, 40282–40292 10.1074/jbc.M508786200 [DOI] [PubMed] [Google Scholar]
- 38. Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., Padmanabhan R., Hild M., Berry D. L., Garza D., Hubbert C. C., et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 [DOI] [PubMed] [Google Scholar]
- 39. Bhat K. P., Turner J. D., Myers S. E., Cape A. D., Ting J. P., and Greer S. F. (2008) The 19S proteasome ATPase Sug1 plays a critical role in regulating MHC class II transcription. Mol. Immunol. 45, 2214–2224 10.1016/j.molimm.2007.12.001 [DOI] [PubMed] [Google Scholar]
- 40. Brooks S. A. (2010) Functional interactions between mRNA turnover and surveillance and the ubiquitin proteasome system. Wiley Interdiscip. Rev. RNA 1, 240–252 10.1002/wrna.11 [DOI] [PubMed] [Google Scholar]
- 41. Wu X., and Brewer G. (2012) The regulation of mRNA stability in mammalian cells: 2.0. Gene 500, 10–21 10.1016/j.gene.2012.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Alarcon C., Verchere C. B., and Rhodes C. J. (2012) Translational control of glucose-induced islet amyloid polypeptide production in pancreatic islets. Endocrinology 153, 2082–2087 10.1210/en.2011-2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brender J. R., Krishnamoorthy J., Sciacca M. F., Vivekanandan S., D'Urso L., Chen J., La Rosa C., and Ramamoorthy A. (2015) Probing the sources of the apparent irreproducibility of amyloid formation: drastic changes in kinetics and a switch in mechanism due to micellelike oligomer formation at critical concentrations of IAPP. J. Phys. Chem. B 119, 2886–2896 10.1021/jp511758w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., and Newgard C. B. (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 10.2337/diabetes.49.3.424 [DOI] [PubMed] [Google Scholar]
- 45. Kitiphongspattana K., Mathews C. E., Leiter E. H., and Gaskins H. R. (2005) Proteasome inhibition alters glucose-stimulated (pro)insulin secretion and turnover in pancreatic β-cells. J. Biol. Chem. 280, 15727–15734 10.1074/jbc.M410876200 [DOI] [PubMed] [Google Scholar]
- 46. Welsh M., Nielsen D. A., MacKrell A. J., and Steiner D. F. (1985) Control of insulin gene expression in pancreatic beta-cells and in an insulin-producing cell line, RIN-5F cells. II. Regulation of insulin mRNA stability. J. Biol. Chem. 260, 13590–13594 [PubMed] [Google Scholar]
- 47. Shepherd L. M., Campbell S. C., and Macfarlane W. M. (2004) Transcriptional regulation of the IAPP gene in pancreatic beta-cells. Biochim. Biophys. Acta 1681, 28–37 10.1016/j.bbaexp.2004.09.009 [DOI] [PubMed] [Google Scholar]
- 48. Friedman J. R., and Kaestner K. H. (2006) The Foxa family of transcription factors in development and metabolism. Cell. Mol. Life Sci. 63, 2317–2328 10.1007/s00018-006-6095-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu G., Chen J., Jing G., and Shalev A. (2013) Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 19, 1141–1146 10.1038/nm.3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trikha S., and Jeremic A. M. (2013) Distinct internalization pathways of human amylin monomers and its cytotoxic oligomers in pancreatic cells. PLoS ONE 8, e73080 10.1371/journal.pone.0073080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]