

Abstract
Deficiency in subunits of the conserved oligomeric Golgi (COG) complex results in pleiotropic defects in glycosylation and causes congenital disorders in humans. Insight regarding the functional consequences of this defective glycosylation and the identity of specific glycoproteins affected is lacking. A chemical glycobiology strategy was adopted to identify the surface glycoproteins most sensitive to altered glycosylation in COG-deficient Chinese hamster ovary (CHO) cells. Following metabolic labeling, an unexpected increase in GalNAz incorporation into several glycoproteins, including α-dystroglycan (α-DG), was noted in cog1-deficient ldlB cells. Western blotting analysis showed a significantly lower molecular weight for α-DG in ldlB cells compared with WT CHO cells. The underglycosylated α-DG molecules on ldlB cells are highly vulnerable to bacterial proteases that co-purify with V. cholerae neuraminidase, leading to rapid removal of the protein from the cell surface. The purified bacterial mucinase StcE can cleave both WT and ldlB α-DG but did not cause rapid degradation of the fragments, implicating other V. cholerae proteases in the final proteolysis of the fragments. Extending terminal glycosylation on the existing mucin-type glycans of ldlB α-DG stabilized the resulting fragments, indicating that fragment stability, but not the initial fragmentation of the protein, is influenced by the glycosylation status of the cell. This discovery highlights a functional importance for mucin-type O-glycans found on α-DG and reinforces a growing role for these glycans as regulators of extracellular proteolysis and protein stability.
Keywords: glycobiology, dystroglycan, neuraminidase, click chemistry, protease, conserved oligomeric Golgi complex, mucin-type glycan
Introduction
The congenital disorders of glycosylation are inherited disorders that are caused by defects in the glycosylation machinery of the cell (1–3). The number of CDG2 subtypes identified is rapidly expanding. These disorders are associated with a wide range of clinical manifestations, including neurological and muscle deficits, gastrointestinal complications, and dysmorphia (4–9). The underlying basis for CDGs includes proteins not directly involved in glycosylation such as the subunits of the conserved oligomeric Golgi (COG) complex (1, 10, 11). The eight-subunit COG complex acts as a vesicle tether for COPI-coated vesicles during intra-Golgi retrograde transport (12, 13). Loss of COG integrity impairs the retrograde transport of glycosylation enzymes, resulting in their mislocalization and/or altered activity (14, 15). COG-deficient cells exhibit defects in both protein and lipid glycosylation, with pronounced alterations in N- and O-glycan galactosylation and sialylation (11, 16–19).
Although glycan profiles in COG-deficient cells, patient samples, and animal models are described (16, 18–20), there is little information regarding which glycoproteins are most sensitive to COG-associated glycosylation defects, with the notable exception of the LDL receptor. The sensitivity of this receptor to COG-driven glycosylation defects served as the basis for identifying the CHO cell mutants, ldlB and ldlC, and highlighted the functional importance of proper glycosylation on cell-surface receptor stability (21–23). Identifying additional glycoproteins impacted by the altered glycosylation associated with COG deficiency and other different CDG subtypes will better inform our understanding of how global glycosylation defects influence glycoprotein function and may also yield new insight into the tissue-specific pathogenesis of these disorders.
Here we took advantage of metabolic engineering of COG-deficient ldlB cells and identified α-dystroglycan as a protein profoundly affected by the glycosylation defects found in these cells. A glycoprotein normally associated with muscle cells, α-dystroglycan is also expressed in follicular epithelial cells where it localizes to the basal surface and plays a role in establishing polarity during oogenesis (24, 26). Our data show that the mucin-type glycosylation on α-DG is strongly affected by COG deficiency, leading to its increased susceptibility to proteases that co-purify with V. cholera neuraminidase. Although WT and ldlB α-DG are equally susceptible to fragmentation by the bacterial mucinase, the altered glycosylation on ldlB α-DG enhances the turnover of these fragments by other proteases that co-purify with V. cholerae neuraminidase (Fig. 1). Restoring the terminal glycosylation of existing O-glycans on ldlB α-DG stabilizes the fragments. This work demonstrates for the first time that α-DG is a substrate for bacterial mucinases and highlights a stabilizing role for the mucin-type O-glycans found on this glycoprotein.
Figure 1.

Domain structure of α-dystroglycan and proposed role of mucin-type glycans in protein stability. The mucin region of α-dystroglycan bears both GalNAc- and mannose-initiated glycans flanked by N- and C-terminal domains that contain N-glycans. ManNAz is incorporated into sialic acid on both N- and O-glycans, whereas GalNAz is mainly incorporated into GalNAc-containing O-glycans. The cleavage of α-dystroglycan by bacterial mucinases occurs in both WT and ldlB CHO cells despite major effects on the mucin-type glycans of this glycoprotein. The susceptibility of the resulting fragments toward other bacterial proteases, however, is influenced by the extent of mucin-type glycosylation on the fragments.
Results
Increased labeling of specific glycoproteins with GalNAz in ldlB CHO cells
We envisioned that metabolic labeling of cells with Ac4ManNAz or Ac4GalNAz could be an effective way to identify changes in the glycosylation status and abundance of cell-surface glycoproteins in the glycosylation-deficient cells. By analyzing labeled glycoproteins by SDS-PAGE and Western blotting, glycoprotein profiles can be assessed to determine whether the underlying glycosylation defect alters the mobility or intensity of individual glycoproteins. Lec2 and ldlB CHO cells, deficient in the CMP-sialic acid transporter and COG complex subunit 1, respectively, were chosen as representative cell lines to determine the impact of impaired sialylation alone (Lec2) or broader glycan alterations (ldlB) on labeled glycoproteins. Although ManNAz-treated cells largely display azide-modified sialic acids on their cell surface, GalNAz-treated cells incorporate azido sugars into both N- and O-glycans, as well as nucleocytoplasmic O-GlcNAc-bearing proteins. Thus, the biorthogonal step was performed using S-DIBO-biotin, which is membrane-impermeable, to selectively add biotin to the cell-surface glycoproteins (27). WT CHO cells showed substantial incorporation of SiaNAz after Ac4ManNAz and S-DIBO-biotin labeling (which increased after 48 h), but there was no detectable incorporation of this label in either Lec2 or ldlB cells (Fig. 2). The lack of ManNAz labeling in Lec2 and ldlB was anticipated based on the underlying glycosylation defects in these cell lines. WT CHO cells incorporated GalNAz into multiple cell-surface glycoproteins. This labeling was decreased in the Lec2 cells. Overall GalNAz incorporation increased following 48 h of labeling in both WT and Lec2 cells. GalNAz labeling was unexpectedly higher in the ldlB cells. These cells showed a strongly labeled band at 60 kDa following 24 h of GalNAz treatment. The intensity of this band decreased after longer GalNAz treatment, possibly reflecting higher turnover of this glycoprotein in ldlB cells.
Figure 2.
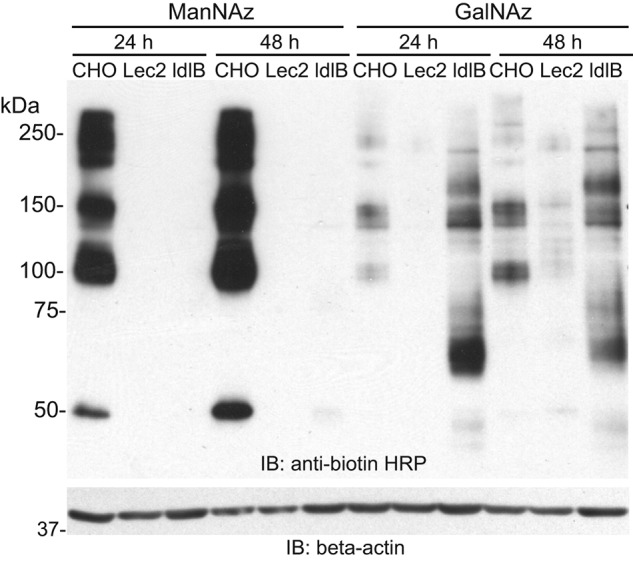
Labeling of cell-surface glycoproteins with GalNAz is increased in ldlB cells. WT CHO, Lec2, and ldlB cells were labeled with Ac4ManNAz or Ac4GalNAz for 24 or 48 h followed by reaction with S-DIBO-biotin and analysis by SDS-PAGE and Western blotting. A representative blot from three independent experiments is shown. IB, immunoblot.
The major GalNAz-labeled 60-kDa band in ldlB cells is an O-glycosylated protein susceptible to V. cholerae proteases that co-purify with its neuraminidase
To determine the nature of the major GalNAz-labeled glycoproteins in ldlB cells, GalNAz-treated WT CHO and ldlB cells were further treated with or without neuraminidase from Vibrio cholerae (VC) and analyzed by SDS-PAGE and Western blotting. Cell lysates from each condition were further treated with PNGase F to assess whether ldlB cells incorporated GalNAz into N- or O-linked glycans of the major labeled glycoproteins including the prominent 60-kDa band. Neuraminidase treatment of WT CHO cells resulted in a shift of the major labeled proteins to a lower molecular weight, consistent with the loss of sialic acid. Surprisingly, neuraminidase treatment of GalNAz-labeled ldlB cells resulted in complete loss of the 60-kDa major labeled band (Fig. 3A). This glycoprotein contains both N- and O-glycans but bears the majority of the GalNAz label within O-glycans because PNGase F reduces the apparent molecular weight but did not result in obvious loss of any biotin label.
Figure 3.

The major GalNAz-labeled 60-kDa band in ldlB cells is an O-glycosylated protein susceptible to V. cholerae proteases that co-purify with its neuraminidase. A, WT CHO and ldlB cells were labeled with Ac4GalNAz, treated with or without V. cholerae neuraminidase, and then reacted with S-DIBO-biotin. The resulting cells were lysed, and the lysates from each condition were treated with or without PNGase F following by SDS-PAGE and Western blotting analysis using an anti-biotin–HRP antibody. A representative blot is depicted from three separate analyses. B, WT CHO and ldlB cells were labeled with ManNAz or GalNAz, and the labeled cells were treated with either V. cholerae or A. ureafaciens neuraminidase, followed by S-DIBO-biotin reaction and Western blotting using anti-biotin–HRP. Note that both neuraminidases can cleave sialic acids, but only the V. cholerae neuraminidase removes the 60-kDa band. The relative intensity of this band was quantified (average ± standard error of the mean; n = 2). C, ManNAz-labeled WT CHO and GalNAz-labeled ldlB cells were treated with V. cholerae neuraminidase in the presence of increasing amounts of the general neuraminidase inhibitor DANA. Following S-DIBO-biotin reaction on whole cells, resulting lysates were subjected to SDS-PAGE and Western blotting using anti-biotin–HRP. A representative blot from two separate experiments is shown. D, GalNAz-labeled ldlB cells were treated with or without V. cholerae neuraminidase in the presence or absence of 50 μm GM6001, a general metalloproteinase inhibitor. Following S-DIBO-biotin reaction, the resulting cell lysates were subjected to Western blotting with anti-biotin–HRP. Quantification of the intensity of the 60-kDa band relative to untreated ldlB cells was performed from three independent experiments (errors bars represent standard error). IB, immunoblot.
V. cholerae contains a hydrolytic complex of enzymes including a neuraminidase and several proteases (28–31). This complex is highly active toward mucins typically found in the airways and intestines. Commercial preparations of V. cholerae neuraminidase contain residual protease activity, because the enzyme is purified from V. cholerae extracts. Arthrobacter ureafaciens (AU) neuraminidase, however, is prepared as a recombinant protein and thus devoid of the protease contaminants found in V. cholerae extracts. We compared VC neuraminidase and AU neuraminidase to address the possibility that the loss of the 60-kDa band in ldlB cells is specific to a certain neuraminidase and/or its substituents. Although both neuraminidases were able to remove azido-sialic acid from ManNAz-treated CHO cells, only the VC neuraminidase was capable of removing the 60-kDa band in GalNAz-treated ldlB cells (Fig. 3B). To verify the removal of the 60-kDa band by the VC-associated protease activity, and not by the neuraminidase activity, we tested co-treatment of a neuraminidase inhibitor, N-acetyl-2,3-dehydro-2-deoxyneuraminic acid (DANA), together with VC neuraminidase after metabolic labeling of ldlB cells with GalNAz. The inhibitory activity of DANA toward neuraminidase was confirmed using CHO cells that were metabolically labeled with ManNAz. This labeling showed effective inhibition of neuraminidase activity at >0.5 mm of DANA (Fig. 3C).
Removal of the 60-kDa band is likely a function of its proteolytic degradation by the VC neuraminidase contaminants, so we tested whether a broad-spectrum matrix metalloproteinase inhibitor, GM6001 (galadin or ilomastat) could impair the removal of this band following treatment of VC neuraminidase. An increase in the level of the 60-kDa band was observed in the presence of GM6001 following neuraminidase treatment of ldlB cells, although this was not sufficient to fully restore the steady-state level seen in non-neuraminidase-treated cells. The abundance of the 60-kDa band was also increased in non-neuraminidase-treated ldlB cells in the presence of GM6001, suggestive of some inherent susceptibility to a metalloproteinase in this cell line (Fig. 3D).
The protease-sensitive 60-kDa glycoprotein in ldlB cells is identified as α-dystroglycan
The removal of the 60-kDa band in the ldlB cells following VC neuraminidase treatment indicated that the surface stability of this glycoprotein is sensitive to the proteolytic components that co-purify with VC neuraminidase. We next sought to identify this 60-kDa glycoprotein using a proteomics-based strategy that takes advantage of this sensitivity. Metabolic labeling of ldlB cells with GalNAz was followed by treatment with or without neuraminidase at 4 or 37 °C (Fig. 4). The 4 °C condition was used to ask whether lowering the temperature of the VC neuraminidase step would slow the loss or turnover of this glycoprotein. Indeed, the abundance of the 60-kDa labeled band was reduced but not absent at 4 °C following neuraminidase treatment, whereas it was removed completely at 37 °C, indicating temperature sensitivity (Fig. 4A). Biotinylated proteins were enriched by immunoprecipitation with anti-biotin antibody, resolved by SDS-PAGE, and subjected to proteomic analysis as described under “Experimental procedures.” The relative spectral counts of identified proteins among three different conditions (4 °C with and without VC neuraminidase and 37 °C with VC neuraminidase) were compared (Fig. 4B). Among the most abundant protein hits, α-dystroglycan (α-DG; DAG) and CD44 showed significantly decreased spectral counts in a temperature-dependent manner upon neuraminidase treatment. To verify the candidates, ldlB cells were labeled with GalNAz and S-DIBO-biotin, and immunoprecipitation performed with either an anti-CD44 antibody (followed by immunoblotting with anti-biotin–HRP) or an anti-biotin antibody (followed by immunoblotting with anti α-DG (core) antibody). The GalNAz labeling of CD44 in ldlB cells was reduced upon neuraminidase treatment, but the molecular weight of the labeled CD44 did not match with the 60-kDa band (Fig. 4C). In contrast, the major GalNAz-labeled 60-kDa band clearly corresponded to the glycoprotein detected by the α-DG core antibody following anti-biotin immunoprecipitation, both with regard to its molecular weight and sensitivity to VC neuraminidase (Fig. 4D).
Figure 4.

Identification of α-dystroglycan as the neuraminidase-sensitive O-glycoprotein in ldlB cells. A, Western blotting analysis of Ac4GalNAz-labeled WT CHO and ldlB cells with or without post-treatment of V. cholerae neuraminidase (n = 2). Note that ldlB cells were treated with neuraminidase at 4 °C or 37 °C. Note that the major labeled 60-kDa glycoprotein is partially spared from neuraminidase-induced loss at the lower temperature. B, normalized spectral counts of a subset of assigned proteins in ldlB cells under the conditions shown. Both DAG1 and CD44 demonstrated decreased abundance following neuraminidase treatment at 37 °C but partial recovery when treated at 4 °C. C, Western blotting analysis of input and eluted fractions with anti-biotin–HRP following immunoprecipitation of GalNAz and S-DIBO-biotin–labeled cell lysates using an anti-CD44 antibody. The cells were treated with or without V. cholerae neuraminidase prior to lysis. D, Western blotting analysis of input, flowthrough, and eluted fractions with anti-DG (core) antisera following immunoprecipitation of lysates from GalNAz and S-DIBO-biotin labeled cells using an anti-biotin antibody. The cells were treated with or without V. cholerae neuraminidase prior to lysis. The asterisk denotes a nonspecific band detected using the anti-DG (core) antibody. The blots shown in C and D are representative of three independent experiments. IB, immunoblot.
Altered mobility of α-DG in COG-deficient CHO cells is a function of COG-dependent defects in O-glycosylation
The electrophoretic mobility of α-DG in WT CHO cells is much different from the one observed in ldlB cells. In WT CHO, the protein exists in multiple bands ranging in size between 110 and 150 kDa, whereas α-DG in ldlB cells shows a single band at 60 kDa. The basis for this large molecular mass shift in α-DG in the ldlB cells was investigated. PNGase F treatment of the lysate showed only a slight decrease of molecular weight of α-DG (5–10 kDa), suggesting that the major portion of molecular weight difference is not due to defects in N-glycosylation, but rather a defect in its O-glycosylation (Fig. 5A). The large molecular weight difference could arise if N-terminal region of α-DG is retained in WT CHO but not in ldlB cells (32, 33). Thus, WT CHO and ldlB cells were treated with an inhibitor of the proprotein convertase furin (one of the convertases responsible for α-DG maturation) to see whether altered processing of the N-terminal region occurred in either cell line. Inhibitor treatment resulted in a similar, dose-dependent shift in the mobility of α-DG in both cell types (Fig. 5B), suggesting that the molecular weight difference of α-DG is not due to a block in its proteolytic maturation by furin-like convertases. Overall, these results strongly suggest that the molecular weight difference of α-DG in between WT CHO and ldlB cells is due to the defect in its O-glycosylation. The abundance and mobility of α-DG was examined in other glycosylation-deficient CHO cell lines (Fig. 5C). Of note, the steady-state level of α-DG was significantly higher in untreated ldlB and ldlC cells compared with WT CHO. Lec2 cells showed similar but nonsialylated forms of α-DG, and its expression level is even lower than WT CHO cells. The cog2-deficient ldlC cells showed the same behavior following VC neuraminidase/protease treatment as ldlB cells. (Fig. 5C). Finally, analysis of the α-DG protein in cog1-corrected ldlB cells showed near complete rescue of the decreased molecular weight of α-DG, suggesting that the altered mobility of α-DG is a direct consequence of COG-related alterations in glycosylation (Fig. 5D).
Figure 5.

Altered mobility of α-DG in COG-deficient CHO cells is a function of COG-dependent defects in O-glycosylation. A, representative Western blotting of WT CHO and ldlB cell lysates with or without PNGase F digestion (n = 2). The cells were treated with or without V. cholerae neuraminidase (120 milliunits/ml) prior to lysis. B, cells were cultured with increasing concentrations of a furin inhibitor, and Western blotting analysis was performed on lysates using an anti-DG (core) antibody (n = 2). Furin inhibition results in a comparable increase in the apparent molecular weight of α-DG in both WT CHO and ldlB cells. C, cells were treated with two different amounts (60 or 120 milliunits/ml) of V. cholerae neuraminidase, and Western blotting analysis was performed. D, Western blotting of α-DG in WT, ldlB, and cog1-corrected ldlB cells treated with or without V. cholerae neuraminidase (n = 2). IB, immunoblot.
StcE mucinase digestion fragments both WT and ldlB α-DG
In light of the fact that multiple hydrolytic activities are known to co-purify with VC neuraminidase, we asked whether bacterial mucinase activity or another protease was responsible for the fragmentation and degradation of α-DG from the surface of ldlB cells. The effects of VC neuraminidase treatment on cell lysates containing a protease inhibitor mixture was first tested, revealing intact neuraminidase activity but no significant degradation of α-DG (Fig. 6A). This experiment confirms the prior observation that the degradation of α-DG in ldlB cells is due to proteolysis and not triggered by desialylation. To ask whether bacterial mucinase activity is specifically responsible for the fragmentation and degradation of the protein, we expressed and purified the mucinase enzyme StcE, a sequelogue of the TagA mucinase found in V. cholerae (34), and assessed its ability to remove cell-surface α-DG in both WT and ldlB cells. The purified mucinase was equally capable of cleaving both WT and ldlB α-DG into distinct fragments (at 45 and 35 kDa for WT CHO and at 35 and 25 kDa for ldlB cells (Fig. 6, B and C). Notably, the resulting fragments were stable even at the highest concentrations of mucinase, consistent with the more selective action of this protease. These data indirectly suggest that the mucinase in the VC neuraminidase preparations is likely responsible for the fragmentation of α-DG but is not the enzyme that completes the degradation of the α-DG fragments. Comparing the purified mucinase and neuraminidase preparations was not possible because of the unknown concentration of contaminating proteases in the neuraminidase preparations. Using ranges for each treatment, we were able to at least qualitatively compare the fragmentation patterns produced by StcE and VC neuraminidase on the same blot (Fig. 6D). Both treatments caused fragmentation of α-DG in ldlB cells, but the pattern of these fragments was distinct, which reinforces the notion that there are likely multiple proteases that co-purify with VC neuraminidase that are capable to cleaving α-DG and ultimately degrading the fragments produced.
Figure 6.

StcE mucinase digestion fragments both WT and ldlB α-DG. A, CHO and ldlB cells were labeled with either ManNAz or GalNAz followed by cell lysis in the presence of a protease inhibitor mixture. Lysates were then treated with or without V. cholerae neuraminidase, and Western blotting was performed using an anti–α-DG core antibody. Relative intensity of the α-DG band was quantifying from three separate experiments. Error bars represent standard error; **, p < 0.01. B and C, CHO (B) and ldlB (C) cells were treated with various concentrations of purified StcE mucinase (1 h), and lysates were resolved by SDS-PAGE prior to Western blotting using the anti–α-DG core antibody. A representative image of two independent experiments is shown. Note the relative stability of the fragments following mucinase cleavage in both cell lines. D, representative Western blotting comparing VC neuraminidase and StcE (2 h) treatment in ldlB cells using the anti–α-DG core antibody. IB, immunoblot.
Terminal extension of existing O-glycans partially inhibits α-DG fragmentation and prolongs fragment half-life
The large difference in molecular weight in ldlB α-DG indicates a loss of O-glycan occupancy or failure to extend existing structures with more complex termini. We tested whether exogenously adding sugars back onto cell-surface α-DG in ldlB cells would result in stabilization of the protein and protection from turnover and/or proteolysis. Enzymatic labeling using ST3Gal1 and its natural substrate CMP-Neu5Ac to extend existing galactose-terminated glycans did not show any molecular weight shift of α-DG (Fig. 7A). However, labeling using Drosophila C1GalT1 and UDP-galactose shifted α-DG to a higher molecular weight and substantially reduced the diversity of different MW species. These data suggest the presence of some O-glycans on ldlB-derived α-DG that were initiated but not extended. This is consistent with the earlier demonstration of weak or absent ManNAz incorporation in these cells. When labeling was performed using C1GalT1, ST3Gal1, UDP-galactose, and CMP-Neu5Ac together, the MW shift of the α-DG band increased further (Fig. 7A). This result indicates that α-DG in ldlB cells bears Tn antigen (Ser/Thr–O-GalNAc), which cannot accept sialic acid directly by ST3Gal1 without prior galactosylation. Nonetheless, these shifts noted are relatively small compared with the large difference in molecular weight between ldlB and WT derived α-DG, which can be interpreted as either the substantial loss of O-glycan occupancy on this glycoprotein or the inefficiency of the enzymatic labeling at the cell surface. The exogenous modification of α-DG with C1GalT1 and ST3Gal1 weakly inhibited the fragmentation of this protein following VC neuraminidase treatment but resulted in significantly greater stability of the 45-kDa α-DG fragment (Fig. 7, B and C). These findings point to a role for the mucin-type glycans in stabilizing the fragments produced by mucinase digestion but not in preventing the initial fragmentation by this enzyme.
Figure 7.

Terminal modification of existing mucin-type O-glycans or increasing O-glycan occupancy inhibits the degradation of surface α-DG and stabilizes the fragments produced by residual V. cholerae protease. A, ldlB cells were SEEL-labeled using the human sialyltransferase ST3Gal1, Drosophila C1GalT1, or a combination of the two enzymes with their natural nucleotide-sugar substrates. Labeled cells were incubated in the presence or absence of A. ureafaciens neuraminidase, and lysates were resolved by SDS-PAGE and blotted with the anti-DG (core) antibody. B, ldlB cells labeled with C1GalT1 alone or in combination with ST3Gal1 were treated with or without V. cholerae neuraminidase, followed by SDS-PAGE and Western blotting with the anti-DG (core) antibody. C, quantification of the abundance of the 60-kDa α-DG band and the 45-kDa fragment in neuraminidase-treated cells relative to the abundance of the 60-kDa α-DG band in untreated cells (n = 3). Error bars represent standard deviations. *, p < 0.05. Cntrl, control; IB, immunoblot; neur, neuraminidase.
Discussion
We took advantage of a metabolic engineering strategy to identify α-DG as a glycoprotein that is strongly sensitive to the glycosylation defects associated with COG deficiency. The profound loss of O-glycan glycosylation on α-DG in the cog1-deficient ldlB CHO mutant results in a form of the protein that is highly susceptible to degradation by bacterial proteases that co-purify with V. cholerae neuraminidase. These findings highlight another possible route whereby altered glycosylation can influence the critical function of this cell-surface glycoprotein. The implications of these findings with regard to the function of mucin-type glycans and disease pathogenesis are considered below.
The rapid loss of α-DG from the surface of ldlB cells upon treatment with commercial VC neuraminidase is a function of the proteases that co-purify with the neuraminidase. V. cholerae is known to contain a complex of hydrolases including the neuraminidase, a hexosaminidase, and several proteases including a mucinase that allow the bacteria to invade epithelial layers in the lung and intestines (28, 30, 31). The specific role of a bacterial mucinase in the degradation of α-DG was tested in both WT and ldlB cells using purified StcE, an enzyme with high similarity to the V. cholerae mucinase TagA. We found that both WT and ldlB α-DG is efficiently fragmented by StcE, suggesting that other VC proteases are likely responsible for the rapid degradation of α-DG in ldlB cells compared with WT CHO cells. Augmenting the existing mucin-type O-glycans on ldlB cells stabilized the α-DG fragments (Fig. 7, B and C), supporting the conclusion that fragment stability is influenced by the glycosylation state of the glycoprotein.
Krieger and co-workers (21–23) identified cog1-deficient ldlB cells as one of four complementation groups that exhibited greatly decreased LDL receptor on the cell surface. Subsequent work lead to the conclusion that altered O-glycosylation of this receptor in the three of these mutants causes its rapid internalization and turnover. The present study identifies α-DG as another heavily O-glycosylated glycoprotein that is affected in these cells and reinforces a functional role for mucin-type (O-GalNAc) glycosylation in protein stability. In ldlB cells, the LDL receptor is made at a normal rate but turned over 5–10-fold faster in ldlB cells, resulting in only residual levels of receptor activity at the cell surface (21). In contrast, the steady-state level of α-DG is increased in ldlB cells compared with WT CHO cells as assessed by the core antibody. It is noteworthy that α-DG is so well labeled with GalNAz despite the loss of O-glycan structures that would typically incorporate the bulk of this sugar. We interpret the heavy GalNAz labeling to likely reflect that fact that the S-DIBO molecule used for the bio-orthogonal step can better access and react with incorporated GalNAz if the terminal structures are devoid of sialic acid. This interpretation is bolstered by the overall increased GalNAz labeling that was noted following removal of sialic acids (Figs. 3B and 4, A and C).
Screens performed in haploid cells identified COG complex subunits as contributors to the α-DG–dependent entry of viruses such lassa virus into the cells (35, 36). Although yet to be formally proven, it is likely that loss of COG complex function impacts multiple glycosyltransferases responsible for the biosynthesis of the M3 glycan (matriglycan) that is assembled on α-DG in WT cells because many of these enzymes required for matriglycan synthesis reside in the COG-sensitive Golgi complex. These new findings, however, suggest that COG-associated defects in glycosylation may also compromise α-DG function at the cell surface through changes in mucin-type glycosylation, as well as O-mannose glycosylation. Prior studies in CHO cells demonstrated that little if any of the M3 glycan is made in this cell type, although overexpression of the LARGE enzyme can lead to the production of higher molecular weight forms of α-DG (37). We were also not able to detect any appreciable IIH6 reactivity in this cell type. Nonetheless, loss of a limited number of O-mannose–initiated glycans would not account for the dramatic shift in molecular weight in the ldlB cells. Moreover, O-mannose glycans are initiated in the ER, whereas mucin-type glycosylation begins in the Golgi. Effects of COG deficiency may be restricted to glycan initiation and processing events in the Golgi, because the primary action of this complex is maintaining the fidelity of retrograde transport of glycosyltransferase-bearing COPI vesicles in Golgi and post-Golgi compartments. Future studies will be aimed at defining how COG complex deficiency alters the biosynthesis of the M3 glycan in muscle cells and whether the early steps in this process are affected.
COG-deficient human patients have multisystem involvement, but muscle phenotypes appear to be limited compared with other CDGs that directly impact the O-mannosylation of α-DG (11, 16, 17, 38–42). Overt muscular dystrophy is not typically noted, suggesting that α-DG glycosylation may not be as affected in muscle as what was observed in the CHO cells. The present study implies that O-glycan occupancy may be a bigger factor in the protection and/or stability of α-DG than the extent to which the O-GalNAc glycans in the mucin region are elaborated. The data in Fig. 7A clearly demonstrate the presence of Tn antigen on α-DG and the ability to extend these glycans to form sialyl T antigen. Attempts were made to fill in the likely loss of O-glycan occupancy within this region using a selective exo-enzymatic labeling (SEEL) approach with ppGalNAcT1/ppGalNAcT2 and UDP-GalNAc, but we were unable to detect any molecular weight shift in α-DG or any protection from V. cholerae proteases. The polypeptide GalNAcTs may need to be used in combination to re-establish the occupancy of the mucin region, but it is equally possible that the conformation of the α-DG protein at the cell surface presents a steric barrier, precluding the use of a “corrective SEEL” approach.
Experimental procedures
Reagents
Ac4ManNAz, Ac4GalNAz, and S-DIBO-biotin were prepared as reported (27, 43). Recombinant enzymes including α-(2,3)-sialyltransferase (ST3Gal1) or Drosophila C1GalT1 were prepared as previously described (44), UDP-GalNAc (U5252), V. cholerae neuraminidase (N6514), DANA (D9050), and protein G beads (protein G–Sepharose, Fast Flow, P3296) were purchased from Sigma–Aldrich. A. ureafaciens neuraminidase (P0722) PNGase F (P0704S) were purchased from New England Biolabs. Alkaline phosphatase (FastAP, EF0651), protease inhibitor mixture tablet (88666), and a MS compatible silver staining kit (24600) were purchased from Thermo Scientific. Mouse monoclonal anti-biotin antibodies (200-032-211, HRP-conjugated from or 200–002-211, nonconjugated form) were purchased from Jackson ImmunoResearch Laboratories. HRP-conjugated anti-β-actin antibody (ab20272) and anti-CD44 antibody (KM81, ab112178) were from Abcam. UDP-galactose (670111), CMP-sialic acid (233264), GM-6001 (CC1010), and furin inhibitor I (344930, decanoyl-RVKR-CMK) were purchased from EMD Millipore.
Cell lines and culture
WT CHO cells (clone K1, ATCC), mutant CHO cells (Lec2, ldlB, or ldlC) and a Cog1-transfected ldlB cells, ldlB[ldlB] were cultured in minimum essential medium α 1X (Cellgro) with Earle's salts, without ribonucleosides, deoxyribonucleosides, and l-glutamine with 10% fetal bovine serum (FBS, BenchMark) and penicillin (100 IU/ml)/streptomycin (100 μg/ml, MediaTech). Cells were cultured in a 5% CO2 atmosphere, 37 °C humid incubator.
Metabolic labeling and enzymatic treatment of cells
Metabolic labeling of cell surface was typically done by incubation of cells with 50–60% confluency in 12-well, 6-well, or 10-cm dishes with 30 μm of Ac4ManNAz or 100 μm of Ac4GalNAz for 24 h in the same cell culture medium at 37 °C. In case of 48 h of labeling, 25–30% confluent cells were used to obtain confluent cells at the end of labeling. After metabolic labeling, the cells were washed with Dulbecco's PBS, and then cells were further incubated with 30 μm of S-DIBO-biotin in Dulbecco's PBS containing 2% FBS for 1 h at room temperature. Treatment of (±) sialidase, in general, was made right after metabolic labeling and before S-DIBO-biotin reaction. Typically, 50 milliunits/ml of V. cholerae neuraminidase or 67 units/ml of A. ureafaciens neuraminidase (determined empirically as equivalent concentration to 50 milliunits/ml of V. cholerae neuraminidase for similar removal of azide-containing sialic acid from metabolically labeled cells with Ac4ManNAz) was treated in serum-free cell culture medium for 2 h at 37 °C. The labeled cells were directly lysed on plate with radioimmune precipitation assay buffer (1.0% NP-40, 50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, and 0.5% sodium deoxycholate), and the resulting lysate was analyzed by immunoblot using anti-biotin–HRP.
Immunoprecipitation and LC-MS/MS proteomics
For identification of the 60-kDa protein(s), ldlB cells were labeled by Ac4GalNAz, and then the labeled cells were further treated with (±) V. cholerae neuraminidase at 37 °C or 4 °C. When V. cholerae neuraminidase was treated at 4 °C, 460 mm sucrose was used. The resulting cells were then treated with S-DIBO-biotin at room temperature for 1 h. The cells were then lysed with radioimmune precipitation assay buffer, and each 2.8 mg of lysate from was used to pull down the biotinylated proteins. Briefly, each lysate was precleared for 1 h with protein G beads (20 μl), and then the precleared lysate was further incubated with protein G beads (20 μl) preconjugated with anti-biotin antibody (13.3 μl) overnight at 4 °C. Elution of biotinylated proteins from beads, SDS-PAGE, silver staining, in gel digestion of gel pieces between 50 and 75 kDa, and LC-MS-MS analysis were done as previously reported (44). The gel bands were destained, denatured by incubating with 10 mm of DTT at 56 °C for an hour, and alkylated by 55 mm of iodoacetamide for 45 min in dark prior to digestion with trypsin overnight. The resulting peptides were separated on a 75-μm (inner diameter) × 15-cm C18 capillary column (packed in house, YMC GEL ODS-AQ120ÅS-5, Waters) and eluted into the nano-electrospray ion source of an Orbitrap FusionTM TribridTM mass spectrometer (Thermo Fisher Scientific) with a 180-min linear gradient consisting of 0.5–100% solvent B over 150 min at a flow rate of 200 nl/min. The spray voltage was set to 2.2 kV, and the temperature of the heated capillary was set to 280 °C. Full MS scans were acquired from m/z 300 to 2000 at 120k resolution, and MS2 scans following collision-induced fragmentation were collected in the ion trap for the most intense ions in the Top-Speed mode within a 3-s cycle using Fusion instrument software (v1.1, Thermo Fisher Scientific). The raw spectra were searched against the human protein database (UniProt, Oct. 2014) using SEQUEST (Proteome Discoverer 1.4, Thermo Fisher Scientific) with full MS peptide tolerance of 20 ppm and MS2 peptide fragment tolerance of 0.5 Da, and filtered using ProteoIQ (v2.7, Premier Biosoft) at the protein level to generate a 1% false discovery rate for protein assignments. UniProt was used to determine cellular localization of the identified proteins. Quantification was performed by normalizing the spectral counts generated in ProteoIQ (v2.7, Premier Biosoft). For verification of candidate proteins, CHO and ldlB cells were labeled again with Ac4GalNAz and then treated with V. cholerae neuraminidase at 37 °C for 2 h followed by biotinylation reaction with S-DIBO. Immunoprecipitation of CD44 was done from each lysate (600 μg) by using 2.2 μg of anti-CD44 [KM81] antibody and 10 μl of protein G beads. Western blotting of the immunoprecipitate fractions was done with anti-biotin–HRP (1:50,000, 4 °C overnight), or biotinylated proteins from each lysate (250 μg each) were pulled down by anti-biotin antibody, and the eluted proteins were analyzed by immunoblot with anti–α-DG (core) antibody (clone 5-2, 1:7 dilution, 3 days) (45).
Treatment of cells with GM6001, DANA, and furin inhibitor I
Metalloprotease inhibitor GM6001 (50 μm) was co-treated with V. cholerae neuraminidase (50 milliunits/ml) in the serum-free culture media to ldlB cells that were prelabeled by Ac4GalNAz (100 μm) for 24 h in 10% FBS-containing medium. After 2 h of the treatment, the cells were then biotinylated by S-DIBO-biotin for 1 h at room temperature followed by lysis for immunoblotting. DANA, a sialidase inhibitor, with various concentrations (0, 0.5, 1, 2, and 4 mm) was co-treated with V. cholerae neuraminidase (typically 50 milliunits/ml if not specified) in serum-free culture medium for 2 h to CHO cells that were prelabeled with Ac4ManNAz (30 μm, 24 h, 10% FBS-containing culture medium) or ldlB cells that were prelabeled by Ac4GalNAz (100 μm, 24 h, 10% FBS-containing culture medium). Then cells were biotinylated by S-DIBO-biotin and then lysed for immunoblot. Furin inhibitor I (decanoyl-PVKR-CMK) with various concentration (0, 5, 10, 20, 50, and 100 μm) were treated to CHO or ldlB cells for 24 h, and then the resulting cells were lysed and analyzed by immunoblot with anti–α-DG (core) antibody.
Expression and purification of the StcE mucinase
Recombinant StcE was expressed using ER2566 cells (IMPACT system, New England Biolabs) containing StcE expression vector (pTEG11) by following the New England Biolabs manual (New England Biolabs catalog no. E69015 V. 3.1). The ER2566 cells expressing StcE was obtained as a generous gift from Dr. Rodney A. Welch's laboratory. Briefly, the ER2566 cells were grown in 100 mm LB +Amp medium until A600 reached ∼0.43 at 37 °C, and then 0.4 mm isopropyl β-d-thiogalactopyranoside was added followed by overnight incubation at room temperature. The resulting cells were harvested and lysed by sonication in 10 ml of column buffer containing 20 mm Tris-HCl, pH 8.0, 500 mm NaCl, 0.1% Triton X-100 on ice. The lysed cells were centrifuged at 15,000 × g for 30 min at 4 °C. The resulting supernatant was collected and loaded on the chitin column (New England Biolabs, catalog no. S6651S, 2 ml of bed volume) for purification. After the loading and washing steps, the resulting chitin column was quickly flushed with column buffer containing 50 mm DTT and then incubated overnight at room temperature. Next, StcE was eluted with column buffer (400 μl for each fraction), and StcE-containing fractions (1∼5) were determined by Coomassie Blue staining. The resulting StcE-containing fractions were dialyzed and concentrated via spin column (molecular mass cutoff, 30 kDa) to give final volume of ∼1.0 ml in 20 mm Tris-HCl, 50 mm NaCl, and 5% glycerol. Protein concentration of StcE (3.1 mg/ml) was determined by using Bio-Rad protein assay dye reagent (Bio-Rad, catalog no. 5000006).
Treatment of StcE on CHO and ldlB cells
Confluent CHO or ldlB cells cultured in 12-well dishes were incubated with various amount of StcE (0, 0.03, 0.06, 0.13, 0.26, 0.53, 1.05, and 2.10 μg/ml) in serum-free cell culture medium for 1 h at 37 °C. The resulting cells were lysed and then analyzed by immunoblot with anti–α-DG (core) antibody. The comparison of V. cholerae neuraminidase and StcE treatment on ldlB cells was made by treating confluent ldlB cells with various amount of StcE (0, 0.26, 0.53, 1.05, and 2.10 μg/ml) or V. cholerae neuraminidase (18, 35, 70, and 140 milliunits/ml) in serum cell culture medium for 2 h at 37 °C. The resulting cells were lysed and then analyzed by immunoblot with anti–α-DG (core) antibody.
Corrective SEEL
Corrective SEEL reaction of ldlB cells were typically done in 6-well dishes with 600 μl of final volume. For SEEL with ST3Gal1, the cells were incubated with ST3Gal1 (25 μg), FastAP (4U), BSA (4 μl of 2 mg/ml), and CMP-Neu5Ac (220 μm as final concentration) in serum-free culture medium for 2 h at 37 °C. For SEEL with C1GalT1, the cells were incubated with C1GalT1 (30 μg), FastAP (8U), BSA (4 μl of 2 mg/ml), MnCl2 (2 mm), and UDP-galactose (2 mm) in serum-free cell culture medium for 2h at 37 °C. For SEEL with both enzymes, the cells were incubated with ST3Gal1 (25 μg), C1GalT1 (30 μg), FastAP (8U), BSA (4 μl of 2 mg/ml), MnCl2 (2 mm), CMP-Neu5Ac (220 μm), and UDP-galactose (2 mm) in serum-free medium for 2 h at 37 °C. For control, the cells were incubated with serum-free medium with BSA only or treated with the same SEEL reaction mixture without UDP-galactose. After the SEEL reaction was done, optionally, the cells were further treated with V. cholerae neuraminidase for 2h at 37 °C before lysis. After lysis, the cells were analyzed by immunoblot with anti–α-DG (core) antibody. Recombinant forms of ST3Gal1 and C1GalT1 were generated as previously described (25) except that a truncated form of Drosophila melanogaster C1GalT1 (residues 43–388, UniProt Q7K237) was generated in the pGEn2 mammalian expression vector as a GFP fusion protein. Both fusion proteins were purified from the conditioned HEK293 cell conditioned medium by Ni2+–nitrilotriacetic acid chromatography and gel filtration prior to use.
Author contributions
S.-H. Y., G.-J. B., K. W. M., L. W., and R. S. conceptualization; S.-H. Y., P. Z., and R. S. formal analysis; S.-H. Y. and P. Z. investigation; S.-H. Y., P. Z., G.-J. B., L. W., and R. S. methodology; S.-H. Y. and R. S. writing-original draft; P. K. P., T. S., A. B., G.-J. B., K. W. M., and L. W. resources; G.-J. B., K. W. M., L. W., and R. S. funding acquisition; L. W. data curation; R. S. project administration; R. S. writing-review and editing.
Supplementary Material
Acknowledgments
We are grateful to Pamela Stanley and Monty Krieger for the CHO cell mutants used in this study. We acknowledge Rod Welch and Kevin Schwartz (University of Wisconsin) for providing us with the StcE expression system.
This work was supported by National Institutes of Health Grants P01GM107012 (to R. S., K. W. M., L. W., and G.-J. B.) and R01GM111939 (to L. W. and A. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains a data set.
- CDG
- congenital disorder of glycosylation
- COG
- conserved oligomeric Golgi
- α-DG
- α-dystroglycan
- LDL
- low density lipoprotein
- CHO
- Chinese hamster ovary
- VC
- V. cholera
- AU
- A. ureafaciens
- SEEL
- selective exo-enzymatic labeling
- DANA
- N-acetyl-2,3-dehydro-2-deoxyneuraminic acid
- HRP
- horseradish peroxidase
- FBS
- fetal bovine serum
- PNGase
- peptide:N-glycosidase.
References
- 1. Freeze H. H. (2007) Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr. Mol. Med. 7, 389–396 10.2174/156652407780831548 [DOI] [PubMed] [Google Scholar]
- 2. Jaeken J. (2010) Congenital disorders of glycosylation. Ann. N.Y. Acad. Sci. 1214, 190–198 10.1111/j.1749-6632.2010.05840.x [DOI] [PubMed] [Google Scholar]
- 3. Jaeken J., and Matthijs G. (2007) Congenital disorders of glycosylation: a rapidly expanding disease family. Annu. Rev. Genomics Hum. Genet. 8, 261–278 10.1146/annurev.genom.8.080706.092327 [DOI] [PubMed] [Google Scholar]
- 4. Haeuptle M. A., and Hennet T. (2009) Congenital disorders of glycosylation: an update on defects affecting the biosynthesis of dolichol-linked oligosaccharides. Hum. Mutat. 30, 1628–1641 10.1002/humu.21126 [DOI] [PubMed] [Google Scholar]
- 5. Kim Y. M., Seo G. H., Jung E., Jang J. H., Kim S. Z., and Lee B. H. (2018) Characteristic dysmorphic features in congenital disorders of glycosylation type IIb. J. Hum. Genet. 63, 383–386 10.1038/s10038-017-0386-7 [DOI] [PubMed] [Google Scholar]
- 6. Krasnewich D., O'Brien K., and Sparks S. (2007) Clinical features in adults with congenital disorders of glycosylation type Ia (CDG-Ia). Am. J. Med. Genet. C Semin. Med. Genet. 145C, 302–306 10.1002/ajmg.c.30143 [DOI] [PubMed] [Google Scholar]
- 7. Marques-da-Silva D., Dos Reis Ferreira V., Monticelli M., Janeiro P., Videira P. A., Witters P., Jaeken J., and Cassiman D. (2017) Liver involvement in congenital disorders of glycosylation (CDG): a systematic review of the literature. J. Inherit. Metab. Dis. 40, 195–207 10.1007/s10545-016-0012-4 [DOI] [PubMed] [Google Scholar]
- 8. Marques-da-Silva D., Francisco R., Webster D., Dos Reis Ferreira V., Jaeken J., and Pulinilkunnil T. (2017) Cardiac complications of congenital disorders of glycosylation (CDG): a systematic review of the literature. J. Inherit. Metab. Dis. 40, 657–672 10.1007/s10545-017-0066-y [DOI] [PubMed] [Google Scholar]
- 9. Barone R., Fiumara A., and Jaeken J. (2014) Congenital disorders of glycosylation with emphasis on cerebellar involvement. Semin. Neurol. 34, 357–366 10.1055/s-0034-1387197 [DOI] [PubMed] [Google Scholar]
- 10. Hennet T. (2012) Diseases of glycosylation beyond classical congenital disorders of glycosylation. Biochim. Biophys. Acta 1820, 1306–1317 10.1016/j.bbagen.2012.02.001 [DOI] [PubMed] [Google Scholar]
- 11. Wu X., Steet R. A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., and Freeze H. H. (2004) Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 10, 518–523 10.1038/nm1041 [DOI] [PubMed] [Google Scholar]
- 12. Ungar D., Oka T., Brittle E. E., Vasile E., Lupashin V. V., Chatterton J. E., Heuser J. E., Krieger M., and Waters M. G. (2002) Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 157, 405–415 10.1083/jcb.200202016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zolov S. N., and Lupashin V. V. (2005) Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J. Cell Biol. 168, 747–759 10.1083/jcb.200412003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oka T., Ungar D., Hughson F. M., and Krieger M. (2004) The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell 15, 2423–2435 10.1091/mbc.e03-09-0699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shestakova A., Zolov S., and Lupashin V. (2006) COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic 7, 191–204 10.1111/j.1600-0854.2005.00376.x [DOI] [PubMed] [Google Scholar]
- 16. Foulquier F., Vasile E., Schollen E., Callewaert N., Raemaekers T., Quelhas D., Jaeken J., Mills P., Winchester B., Krieger M., Annaert W., and Matthijs G. (2006) Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. U.S.A. 103, 3764–3769 10.1073/pnas.0507685103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kranz C., Ng B. G., Sun L., Sharma V., Eklund E. A., Miura Y., Ungar D., Lupashin V., Winkel R. D., Cipollo J. F., Costello C. E., Loh E., Hong W., and Freeze H. H. (2007) COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 16, 731–741 10.1093/hmg/ddm028 [DOI] [PubMed] [Google Scholar]
- 18. Palmigiano A., Bua R. O., Barone R., Rymen D., Régal L., Deconinck N., Dionisi-Vici C., Fung C. W., Garozzo D., Jaeken J., and Sturiale L. (2017) MALDI-MS profiling of serum O-glycosylation and N-glycosylation in COG5-CDG. J. Mass Spectrom. 52, 372–377 10.1002/jms.3936 [DOI] [PubMed] [Google Scholar]
- 19. Xia B., Zhang W., Li X., Jiang R., Harper T., Liu R., Cummings R. D., and He M. (2013) Serum N-glycan and O-glycan analysis by mass spectrometry for diagnosis of congenital disorders of glycosylation. Anal. Biochem. 442, 178–185 10.1016/j.ab.2013.07.037 [DOI] [PubMed] [Google Scholar]
- 20. Frappaolo A., Sechi S., Kumagai T., Robinson S., Fraschini R., Karimpour-Ghahnavieh A., Belloni G., Piergentili R., Tiemeyer K. H., Tiemeyer M., and Giansanti M. G. (2017) COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes. J. Cell Sci. 130, 3637–3649 10.1242/jcs.209049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kingsley D. M., Kozarsky K. F., Segal M., and Krieger M. (1986) Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 102, 1576–1585 10.1083/jcb.102.5.1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kingsley D. M., and Krieger M. (1984) Receptor-mediated endocytosis of low density lipoprotein: somatic cell mutants define multiple genes required for expression of surface-receptor activity. Proc. Natl. Acad. Sci. U.S.A. 81, 5454–5458 10.1073/pnas.81.17.5454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kozarsky K. F., Brush H. A., and Krieger M. (1986) Unusual forms of low density lipoprotein receptors in hamster cell mutants with defects in the receptor structural gene. J. Cell Biol. 102, 1567–1575 10.1083/jcb.102.5.1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Deng W. M., Schneider M., Frock R., Castillejo-Lopez C., Gaman E. A., Baumgartner S., and Ruohola-Baker H. (2003) Dystroglycan is required for polarizing the epithelial cells and the oocyte in Drosophila. Development 130, 173–184 10.1242/dev.00199 [DOI] [PubMed] [Google Scholar]
- 25. Moremen K. W., Ramiah A., Stuart M., Steel J., Meng L., Forouhar F., Moniz H. A., Gahlay G., Gao Z., Chapla D., Wang S., Yang J. Y., Prabhakar P. K., Johnson R., Rosa M. D., et al. (2018) Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 14, 156–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schneider M., Khalil A. A., Poulton J., Castillejo-Lopez C., Egger-Adam D., Wodarz A., Deng W. M., and Baumgartner S. (2006) Perlecan and Dystroglycan act at the basal side of the Drosophila follicular epithelium to maintain epithelial organization. Development 133, 3805–3815 10.1242/dev.02549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Friscourt F., Ledin P. A., Mbua N. E., Flanagan-Steet H. R., Wolfert M. A., Steet R., and Boons G. J. (2012) Polar dibenzocyclooctynes for selective labeling of extracellular glycoconjugates of living cells. J. Am. Chem. Soc. 134, 5381–5389 10.1021/ja3002666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Burnet F. M., and Stone J. D. (1947) Desquamation of intestinal epithelium in vitro by V. cholerae filtrates: characterization of mucinase and tissue disintegrating enzymes. Aust. J. Exp. Biol. Med. Sci. 25, 219–226 10.1038/icb.1947.32 [DOI] [PubMed] [Google Scholar]
- 29. Crennell S., Garman E., Laver G., Vimr E., and Taylor G. (1994) Crystal structure of Vibrio cholera neuraminidase reveals dual lectin-like domains in addition to the catalytic domain. Structure 2, 535–544 10.1016/S0969-2126(00)00053-8 [DOI] [PubMed] [Google Scholar]
- 30. Schneider D. R., and Parker C. D. (1982) Purification and characterization of the mucinase of Vibrio cholerae. J. Infect. Dis. 145, 474–482 10.1093/infdis/145.4.474 [DOI] [PubMed] [Google Scholar]
- 31. Stewart-Tull D. E., Ollar R. A., and Scobie T. S. (1986) Studies on the Vibrio cholerae mucinase complex: I. enzymic activities associated with the complex. J. Med. Microbiol. 22, 325–333 10.1099/00222615-22-4-325 [DOI] [PubMed] [Google Scholar]
- 32. Brancaccio A., Schulthess T., Gesemann M., and Engel J. (1997) The N-terminal region of α-dystroglycan is an autonomous globular domain. Eur. J. Biochem. 246, 166–172 10.1111/j.1432-1033.1997.00166.x [DOI] [PubMed] [Google Scholar]
- 33. Heng S., Paule S. G., Li Y., Rombauts L. J., Vollenhoven B., Salamonsen L. A., and Nie G. (2015) Posttranslational removal of α-dystroglycan N terminus by PC5/6 cleavage is important for uterine preparation for embryo implantation in women. FASEB J. 29, 4011–4022 10.1096/fj.14-269456 [DOI] [PubMed] [Google Scholar]
- 34. Szabady R. L., Yanta J. H., Halladin D. K., Schofield M. J., and Welch R. A. (2011) TagA is a secreted protease of Vibrio cholerae that specifically cleaves mucin glycoproteins. Microbiology 157, 516–525 10.1099/mic.0.044529-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jae L. T., Raaben M., Herbert A. S., Kuehne A. I., Wirchnianski A. S., Soh T. K., Stubbs S. H., Janssen H., Damme M., Saftig P., Whelan S. P., Dye J. M., and Brummelkamp T. R. (2014) Virus entry: lassa virus entry requires a trigger-induced receptor switch. Science 344, 1506–1510 10.1126/science.1252480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jae L. T., Raaben M., Riemersma M., van Beusekom E., Blomen V. A., Velds A., Kerkhoven R. M., Carette J. E., Topaloglu H., Meinecke P., Wessels M. W., Lefeber D. J., Whelan S. P., van Bokhoven H., and Brummelkamp T. R. (2013) Deciphering the glycosylome of dystroglycanopathies using haploid screens for lassa virus entry. Science 340, 479–483 10.1126/science.1233675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Aguilan J. T., Sundaram S., Nieves E., and Stanley P. (2009) Mutational and functional analysis of Large in a novel CHO glycosylation mutant. Glycobiology 19, 971–986 10.1093/glycob/cwp074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lübbehusen J., Thiel C., Rind N., Ungar D., Prinsen B. H., de Koning T. J., van Hasselt P. M., and Körner C. (2010) Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum. Mol. Genet. 19, 3623–3633 10.1093/hmg/ddq278 [DOI] [PubMed] [Google Scholar]
- 39. Paesold-Burda P., Maag C., Troxler H., Foulquier F., Kleinert P., Schnabel S., Baumgartner M., and Hennet T. (2009) Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 18, 4350–4356 10.1093/hmg/ddp389 [DOI] [PubMed] [Google Scholar]
- 40. Rymen D., Keldermans L., Race V., Régal L., Deconinck N., Dionisi-Vici C., Fung C. W., Sturiale L., Rosnoblet C., Foulquier F., Matthijs G., and Jaeken J. (2012) COG5-CDG: expanding the clinical spectrum. Orphanet J. Rare Dis. 7, 94 10.1186/1750-1172-7-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rymen D., Winter J., Van Hasselt P. M., Jaeken J., Kasapkara C., Gokçay G., Haijes H., Goyens P., Tokatli A., Thiel C., Bartsch O., Hecht J., Krawitz P., Prinsen H. C., Mildenberger E., et al. (2015) Key features and clinical variability of COG6-CDG. Mol. Genet. Metab. 116, 163–170 10.1016/j.ymgme.2015.07.003 [DOI] [PubMed] [Google Scholar]
- 42. Lefeber D. J., Schönberger J., Morava E., Guillard M., Huyben K. M., Verrijp K., Grafakou O., Evangeliou A., Preijers F. W., Manta P., Yildiz J., Grünewald S., Spilioti M., van den Elzen C., Klein D., et al. (2009) Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am. J. Hum. Genet. 85, 76–86 10.1016/j.ajhg.2009.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mbua N. E., Guo J., Wolfert M. A., Steet R., and Boons G. J. (2011) Strain-promoted alkyne-azide cycloadditions (SPAAC) reveal new features of glycoconjugate biosynthesis. Chembiochem 12, 1912–1921 10.1002/cbic.201100117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yu S. H., Zhao P., Sun T., Gao Z., Moremen K. W., Boons G. J., Wells L., and Steet R. (2016) Selective exo-enzymatic labeling detects increased cell surface sialoglycoprotein expression upon megakaryocytic differentiation. J. Biol. Chem. 291, 3982–3989 10.1074/jbc.M115.700369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fortunato M. J., Ball C. E., Hollinger K., Patel N. B., Modi J. N., Rajasekaran V., Nonneman D. J., Ross J. W., Kennedy E. J., Selsby J. T., and Beedle A. M. (2014) Development of rabbit monoclonal antibodies for detection of α-dystroglycan in normal and dystrophic tissue. PLoS One 9, e97567 10.1371/journal.pone.0097567 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.