

Abstract
Biofilms are communities of microbes embedded in a microbial extracellular matrix. Their formation is considered the main virulence mechanism enabling the opportunistic bacterial pathogen Staphylococcus epidermidis to cause devastating nosocomial, implant-associated infections. Biofilms often contain proteins, and an 18-kDa protein called small basic protein (Sbp) recently was discovered in the S. epidermidis biofilm matrix and may serve as a scaffolding protein in both polysaccharide intercellular adhesin (PIA)-dependent and accumulation-associated protein (Aap)-dependent biofilm formations. In Aap-mediated biofilm formation, Sbp colocalizes with Domain-B of Aap, implying that Sbp directly interacts with Aap's Domain-B. However, the structure of Sbp and its interaction with Aap, as well as the molecular mechanism underlying Sbp's roles in biofilm formation, are incompletely understood. In this work, we used small-angle X-ray scattering (SAXS), NMR, analytical size-exclusion chromatography, and isothermal titration calorimetry analyses to determine the Sbp structure and characterize its interaction with Aap's Domain-B. We found that Sbp is monomeric and partially folded in solution, and, unexpectedly, we observed no direct interactions between Sbp and Aap Domain-B. Instead, we noted that Sbp forms amyloid fibrils both in vitro and in vivo. Atomic force, transmission electron, and confocal fluorescence microscopy methods confirmed the formation of Sbp amyloid fibrils and revealed their morphology. Taken together, the Sbp amyloid fibril structures identified here may account for Sbp's role as a scaffolding protein in the S. epidermidis biofilm matrix.
Keywords: amyloid, small-angle X-ray scattering (SAXS), electron microscopy (EM), atomic force microscopy (AFM), confocal microscopy, accumulated associated protein, biofilm formation, extracellular matrix protein, small basic protein, Staphylococcus epidermidis
Introduction
Previously regarded as an innocuous human commensal that normally colonized the human skin and mucous membrane, Staphylococcus epidermidis has emerged as a leading opportunistic pathogen causing nosocomial infection associated with permanent or intermittent indwelling medical devices (1, 2). In contrast with its more virulent cousin Staphylococcus aureus, which produces an abundance of toxins, adherence factors, and cytolysins, the main virulence mechanism through which S. epidermidis evade the host immune response relates to its capability of adhering and forming biofilms on biotic and abiotic surfaces (3). Staphylococcal biofilms are adherent multilayered communities of multicellular organisms embedded in self-produced extracellular matrix that are functionally resistant to antibiotics and components of the immune system (4). Therefore, S. epidermidis–mediated infections are difficult to treat and affect millions of patients worldwide annually, causing significant morbidity and mortality and a high burden for the public health system (5).
Biofilm formation is a complex, multifactorial process and can be divided into at least three stages: initial attachment to abiotic surfaces, protein-coated materials, and host cells; subsequent cell proliferation and accumulation via cell–cell adhesion leading to a mature biofilm; and finally detachment/dispersal (4, 5). A variety of extracellular matrix components involved in biofilm formation, including proteinaceous adherence factors, polysaccharides, extracellular DNA, teichoic acids, nucleases/proteases, and phenol-soluble modulin factors, have been extensively studies and recently reviewed (6, 7). Although polycationic polysaccharide intercellular adhesin (PIA),3 which is synthesized by the icaADBC operon that is widely distributed in clinical S. epidermidis isolates, has long been identified as a major component of the biofilm matrix and considered as the major intercellular adhesin in staphylococci biofilm formation, not all strains of S. epidermidis carry the icaADBC operon, and it is now clear that protein-based matrices also have the ability to mediate biofilm accumulation and cause infection in an ica-independent manner (6, 8). In S. epidermidis, among these biofilm matrix proteins, is the well-studied accumulation-associated protein (Aap), which is a cell wall–anchored, multidomain, multifunctional surface protein with significant roles in biofilm formation (3, 4, 9). Aap consists of an N-terminal Domain-A with a variable number of 16-aa repeats located N terminally and a putative L-type lectin subdomain, a C-terminal Domain-B composed of between 3 and 17 nearly identical 128-aa B repeats terminating in a conserved “half-repeat” motif. Domain-B in Aap is followed by a collagen-like domain and an LPXTG cell wall anchor sequence. Each of these B repeats is composed of two subdomains, G5 and E (10). Recent structural studies revealed that the G5-E repeats can undergo a Zn2+-dependent homophilic interaction, suggesting that two identical Aap molecules expressed on the surface of neighboring cells may interact with each other and promote cell–cell accumulation during biofilm development (11, 12). Besides Aap, specific homophilic interactions have been identified for SraP (13)-, IsdC (14)-, SasG (15)-, and SdrC (16)-mediated biofilm accumulation; thus, homophilic interactions appear to be a common mechanism by which the matrix proteins promote biofilm formation. Clearly, understanding the molecular mechanism of biofilm matrix proteins involved in biofilm formation is key to combat the significant nosocomial infectious diseases (3, 9).
There is increasing evidence showing that in some bacteria one of the extracellular matrix components includes amyloid fibers (17–19). Amyloids are highly organized protein aggregates that form unbranched protein fibers composed of cross-β-sheet quaternary structures, which have long been viewed as pathological entities traditionally associated with several incurable neurodegenerative diseases, including Alzheimer's disease, Parkinson's disease, Huntington's disease, and prion diseases (20). Growing evidence, however, indicates that most proteins can adopt the amyloid fold given the correct environment and that some amyloids carry out nonpathological biological roles and are known as functional amyloids (21, 22). Functional amyloids have been found in a wide range of organisms, from bacteria to mammals, with functions as diverse as biofilm formation, development of aerial structures, scaffolding, regulation of melanin synthesis, epigenetic control of polyamines, and information transfer (21). There is a limited but growing number of examples, including the curli homolog in Salmonella (23), FapC in many Pseudomonas species (24), TasA in Bacillus subtilis (25), the phenol-soluble modulins (26) and the biofilm-associated surface protein (27) in Staphylococcus aureus, and the adhesin protein P1 in Streptococcus mutans (28), found to be able to form amyloid fibrils and that are implicated in biofilm formation (29).
Recently, an 18-kDa extracellular protein called small basic protein (Sbp) was identified in S. epidermidis biofilm matrix. Sbp was implicated to serve as a scaffold protein to provide support for cell–cell adhesion and aggregation for both PIA- and Aap-mediated biofilm accumulation (30). Sbp has been found to colocalize with Domain-B of Aap. Besides the probable direct Sbp–Aap Domain-B interactions involved in Aap-mediated biofilm formation, Sbp could also have indirect impact by recruiting additional and yet unknown factors for biofilm formation (30). So far, little is known about the structure of Sbp and its interaction with Aap, and the precise molecular mechanism underlying its roles in biofilm formation remains largely unexplored.
Here, we report a detailed structural and biophysical characterization of Sbp and its interaction with Aap Domain-B. Sbp is monomeric and partially folded in solution, and unexpectedly, no direct interactions were observed between Sbp and Aap Domain-B under our in vitro experimental conditions. Instead, we found that Sbp assembles into amyloid-like fibrils both in vitro and in vivo. Biochemical, kinetic, and microscopy studies indicate that these fibrils have characteristics of bacterial functional amyloids (18, 31). We propose that amyloid fibril formation of Sbp is responsible for its role as a scaffold protein in S. epidermidis biofilm formation.
Results
Sbp is monomeric and partially folded in solution
Previous bioinformatics analysis identified an N-terminal export signal (aa 1–20) for Sbp (30). To study its structure and interaction in vitro, a recombinant Sbp (rSbp; aa 21–169) without its export signal was expressed in BL21(DE3) and purified by affinity, ion-exchange, and size-exclusion chromatography. The His6 tag was removed by tobacco etch virus (TEV) protease cleavage. Purified rSbp eluted as a single peak in size-exclusion chromatography (SEC) and was shown as a single band in SDS-PAGE, indicting high homogeneity of the rSbp protein in solution (Fig. S1).
To study the overall conformation of rSbp in solution, we performed small-angle X-ray scattering (SAXS) analysis. The scattering profile, with scattering intensity I(q) plotted against momentum transfer q, and pair distance distribution function (PDDF) transformed from the scattering profile for rSbp are shown in Fig. 1, A and B. The Guinier plot is linear in the Guinier region (qmax × Rg < 1.3) of the scattering profile, indicating that the rSbp is monodisperse and homogeneous in solution. The PDDF is asymmetric with maximum frequency at a distance shorter than half of the maximum distance within the molecule (Dmax), indicating that rSbp is rather open and elongated in solution. The overall structural parameters, including Rg calculated from Guinier slops, Rg and Dmax from PDDF functions, and molecular weights derived from the power law of volume-of-correlation (Vc) (32) and the SAXSMoW package (33), are summarized in Table 1 where the molecular weights from SAXS data are compared with the predicted molecular weight, indicating that rSbp is monomeric in our solution condition. The dimensionless Kratky and Porod–Debye plots (Fig. 1, C and D), plotted as (qRg)2I(q)/I(0) versus qRg and as I(q)q4 versus q4, respectively, are useful methods to infer the folding, compactness, and flexibility of biomolecules in solution (34). For a compact globular protein, the Kratky plot has a typical bell shape with a clear maximum. For a completely unfolded protein or protein in a premolten globule conformation, no such maximum in the Kratky plot can be observed, and the curve displays a plateau at elevated q. As shown in Fig. 1C, the dimensionless Kratky plot of rSbp is found between these two extremes, indicating that rSbp is a partially folded protein with some flexibility. This was further supported by the Porod–Debye plot (Fig. 1D), which shows a plateau for the protein lysozyme that is lost for rSbp, suggesting increased flexibility of rSbp in solution.
Figure 1.

Folding and flexibility analysis of rSbp by SAXS and NMR. A and B, scattering profile (A) and PDDF (B) of Sbp. The inset in A is the Guinier region with fitting line of the scattering profile. C and D, dimensionless Kratky plot (C) and Porod-Debye plot (D) of Sbp (black line) and the protein lysozyme (red line). Lysozyme is a well-folded protein serving here as a reference. E and F, 2D 1H-15N HSQC of rSbp in 20 mm KiPO4, 100 mm KCl, pH 7.20, collected at 25 (E) and 15 °C (F). Many peaks are clustered within the boxed central region. a.u., arbitrary units.
Table 1.
Data collection and structural parameters derived from SAXS experiments

a Rg derived from Guinier fitting.
b Rg derived from GNOM analysis.
c Molecular weight was calculated from the primary protein sequence.
d Molecular weight was calculated using the Rg/Vc power law developed by Rambo and Tainer (32).
e Molecular weight was calculated using the web portal SAXSMoW.
The overall folding of rSbp was further assessed by 2D 1H-15N heteronuclear single quantum coherence (HSQC) NMR spectra. For properly folded proteins of medium sizes (smaller than 200 aa), peaks in the HSQC spectra are usually of similar intensities and well-resolved, but for intrinsically disordered proteins, peaks in HSQC are usually heavily overlapped. As shown in Fig. 1E, HSQC of rSbp at 25 °C shows some well-dispersed peaks with nonuniform intensities, but significant overlap also exists between 8.0 and 8.8 ppm in the 1H dimension, indicative of unstructured domains (35), suggesting that rSbp is only partially folded. Lowering the temperature to 15 °C slightly improves the spectrum (Fig. 1F) such that some well-resolved new peaks appear and the intensities of peaks become more uniform. However, further lowering temperature to 5 °C or increasing to 35 °C worsens the spectra (Fig. S2, A and B), suggesting that rSbp has an optimal folding temperature around 15–25 °C. In the spectra at 37 °C, those well-dispersed peaks disappear possibly due to broadening exchange. The majority of amide proton frequencies are clustered within an area of 1 ppm (8.6–7.6-ppm region) (Fig. S2B), characteristic of intrinsically disordered proteins (IDPs) (35). Increasing the salt concentration up to 400 mm does not improve the spectra significantly; the central region is still heavily overlapped (Fig. S2, C and D), preventing further high-resolution three-dimensional (3D) structure determination of rSbp by NMR.
Determination of low-resolution model for Sbp
So far, no experimentally determined high-resolution or homologous 3D structural information is available for Sbp. We therefore first predicted the secondary structure of Sbp (aa 1–169) using the PSIPRED web server (36), which reveals extensive β-sheet regular secondary structure interspersed with several α-helices for Sbp (Fig. 2). To verify the secondary structure composition, a circular dichroism (CD) spectrum was recorded for rSbp in solution. As shown in Fig. 3A, a minimum at 200 nm and positive ellipticity below 192 nm were observed, suggesting that β-sheets are predominant in Sbp along with regions of low structural complexity. Based on the CD data, an estimation of the secondary structure composition according to Reed's algorithm (37) is 13% α-helices, 45% β-sheets, and 31% random coil. The tertiary structure of rSbp was further predicted using the I-TASSER web server, resulting in a compact structure with three α-helices packed against a β-sheet (Fig. 3B).
Figure 2.

The primary sequence, predicted secondary structure, and amyloidogenic hot spot of Sbp. The secondary structure predicted by PSIPRED server is shown above the primary sequence of Sbp (aa 1–169), indicating high richness of β-strand content. Helical and β-strand secondary structure elements are depicted by pink barrels and yellow arrows, respectively. Below the primary sequence are the putative aggregation hot spots predicted (Pred) by AMYLPRED analysis of the primary sequence. Regions comprising residues 4–19, 84–90, 102–105, and 137–139, respectively, having the highest consensus and recognized as proamyloidogenic by six different algorithms are colored in red bars. Pafig, Prediction of amyloid fibril-forming segments; SecStr, Secondary Structure Prediction.
Figure 3.

Secondary structure and low-resolution 3D structure of rSbp. A, the far UV CD spectrum of rSbp. B, the 3D atomic model of rSbp predicted by the I-TASSER online server, revealing three helix bundles packed against a β-sheet. C, the ab initio shape envelope in two views for rSbp, reconstructed by DAMMIN, is elongated. The atomic model in B was superimposed onto the shape envelope. mdeg, millidegrees.
To gain more specific information on the overall 3D structures of rSbp, a low-resolution ab initio shape envelope was built using the program DAMMIN and auxiliary programs. This strategy models a macromolecule as an assembly of scattering beads arranged in space such that a calculated scattering curve reproduces the experimental curve. As shown in Fig. 3C, the bead model has an elongated ellipsoidal shape, which is much larger than the predicted compact atomic model, indicating that rSbp is much extended and open in solution.
No direct interaction between Sbp and Aap Domain-B in vitro
Previously, Sbp was found to be colocalized with Aap Domain-B, suggesting that Sbp may interact with Aap Domain-B directly (30). Domain-B of Aap is a Zn2+-binding protein that can mediate intercellular adhesion through Zn2+-dependent self-assembly (11, 12). Interestingly, a dose-dependent increase in Sbp binding in the presence of elevated ZnCl2 concentration has also been observed for immobilized Aap Domain-B in solid-phase binding assays (30). To validate and characterize the interactions between Sbp and Aap Domain-B on a biochemical level, a construct consisting of the terminal intact B repeat along with the C-terminal G5 domain (called Brpt1.5), which represents Aap Domain-B (11), was expressed and purified. The interaction between Brpt1.5 and rSbp was studied in the absence and presence of ZnCl2 using analytical SEC (Fig. 4). Unexpectedly, no interactions between rSbp and Brpt1.5 were observed either in the absence or presence of Zn2+. As shown in Fig. 4A, both protein components in the sample mixtures of Brpt1.5 and rSbp (with molar ratios of 1:2, 2:2, and 4:2, respectively) eluted at their respective retention volumes as for each protein alone through the SuperdexTM 75 SEC column. The rSbp binding property of Brpt1.5 was further verified using isothermal titration calorimetry (ITC). Again, as shown in Fig. 4, B and C, rSbp does not bind with Brpt1.5 either in the absence or presence of Zn2+.
Figure 4.

Interaction of rSbp and Aap Domain-B (Brpt1.5) by SEC and ITC. A, SEC elution profiles of rSbp alone, Brpt1.5 alone, and mixtures of Brpt1.5 and rSbp at ratios of 1:2, 2:2, and 4:2, respectively. The running buffer is 20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm DTT. B and C, raw calorimetric data and binding isotherms by ITC measurement of the interaction of Brpt1.5 and rSbp in the absence (B) and presence (C) of Zn2+, respectively. mAU, milliabsorbance units.
Sbp forms amyloid fibrils in vitro
Because no direct interaction between rSbp and Aap Domain-B was observed, Sbp may utilize a different modality to contribute biofilm formation. Given its richness of β-sheet secondary structure and partial folding characteristic, we became interested in its propensity to form amyloid fibrils. A variety of bioinformatics tools have been developed to identify amyloidogenic hot spots within proteins that are prone to aggregation and may induce amyloid formation. The propensity of Sbp to form amyloid fibrils was analyzed using AMYLPRED2 (38, 39), which determines amyloidogenic regions based on the consensus score of 11 such tools and thus provides a reliable estimate of the amyloid propensity. Consensus short peptide segments that might be responsible for amyloid fibril formation were predicted for Sbp (Fig. 2), suggesting that Sbp has high potential to form amyloid fibrils.
The amyloidogenic nature of rSbp was first confirmed using Congo red (CR) binding assays specific for amyloids (Fig. 5A). In in vitro assays, fibrillization is routinely triggered by mechanical agitation to increase the probability of the intermolecular collisions necessary to form the initial nucleus. The binding of CR to rSbp that had been agitated at 37 °C for 12 h was evaluated. As shown in Fig. 5A, compared with CR alone, the spectra of the agitated rSbp sample demonstrated dye binding and a shift in the CR absorbance maxima that is characteristic of amyloid fibrils. We speculated that, under this condition, rSbp assembled into an amyloid fibrillar state, hereafter named as rSbpfib (as opposed to the native, soluble Sbpsol). Based on Fourier transform IR (FTIR) spectroscopy data, distinct structural differences were observed between rSbpsol and rSbpfib (Fig. 5B). The amide I region (1,600–1,700 cm−1) reflects the vibrational stretching of the amide backbone and is indicative of secondary structure (40). The shift in the β-sheet peaks to lower wavenumber (from 1631 cm−1 in Sbpsol to 1624 cm−1 in Sbpfib) is indicative of rearrangement of globular β-sheets to extended β-sheets, which is consistent with a previous report of amyloid fibrils (40).
Figure 5.
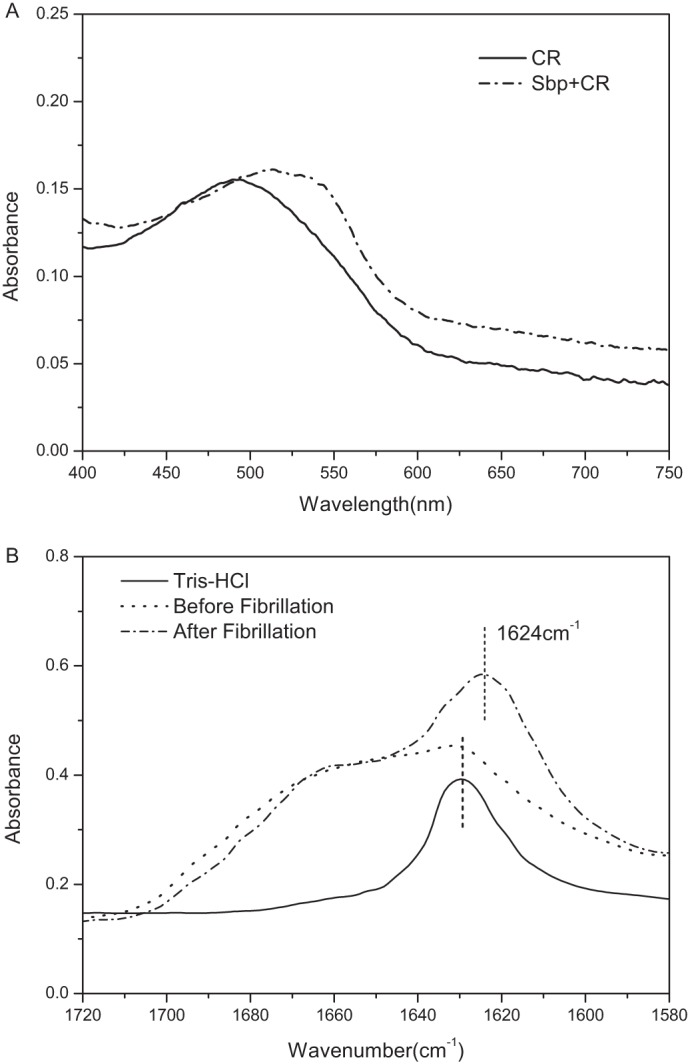
In vitro amyloid fibrils formation by rSbp. A, absorption spectra of Congo red binding to Sbp amyloid fibrils (dashed line). The Congo red–only spectrum (solid line) was included for comparison. B, FTIR spectra of native (dashed dot) and aggregated (dashed line) Sbp at pH 7.20. The FTIR spectrum of Tris buffer (solid line) was included for comparison.
The kinetics of Sbpfib formation was monitored by thioflavin T (ThT) fluorescence at 37 °C. As shown in Fig. 6A, Sbpfib formation follows a typical sigmoidal time profile, suggesting that Sbpfib undergoes a nucleation-dependent polymerization process characterized by an initial lag phase reflecting nucleation and a subsequent elongation/growth phase culminating in a steady plateau (20). We also performed time-resolved atomic force microscopy (AFM) imaging of the system to ascertain that rSbp forms amyloid fibrils. Starting from the homogeneous solution of rSbp monomer, rSbp showed amorphous structure at times 0 and 2 h; isolated protofilament or short fibers were not seen until at 4 h. Upon progression of time, more and longer fibers were formed, and fiber branching was observed at 8 h of incubation (Fig. 6, B–E). The aspect ratio, which was defined as the ratio of fibril length with respect to fibril diameter (41), varies significantly for rSbp fibrils. As shown in Fig. 6E, the diameter of the fibril by AFM is about 3 nm; the aspect ratio of one short and long fibril can be about 84–373. After 12 h of incubation, rSbp formed a mature, intertwined fibril network (Fig. 6F). The formation and morphology of rSbp fibrils were further confirmed with transmission electron microscopy (TEM) analysis of mature rSbp fibrils at 12, 24, and 48 h, respectively (Fig. 6, G–I). The mature fibril structure usually exhibits a width of about 5 nm and a length of 100–5,000 nm.
Figure 6.

Kinetics of amyloid fibrillization of rSbp at 37 °C. A, kinetics of rSbp amyloid fibril formation was monitored by in situ measurement of ThT fluorescence. B–F, kinetics of rSbp amyloid fibril formation probed by AFM. One short (green) and one long (red) fibril in E selected for aspect ratio analysis are marked with arrows. The images are shown as height traces, and the scale bar corresponds to 500 nm. G-I, transmission electron micrographs of negatively stained rSbp amyloid fibrils. Scale bars were set to 100 nm.
Sbp undergoes further unfolding in lag phase of fibrillation
To characterize the structural changes of rSbp in lag phase, we performed in situ SAXS measurement of the fibrillation reaction of rSbp at 37 °C. The respective time-dependent scattering profiles and the PDDFs are shown in Fig. 7, A and B. To roughly evaluate changes in the profiles, apparent molecular weights were calculated using the scattering profiles as input by the SAXSMoW package, which reveals gradually increased molecular weight over time but that are still smaller than the molecular weight of a dimer (Table S2), suggesting that the monomeric rSbp dominates during the lag phase. The Dmax as well as the dominant peaks in PDDFs shift to larger distances over time, implying obvious structural changes, which were supported by the Kratky plots (Fig. 7C). At the starting point time, the Kratky plot of rSbp has a curving-down feature, suggesting a partial folding structure; however, at a time of 2 h, the Kratky plot has a clear plateau and continues to increase over the q range 0.18–0.3 Å−1, reflecting the predominantly extended conformation characteristic of unfolded rSbp. Therefore, rSbp undergoes further unfolding during the lag phase of fibrillation.
Figure 7.
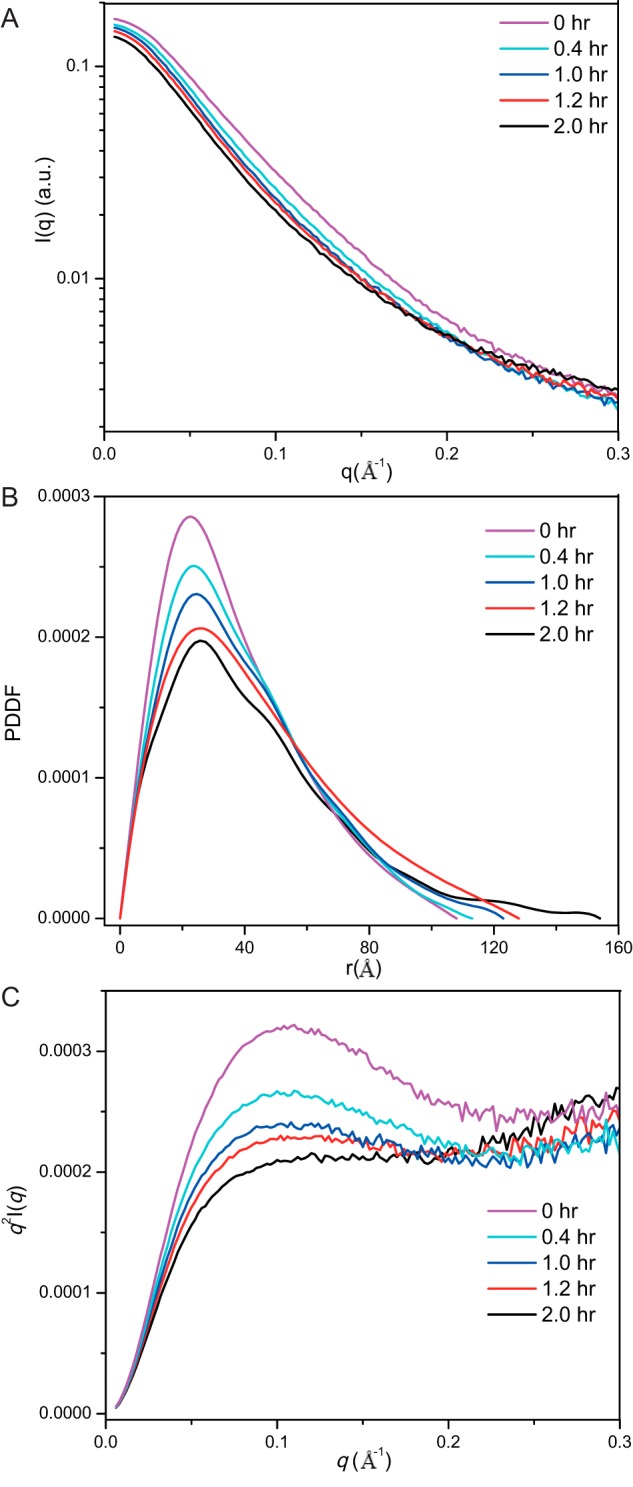
SAXS analysis of rSbp fibrillization at lag phase. A–C, the scattering profiles (A), PDDF (B), and Kratky plots (C) of rSbp at 0-, 0.4-, 1.0-, 1.2-, and 2.0-h incubation time in buffer containing 20 mm Tris-HCl, pH 7.4, 300 mm NaCl, 1 mm DTT at 37 °C. a.u., arbitrary units.
Sbp forms intracellular amyloid aggregates in Escherichia coli cells
It has been shown that amyloidogenic proteins from different origins, when expressed in E. coli, are accumulated as cytoplasmic inclusions of amyloid nature (42). To investigate whether Sbp can form amyloid aggregates in vivo, E. coli cells carrying pSbp plasmid were grown at 37 and 16 °C, respectively, until early exponential phase was reached, and then the expression of Sbp was induced. At defined time points (i.e. 4, 8, and 12 h), aliquots of induced cells were sampled and fixed with 4% PFA, stained with the probes BTA-1 and thioflavin S (ThS) that specifically bind to amyloid aggregates, and visualized by confocal microscopy. As shown in Figure 8, a variable number of inclusions were observed in cells grown at 37 °C at specific induction time intervals that were recognized by both amyloidophilic dyes as intense fluorescent foci. In contrast, cells grown at 16 °C do not accumulate inclusions.
Figure 8.

Formation of intracellular amyloid inclusions in E. coli cells expressing rSbp at 16 and 37 °C, respectively. Cells sampled after 4, 8, and 12 h of induction, respectively, were fixed with PFA, then stained with either BTA-1 or ThS, and visualized by confocal microscopy. BF, bright field. Scale bar, 1 μm.
Sbp is essential for S. epidermidis biofilm formation
Following the observation of amyloid fibril formation of overexpressed Sbp in E. coli at 37 °C, to test the relevance of Sbp amyloid fibrils to S. epidermidis biofilm formation, we knocked out sbp in S. epidermidis 1457 using the Gateway system (Fig. 9, A and B). S. epidermidis 1457 WT and 1457 Δsbp were grown at 37 °C overnight, stained with ThS, and visualized by confocal microscopy. As shown in Fig. 9C, S. epidermidis 1457 WT could form a biofilm in which cells are clustered together showing clear ThS fluorescent foci. In contrast, S. epidermidis 1457 Δsbp do not form biofilm: the cells were detached, and no fluorescent foci were observed (Fig. 9D). The results support a close interconnection of Sbp amyloid fibril formation and S. epidermidis biofilm formation in vivo.
Figure 9.

Sbp amyloid fibrils and S. epidermidis biofilm formation in vivo. A, strategy of allele replacement for construction of S. epidermidis 1457 mutant in which sbp gene was knocked out (KO) using the Gateway system. B, the mutant was verified with PCR analysis on the 1457 WT and 1457 Δsbp. S. epidermidis 1457 WT (C) and 1457 Δsbp (D) cells were grown overnight at 37 °C, stained with ThS, and visualized by confocal microscopy (right, bright field image; middle, ThS fluorescence; left, merged).
Discussion
In this study, we provide a detailed structural characterization of an extracellular protein called Sbp in S. epidermidis and demonstrate its ability to form amyloid fibrils using different biophysical techniques. CD, SAXS, and NMR analyses show that Sbp is a β-sheet–rich protein adopting a monomeric and partially folded structure at physiological condition. Alternatively, Sbp can aggregate into amyloid fibrils, which was confirmed and characterized by fluorescence spectroscopy and microscopy both in vitro and in vivo.
We initially attempted to determine the high-resolution structure of Sbp using NMR. If well-folded, Sbp should be suitable for NMR study given its medium size. Both secondary and tertiary structure predictions suggest that Sbp could be a well-folded protein consisting of regular secondary structures. However, our NMR and SAXS studies indicate that Sbp is partially folded at near physiological condition. At elevated temperature (37 °C), Sbp shows characteristics of IDPs. This property may relate to its high propensity to form amyloid fibrils at elevated temperature in vitro, which seems relevant to S. epidermidis biofilm formation in vivo. Many amyloidogenic proteins such as α-synuclein, Aβ(1–42), SOD1, and TDP-43, which are involved in neurodegenerative diseases, are partially folded or intrinsically disordered proteins that lack definite ordered 3D structure (43). Structural flexibility and plasticity originating from the lack of a definite ordered 3D structure are believed to represent the major functional advantages for these proteins, enabling them to interact with a broad range of binding partners, including self-aggregation to form secondary structures and subsequently fibers with other proteins, membranes, nucleic acids, and various small molecules. One example is the human hormone islet amyloid polypeptide (hIAPP), an IDP that functions in glucose homeostasis (44). Monomeric hIAPP is primarily, but not completely, unstructured. The interaction of hIAPP with membrane promotes formation of secondary structures that nucleate aggregation; this process in turn induces membrane disruption via a two-step mechanism (44, 45). Sbp appears to be a partially unfolded protein with an extended coil-like conformation. As our time-resolved SAXS measurement shows, in the lag phase of fibrillation, Sbp undergoes a further unfolding process, which may expose the amyloidogenic hot spot to form the nucleation sites leading to polymerization.
It was proposed that Sbp fosters PIA- and Aap-mediated biofilm accumulation by forming a biofilm scaffold instead of directly inducing cell aggregation. Recently, Sbp was identified by affinity purification from S. epidermidis crude biofilm matrix preparations using recombinant Aap Domain-B as a ligand, suggesting a direct Sbp–Aap Domain-B interaction (30). However, the exact modalities of Sbp–Aap Domain-B interaction remain unclear. It was reported that Aap Domain-B is a zinc-binding protein and undergoes Zn2+-induced self-association, suggesting the importance of Zn2+ in Aap function. We expressed and purified both Sbp and the Aap Domain-B consisting of 1.5 G5 repeats and characterized the interactions using SEC and ITC both in the absence and presence of Zn2+. Unexpectedly, no obvious interaction of Sbp and Aap Domain-B was observed in our condition. Given that Sbp can adopt different folding states, either the partially folded monomer or the amyloid fibril state, there is a possibility that the Sbp monomeric state is not the active form. Notably, PAP(248–286), a peptide fragment of prostatic acid phosphatase, a protein abundant in human semen, is inactive in the monomeric state and must first aggregate to form the active form, semen enhancer of viral infection, with characteristics of amyloid fibrils to promote viral infection. Bacterial amyloid proteins like curli can promote the aggregation of PAP(248–286) into semen enhancer of viral infection (46). It is of interest in future studies to determine whether Sbp in crude biofilm matrix preparations forms amyloid fibrils, therefore presenting the active form for Aap Domain-B binding, or whether Aap Domain-B may interact with Sbp by enhancing the aggregation of Sbp into functional amyloid.
Sbp is the first structural S. epidermidis extracellular biofilm matrix protein with significant relevance for both Aap- and PIA-dependent biofilm formation. It is now recognized that several staphylococcal surface proteins, including Aap, can promote biofilm accumulation in a PIA-independent manner through specific homophilic interactions between proteins expressed on different cells (9). Recent studies have recognized amyloid fibers as common functional components in bacterial biofilm matrix. There is a growing number of examples demonstrating the direct implication of amyloids in biofilm formation, including the phenol-soluble modulin and Bap protein from Staphylococcus aureus (29). Bap is a large protein consisting of multiple domains whose function is regulated post-translationally (47). It was found that Bap forms an amyloid scaffold under acidic solution conditions to promote biofilm development (18). To our knowledge, Sbp is the first S. epidermidis protein identified to have the propensity to assemble into amyloids at near physiological conditions both in vitro and in vivo. We propose that the formation of functional amyloid fibrils by Sbp may account for its role as a scaffolding protein in S. epidermidis biofilm formation. Amyloid formation may represent another common mechanism in protein-mediated cell–cell accumulation. We expect more such proteins to be identified.
Taken together, we report that Sbp has an intrinsic amyloid-forming ability. This is the first report about a functional amyloid in S. epidermidis. The well-organized Sbp amyloid fibers may be involved in both PIA- and Aap-mediated biofilm formation as a matrix component.
Experimental procedures
Protein expression and purification
The genomic DNA of S. epidermidis 1457 was extracted for gene cloning using the EasyPure Bacteria Genomic DNA kit (TransGen Biotech). The DNA fragments encoding rSbp (aa 21–169) and Brpt1.5 were amplified from the genomic DNA by PCR using the appropriate primers (Table S1). The PCR products were then subcloned into pET28a (for rSbp) and pGEX-4T-2 (for Brpt1.5), respectively. The positive clones were confirmed by DNA sequencing and subsequently transformed into E. coli BL21(DE3).
For purification of rSbp, cells were grown in LB culture medium at 37 °C until A600 nm reached ∼0.8. Expression of the rSbp recombinant protein was induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) at 16 °C for another 20 h before harvesting. Bacteria were collected by centrifugation at 4,500 × g for 30 min. The cell pellet was suspended in lysis buffer (20 mm Tris-HCl, pH 7.4, 500 mm NaCl) and then homogenized by a high-pressure cell disruptor system. After centrifugation at 18,500 × g for 30 min, the supernatant containing the His-tagged protein was collected and loaded onto a nickel-nitrilotriacetic acid affinity resin that was pre-equilibrated with binding buffer (20 mm Tris-HCl, pH 7.4, 500 mm NaCl). The resin was extensively washed by two different buffers (20 mm Tris-HCl, pH 7.4, 1 m NaCl, 20 mm imidazole and 20 mm Tris-HCl, pH 7.4, 200 mm NaCl, 20 mm imidazole) sequentially to remove nucleic acid contamination. The Sbp protein was then eluted with 20 mm Tris-HCl, pH 7.4, 200 mm NaCl, 250 mm imidazole. The protein fractions were mixed and digested by TEV protease to remove the His tag. Next, the protein was further purified using an ion-exchange HiTrap S column and a HiLoad16/600 Superdex 75 PG column pre-equilibrated with gel filtration buffer (20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm DTT). The final sample purity was checked by SDS-PAGE and MS (MALDI-TOF).
For purification of Brpt1.5, cells were grown in LB culture medium supplemented with 100 μg/ml ampicillin at 37 °C until A600 nm reached 0.8. Expression of Brpt1.5 was induced with 0.5 mm IPTG at 37 °C for 5 h. The cells were harvested, suspended, and lysed using a similar protocol as described above. After centrifugation at 18,500 × g for 30 min, the supernatant was loaded onto a GST column. The resin was extensively washed with binding buffer. The target protein was eluted with 20 mm Tris-HCl, pH 7.4, 200 mm NaCl, 25 mm GSH. The GST tag was removed with TEV protease treatment. After cleavage, the protein was loaded onto a GST column to remove uncleaved protein and the free cleaved GST tag. The flow-through was collected, concentrated, and injected onto a HiLoad16/600 Superdex 75 PG column pre-equilibrated with gel filtration buffer. The final sample purity was checked by SDS-PAGE.
Structural analysis by small angle X-ray scattering
Conventional and in situ time-resolved SAXS measurements were carried out at room temperature and 37 °C, respectively, at beamline 12 ID-B of the Advanced Photon Source, Argonne National Laboratory, equipped with a temperature controller. The scattered X-ray photons were recorded with a PILATUS 1M detector (Dectris). The setups were adjusted to achieve scattering q values of 0.005 < q < 0.89 Å−1 where q = (4π/λ)sinθ and 2θ is the scattering angle. Thirty two-dimensional images were recorded for each buffer or sample solution using a flow cell with an exposure time of 0.5–2 s to minimize radiation damage and obtain good signal-to-noise ratio. No radiation damage was observed as confirmed by the absence of systematic signal changes in sequentially collected X-ray scattering images. The 2D images were reduced to one-dimensional scattering profiles using Matlab scripts on site. Scattering profiles of the protein were calculated by subtracting the background buffer contribution from the sample buffer profile using the program PRIMUS (48) following standard procedures (49). Concentration series measurements (4- and 2-fold dilution and stock solutions) for the same sample were carried out to remove the scattering contribution due to interparticle interactions and to extrapolate the data to infinite dilution. The forward scattering intensity I(0) and the radius of gyration (Rg) were calculated from the data of infinite dilution at low q values in the range of qRg < 1.3 using the Guinier approximation: lnI(q) ≈ ln(I(0)) − Rg2q2/3. These parameters were also estimated from the scattering profile with a broader q range of 0.006–0.30 Å−1 using the indirect Fourier transform method implemented in the program GNOM (50) along with the PDDF, p(r), and Dmax. The parameter Dmax (the upper end of distance r) was chosen so that the resulting PDDF has a short, near-zero-value tail to avoid underestimation of the molecular dimension and consequent distortion in low-resolution structural reconstruction. The Vc values were calculated using the program Scatter, and the molecular weights of solutes were calculated on a relative scale using the Rg/Vc power law developed by Rambo and Tainer (32), independent of protein concentration and with minimal user bias.
Low-resolution ab initio bead models were built up with the program DAMMIN, which generates models represented by an ensemble of densely packed beads (51), using scattering data within the q range of 0.006–0.30 Å−1. Thirty-two independent runs were performed, and the resulting models were subjected to averaging by DAMAVER (52), superimposed by SUPCOMB (53) based on the normalized spatial discrepancy (NSD) criteria, and filtered using DAMFILT to generate the final model. NSD is a measure of quantitative similarity between sets of three-dimensional models: if two models systematically differ from each other, their NSD exceeds 1; for identical objects, it is 0.
NMR spectroscopy
A similar preparation protocol as above was followed to prepare 13C,15N-labeled rSbp except that cells were grown and induced in M9 minimal medium with 15NH4Cl and [13C6]glucose as the sole nitrogen and carbon sources, respectively. The samples used for NMR experiment were as follows: 0.5 mm 13C,15N-labeled-rSbp in 90% H2O, 10% D2O containing 20 mm K2HPO4/NaH2PO4, 100 mm NaCl, 50 μm NaN3, pH 7.20, and additives. 2D 1H-15N HSQC spectra were recorded on Bruker Avance 600- or 800-MHz spectrometer at variable temperature, all equipped with four radio frequency channels and a triple-resonance cryoprobe with pulsed field gradients. Each spectrum comprised 1,024 complex points in the t2 dimension and 160 complex points in the t1 dimension. All spectra were processed using the software package NMRPipe (54) and analyzed with NMRView (55).
CD spectroscopy
The secondary structure of rSbp in solution was checked by CD on an Applied Photophysics Chirascan-plus (Leatherhead) controlled by Pro-Data Chirascan v4 software. Far-UV CD spectra were recorded between 190 and 260 nm with a step resolution of 1 nm, a slit width of 0.6 nm, and an integration time of 5 s. Acquisition was performed at 25 °C using a 0.2-mm-path length cuvette with a protein concentration of ∼0.3 mg/ml. The spectra were averaged over three scans and corrected by subtraction of the buffer signal.
Secondary and tertiary structure prediction
The secondary structure of Sbp was predicted using the PSIPRED web server (36). The tertiary structure of Sbp was predicted using the I-TASSER online server (56).
Analytical size-exclusion chromatography
Samples of Sbp alone, Brpt1.5 alone, and the mixture of Sbp and Brpt1.5 with molar ratios of 1:1 and 2:1 were injected onto a Superdex 75 10/300 GL column (24.0 ml; GE Healthcare) pre-equilibrated with binding buffer (20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm DTT or 20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm DTT, 1 mm ZnCl2), respectively. Concentrations of Brpt1.5 protein were kept constant at 30 μm.
Isothermal titration calorimetry
Purified Sbp and Brpt1.5 samples were extensively exchanged into buffer (20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm DTT or 20 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm DTT, 1 mm ZnCl2, respectively) using a Superdex 75 10/300 GL column and then concentrated to 30 μm for Brpt1.5 and 300 μm for Sbp. The ITC measurements were performed with a MicroCal iTC 200 calorimeter (GE Healthcare) at 25 °C. The background data obtained from the buffer sample were subtracted before the data analysis. The data were fitted using the Origin7 software package (MicroCal). Measurements were repeated twice, and similar results were obtained.
In vitro amyloid fibril formation
For amyloid fibril formation, rSbp at a concentration of 250 μm in 20 mm Tris-HCl, pH 7.2, containing varied concentrations of NaCl and 0.01% sodium azide was incubated at 37 °C with rotary agitation at 220 rpm. The rSbp solution after salt-induced fibrillation was subjected to CR assay, FTIR characterization, ThT fluorescence assay, AFM, and TEM characterization at certain time points.
CR assay
The assay was performed with an Implen UV-visible nanophotometer (München, Germany). The concentration of CR solution was 5 μm. The buffer was 10 mm phosphate saline buffer, pH 7.4. The assay was performed ex situ. 20 μl of rSbp solution was added into 980 μl of CR solution in a 1.0-cm quartz cuvette. The solution was incubated in the dark for 30 min. The solution in the cuvette was shaken first before each spectral acquisition.
FTIR spectroscopy
The FTIR spectra were recorded using a Bruker Vertex 70 FTIR spectrometer equipped with a DLaTGS detector. A Bruker-made sample shuttle was installed inside the sample compartment, which has two sample holders: one for sample and the other for reference. The FTIR spectra of the samples were collected with 4 cm−1 resolution and 32 scans. 100 μl of solution was deposited onto the CaF2 windows, and then the CaF2 window surface was dried with a dryer.
ThT binding assay
The ThT binding assay was performed with a Hitachi F-7000 fluorescence spectrophotometer (Tokyo, Japan). An excitation wavelength of 450 nm with a slit width of 5 nm was used. The emission spectrum in the region of 460–600 nm was measured using a slit width of 10 nm. The concentration of ThT solution was 10 μm. The buffer was 20 mm Tris-HCl, 100 mm NaCl, pH 7.2. The assay was performed ex situ. For each measurement, 1 μl of rSbp solution was added into 1 ml of ThT solution in a 1.0-cm quartz cuvette. The solution in the cuvette was shaken first before each spectral acquisition.
Atomic force microscopy
Time-dependent morphological changes during rSbp aggregation were measured using an atomic force microscope (SPM P47 Solver, NT-MDT Co., Zelenograd, Russia) in tapping mode. At regular time intervals during aggregation, small aliquots of the incubated rSbp samples were taken out and diluted with deionized water to a final concentration of 40 μm. A 50-μl aliquot of the diluted solution was deposited onto a freshly cleaved mica surface followed by incubation at room temperature for 5 min. Washing steps were carried out twice with deionized water to remove unbound proteins/aggregates followed by drying in a desiccator. All AFM images were taken on dried samples in air, and 5 × 5 μm scanning was used throughout the AFM experiment.
Transmission electron microscopy
The formation and morphology of amyloid fibrils by Sbp were confirmed by negatively staining samples. rSbp fibrils were diluted to 0.5 mg/ml, and 3 μl of sample was directly deposited onto a glow-discharged 300 mesh carbon-coated copper grid (BZ10023a, Zhongjingkeyi, China) followed by staining with 2% (w/v) uranyl acetate and blotted dry with filter paper three times. The stained samples were imaged using a Tecnai Spirit with iCorr D1319 (FEI) operating at 120 kV at a magnification of ×52,000.
Knockout of Sbp by Gateway system in S. epidermidis 1457
An allele replacement strategy was used for construction of a specific Sbp knockout mutant in S. epidermidis 1457 as reported previously (30). Briefly, two primers flanking regions upstream (sbp_ko_att, 5′GGGGACAAGTTTGTACAAAAAAGCAGGCTTATATCCTGTCGTACTCGTG-3′ and sbp_eco_r, 5′-ACCGCCGAATTCTCACTCCTTTGATTCTTTATGTCTTCTG-3′) and reverse primers covering downstream regions (sbp_ko_eco, 5′-ACCGCCGAATTCAAAGATAAAAATGTGAAGTTATATCGTA-3′ and sbp_att_r, 5′-GGGGACCACTTTGTACAAGAAAGCTGGGTAGTACGTGCAGATAAACGT-3′) of the Sbp-coding region were amplified following a standard PCR protocol. Purified PCR products were cleaved using EcoRI, and the resulting, sbp-flanking fragments were ligated with T4 DNA ligase. The 5′ and 3′ attB site ligation products were introduced into pKOR1 using BP clones (Invitrogen, Karlsruhe, Germany) and transformed into E. coli DC10B. Recombinant plasmid pKOR1Δsbp was verified by PCR and DNA sequencing.
For electrocompetent cells, S. epidermidis 1457 were grown in tryptic soy agar medium until A600 nm reached 0.8. Cells were harvested and washed three times with double-distilled H2O by centrifugation at 3,000 × g for 10 min at room temperature. Cells were resuspended and washed with 2 ml of 2% glycerol twice and finally resuspended with 10% glycerol. Electroporation was performed using a Bio-Rad Gene Pulser and Pulse Controller using a standard protocol (57). Briefly, 10 μg of pKOR1 was added into 50 μl of competent cells and incubated on ice for 30 min before transfer to a prechilled 0.2-cm-gap electroporation cuvette. The cells were immediately pulsed with 2.1-kV voltage and diluted in 800 μl of B2 culture medium followed by shaking at 30 °C for 3 h. Transformation products were plated on a TSBKan plate and grown at 37 °C for 48 h. Clones verified correct by PCR were transferred into 3 ml of TSBcm10 medium at 30 °C overnight followed by dilution at a 1:100 volume ratio into 50 ml of TSBcm10 medium and shaking at 42 °C overnight. Cells were diluted at a 1:100 volume ratio into 5 ml of TSBcm5 medium and shaken at 42 ° overnight, twice followed by plating on a TSBcm5 plate, and then incubated at 2 ° overnight. Single clones were picked into 5 ml of TSB medium and shaken at 30 °C overnight. Cell were diluted 1:10,000, plated on a TSBATc plate, and grown at 37 °C overnight. Ten clones were picked and inoculated into both a TSB plate and a TSBcm10 plate. Three clones that only grew on the TSB plate were picked, and genomic DNA was extracted for PCR verification and sequencing.
Cell fixation and staining for confocal microscopy
Cells were grown at 37 or 16 °C with shaking at 220 rpm until A600 nm reached 0.8 and then induced with 0.5 mm IPTG for another 12 h. At regular time intervals, 500 μl of cell cultures was sampled and centrifuged at 3,000 × g for 4 min at room temperature, and the pellets were washed twice with the same volume of PBS buffer for the following fixation and staining steps. For BTA-1 staining, the washed cells were resuspended in 250 μl of 4% PFA, incubated for 30 min at room temperature, then washed twice with 250 μl of PBS buffer, resuspended in 1 mm BTA-1 prepared in 100% ethanol, and incubated for 30 min at room temperature. Finally, the stained cells were washed twice with PBS again. For ThS staining, the washed cells were suspended in 250 μl of 0.05% (w/v) ThS in 12.5% ethanol, incubated for 1 h at room temperature, and then washed three times with 250 μl of PBS buffer. Stained cells were suspended in the appropriate volume of PBS buffer and observed under confocal microscopy.
Confocal microscopy
Microscopy observations were performed using an LSM 780 (Zeiss) inversion confocal microscope with a 63×/numerical aperture 1.40 oil immersion objective. BTA-1 fluorescence was excited with a 405-nm laser diode, and the emission was registered between 410 and 495 nm. ThS fluorescence was excited using a 488-nm argon laser, and the emission was registered in a range from 493 to 630 nm.
Structural illustration
All of the structural illustrations were generated using the PyMOL Molecular Graphics System, version 1.3 (Schrödinger, LLC).
Author contributions
Y. Wang, J. J., Y. G., and X. F. formal analysis; Y. Wang, J. J., Y. G., Y. S., and X. F. investigation; J. D., Y. Wu, D. Q., G. M., and X. F. resources; G. M. methodology; X. F. conceptualization; X. F. supervision; X. F. funding acquisition; X. F. visualization; X. F. writing-original draft; X. F. project administration; X. F. writing-review and editing.
Supplementary Material
Acknowledgments
This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory (ANL) under Contract DE-AC02-06CH11357. We thank Dr. Xiaobing Zuo (ANL) and Dr. Lixin Fan (National Cancer Institute) for expert support in the SAXS experiment. Use of the shared scattering beamline 12-ID-B resource is allocated under the PUP-24152 agreement between the National Cancer Institute and Argonne National Laboratory.
This work was supported by National Key Research and Development Program of China Grant 2016YFA0500700, the China Youth 1000-Talent Program of the State Council of China, the Beijing Advanced Innovation Center for Structural Biology, and the Tsinghua-Peking Joint Center for Life Sciences (to X. F.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1 and S2 and Tables S1 and S2.
- PIA
- polysaccharide intercellular adhesin
- 2D
- two-dimensional
- 3D
- three-dimensional
- Sbp
- small basic protein
- rSbp
- recombinant Sbp
- ThT
- thioflavin T
- BTA-1
- 2-(4′-methylaminophenyl)benzothiazole
- ThS
- thioflavin S
- SAXS
- small-angle X-ray scattering
- PDDF
- pair distance distribution function
- NSD
- normalized spatial discrepancy
- SEC
- size-exclusion chromatography
- ITC
- isothermal titration calorimetry
- AFM
- atomic force microscopy
- TEM
- transmission electron microscopy
- Rg
- radius of gyration
- Aap
- accumulation-associated protein
- aa
- amino acid(s)
- TEV
- tobacco etch virus
- Dmax
- maximum distance within the molecule
- Vc
- volume-of-correlation
- HSQC
- heteronuclear single quantum coherence
- IDP
- intrinsically disordered protein
- CR
- Congo red
- PFA
- paraformaldehyde
- hiAPP
- hormone islet amyloid polypeptide
- PAP
- prostatic acid phosphatase
- IPTG
- isopropyl β-d-1-thiogalactopyranoside
- GST
- glutathione S-transferase
- I(0)
- forward scattering intensity
- TSB
- tryptic soy broth.
References
- 1. Otto M. (2009) Staphylococcus epidermidis—the ‘accidental’ pathogen. Nat. Rev. Microbiol. 7, 555–567 10.1038/nrmicro2182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Campoccia D., Montanaro L., and Arciola C. R. (2006) The significance of infection related to orthopedic devices and issues of antibiotic resistance. Biomaterials 27, 2331–2339 10.1016/j.biomaterials.2005.11.044 [DOI] [PubMed] [Google Scholar]
- 3. Büttner H., Mack D., and Rohde H. (2015) Structural basis of Staphylococcus epidermidis biofilm formation: mechanisms and molecular interactions. Front. Cell. Infect. Microbiol. 5, 14 10.3389/fcimb.2015.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paharik A. E., and Horswill A. R. (2016) The staphylococcal biofilm: adhesins, regulation, and host response. Microbiol. Spectr. 4, VMBF-0022-2015 10.1128/microbiolspec.VMBF-0022-2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Van Mellaert L., Shahrooei M., Hofmans D., and Eldere J. V. (2012) Immunoprophylaxis and immunotherapy of Staphylococcus epidermidis infections: challenges and prospects. Expert Rev. Vaccines 11, 319–334 10.1586/erv.11.190 [DOI] [PubMed] [Google Scholar]
- 6. Schaeffer C. R., Hoang T. N., Sudbeck C. M., Alawi M., Tolo I. E., Robinson D. A., Horswill A. R., Rohde H., and Fey P. D. (2016) Versatility of biofilm matrix molecules in Staphylococcus epidermidis clinical isolates and importance of polysaccharide intercellular adhesin expression during high shear stress. mSphere 1, e00165–16 10.1128/mSphere.00165-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Flemming H. C., and Wingender J. (2010) The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633 10.1038/nrmicro2415 [DOI] [PubMed] [Google Scholar]
- 8. O'Gara J. P. (2007) ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol. Lett. 270, 179–188 10.1111/j.1574-6968.2007.00688.x [DOI] [PubMed] [Google Scholar]
- 9. Speziale P., Pietrocola G., Foster T. J., and Geoghegan J. A. (2014) Protein-based biofilm matrices in staphylococci. Front. Cell. Infect. Microbiol 4, 171 10.3389/fcimb.2014.00171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaeffer C. R., Woods K. M., Longo G. M., Kiedrowski M. R., Paharik A. E., Büttner H., Christner M., Boissy R. J., Horswill A. R., Rohde H., and Fey P. D. (2015) Accumulation-associated protein enhances Staphylococcus epidermidis biofilm formation under dynamic conditions and is required for infection in a rat catheter model. Infect. Immun. 83, 214–226 10.1128/IAI.02177-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conrady D. G., Wilson J. J., and Herr A. B. (2013) Structural basis for Zn2+-dependent intercellular adhesion in staphylococcal biofilms. Proc. Natl. Acad. Sci. U.S.A. 110, E202–E211 10.1073/pnas.1208134110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Conrady D. G., Brescia C. C., Horii K., Weiss A. A., Hassett D. J., and Herr A. B. (2008) A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc. Natl. Acad. Sci. U.S.A. 105, 19456–19461 10.1073/pnas.0807717105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanchez C. J., Shivshankar P., Stol K., Trakhtenbroit S., Sullam P. M., Sauer K., Hermans P. W., and Orihuela C. J. (2010) The pneumococcal serine-rich repeat protein is an intra-species bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog. 6, e1001044 10.1371/journal.ppat.1001044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Missineo A., Di Poto A., Geoghegan J. A., Rindi S., Heilbronner S., Gianotti V., Arciola C. R., Foster T. J., Speziale P., and Pietrocola G. (2014) IsdC from Staphylococcus lugdunensis induces biofilm formation under low-iron growth conditions. Infect. Immun. 82, 2448–2459 10.1128/IAI.01542-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geoghegan J. A., Corrigan R. M., Gruszka D. T., Speziale P., O'Gara J. P., Potts J. R., and Foster T. J. (2010) Role of surface protein SasG in biofilm formation by Staphylococcus aureus. J. Bacteriol. 192, 5663–5673 10.1128/JB.00628-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbu E. M., Mackenzie C., Foster T. J., and Höök M. (2014) SdrC induces staphylococcal biofilm formation through a homophilic interaction. Mol. Microbiol. 94, 172–185 10.1111/mmi.12750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. DePas W. H., and Chapman M. R. (2012) Microbial manipulation of the amyloid fold. Res. Microbiol. 163, 592–606 10.1016/j.resmic.2012.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Taglialegna A., Lasa I., and Valle J. (2016) Amyloid structures as biofilm matrix scaffolds. J. Bacteriol. 198, 2579–2588 10.1128/JB.00122-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dragoš A., Kovács Á. T., and Claessen D. (2017) The role of functional amyloids in multicellular growth and development of Gram-positive bacteria. Biomolecules 7, E60 10.3390/biom7030060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cremades N., and Dobson C. M. (2018) The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol. Dis. 109, 178–190 10.1016/j.nbd.2017.07.009 [DOI] [PubMed] [Google Scholar]
- 21. Maury C. P. (2009) The emerging concept of functional amyloid. J. Intern. Med. 265, 329–334 10.1111/j.1365-2796.2008.02068.x [DOI] [PubMed] [Google Scholar]
- 22. Chiti F., and Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 10.1146/annurev.biochem.75.101304.123901 [DOI] [PubMed] [Google Scholar]
- 23. Gibson D. L., White A. P., Rajotte C. M., and Kay W. W. (2007) AgfC and AgfE facilitate extracellular thin aggregative fimbriae synthesis in Salmonella enteritidis. Microbiology 153, 1131–1140 10.1099/mic.0.2006/000935-0 [DOI] [PubMed] [Google Scholar]
- 24. Dueholm M. S., Petersen S. V., Sønderkær M., Larsen P., Christiansen G., Hein K. L., Enghild J. J., Nielsen J. L., Nielsen K. L., Nielsen P. H., and Otzen D. E. (2010) Functional amyloid in Pseudomonas. Mol. Microbiol. 77, 1009–1020 10.1111/j.1365-2958.2010.07269.x [DOI] [PubMed] [Google Scholar]
- 25. Romero D., Aguilar C., Losick R., and Kolter R. (2010) Amyloid fibers provide structural integrity to Bacillus subtilis biofilms. Proc. Natl. Acad. Sci. U.S.A. 107, 2230–2234 10.1073/pnas.0910560107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwartz K., Syed A. K., Stephenson R. E., Rickard A. H., and Boles B. R. (2012) Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. 8, e1002744 10.1371/journal.ppat.1002744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taglialegna A., Navarro S., Ventura S., Garnett J. A., Matthews S., Penades J. R., Lasa I., and Valle J. (2016) Staphylococcal Bap proteins build amyloid scaffold biofilm matrices in response to environmental signals. PLoS Pathog. 12, e1005711 10.1371/journal.ppat.1005711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oli M. W., Otoo H. N., Crowley P. J., Heim K. P., Nascimento M. M., Ramsook C. B., Lipke P. N., and Brady L. J. (2012) Functional amyloid formation by Streptococcus mutans. Microbiology 158, 2903–2916 10.1099/mic.0.060855-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Romero D., and Kolter R. (2014) Functional amyloids in bacteria. Int. Microbiol. 17, 65–73 10.2436/20.1501.01.208 [DOI] [PubMed] [Google Scholar]
- 30. Decker R., Burdelski C., Zobiak M., Büttner H., Franke G., Christner M., Saß K., Zobiak B., Henke H. A., Horswill A. R., Bischoff M., Bur S., Hartmann T., Schaeffer C. R., Fey P. D., et al. (2015) An 18 kDa scaffold protein is critical for Staphylococcus epidermidis biofilm formation. PLoS Pathog. 11, e1004735 10.1371/journal.ppat.1004735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dutta A., Bhattacharyya S., Kundu A., Dutta D., and Das A. K. (2016) Macroscopic amyloid fiber formation by staphylococcal biofilm associated SuhB protein. Biophys. Chem. 217, 32–41 10.1016/j.bpc.2016.07.006 [DOI] [PubMed] [Google Scholar]
- 32. Rambo R. P., and Tainer J. A. (2013) Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481 10.1038/nature12070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fischer H., de Oliveira Neto M., Napolitano H. B., Polikarpov I., and Craievich A. F. (2010) Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109 10.1107/S0021889809043076 [DOI] [Google Scholar]
- 34. Rambo R. P., and Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 10.1002/bip.21638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kosol S., Contreras-Martos S., Cedeño C., and Tompa P. (2013) Structural characterization of intrinsically disordered proteins by NMR spectroscopy. Molecules 18, 10802–10828 10.3390/molecules180910802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Buchan D. W., Minneci F., Nugent T. C., Bryson K., and Jones D. T. (2013) Scalable web services for the PSIPRED Protein Analysis Workbench. Nucleic Acids Res. 41, W349–W357 10.1093/nar/gkt381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Greenfield N. J. (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 1, 2876–2890 10.1038/nprot.2006.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Espargaró A., Busquets M. A., Estelrich J., and Sabate R. (2015) Predicting the aggregation propensity of prion sequences. Virus Res. 207, 127–135 10.1016/j.virusres.2015.03.001 [DOI] [PubMed] [Google Scholar]
- 39. Frousios K. K., Iconomidou V. A., Karletidi C. M., and Hamodrakas S. J. (2009) Amyloidogenic determinants are usually not buried. BMC Struct. Biol. 9, 44 10.1186/1472-6807-9-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sarroukh R., Goormaghtigh E., Ruysschaert J. M., and Raussens V. (2013) ATR-FTIR: a “rejuvenated” tool to investigate amyloid proteins. Biochim. Biophys. Acta 1828, 2328–2338 10.1016/j.bbamem.2013.04.012 [DOI] [PubMed] [Google Scholar]
- 41. Lee G., Lee W., Lee H., Lee C. Y., Eom K., and Kwon T. (2015) Self-assembled amyloid fibrils with controllable conformational heterogeneity. Sci. Rep. 5, 16220 10.1038/srep16220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aguilera P., Marcoleta A., Lobos-Ruiz P., Arranz R., Valpuesta J. M., Monasterio O., and Lagos R. (2016) Identification of key amino acid residues modulating intracellular and in vitro microcin E492 amyloid formation. Front. Microbiol. 7, 35 10.3389/fmicb.2016.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Uversky V. N. (2010) Targeting intrinsically disordered proteins in neurodegenerative and protein dysfunction diseases: another illustration of the D(2) concept. Expert Rev. Proteomics 7, 543–564 10.1586/epr.10.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nanga R. P., Brender J. R., Vivekanandan S., and Ramamoorthy A. (2011) Structure and membrane orientation of IAPP in its natively amidated form at physiological pH in a membrane environment. Biochim. Biophys. Acta 1808, 2337–2342 10.1016/j.bbamem.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sciacca M. F., Brender J. R., Lee D. K., and Ramamoorthy A. (2012) Phosphatidylethanolamine enhances amyloid fiber-dependent membrane fragmentation. Biochemistry 51, 7676–7684 10.1021/bi3009888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hartman K., Brender J. R., Monde K., Ono A., Evans M. L., Popovych N., Chapman M. R., and Ramamoorthy A. (2013) Bacterial curli protein promotes the conversion of PAP248–286 into the amyloid SEVI: cross-seeding of dissimilar amyloid sequences. PeerJ 1, e5 10.7717/peerj.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fong J. N., and Yildiz F. H. (2015) Biofilm matrix proteins. Microbiol. Spectr. 3, MB-0004–2014 10.1128/microbiolspec.MB-0004-2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., and Svergun D. I. (2003) PRIMUS—a Windows-PC based system for small-angle scattering data analysis. J. Appl Crystallogr. 36, 1277–1282 10.1107/S0021889803012779 [DOI] [Google Scholar]
- 49. Wang J., Zuo X., Yu P., Xu H., Starich M. R., Tiede D. M., Shapiro B. A., Schwieters C. D., and Wang Y. X. (2009) A method for helical RNA global structure determination in solution using small-angle x-ray scattering and NMR measurements. J. Mol. Biol. 393, 717–734 10.1016/j.jmb.2009.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 10.1107/S0021889892001663 [DOI] [Google Scholar]
- 51. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 10.1016/S0006-3495(99)77443-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Volkov V. V., and Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864 10.1107/S0021889803000268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kozin M. B., and Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 10.1107/S0021889800014126 [DOI] [Google Scholar]
- 54. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., and Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 55. Johnson B. A. (2004) Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol. Biol. 278, 313–352 10.1385/1-59259-809-9:313 [DOI] [PubMed] [Google Scholar]
- 56. Yang J., Yan R., Roy A., Xu D., Poisson J., and Zhang Y. (2015) The I-TASSER Suite: protein structure and function prediction. Nat. Methods 12, 7–8 10.1038/nmeth.3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brückner R. (1997) Gene replacement in Staphylococcus carnosus and Staphylococcus xylosus. FEMS Microbiol. Lett. 151, 1–8 10.1016/S0378-1097(97)00116-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.