

Abstract
Voltage-gated Cav Ca2+ channels play crucial roles in regulating gene transcription, neuronal excitability, and synaptic transmission. Natural or pathological variations in Cav channels have yielded rich insights into the molecular determinants controlling channel function. Here, we report the consequences of a natural, putatively disease-associated mutation in the CACNA1D gene encoding the pore-forming Cav1.3 α1 subunit. The mutation causes a substitution of a glutamine residue that is highly conserved in the extracellular S1–S2 loop of domain II in all Cav channels with a histidine and was identified by whole-exome sequencing of an individual with moderate hearing impairment, developmental delay, and epilepsy. When introduced into the rat Cav1.3 cDNA, Q558H significantly decreased the density of Ca2+ currents in transfected HEK293T cells. Gating current analyses and cell-surface biotinylation experiments suggested that the smaller current amplitudes caused by Q558H were because of decreased numbers of functional Cav1.3 channels at the cell surface. The substitution also produced more sustained Ca2+ currents by weakening voltage-dependent inactivation. When inserted into the corresponding locus of Cav2.1, the substitution had similar effects as in Cav1.3. However, the substitution introduced in Cav3.1 reduced current density, but had no effects on voltage-dependent inactivation. Our results reveal a critical extracellular determinant of current density for all Cav family members and of voltage-dependent inactivation of Cav1.3 and Cav2.1 channels.
Keywords: calcium channel, gating, ion channel, autism, epilepsy, CACNA1D, calcium voltage-gated channel subunit alpha1 D, Ca2+ channel, modulation, inactivation, intellectual disability, hearing impairment
Introduction
Within the human genome, hundreds of genes encode ion channels, mutations which cause diseases affecting a variety of organ systems (1). In many cases, electrophysiological analysis of the mutations has uncovered mechanistic insights into channel function. Mutations causing long QT syndrome have revealed molecular determinants controlling voltage-dependent activation (2) and inactivation (3). In addition, the functional characterization of a mutation causing congenital stationary night blindness in the CACNA1F gene encoding voltage-gated Cav1.4 Ca2+ channels uncovered a role for a C-terminal automodulatory domain that suppresses Ca2+-dependent inactivation of these channels (4). Thus, the study of channelopathy-causing mutations can advance understanding of the structure–function relationships of ion channel proteins as well as inform about the physiological roles of these channels in humans.
In addition to congenital stationary night blindness, mutations affecting voltage-gated Ca2+ channels cause migraine, ataxia, malignant hypothermia, and cardiac arrhythmia (5, 6). Within the Cav channel2 superfamily, Cav1.3 L-type channels have emerged as targets of heterogeneous disorders including deafness and autism (7, 8). In the ear, Cav1.3 channels are localized at inner hair cell synapses where they mediate Ca2+ influx required for transmission of auditory information into the brain via the auditory nerve (9, 10). A mutation involving a glycine insertion in the domain IS6 transmembrane helix prevents voltage-dependent gating of Cav1.3 channels and causes deafness in homozygous individuals carrying this mutation (11). Cav1.3 channels are also expressed in the brain, where they regulate mood-related behaviors (12) and fear consolidation (13). De novo mutations causing a gain of function in Cav1.3 are associated with intellectual disability and autism spectrum disorders (7, 8).
Here, we report the functional effects of a mutation in the CACNA1D gene encoding Cav1.3 found by exome sequencing of an individual with intellectual disability, developmental delay, and mild hearing impairment. We show that this mutation, which causes a histidine substitution for a glutamine residue in the extracellular loop connecting the S1 and S2 helices in domain II (IIS1–S2L), causes a significant reduction in Cav1.3 current density and yet suppresses voltage-dependent inactivation of these channels in transfected cells. The glutamine residue is conserved in all Cav family members and its mutation to histidine has similar effects when introduced into Cav2.1 and Cav3.1 channels. Our findings implicate IIS1–S2L as a key determinant for regulating the cell-surface density and inactivation gating of Cav channels.
Results
Q567H mutation in an individual with moderate hearing impairment and intellectual disability
The male proband was the first-born child of healthy parents who were first cousins of Arabic descent. The father had a history of febrile seizures since childhood. The proband was born after an uneventful pregnancy and delivered at 38 weeks of gestation by cesarean section because of fetal distress. He failed the hearing test at birth and had seizures beginning at 4 months of age. At the age of 4 ½ years, he was reassessed at the genetic clinic for the purpose of family planning. He did not speak and his comprehension was limited. He exhibited autistic behaviors including very limited eye contact and hyperactivity. He wore hearing aids from the age of 11 months. At 5 years of age, he underwent hearing tests which revealed moderate hearing loss (40–50 db SPL across all frequencies from 500 Hz to 40 kHz).
Whole-exome sequencing and filtering for homozygous variants revealed two potential candidate genes: A homozygous mutation in the otogelin gene (OTOG): Chr11(GRCh37): g.17631250C>T; NM_001277269.1: c.4439C>T (p.Thr1480Ile); and a homozygous missense variant in CACNA1D: Chr3 (GRCh37): g.53756476G>C; NM_000720.3: c.1701G>C (p.Gln567His). The OTOG variant was present in a heterozygous state in the gnomAD browser (0.00003348), whereas the CACNA1D was found neither in gnomAD nor in an in-house exome database. Both parents were heterozygous for the OTOG and CACNA1D variants. The high degree of consanguinity within the family was the basis for assuming a recessive model of inheritance. A full pedigree molecular analysis was not possible because of difficulties in obtaining consent from other family members.
OTOG encodes otogelin, an N-glycosylated protein that is enriched in cellular membranes of the inner ear (14). Mutations in OTOG cause autosomal-recessive moderate hearing impairment (15). CACNA1D encodes the pore-forming subunit of Cav1.3 Ca2+ channels (Fig. 1A), which mediate the presynaptic release of glutamate from inner hair cells in the cochlea (9, 16). A mutation in CACNA1D that causes a complete loss of Cav1.3 function causes congenital deafness and sinoatrial node dysfunction (11). CACNA1D variants have also been associated with autism spectrum disorders (8) and epilepsy (8, 17, 18). Because of the coincident involvement of CACNA1D in the proband's symptoms (i.e. hearing loss, epilepsy, autism), we analyzed the functional consequences of the CACNA1D in a heterologous expression system.
Figure 1.
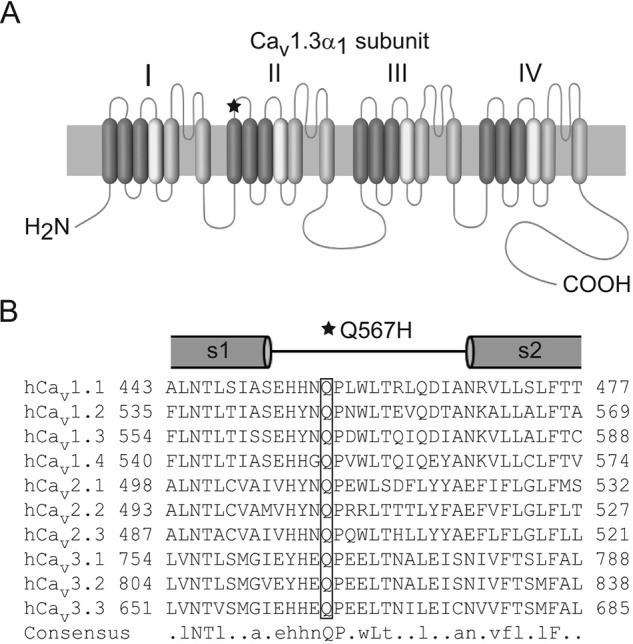
Q567H is located in the extracellular S1–S2 loop of domain II in the CaV1.3 α1 subunit. A, schematic of CaV1.3 α1 subunit with the mutation indicated (star). B, alignment of amino acids corresponding to this extracellular loop flanked by portions of S1 and S2. Box indicates conserved glutamine that is substituted with histidine because of the mutation. Capital letters indicate amino acids that are conserved in all sequences. Lowercase letters indicate conserved amino acids in at least six sequences.
The Gln to His mutation decreases current density and cell-surface levels of Cav1.3
The mutation in the affected individual causes substitution of a histidine for a glutamine residue in the proximal third of the extracellular loop connecting transmembrane helices S1 and S2 in domain II (Q567H) (Fig. 1B). To elucidate the functional consequences of Q567H, we introduced this mutation in the corresponding region of the rat Cav1.3 channel (Q558H). We used whole-cell patch clamp electrophysiology to compare the properties of the wild-type (WT) and mutant channels coexpressed with the auxiliary β2a and α2δ1 subunits in human embryonic kidney cells transformed with T-antigen (HEK293T). In current-voltage (I-V) relationships, the most prominent difference between cells transfected with WT and Q558H was a significant reduction in the peak current density of the mutant channel as compared with the WT channel (Fig. 2A; Table 1). To determine whether this was because of alterations in the number of functional Q558H channels and/or their open probability, we analyzed gating currents evoked by depolarization to the reversal potential (Igating) and tail currents (Itail) upon repolarization to the holding voltage (Fig. 2B). As was described previously (19, 20), the time integral of Igating (Qmax) is a measure of the number of channels in the cell membrane, and the ratio of Itail and Qmax describes the relative channel open probability. Q558H caused a significant reduction in Qmax (56.5 ± 6.4 fC for WT versus 16 ± 2.1 fC for Q558H; p < 0.01 by t test), but no change in the slope of the Itail versus Qmax relationship (9.8 ± 0.8 for WT versus 12.4 ± 1.2 for Q558H; p = 0.62 by F-test) (Fig. 2C). Thus, Q558H causes a decrease in the number of functional channels, but not their open probability.
Figure 2.
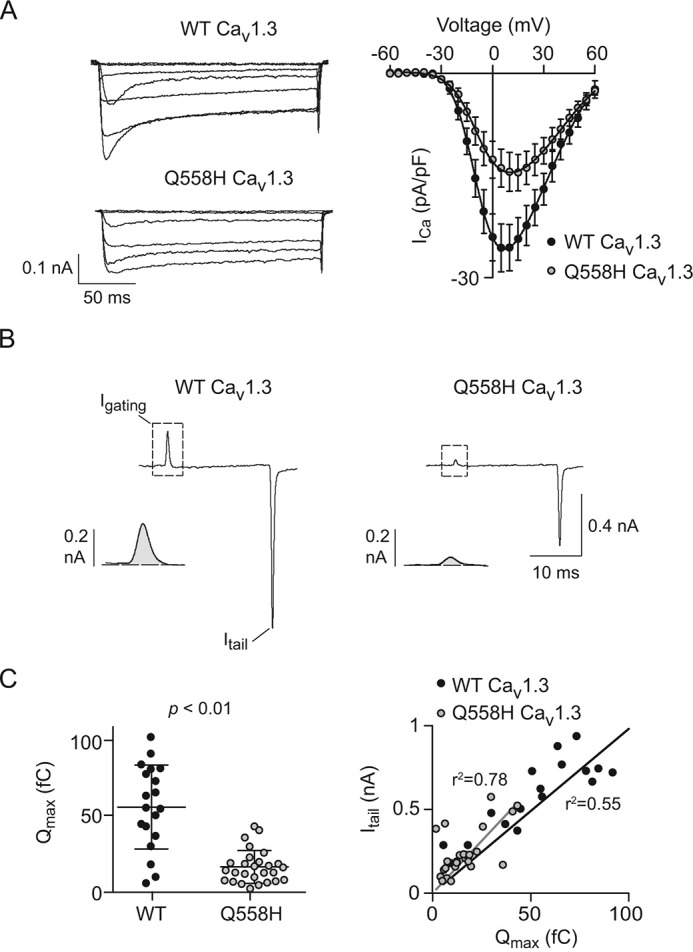
Q558H reduces current density and the number of functional Cav1. 3 channels. A, left, family of representative current traces obtained using 200-ms test pulses to various voltages from a holding voltage of −80 mV. Right, current-voltage (I-V) plots. n values and I-V parameters are listed in Table 1. Results are from four independent experiments. B, representative traces showing Igating evoked by a 20-ms step depolarization from −80 mV to +70 mV and Itail upon repolarization to −80 mV. Insets show Igating and Itail on expanded time scale. C, left, average maximal gating charge (Qmax) for WT and mutant; p value was determined by t test. Right, Itail for each cell is plotted against Qmax. The fit by linear regression and coefficient of determination (r2) are shown for WT (n = 19 cells) and Q558H (n = 27 cells). Results are from four independent experiments. Error bars represent S.E.
Table 1.
Parameters of I-V relationships
Values for V½ and k were obtained from Boltzmann fits of data in Fig. 2A. Results were obtained from six independent experiments, and p values were determined by t test. pA/pF, picoamperes/picofarads.
| Channel | WT | n | Mutant | n | p value |
|---|---|---|---|---|---|
| CaV1.3 | |||||
| V½ (mV) | −9.9 ± 1.4 | 10 | −6.8 ± 1.2 | 11 | 0.12 |
| k | 7.1 ± 0.5 | 10.2 ± 1.2 | 0.03 | ||
| ICa density (pA/pF) | −25.6 ± 3.3 | −14.5 ± 2.8 | 0.02 | ||
| CaV2.1 | |||||
| V½ (mV) | −2.4 ± 1.1 | 8 | 0.7 ± 1.4 | 9 | 0.11 |
| k | 3.1 ± 0.5 | 3.6 ± 0.5 | 0.51 | ||
| ICa density (pA/pF) | −72.8 ± 12 | −30.1 ± 7.8 | 0.03 | ||
| CaV3.1 | |||||
| V½ (mV) | −41.8 ± 1.2 | 11 | −40.7 ± 0.8 | 14 | 0.43 |
| k | 3.3 ± 0.4 | 3.4 ± 0.4 | 0.83 | ||
| ICa density (pA/pF) | −165.5 ± 14 | −67.9 ± 13 | <0.01 | ||
Q558H could cause greater degradation of the channel because of improper folding. Alternatively, the mutation could alter the trafficking of the channel to or from the plasma membrane. To distinguish between these possibilities, we compared the amount of plasma membrane–associated WT and mutant channel protein in cell-surface biotinylation assays. Cell-surface proteins were subject to biotinylation and pulldown by streptavidin beads, and the levels of WT and mutant channels were compared by Western blotting with Cav1.3 antibodies. Blotting with antibodies of Na+/K+ ATPase was used as a positive control for the cell-surface biotinylation reaction. Expression of the WT or mutant channels had no effect on the amount of cell-surface Na+/K+ ATPase as the corresponding signals measured by densitometry were not significantly different in untransfected control cells (0.28 ± 0.1 arbitrary units (a.u.)) or those transfected with WT (0.33 ± 0.11 a.u.) or Q558H (0.24 ± 0.07 a.u.; p = 0.83 by analysis of variance (ANOVA)).
Although there was no difference in the total levels of WT and mutant channel protein in whole-cell lysates, there was a significant reduction in the levels of Q558H channel protein at the cell surface compared with WT channels (Fig. 3, A and B). To confirm these results, we also compared the cell-surface localization of WT and Q558H mutant channels containing an extracellular FLAG epitope between S5 and S6 of domain II. Consistent with the lack of effect of epitopes in this region on channel function (21), these FLAG-tagged channels exhibited similar electrophysiological properties as the untagged channels (data not shown). Cells transfected with FLAG-tagged WT and mutant channels were labeled first with FLAG antibodies under nonpermeabilizing conditions followed by permeabilization and labeling with Cav1.3 antibodies to mark the distribution of cell-surface and total channels, respectively. Because of our transient transfection strategy, channel expression levels varied considerably between cells as detected by Cav1.3 immunofluorescence, and this impacted the intensity of cell-surface FLAG-labeled channels. However, in cells selected for a similar level of total Cav1.3 labeling, FLAG labeling was less apparent at the plasma membrane when transfected with Q558H than with the WT channel (Fig. 3C). Taken together, our results show that Q558H causes a loss of function because of improper cell-surface trafficking of Cav1.3.
Figure 3.

Q558H reduces the cell-surface density of Cav1.3. A, Western blotting of lysates of untransfected HEK293T cells (Control) or cells transfected with β2a, α2δ1, and WT Cav1.3 or Q567H mutant channels. Blots were probed with Cav1.3 antibodies showing levels of total (Input) or surface protein isolated by exposing transfected cells to biotinylation reagents prior to lysis and pulldown on Neutravidin beads. Blots were also probed with antibodies against Na+/K+ ATPase and GAPDH as normalization controls. B, densitometric analysis of total and surface Cav1.3 protein. From Western blots obtained as in A, signals corresponding to surface Cav1.3 protein were normalized to those for Na+/K+ ATPase (Surface), and the signals for total Cav1.3 protein were normalized to those for GAPDH. Results are from seven independent experiments. Error bars represent S.E. C, representative confocal images from HEK293T cells transfected with β2a, α2δ1, and WT FLAG-Cav1.3 (top) or FLAG-Q558H mutant channels (bottom) labeled under nonpermeabilized conditions with FLAG antibodies to mark cell-surface channels (green), and following detergent permeabilization, with Cav1.3 antibodies to label the total population of intracellular and cell-surface channels (red). The degree of cell-surface labeling is indicated by regions of overlap of the red and green channels (yellow, Merge). Boxed region is shown at higher magnification in far left panel, and indicates weaker cell-surface labeling of the mutant compared with WT channel. Scale bars: 20 μm in initial three panels, 10 μm in far right panel. Results are representative of two independent experiments.
Q558H alters voltage-dependent inactivation of Cav1.3
Compared with WT currents, Ca2+ currents (ICa) mediated by Q558H channels were more prolonged (Fig. 2A), suggesting defects in inactivation kinetics. To characterize this more thoroughly, we performed exponential curve fitting of ICa evoked by sustained (3-s) pulses to +10 mV from a holding voltage of −80 mV. Traces corresponding to WT and mutant ICa were best fit with a double-exponential function, indicating the presence of a fast and slow phase of inactivation. Compared with WT channels, the time constant (τfast) and fractional amplitude of the fast phase of inactivation (Ffast) were significantly reduced in mutant channels (Fig. 4, A–C). Conversely, the time constant (τslow) and fractional amplitude of slow inactivation (Fslow) for the mutant channels were significantly greater than for WT channels (Fig. 4, A–C). These results indicate that Q558H prolongs Cav1.3 currents by stabilizing slow inactivation.
Figure 4.
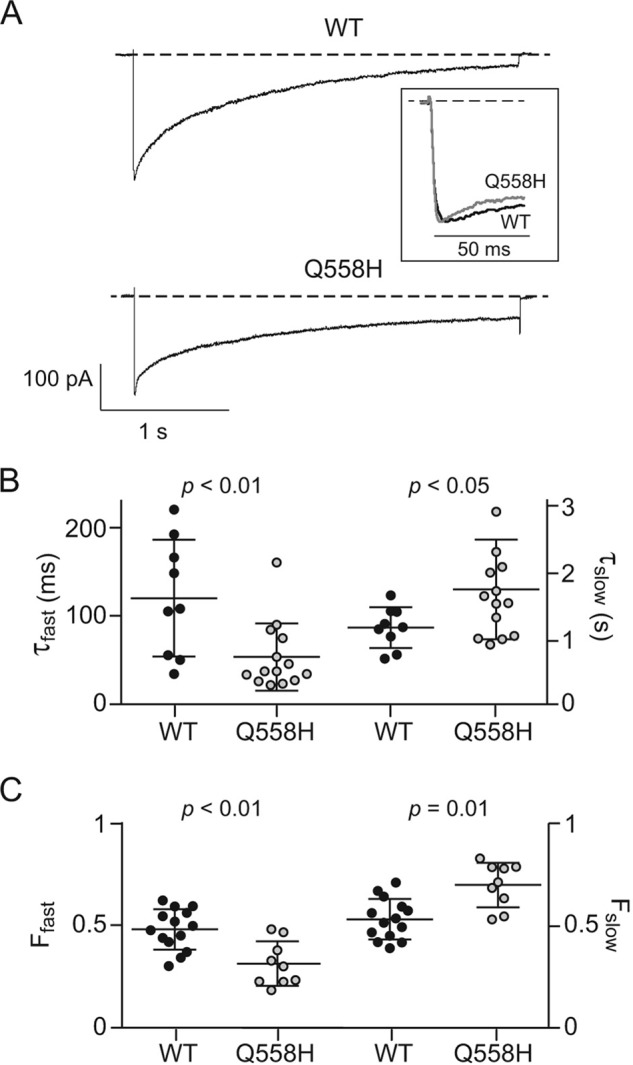
Q558H modifies inactivation of Cav1. 3 during sustained depolarizations. A, representative traces for currents evoked by a 3-s test pulse from −80 mV to +10 mV in cells transfected with WT or mutant channels. Inset, normalized traces of the displayed currents with expanded time scale showing faster initial inactivation of Q558H compared with WT. B and C, comparison of kinetics of inactivation of WT (n = 9 cells) and Q558H channels (n = 14 cells). Time constants (τfast, τslow) (B) and fractional amplitudes (Ffast and Fslow) (C) were obtained from double exponential fits of current traces obtained as in (A). Error bars represent S.E. p values were determined by t test. Results are from five independent experiments.
To investigate effects of Q558H on the voltage-dependence of inactivation, we measured the impact of a 5-s conditioning prepulse to various voltages on inactivation of a test current evoked by a 40-ms pulse to +10 mV. The amplitude of the test current was normalized to that with a −60 mV prepulse (Normalized ICa) and plotted against prepulse voltage. Note that although we used Ca2+ as the charge carrier, this voltage protocol measures voltage-dependent inactivation (VDI) rather than Ca2+-dependent inactivation given the relatively long duration of the conditioning prepulse (22). Consistent with a voltage-dependent process, Boltzmann fits of the data showed a sigmoidal dependence on prepulse voltage. Parameters from these fits indicated no difference in the voltage of half-maximal inactivation (V½) but a significantly greater slope (k) for Q558H compared with WT channels (Fig. 5A; Table 2). To determine whether this effect of Q558H might be secondary to effects on voltage-dependent activation, we analyzed the amplitude of tail currents measured by 2-ms repolarizations to −60 mV from various test voltages. The amplitudes of tail currents were normalized to that using a +40 mV test pulse and plotted against test voltage. Boltzmann fits of the data yielded similar parameters for WT and Q558H channels in terms of Vh and k (Fig. 5B; Table 3), which argued against an effect of Q558H on voltage-dependent activation. Overlay of the inactivation and activation curves revealed a sizeable increase in window currents generated by Q558H (Fig. 5C). Thus, despite the decrease in peak current density caused by Q558H, the mutation also has a gain of function effect in allowing Ca2+ entry at voltages at which WT channels are inactivated.
Figure 5.
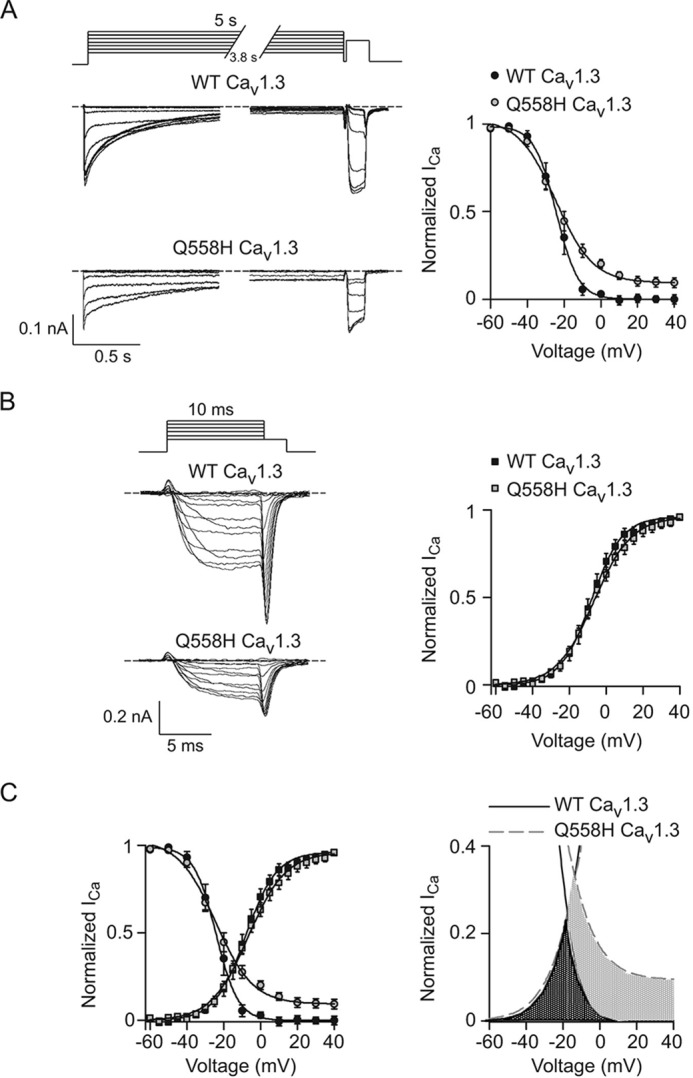
Q558H suppresses voltage-dependent inactivation of Cav1. 3. A, left, representative traces of currents evoked by 5-s prepulses to various voltages prior to a 40-ms test pulse to +10 mV from a holding voltage of −80 mV. Right, test current amplitudes were normalized with the test current amplitude obtained to −60 mV and plotted against prepulse voltage for WT (n = 7 cells) and Q558H mutant channels (n = 7 cells). B, left, representative traces of currents evoked by 10-ms steps to various voltages prior to 2-ms repolarization to −60 mV to better resolve tail currents. The amplitudes of tail currents were normalized to those obtained with +40 mV test pulse and plotted against test voltage for WT (n = 9 cells) and Q558H mutant channels (n = 10 cells). C, left, overlay of activation and inactivation curves for WT and mutant channels from data in A and B. Right, data from C are shown on expanded vertical scale. Window current of Q558H (gray shading) extends to depolarized voltages at which WT window current (dark shading) is absent. Results are from six independent experiments. Error bars represent S.E.
Table 2.
Parameters for voltage-dependent inactivation
Values for V½ and k were obtained from Boltzmann fits of data in Fig. 5A. Results were obtained from nine independent experiments, and p values were determined by t test.
| Channel | WT | n | Mutant | n | p value |
|---|---|---|---|---|---|
| CaV1.3 | |||||
| V½ (mV) | −24.6 ± 2.1 | 7 | −21.5 ± 1.9 | 7 | 0.31 |
| k | −4.9 ± 0.5 | −9.5 ± 0.8 | <0.01 | ||
| CaV2.1 | |||||
| V½ (mV) | −18 ± 0.8 | 8 | −6.1 ± 2.5 | 8 | <0.01 |
| k | −3.0 ± 0.4 | −4.3 ± 0.9 | 0.27 | ||
| CaV3.1 | |||||
| V½ (mV) | −62.5 ± 0.8 | 7 | −60.5 ± 1 | 7 | 0.24 |
| k | −3.9 ± 0.1 | −3.4 ± 0.1 | 0.04 | ||
Table 3.
Parameters for voltage-dependent activation
Values for V½ and k were obtained from Boltzmann fits of data in Fig. 5B; n represents number of cells. Results were obtained from nine independent experiments and p values were determined by t test.
| Channel | WT | n | Mutant | n | p value |
|---|---|---|---|---|---|
| CaV1.3 | |||||
| V½ (mV) | −7.8 ± 1.6 | 9 | −5.6 ± 1.5 | 10 | 0.36 |
| k | 8 ± 0.9 | 10.3 ± 0.8 | 0.10 | ||
| CaV2.1 | |||||
| V½ (mV) | 8.6 ± 1.6 | 7 | 11.8 ± 1.4 | 8 | 0.21 |
| k | 7.5 ± 0.5 | 5.7 ± 1 | 0.24 | ||
| CaV3.1 | |||||
| V½ (mV) | −37.1 ± 0.9 | 7 | −32.4 ± 0.9 | 8 | <0.01 |
| k | 3.3 ± 0.3 | 4.7 ± 0.4 | 0.03 | ||
Alterations in Cav2.1 and Cav3.1 properties by similar Gln to His mutations
The strong conservation of Gln-567 (Fig. 1B) suggests that it may play a key role in regulating current density and steady-state inactivation of all Cav channel family members. To test this, we introduced the Gln to His mutation in the corresponding sites of Cav2.1 (Q511H) and the more distantly related Cav3.1 (Q767H). Similar to the effects of Q558H in Cav1.3 (Fig. 2), Q511H and Q767H caused significant reductions in peak current densities but without a change in Boltzmann parameters for the I-V relationship (Fig. 6, A and B; Table 1). We were unable to test whether the reduced peak current density for Q511H and Q767H was due to decreased cell-surface channel protein, as was done for Cav1.3 and Q567H (Fig. 3, A and B) because of low levels of cell-surface biotinylation of the mutant channels. However, gating current analyses revealed a significant reduction in Qmax because of the Gln to His mutations (21.9 ± 1.8 fC for WT Cav2.1 versus 10.2 ± 1.9 fC for Q511H, p < 0.01 by t test; 95.1 ± 16.9 fC for WT Cav3.1 versus 26.2 ± 4.6 fC for Q767H, p < 0.01 by t test), but no change in the slope of the Itail versus Qmax relationship (42.9 ± 3.8 for WT Cav2.1 versus 41 ± 5.6 for Q511H, p = 0.76 by F-test; 51.5 ± 3.7 for WT Cav3.1 versus 69.3 ± 8.6 for Q767H, p = 0.45 by F-test) (Fig. 7). Thus, the Gln to His mutations in all three classes of Cav channel decrease the number of functional channels, but not their open probability, which could be explained by decreased levels of channels in the plasma membrane.
Figure 6.
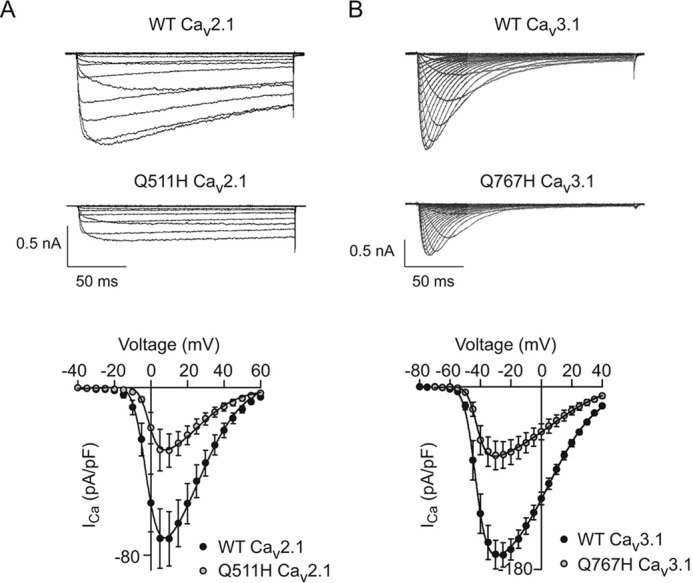
The corresponding mutation of Q511H in Cav2.1 and Q767H Cav3.1 suppress current density. A and B, representative traces (top) and I-V relationships (B) for WT (n = 8 cells) and mutant Cav2.1 channels (n = 9 cells, A) or WT (n = 11 cells) and mutant Cav3.1 channels (n = 14 cells, B). Voltage protocol was the same as in Fig. 2A. Results are from four independent experiments. Error bars represent S.E.
Figure 7.
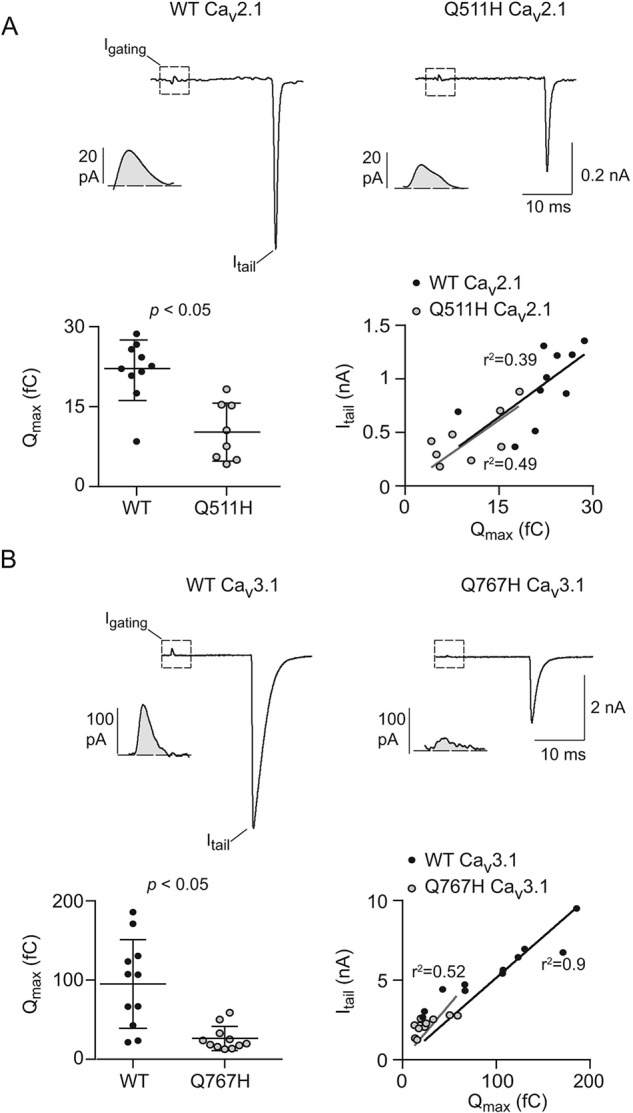
Q511H in Cav2.1 and Q767H in CaV3.1 decrease the number of functional channels. A, top, representative current traces from WT (left) or Q511H mutant (right) Cav2.1 channels showing Igating evoked by a 20-ms step depolarization from −80 mV to +70 mV and Itail upon repolarization to −80 mV. Insets show Igating on expanded time scale. Bottom left, average maximal gating charge (Qmax) for WT (n = 10 cells) and Q511H mutant Cav2.1 channels (n = 8 cells); p value was determined by t test. Bottom right, Itail for each cell is plotted against Qmax. The fit by linear regression and coefficient of determination (r2) are shown for WT and mutant channels. Results are from four independent experiments. Error bars represent S.E. B, same as in A except for WT (n = 11 cells) and Q767H mutant CaV3.1 channels (n = 11 cells). Results are from four independent experiments.
Steady-state inactivation was also suppressed by Q511H as compared with WT Cav2.1, but was more severe than Q558H in Cav1.3 in that there was a strong positive shift in V½ because of Q511H (Fig. 8A; Table 2). Although voltage-dependent activation of Q511H was not affected (Fig. 8B; Table 3), the window current generated by the mutant channels was greater than for WT channels because of lack of inactivation at positive voltages (Fig. 8C). In contrast to the robust effects of Q558H and Q511H on inactivation in Cav1.3 and Cav2.1, respectively, VDI of Cav3.1 was not affected by Q767H (Fig. 9A; Table 2). Although Q767H modestly suppressed voltage-dependent activation of Cav3.1 (Fig. 9B; Table 3), there was limited impact of this mutation on the window current (Fig. 9C). We conclude that Gln-558 and Gln-511 are determinants regulating VDI of high-voltage activated (i.e. Cav1 and Cav2) Ca2+ channels, and of the cell-surface density of all Cav channel family members.
Figure 8.
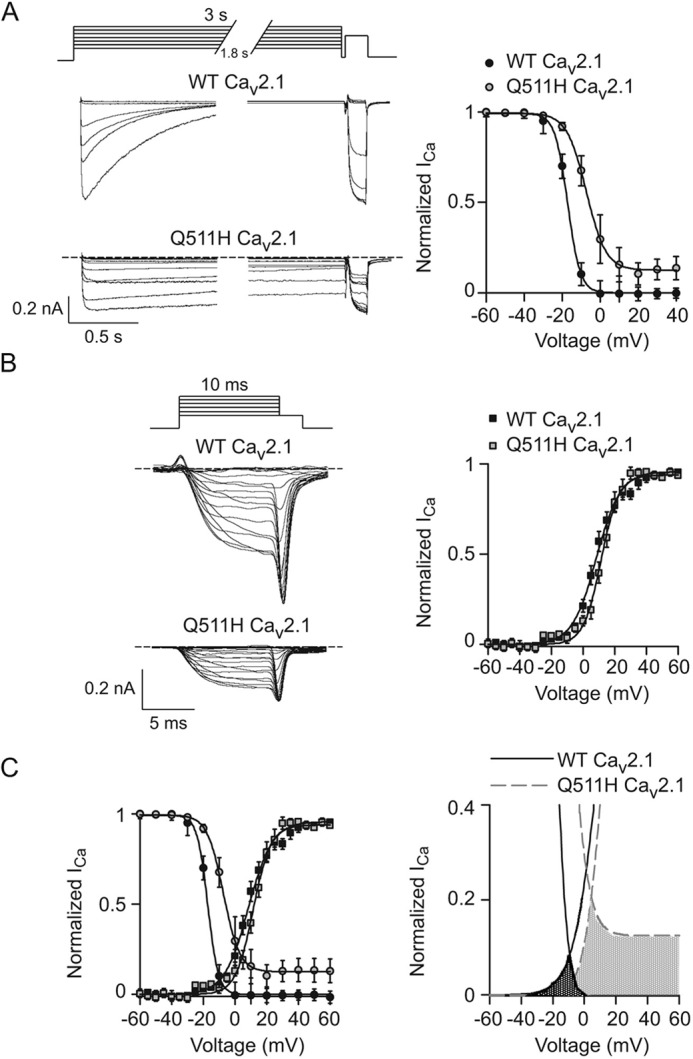
Q511H suppresses voltage-dependent inactivation of Cav2.1. A–C, same as in Fig. 5 except that the inactivation protocol was essayed with 3-s prepulses in cells transfected with WT (n = 15 cells) or Q511H mutant Cav2.1 channels (n = 16 cells). Results are from six independent experiments. Error bars represent S.E.
Figure 9.
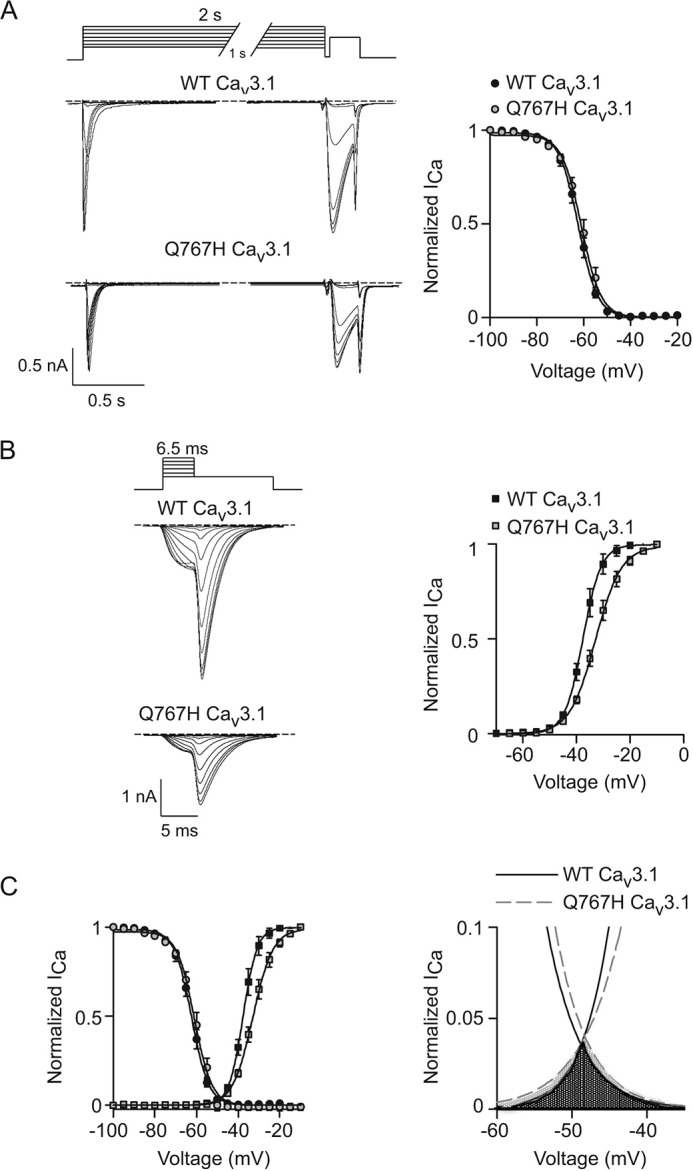
Q767H does not affect voltage-dependent inactivation of Cav3.1. A–C, same as in Fig. 5 except that inactivation protocol was essayed with 2-s prepulses and the tail currents were evoked by 6.5-ms steps to various voltages prior to 30-ms repolarization to −100 mV in cells transfected with WT (n = 14 cells) or Q767H mutant Cav3.1 channels (n = 15 cells). Results are from six independent experiments. Error bars represent S.E.
Discussion
VDI is a crucial feature of most Cav channels, limiting Ca2+ influx that could otherwise cause aberrant cellular excitability (23, 24). A variety of factors regulate VDI of Cav channels, including the auxiliary Cavβ subunit (reviewed in Ref. 25). Within the main α1 subunit, multiple elements contributing to VDI of Cav1 and Cav2 channels have been identified in cytoplasmic regions (26–28) or transmembrane segments (29, 30). Considering that Gln-558 and Gln-511 are located in the extracellular S1–S2 loop of domain II (IIS1–S2L) (Fig. 1), how these residues might promote VDI (Figs. 5 and 7) is unclear. Together with S3 and S4, S1 and S2 form the voltage sensor domain (VSD), a conserved module in voltage-gated ion channels that senses changes in the cell membrane potential in ways that alter the opening, closing, and inactivation of the channel (31). Within Cav1 and Cav2 channels, VSDs in domains I through IV may be functionally heterogeneous, with domains II and III playing the most prominent roles in voltage-dependent activation (32) and VDI (33). Thus, Gln-558 and Gln-511 may modulate the contribution of the VSD in domain II (VSDII) to gating of VDI. In the structure of the skeletal muscle Cav1.1 complex by cryo-EM, S1–S2 loops are closely associated with the pore-lining S5–S6 loop (34). The substitution of a charged histidine residue at this site might produce electrostatic alterations that destabilize VDI by maintaining the pore in an open rather than inactivated conformation (Figs. 5 and 7). Determinants for VDI in Cav3 channels differ from those in Cav1 and Cav2 channels (35), which may explain the lack of effect of the Gln to His mutation on Cav3.1 VDI (Fig. 9).
At first glance, the inhibitory effects of the Gln to His mutations on current density in all three classes of Cav channels (Figs. 2 and 6) might simply result from a decrease in the stability of the channel protein. However, in the context of Cav1.3, Q558H diminished the amount of Cav1.3 protein on the cell surface but had no effect on total protein Cav1.3 levels (Fig. 3). Moreover, gating current analyses indicate a decrease in the number of functional Cav1.3 channels with the Q558H mutation (Fig. 2, B and C). Taken together, these results suggest that the decrease in current density caused by the Gln to His mutation results from an impairment in the trafficking and/or maintenance of channels in the plasma membrane. Although auxiliary α2δ subunit promotes the cell-surface trafficking of these channels through interactions with extracellular sites in the α1 subunit (reviewed in Ref. 36), the Gln to His mutations are unlikely to disrupt the interaction of Cav1.3 and Cav2.1 with α2δ given that domain II S1–S2 is distant from sites in Cav α1 that are found in contact with α2δ based on the cryo-EM structure of Cav1.1 (34). Moreover, Q767H also significantly reduces the current density of Cav3.1 expressed without α2δ (Fig. 6B).
Within Kv channels, a conserved threonine in the S1–S2 linker (S1–S2L) is required for efficient trafficking of the channel to the plasma membrane (37, 38). This residue is thought to interact with a glutamate in the S5–S6L (39), which may stabilize VSDs and gating domains in ways that support Kv channel trafficking out of the endoplasmic reticulum and into the plasma membrane (37). In the cryo-EM structure of Cav1.1, the S1–S2L in all the VSDs is well resolved, indicative of rigidity that may be important for its interaction with LS5–S6 (34). Similar to the role of S1–S2L in Kv channels (37, 38), efficient trafficking of Cav channels may depend on the contact between S1–S2L and S5–S6L, and this is disrupted by the Gln to His mutation.
It is important to note that both the proband and his parents possess a variant in OTOG, a gene that is associated with autosomal-recessive moderate hearing impairment (15). Thus we cannot conclude whether it is the OTOG and/or CACNA1D variant that contributes to the hearing impairment of the affected individual. However, there are 12 reported mutations in OTOG of which 11 are lack of function, and only 1 reported missense mutation, making the mutation in this gene less of a candidate for the individual's symptoms. Considering the additional phenotypes of the proband, it is noteworthy that gain of function mutations in CACNA1D are associated with autism spectrum disorder (ASD) and epilepsy (7, 8). Like Q567H, these mutations cause an increase in the window current mediated by Cav1.3. However, the mechanism involves a slowing of VDI and/or enhancement in voltage-dependent activation (7, 8), in contrast to the selective effect of Q558H on suppressing VDI (Figs. 4 and 5). The gain of function in Cav1.3 would be expected to cause abnormalities in the morphology of dendritic spines (40), which have been linked to ASD (41) and epilepsy (42). Cav1.3 regulates neuronal excitability and synaptic plasticity in the striatum (43–46), a brain region implicated in the neuropathology of ASD (47). However, we are cautious in noting that the impact of Q558H on Cav1.3 properties may be influenced by additional factors present in neurons that are absent from HEK293T cells. As has been demonstrated for ASD-linked mutations in Cav1.2 (48–50), an understanding of how these gain of function mutations in Cav1.3 contribute neurodevelopmental and neurological disorders will be facilitated by the development of appropriate animal models.
Experimental procedures
Genetic analysis
This study abides by the Declaration of Helsinki principles and was approved by the Institutional Review Board at Hadassah-Hebrew University Hospital. Affected patients' guardians gave informed consent for exome sequencing and to publication of this study.
Whole exome sequencing was performed as described previously (51). Briefly, exonic sequences were enriched using SureSelect Human All Exon 50 megabase kit (Agilent Technologies, Santa Clara, CA). Sequences were determined by HiSeq 2500 (Illumina, San Diego, CA) as 100-bp paired-end runs. Data analysis including read alignment and variant calling was performed by DNAnexus software (Palo Alto, CA) using the default parameters with the human genome assembly hg19 (GRCh37) as reference. Exome analysis of the analyzed individuals yielded 44.1 million mapped reads with a coverage of 86.0×. Following alignment to the reference genome (hg19) and variant calling, variants were filtered out if they were off-target (>8 bp from splice junction), synonymous (>3 bp from splice junction), or had minor allele frequency >0.01 in the ExAC database (Exome Aggregation Consortium, Cambridge, MA; URL: http://exac.broadinstitute.org)3 or in our in-house exome database comprising ∼2500 exomes. All potentially causative variants were confirmed using Sanger sequencing.
Molecular biology
The following cDNAs were used: CaV1.3 (GenBankTM no. AF370009), CaV2.1 (GenBankTM no. NM_001127221), Cav3.1 (AF190860), β2a (GenBankTM no. NC013684), and α2δ1 (GenBankTM no. M76559.1). The Q558H mutation was inserted into the corresponding regions of the domain II S1–S2 linker (IIS1–S2L) in CaV1.3, CaV2.1, and CaV3.1 using the NEBuilder HiFi DNA Assembly cloning system (New England Biolabs). Channel fragments were amplified by PCR with appropriate primers and ligated into the parent plasmid (CaV1.3/pcDNA6, CaV2.1/pcDNA3.1, CaV3.1/pDsRed Express-N1). For the FLAG-tagged CaV1.3 WT and Q567H constructs, a FLAG epitope with spacer sequences (TRH-DYKDDDDK-VTFDEMQT) was added to the extracellular loop just C-terminal to residue Gly-693 by PCR using NEBuilder HiFi DNA Assembly cloning system (New England Biolabs). The vector was linearized by digestion with BsiWI and BamHI enzymes (New England Biolabs). The entire open reading frames of all the plasmids were verified by sequencing to confirm the presence of the mutation.
Cell culture and transfection
Human embryonic kidney cells transformed with SV40 T-antigen (ATCC, cat. no. CRL-3216, RRID: CVCL_0063) were maintained in Dulbecco's modified Eagle medium (Gibco, 11965) supplemented with 10% fetal bovine serum (Atlanta Biologicals, S11150), at 37 °C in 5% CO2–95% air humidified atmosphere. For electrophysiological recordings, the cells were grown at ∼70% confluent in 35-mm Petri dish prior to transient transfection with Cav channel cDNAs using FuGene 6 transfection reagent (Promega, E2691). The following amounts of cDNAs were used: WT or mutant CaV1.3 or CaV2.1 (2.5 μg) and 1 μg each of the auxiliary subunits β2a and α2δ1. Cav3.1 (2.5 μg) was transfected without auxiliary subunit cDNAs. In all cases, cDNA encoding the GFP (pEGFP) was cotransfected to aid fluorescent detection of transfected cells. cDNA was incubated with 6 μl of FuGene transfection reagent in 100 μl of FBS-free DMEM in a sterile microfuge tube (1.5 ml). The transfection solution was added to the dish and cells were maintained at 37 °C for 48 h. Two h before its electrophysiological recording the cells were dissociated using Versene reagent (Thermo Fisher Scientific, 15040066) in 35-mm Petri dishes with fresh medium at low density ∼20%.
For cell-surface biotinylation assays, HEK293T cells were transfected using Lipofectamine 3000 reagent (Life Technologies, L3000-008) following the manufacturer's protocol. Plasmid DNA (WT or mutant Cav1.3 (1.2 μg); β2a and α2δ1 (0.4 μg each)) was diluted in Opti-MEM (50 μl, Life Technologies) and 4 μl of P3000 Reagent, and added to a mixture of Opti-MEM (50 μl) and Lipofectamine 3000 Reagent (3 μl) and allowed to incubate for 10 min at room temperature. The solution was then added to the cells. The cell culture media were changed after 24 h and cell-surface biotinylation assays were performed 48 h after transfection.
Electrophysiology
Transfected HEK293T cells were recorded with the whole-cell patch clamp technique at room temperature (21–24 °C) using an EPC-8 patch clamp amplifier and Patchmaster software (HEKA Elektronik). Electrophysiological data were sampled at 20 kHz and filtered at 5 kHz. Leak current was subtracted using an online P/4 protocol. The external solution contained the following (in mm): CaCl2 10, TEA-Cl 125, HEPES 10, and glucose 10, pH 7.3, with TEA-OH. The internal solution contained (in mm): CsCl 110, MgCl2 5, EGTA 10, HEPES 10, Na-ATP 4, GTP 0.1, pH 7.3, with CsOH. The recording pipettes had a resistance in the bath solution in the range of 4–7 megohms. All reagents were purchased from Sigma-Aldrich.
Current-voltage relationships were obtained from the peak amplitude of currents evoked by 200-ms depolarizations from −80 mV to various voltages in 5-mV increments. I-V data were fit with the Boltzmann equation: I = G·(V − E)/(1 + exp(− (V − V½)/k)), where G is conductance, E is the reversal potential, V½ is the voltage of half-maximal activation, and k is the slope factor for activation. The current density was obtained by normalizing the current amplitude with the membrane capacitance.
The kinetics of inactivation were measured by fitting traces of currents with a double exponential function: I = Afastexp(−t/τfast) + Fslowexp(−t/τslow), where t is time, and Afast and Aslow are the amplitudes of the current decaying with corresponding time constants (τ). The fraction of the total inactivating current (Afast + Aslow) corresponding to fast inactivation was determined as Ffast = Afast/(Afast + Aslow) and that for slow inactivation (Fslow) was determined as Aslow/(Afast + Aslow).
For measuring steady-state inactivation, a conditioning prepulse to various voltages preceded a test depolarization to +10 mV for CaV1.3 and CaV2.1 and to −30 mV for CaV3.1. Inactivation curves were fitted with the Boltzmann function: I = Imax/(1 + exp (V − V½)/k), where I represents test current amplitude, V is test voltage, V½ is the voltage of half-maximal inactivation, and k is the slope factor. Voltage dependence of activation was measured by plotting the normalized tail currents evoked by 2-ms repolarizations to −60 mV from 10-ms pulses to various voltages, followed by fitting with the above Boltzmann function.
Data were analyzed using custom routines in Igor Pro software (WaveMetrics) and SigmaPlot (Systat Software Inc., San Jose, CA). Averaged data are shown as the mean ± S.E. For statistical analysis, data were first analyzed for normality by Shapiro-Wilk test followed by Student's t test with differences considered statically significant when p < 0.05. For gating current analysis, the data were fit by linear regression and the slopes compared by F-test.
Biochemical analysis of cell-surface Cav1.3 protein
HEK293T cells were transfected with WT and mutant Cav1.3 and auxiliary subunits as described above, and 1 day later, cell-surface proteins were labeled with a biotin labeling kit (Thermo Scientific, cat. no.89881) according to the manufacturer's protocol. Briefly, transfected cells were quickly washed with ice-cold phosphate buffered saline containing the following (in mm): 2.5 KCl, 136 NaCl, 1.5 KH2PO4, 6.5 Na2HPO4 (pH 7.4), and incubated with 0.25 mg/ml of the cell membrane impermeable biotinylation reagent sulfo-NHS-SS-biotin for 30 min at 4 °C. To stop the reaction, the cells were then incubated with biotin quenching solution containing (in mm): glycine 50, CaCl2 2.5, MgCl2 1, pH 7.4. The cells were pelleted by centrifugation and resuspended in radioimmune precipitation assay buffer containing (in mm) NaCl 150, PMSF 0.5, Tris-HCl 25, pH 7.6, with 1% Nonidet P-40, 1% Na deoxycholate, 0.1% SDS, and protease inhibitors. After 10 min incubation on ice, cell lysates were subject to centrifugation (16,000 × g for 10 min at 4 °C) and biotinylated proteins recovered with NeutrAvidin gel. The bound proteins were eluted by incubating with SDS-PAGE sample buffer (58 mm Tris-Cl, 50 mm DTT, 1.7% SDS, 5% glycerol, 0.002% bromphenol blue, pH 6.8, and subject to electrophoresis using NovexTM WedgeWellTM 4–20% Tris-Glycine gel (Invitrogen cat. no.105955) and transfer to nitrocellulose blotting membranes (Amersham Biosciences, Protran 0.2 μm NC, cat. no. 10600001).
For Western blotting, the membranes were incubated in blocking buffer containing milk (5%) in TBS-T (100 mm Tris-HCl, 0.15 m NaCl, 0.05% Tween 20) followed by incubation with the following antibodies diluted in blocking buffer: CaV1.3 (1:3000) (52), N+/K+ ATPase (1:700) (Developmental Studies Hybridoma Bank at University of Iowa, cat. no. a6F RRID:AB_2314847), and GAPDH (1:10,000) (Cell Signaling Technology, cat. no. 14C10). Secondary antibodies used were anti-rabbit HRP (1:3000) (GE Healthcare, cat. no. NA934–1ML), and anti-mouse HRP (1:3000) (GE Healthcare, cat. no. NA931V) followed by chemiluminescent detection (Thermo Scientific, SuperSignal West Pico cat. no. 34080). The Western blotting signals were visualized with the Odyssey Fc Imaging System (LI-COR). The results shown were obtained from at least three independent experiments. Densitometric analysis was performed with Image Studio Lite software (LI-COR).
Double-label immunofluorescence
HEK293T cells were transfected as described for cell-surface biotinylation experiments except that FLAG-tagged Cav1.3 and Q567H cDNAs were used, and cells were plated on glass coverslips. Sequential double-labeling for cell-surface and total channels was performed as described previously (53). Twenty-four h post transfection, the cells were rinsed in PBS and incubated with mouse anti-FLAG M2 antibody (Sigma-Aldrich) (1:50 diluted in DMEM media with 10% FBS) at room temperature for 30 min. The cells were then rinsed in PBS and fixed with 4% paraformaldehyde in PBS. After rinsing three times with PBS the cells were then blocked using 5% normal goat serum and 0.3% Triton X-100 in PBS for 30 min at room temperature and incubated with rabbit anti Cav1.3 antibodies to label the total intracellular and cell-surface channels for 1 h at room temperature. After three times with PBS, cells were incubated in secondary antibodies (RhoRed-X 568 anti-rabbit and Alexa Fluor 488 anti-mouse, Jackson ImmunoResearch Laboratories) (1:1000 each diluted in 5% normal goat serum and 0.1% Triton X-100 in PBS) for 1 h at room temperature. Coverslips were rinsed again three times in PBS and one time with water before mounting in Fluoromount G (Electron Microscopy Sciences, 17984–25). Labeled cells were visualized with a 60× oil-immersion objective on a confocal microscope (Fluoview 1000, Olympus). Images were acquired at 510 for 488 channel and 500 for 568 channel laser intensity and processed in ImageJ.
Author contributions
E. G.-L. and A. L. formal analysis; E. G.-L., J. A. L., J. H., R. S., V. M., and A. L. investigation; E. G.-L., R. S., V. M., and A. L. writing-review and editing; V. M. and A. L. conceptualization; V. M. and A. L. resources; V. M. and A. L. supervision; V. M. and A. L. project administration; A. L. funding acquisition; A. L. writing-original draft.
Acknowledgments
We thank Dr. Jianping Wu for helpful insights, and Mackenzie McKnight and Brian Goodell for excellent technical assistance.
This work was supported by Karl Kahane Foundation (to V. M. and R. S.), Conacyt (Mexico) postdoctoral fellowship (to E. G. L.), and National Institutes of Health Grants NS084190 and DC009433 and Carver Research Program of Excellence Award (to A. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- Cav channel
- voltage-gated calcium channel
- I-V
- current-voltage
- a.u.
- arbitrary unit
- ANOVA
- analysis of variance
- VDI
- voltage-dependent inactivation
- VSD
- voltage sensor domain
- ASD
- autism spectrum disorder
- fC
- femtocoulomb.
References
- 1. Hübner C. A., and Jentsch T. J. (2002) Ion channel diseases. Hum. Mol. Genet. 11, 2435–2445 10.1093/hmg/11.20.2435 [DOI] [PubMed] [Google Scholar]
- 2. Grant A. O., Carboni M. P., Neplioueva V., Starmer C. F., Memmi M., Napolitano C., and Priori S. (2002) Long QT syndrome, Brugada syndrome, and conduction system disease are linked to a single sodium channel mutation. J. Clin. Invest. 110, 1201–1209 10.1172/JCI0215570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wehrens X. H., Rossenbacker T., Jongbloed R. J., Gewillig M., Heidbuchel H., Doevendans P. A., Vos M. A., Wellens H. J., and Kass R. S. (2003) A novel mutation L619F in the cardiac Na+ channel SCN5A associated with long-QT syndrome (LQT3): A role for the I–II linker in inactivation gating. Hum. Mutat. 21, 552 10.1002/humu.9136 [DOI] [PubMed] [Google Scholar]
- 4. Singh A., Hamedinger D., Hoda J. C., Gebhart M., Koschak A., Romanin C., and Striessnig J. (2006) C-terminal modulator controls Ca2+-dependent gating of Cav1.4 L-type Ca2+ channels. Nat. Neurosci. 9, 1108–1116 10.1038/nn1751 [DOI] [PubMed] [Google Scholar]
- 5. Pietrobon D. (2010) CaV2.1 channelopathies. Pflugers Arch. 460, 375–393 10.1007/s00424-010-0802-8 [DOI] [PubMed] [Google Scholar]
- 6. Striessnig J., Bolz H. J., and Koschak A. (2010) Channelopathies in Cav1.1, Cav1.3, and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. 460, 361–374 10.1007/s00424-010-0800-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pinggera A., Lieb A., Benedetti B., Lampert M., Monteleone S., Liedl K. R., Tuluc P., and Striessnig J. (2015) CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol. Psychiatry 77, 816–822 10.1016/j.biopsych.2014.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pinggera A., Mackenroth L., Rump A., Schallner J., Beleggia F., Wollnik B., and Striessnig J. (2017) New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum. Mol. Genet. 26, 2923–2932 10.1093/hmg/ddx175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Platzer J., Engel J., Schrott-Fischer A., Stephan K., Bova S., Chen H., Zheng H., and Striessnig J. (2000) Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell 102, 89–97 10.1016/S0092-8674(00)00013-1 [DOI] [PubMed] [Google Scholar]
- 10. Brandt A., Striessnig J., and Moser T. (2003) CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J. Neurosci. 23, 10832–10840 10.1523/JNEUROSCI.23-34-10832.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baig S. M., Koschak A., Lieb A., Gebhart M., Dafinger C., Nürnberg G., Ali A., Ahmad I., Sinnegger-Brauns M. J., Brandt N., Engel J., Mangoni M. E., Farooq M., Khan H. U., Nürnberg P., Striessnig J., and Bolz H. J. (2011) Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 14, 77–84 10.1038/nn.2694 [DOI] [PubMed] [Google Scholar]
- 12. Sinnegger-Brauns M. J., Huber I. G., Koschak A., Wild C., Obermair G. J., Einzinger U., Hoda J. C., Sartori S. B., and Striessnig J. (2009) Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol. Pharmacol. 75, 407–414 10.1124/mol.108.049981 [DOI] [PubMed] [Google Scholar]
- 13. McKinney B. C., and Murphy G. G. (2006) The L-type voltage-gated calcium channel CaV1.3 mediates consolidation, but not extinction, of contextually conditioned fear in mice. Learn. Mem. 13, 584–589 10.1101/lm.279006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Simmler M. C., Cohen-Salmon M., El-Amraoui A., Guillaud L., Benichou J. C., Petit C., and Panthier J. J. (2000) Targeted disruption of Otog results in deafness and severe imbalance. Nat. Genet. 24, 139–143 10.1038/72793 [DOI] [PubMed] [Google Scholar]
- 15. Schraders M., Ruiz-Palmero L., Kalay E., Oostrik J., del Castillo F. J., Sezgin O., Beynon A. J., Strom T. M., Pennings R. J., Zazo Seco C., Oonk A. M., Kunst H. P., Domínguez-Ruiz M., García-Arumi A. M., del Campo M., et al. (2012) Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment. Am. J. Hum. Genet. 91, 883–889 10.1016/j.ajhg.2012.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dou H., Vazquez A. E., Namkung Y., Chu H., Cardell E. L., Nie L., Parson S., Shin H. S., and Yamoah E. N. (2004) Null mutation of α1D Ca2+ channel gene results in deafness but no vestibular defect in mice. J. Assoc. Res. Otolaryngol. 5, 215–226 10.1007/s10162-003-4020-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Scholl U. I., Goh G., Stölting G., de Oliveira R. C., Choi M., Overton J. D., Fonseca A. L., Korah R., Starker L. F., Kunstman J. W., Prasad M. L., Hartung E. A., Mauras N., Benson M. R., Brady T., et al. (2013) Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 45, 1050–1054 10.1038/ng.2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Klassen T., Davis C., Goldman A., Burgess D., Chen T., Wheeler D., McPherson J., Bourquin T., Lewis L., Villasana D., Morgan M., Muzny D., Gibbs R., and Noebels J. (2011) Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 145, 1036–1048 10.1016/j.cell.2011.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takahashi S. X., Miriyala J., and Colecraft H. M. (2004) Membrane-associated guanylate kinase-like properties of β-subunits required for modulation of voltage-dependent Ca2+ channels. Proc. Natl. Acad. Sci. U.S.A. 101, 7193–7198 10.1073/pnas.0306665101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wei X., Neely A., Lacerda A. E., Olcese R., Stefani E., Perez-Reyes E., and Birnbaumer L. (1994) Modification of Ca2+ channel activity by deletions at the carboxyl terminus of the cardiac α1 subunit. J. Biol. Chem. 269, 1635–1640 [PubMed] [Google Scholar]
- 21. Altier C., Dubel S. J., Barrère C., Jarvis S. E., Stotz S. C., Spaetgens R. L., Scott J. D., Cornet V., De Waard M., Zamponi G. W., Nargeot J., and Bourinet E. (2002) Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J. Biol. Chem. 277, 33598–33603 10.1074/jbc.M202476200 [DOI] [PubMed] [Google Scholar]
- 22. Christel C., and Lee A. (2012) Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. Biophys. Acta 1820, 1243–1252 10.1016/j.bbagen.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hennessey J. A., Boczek N. J., Jiang Y. H., Miller J. D., Patrick W., Pfeiffer R., Sutphin B. S., Tester D. J., Barajas-Martinez H., Ackerman M. J., Antzelevitch C., Kanter R., and Pitt G. S. (2014) A CACNA1C variant associated with reduced voltage-dependent inactivation, increased CaV1.2 channel window current, and arrhythmogenesis. PLoS One 9, e106982 10.1371/journal.pone.0106982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barrett C. F., and Tsien R. W. (2008) The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc. Natl. Acad. Sci. U.S.A. 105, 2157–2162 10.1073/pnas.0710501105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Buraei Z., and Yang J. (2013) Structure and function of the β subunit of voltage-gated Ca2+ channels. Biochim. Biophys. Acta 1828, 1530–1540 10.1016/j.bbamem.2012.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herlitze S., Hockerman G. H., Scheuer T., and Catterall W. A. (1997) Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proc. Natl. Acad. Sci. U.S.A. 94,1512–1516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berrou L., Bernatchez G., and Parent L. (2001) Molecular determinants of inactivation within the I–II linker of α1E (CaV2.3) calcium channels. Biophys. J. 80, 215–228 10.1016/S0006-3495(01)76008-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bourinet E., Soong T. W., Sutton K., Slaymaker S., Mathews E., Monteil A., Zamponi G. W., Nargeot J., and Snutch T. P. (1999) Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat. Neurosci. 2, 407–415 10.1038/8070 [DOI] [PubMed] [Google Scholar]
- 29. Stotz S. C., and Zamponi G. W. (2001) Identification of inactivation determinants in the domain IIS6 region of high voltage-activated calcium channels. J. Biol. Chem. 276, 33001–33010 10.1074/jbc.M104387200 [DOI] [PubMed] [Google Scholar]
- 30. Tadross M. R., Ben Johny M., and Yue D. T. (2010) Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of CaV1.3 channels. J. Gen. Physiol. 135, 197–215 10.1085/jgp.200910308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Palovcak E., Delemotte L., Klein M. L., and Carnevale V. (2014) Evolutionary imprint of activation: The design principles of VSDs. J. Gen. Physiol. 143, 145–156 10.1085/jgp.201311103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pantazis A., Savalli N., Sigg D., Neely A., and Olcese R. (2014) Functional heterogeneity of the four voltage sensors of a human L-type calcium channel. Proc. Natl. Acad. Sci. U.S.A. 111, 18381–18386 10.1073/pnas.1411127112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Spaetgens R. L., and Zamponi G. W. (1999) Multiple structural domains contribute to voltage-dependent inactivation of rat brain α1E calcium channels. J. Biol. Chem. 274, 22428–22436 10.1074/jbc.274.32.22428 [DOI] [PubMed] [Google Scholar]
- 34. Wu J., Yan Z., Li Z., Qian X., Lu S., Dong M., Zhou Q., and Yan N. (2016) Structure of the voltage-gated calcium channel CaV1.1 at 3.6 Å resolution. Nature 537, 191–196 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- 35. Hamid J., Peloquin J. B., Monteil A., and Zamponi G. W. (2006) Determinants of the differential gating properties of CaV3.1 and CaV3.3 T-type channels: A role of domain IV? Neuroscience 143, 717–728 10.1016/j.neuroscience.2006.08.023 [DOI] [PubMed] [Google Scholar]
- 36. Dolphin A. C. (2013) The α2δ subunits of voltage-gated calcium channels. Biochim. Biophys. Acta 1828, 1541–1549 10.1016/j.bbamem.2012.11.019 [DOI] [PubMed] [Google Scholar]
- 37. McKeown L., Burnham M. P., Hodson C., and Jones O. T. (2008) Identification of an evolutionarily conserved extracellular threonine residue critical for surface expression and its potential coupling of adjacent voltage-sensing and gating domains in voltage-gated potassium channels. J. Biol. Chem. 283, 30421–30432 10.1074/jbc.M708921200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Füll Y., Seebohm G., Lerche H., and Maljevic S. (2013) A conserved threonine in the S1–S2 loop of KV7.2 and KV7.3 channels regulates voltage-dependent activation. Pflugers Arch. 465, 797–804 10.1007/s00424-012-1184-x [DOI] [PubMed] [Google Scholar]
- 39. Long S. B., Campbell E. B., and Mackinnon R. (2005) Voltage sensor of KV1.2: structural basis of electromechanical coupling. Science 309, 903–908 10.1126/science.1116270 [DOI] [PubMed] [Google Scholar]
- 40. Stanika R., Campiglio M., Pinggera A., Lee A., Striessnig J., Flucher B. E., and Obermair G. J. (2016) Splice variants of the CaV1.3 L-type calcium channel regulate dendritic spine morphology. Sci. Rep. 6, 34528 10.1038/srep34528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joensuu M., Lanoue V., and Hotulainen P. (2018) Dendritic spine actin cytoskeleton in autism spectrum disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 84, 362–381 10.1016/j.pnpbp.2017.08.023 [DOI] [PubMed] [Google Scholar]
- 42. Stawarski M., Stefaniuk M., and Wlodarczyk J. (2014) Matrix metalloproteinase-9 involvement in the structural plasticity of dendritic spines. Front. Neuroanat. 8, 68 10.3389/fnana.2014.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olson P. A., Tkatch T., Hernandez-Lopez S., Ulrich S., Ilijic E., Mugnaini E., Zhang H., Bezprozvanny I., and Surmeier D. J. (2005) G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J. Neurosci. 25, 1050–1062 10.1523/JNEUROSCI.3327-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang H., Fu Y., Altier C., Platzer J., Surmeier D. J., and Bezprozvanny I. (2006) CaV1.2 and CaV1.3 neuronal L-type calcium channels: Differential targeting and signaling to pCREB. Eur. J. Neurosci. 23, 2297–2310 10.1111/j.1460-9568.2006.04734.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang Z., Kai L., Day M., Ronesi J., Yin H. H., Ding J., Tkatch T., Lovinger D. M., and Surmeier D. J. (2006) Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron 50, 443–452 10.1016/j.neuron.2006.04.010 [DOI] [PubMed] [Google Scholar]
- 46. Day M., Wang Z., Ding J., An X., Ingham C. A., Shering A. F., Wokosin D., Ilijic E., Sun Z., Sampson A. R., Mugnaini E., Deutch A. Y., Sesack S. R., Arbuthnott G. W., and Surmeier D. J. (2006) Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 9, 251–259 10.1038/nn1632 [DOI] [PubMed] [Google Scholar]
- 47. Graybiel A. M. (2016) The striatum and decision-making based on value. in Micro-, Meso- and Macro-Dynamics of the Brain (Buzsaki G., and Christen Y., eds) pp. 81–84, Springer, New York, NY: 10.1007/978-3-319-28802-4 [DOI] [PubMed] [Google Scholar]
- 48. Krey J. F., Paca S. P., Shcheglovitov A., Yazawa M., Schwemberger R., Rasmusson R., and Dolmetsch R. E. (2013) Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 16, 201–209 10.1038/nn.3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ehlinger D. G., and Commons K. G. (2017) Altered CaV1.2 function in the Timothy syndrome mouse model produces ascending serotonergic abnormalities. Eur. J. Neurosci. 46, 2416–2425 10.1111/ejn.13707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bett G. C., Lis A., Wersinger S. R., Baizer J. S., Duffey M. E., and Rasmusson R. L. (2012) A mouse model of Timothy syndrome: A complex autistic disorder resulting from a point mutation in CaV1.2. N. Am. J. Med. Sci. 5, 135–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Harel T., Hacohen N., Shaag A., Gomori M., Singer A., Elpeleg O., and Meiner V. (2017) Homozygous null variant in CRADD, encoding an adaptor protein that mediates apoptosis, is associated with lissencephaly. Am. J. Med. Genet. 173, 2539–2544 10.1002/ajmg.a.38347 [DOI] [PubMed] [Google Scholar]
- 52. Gregory F. D., Pangrsic T., Calin-Jageman I. E., Moser T., and Lee A. (2013) Harmonin enhances voltage-dependent facilitation of CaV1.3 channels and synchronous exocytosis in mouse inner hair cells. J. Physiol. 591, 3253–3269 10.1113/jphysiol.2013.254367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang S., Stanika R. I., Wang X., Hagen J., Kennedy M. B., Obermair G. J., Colbran R. J., and Lee A. (2017) Densin-180 controls the trafficking and signaling of L-type voltage-gated CaV1.2 Ca2+ channels at excitatory synapses. J. Neurosci. 37, 4679–4691 10.1523/JNEUROSCI.2583-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]