

Abstract
Acetylcholinesterase (AChE) hydrolyzes the neurotransmitter acetylcholine in neurons. However, AChE has been proposed to also have nonneuronal functions in different cell types. Here, we report that AChE is expressed in melanocytes and melanoma cells, and that the tetrameric (G4) form is the major AChE isoform in these cells. During melanogenesis of B16F10 murine melanoma cells, AChE levels decreased markedly. The differentiation of melanoma cells led to (i) an increase in melanin and tyrosinase, (ii) a change in intracellular cAMP levels, and (iii) a decrease in microphthalmia-associated transcription factor (MITF). We hypothesized that the regulation of AChE during melanogenesis is mediated by two transcription factors: cAMP-response element–binding protein (CREB) and MITF. In melanoma cells, exogenous cAMP suppressed AChE expression and the promoter activity of the ACHE gene. This suppression was mediated by a cAMP-response element (CRE) located on the ACHE promoter, as mutation of CRE relieved the suppression. In melanoma, MITF overexpression induced ACHE transcription, and mutation of an E-box site in human ACHE promoter blocked this induction. An AChE inhibitor greatly enhanced acetylcholine-mediated responses of melanogenic gene expression levels in vitro; however, this enhancement was not observed in the presence of agonists of the muscarinic acetylcholine receptor. These results indicate that ACHE transcription is regulated by cAMP-dependent signaling during melanogenesis of B16F10 cells, and the effect of this enzyme on melanin production suggests that it has a potential role in skin pigmentation.
Keywords: acetylcholinesterase (AChE), melanogenesis, cyclic AMP (cAMP), melanoma, epidermis, melanin, melanocyte, MITF, skin pigment
Introduction
Melanogenesis is a process of melanin production in melanocyte or melanoma cells. Melanocytes originate from neural crest cells and exist in epidermis, hair, and iris (1). It was found in heart (2), nervous system, and inner ear (3, 4). In fact, melanogenesis is a physiological response to protect cells from DNA damage and apoptosis, as melanin has the properties of UV absorption, antioxidation and free radical scavenging (5, 6). Upon UV irradiation, the keratinocytes surrounding melanocytes and melanocytes themselves in epidermis secrete α-melanocyte stimulating hormone (α-MSH).3 In melanocytes, the binding of α-MSH to melanocortin 1 receptor (MC1R) increases adenylyl cyclase activity, resulting in cAMP-induced transcription of the master transcriptional factor MITF (7). This basic helix-loop-helix leucine zipper transcription factor MITF binds to E-box sequence in promoter region of genes coding for melanin synthesis, including tyrosinase (TYR), tyrosinase-related protein 1 (TRP1), and DOPA-chrome tautomerase (DCT). These are the enzymes to convert colorless l-dopa into melanin (8).
The cholinergic system in keratinocyte and melanocyte is possibly correlated with melanin production (9, 10). The epidermal keratinocyte has a full spectrum of cholinergic markers (9). Indeed, autocrine and paracrine acetylcholine (ACh) is required to sustain the viability of keratinocytes in vivo. In human melanocytes, muscarinic acetylcholine receptors (mAChR) on cell membrane regulate the concentration of intracellular free Ca2+ in response to ACh released by keratinocytes affecting skin pigmentation (9, 11). However, the expression profiles of other cholinergic molecules in skin-related cells and their effect in skin pigmentation have not been fully elucidated.
Acetylcholinesterase (AChE) (EC 3.1.1.7) is an enzyme hydrolyzing ACh into acetate and choline. It is located on postsynaptic membrane and plays an important role in terminating the ACh-mediated signal transmission. In mammals, mRNA splicing generates three known variants of AChE transcripts: read-through (AChER), hydrophobic (AChEH), and tailed (AChET) (12). Through posttranslational modifications, AChER forms soluble monomers (G1), which are involved in response to psychological stress (13). AChEH is anchored via phosphatidylinositol to erythrocyte membranes as dimers (G2) (14, 15). AChET is the most abundant form of AChE in brain and muscle. AChET and collagen Q (ColQ) form an asymmetric enzyme at neuromuscular junctions (16); its association with proline-rich membrane anchor (PRiMA) forms the globular tetramer (G4) in brain and muscle (17–19).
The nonneuronal functions of AChE in various tissues have been proposed by different groups (20, 21). AChE is expressed in bone tissue, osteoblast, and osteoblast-like cell lines. The expression of AChE is by Wnt/β-catenin signaling pathway during osteoblastic differentiation, suggesting its function in bone formation (21). The presence of AChE in epidermis was shown in patients suffering from vitiligo (22), implying the possible functional roles of AChE in skin pigmentation (23–25). In this study, we found that AChET was the major variant constituting G4 AChE in melanocytes and melanoma; the activity and expression levels of AChE decreased during the melanogenesis progress; and ACh, the agonist of AChR, inhibited melanin production.
Results
Expression of AChE during melanogenesis
The expression of AChE in B16F10 cells, Melan-A cells, human epidermal melanocytes (HEM), and rat dermal fibroblasts (RDF) was recognized by using anti-AChE antibody in immunostaining (Fig. 1A). AChE activities were measured in these cultures. In sucrose density gradient analysis, G4 was the major form of AChE being found in all tested cell types (Fig. 1B). The G1/G2 forms were at relatively low level. Immunofluorescence staining of mouse skin showed that AChE was highly expressed in melanocytes (indicated by TYR as marker) (Fig. 1C). The G1/G2 and G4 forms of AChE showed similar levels of activity in skin, which could be because of the presence of various cell types in skin, e.g. keratinocytes. The localization of AChE in plasma membrane of cultured B16F10 cells was shown (Fig. S1A). The mRNA encoding AChER, AChEH, and AChET were quantified by qPCR. Both B16F10 and Melan-A cells contained the most copies of AChET variant, at least 2-fold higher than that of AChER or AChEH (Fig. 1D).
Figure 1.

AChE expression in skin-related cells. Mouse skin melanoma (B16F10), mouse skin melanocytes (Melan-A), primary culture HEMs, and primary culture RDF were cultured. A, cells were fixed with 4% PFA for 15 min and then stained with anti-AChE antibody with 0.1% Triton X-100, followed with the Alexa Fluor 488 conjugated antibody. Nucleus were stained with DAPI. B, cell lysates (200 μl) were collected to analyze AChE molecular forms by sucrose density gradient analysis. AChE activity was plotted as a function of the sedimentation (S) value, estimated from the position of the sedimentation markers. Enzymatic activities are expressed in arbitrary units of which 1 is equal to 0.80, 0.56, 0.53, and 0.21 milliunit/milligram, in Melan-A, HEM, B16F10, and RDF, respectively, and a representative result was shown, n = 4. C, the 10-μm section of mice dorsal skin (2 weeks old) was fixed with 4% PFA and embedded in OCT medium for section followed by incubated with anti-AChE antibody, KRT antibody, and anti-TYR antibody at 4 °C overnight (left). The skin lysate was subjected to the sucrose density gradient analysis (right) and enzymatic activities are expressed in arbitrary units of which 1 is equal to 0.93 milliunit/milligram. D, total RNA was extracted from B16F10 and Melan-A cell lines for absolute quantitative PCR analysis to determine AChE splicing variants. The absolute amount of each AChE splicing variant per 50 ng of total RNA was calculated according to the method of quantification with qPCR coupled with standard curves. Data are expressed as number of copy. Values are mean ± S.D., n = 4, each with triplicate samples; *, p < 0.05; **, p < 0.01.
B16F10 murine melanoma cell is an in vitro model for the study of melanogenesis. During the melanogenesis, the amount of melanin in B16F10 cells increased; the plateau of melanin production, which was ∼2.5-fold of increase, was reached at about 4 days after culture (Fig. 2A). On the contrary, the enzymatic activity of AChE decreased at least by 50% during this process (Fig. 2A). The level of intracellular cAMP, a known regulator for melanogenesis, peaked on day 2 before dropping to the basal level (Fig. 2A). According to results from sucrose density gradient analysis, the G4 form was the major AChE form being regulated during melanogenesis (Fig. 2B). The promoter constructs pAChE-Luc and pCRE-Luc were transfected into cultured B16F10 cells, and the promoter-driven luciferase activities were decreased to a low level after 4 days of culture. The promoter activity of pCRE-Luc peaked on day 2, similar to that of cAMP level (Fig. 2C). The AChE and MITF mRNA levels decreased by ∼50% during melanogenesis (Fig. 2D). TYR mRNA peaked on day 3 of culture before a decline. The amount of AChE protein (∼60 kDa) decreased by ∼40% during melanogenesis (Fig. 2E), correlating to the trends in activity and mRNA levels. The amount of TYR protein (∼80 kDa) reached its plateau on day 3 of culture before a decline (Fig. 2E). MITF protein (∼60 kDa) level was markedly reduced by over 70% (Fig. 2E). To conclude, the expression of AChE, TYR, and MITF were in synchronization at both transcriptional and translational levels. The protein kinase A (PKA) and CREB phosphorylation also showed the similar tendencies that they reached their highest levels on day 2 before attenuation (Fig. 2, F and G). These data suggest that during melanogenesis, the regulation of AChE could be triggered by increased intracellular cAMP. Resembling phenomenon was reported during the formation of myotube (26).
Figure 2.

AChE expression during melanogenesis in B16F10 cells. Cultured B16F10 cultures were harvested on different days, and cell lysates were collected. A, AChE activity, intracellular melanin, and cAMP level were determined. Data are expressed in arbitrary units. For AChE activity, 1 is equal to 0.98 milliunit/milligram; for melanin content, 1 is equal to 0.023 mg/mg; and for cAMP, 1 is equal to 4.83 picomole/milligram. B, AChE molecular forms after sucrose density gradient analysis. Data are expressed in arbitrary units of which 1 is equal to 0.34 milliunit/milligram. C, cultured cells transfected with pAChE-Luc and pCRE-Luc were collected for luciferase assay. Data are normalized and expressed as the percentage of the control (day 1). D, total RNA was extracted from B16F10 cells for quantitative PCR to determine the transcriptional levels of ACHE, TYR, and MITF genes. Data are normalized and expressed as the percentage of the control (day 1). E, cell lysates (40 μg) were collected for Western blot analysis. GAPDH was used as loading control. AChE (∼60 kDa), TYR (∼80 kDa), MITF (∼60 kDa), and GAPDH (∼37 kDa) are shown. F, cell lysates (40 μg) were collected for Western blot analysis. Total CREB (t-CREB) was used as loading control; phosphorylated CREB (p-CREB) (∼43 kDa); t-CREB (∼43 kDa). G, PKA activity was determined from cultured B16F10 from day 1 to day 5. Data are normalized and expressed as the percentage of the control (day 1). All values are in mean ± S.D., each with triplicate samples, n = 4; *, p < 0.05; **, p < 0.01.
AChE expression is suppressed by cAMP
The cAMP-dependent signaling is a dominant pathway in regulating melanogenesis (27). To investigate the role of cAMP in AChE expression, cultured B16F10 cells were treated with Bt2-cAMP for 24 h. The Bt2-cAMP treatment reduced AChE activity in a dose-dependent manner; AChE activity decreased to around 60% after 1 mm Bt2-cAMP treatment (Fig. 3A). However, melanin amount increased by over 50% in response to the same Bt2-cAMP treatment (Fig. 3A). AChE mRNA production was suppressed after administration of 1 mm Bt2-cAMP whereas the mRNAs of TYR and MITF increased drastically in the presence of Bt2-cAMP (Fig. 3B). The protein levels of AChE, TYR, and MITF were determined in Bt2-cAMP–treated B16F10 cells after 12 h of Bt2-cAMP administration. TYR expression was transiently induced; AChE was down-regulated by ∼80%; and MITF protein expression was induced by ∼8-fold (Fig. 3C).
Figure 3.

cAMP suppresses AChE expression in B16F10 cells. Cultured B16F10 cells were treated with Bt2-cAMP at various concentrations, and cell lysates were collected. A, AChE activity is expressed as the percentage of control (sample treated with vehicle); intracellular melanin content is expressed as the percentage of increase to control. B, cultured cells were treated with Bt2-cAMP (0.5 mm) for 24 h, and total RNA was extracted for qPCR of ACHE, MITF, and TYR genes. Data are normalized and expressed as the percentage of control. C, cultured cells were treated with Bt2-cAMP (0.5 mm) for different time courses, and the cell lysates (40 μg) were collected for Western blot analysis. GAPDH was used as a loading control. AChE (∼60 kDa), TYR (∼80 kDa), MITF (∼60 kDa), and GAPDH (∼37 kDa) are shown. Data are normalized and expressed as the percentage/fold of control. All values are in mean ± S.D., n = 4, each with triplicate samples; *, p < 0.05; **, p < 0.01.
The role of cAMP in regulating AChE expression was unfolded by measuring luciferase activities of cultured B16F10 cells transfected with plasmids coding fusion genes pAChE-Luc and pCRE-Luc. The suppression of pAChE-Luc activity in transfected B16F10 cells, induced by Bt2-cAMP, was rescued by a PKA inhibitor H89 at 10 μm (Fig. 4A). The suppression was not fully rescued, possibly because of incomplete blockage of PKA activity by H89. CRE site was identified in human ACHE gene (28). Site-directed mutagenesis was conducted to generate a promoter construct pAChEΔCRE-Luc (Fig. 4B). B16F10 cells transfected with this mutant showed no observable response to Bt2-cAMP (Fig. 4C), suggesting an important role of CRE site in AChE regulation during melanogenesis. Similarly, the adenylyl cyclase activator forskolin showed same effects as those of Bt2-cAMP in the activities of pCRE-Luc, pAChE-Luc, and pAChEΔCRE-Luc, as well as AChE activity (Fig. S1B). In summary, the expression of AChE was suppressed by cAMP through CRE-binding site in the promoter of ACHE.
Figure 4.
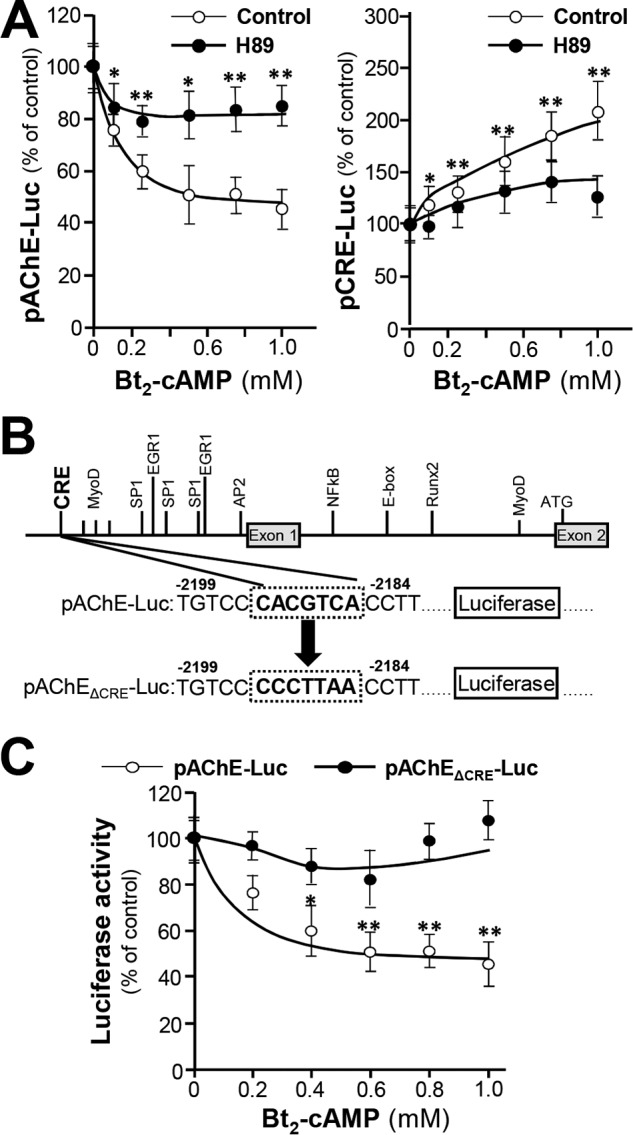
cAMP suppresses AChE expression through CRE site. A, cultured B16F10 cells transfected with pAChE-Luc or pCRE-Luc constructs were treated with various concentrations of Bt2-cAMP for 12 h, and then cell lysates were collected for luciferase assay. B, the mutagenesis of CRE site on pAChE-Luc is shown. C, cultured B16F10 cells transfected with pAChE-Luc or pAChEΔCRE-Luc construction were treated with various concentrations of Bt2-cAMP for 12 h, and then cell lysates were collected for luciferase assay. Data are normalized and expressed as the percentage of control. All values are in mean ± S.D., n = 4, each with triplicate samples; *, p < 0.05; **, p < 0.01.
MITF promotes AChE expression
MITF is the master transcription factor of melanogenesis and development of melanocyte/melanoma. It acts through binding to E-box motifs consisting of a core hexa-nucleotide sequence CAXXTG (X can be A/T/C/G). E-box is identified in genes encoding TYR and its related protein TRP1 (29). Because the E-box motif CAGCTG was found between exons 1 and 2 of mammalian ACHE gene (28), we hypothesized that this motif serves as a binding site for MITF. To determine whether ACHE transcription is influenced by MITF, DNA construct encoding MITF was transiently transfected into cultured B16F10 cells to overexpress MITF. The resultant transcriptional expression of AChE and TYR increased by 50% (Fig. 5A). The protein level of AChE increased by over 70% (Fig. 5A). The internal control protein GAPDH showed no observable change. The binding of MITF to the ACHE gene was further monitored by ChIP assay. In MITF overexpression group, the DNA enrichment by immunoprecipitation with anti-MITF antibody was more than 4-fold, doubling the DNA enrichment in the control group (vehicle only) (Fig. 5B). This result implied that the binding of MITF to E-box motif in ACHE gene is associated to an up-regulation of AChE mRNA and protein. This E-box motif is highly conserved in mammalian species (Fig. 5C). To further confirm the binding of this E-box to MITF, the E-box of human AChE promoter was mutated in a luciferase plasmid pAChEΔE-box-Luc for further transfection in B16F10 cells (Fig. 5C). pAChEΔE-box-Luc did not show response in luciferase activity to MITF overexpression; whereas an induction was observed in the native pAChE-Luc construct (Fig. 5D).
Figure 5.

MITF induces AChE expression through binding to E-box. A, cultured B16F10 cells were transfected with cDNA encoding MITF, and cell lysates were collected for quantitative PCR (left panel) and Western blot analysis (right panel). GAPDH was used as loading control. Data are normalized and expressed as the percentage/fold of control (transfected with empty vector). B, cultured B16F10 cells were transfected with cDNA encoding MITF, then the quantitative ChIP assay was performed. The primers flanking the E-box–binding site of the ACHE promoter were used, and the results were analyzed through the fold enrichment method. The signal intensities were expressed as the fold of basal. C, mutagenesis of the E-box–binding site on the ACHE promoter in pAChE-Luc is shown. D, cultured B16F10 cells were transfected with pAChE-Luc or pAChEΔE-box-Luc, and the cell lysates were collected for luciferase assay. Data are normalized and expressed as the percentage of control (transfected with empty vector). All values are in mean ± S.D., n = 4, each with triplicate samples; *, p < 0.05; **, p < 0.01.
ACh reduces melanin production
During the development of the melanocytes and melanoma cells, α-MSH is produced and activates MC1R (Gs protein couple receptor) thus increasing adenylyl cyclase activity. ACh functions as a ligand agonist of mAChR and nicotinic acetylcholine receptor (nAChR) (30). When activated, mAChR M2 and M4 subtypes coupled with Gi proteins inhibit adenylyl cyclase activity, leading to a decrease in cAMP level in cells (31). To determine the role of ACh in melanogenesis, cultured B16F10 cells were treated with ACh, with or without AChE inhibitor BW284c51 for determination of melanin contents (Fig. 6A). Melanin production in B16F10 cells was slightly affected by ACh but was significantly reduced when treated with AChE inhibitor (BW284c51). This suggests that the surface AChE of melanocyte cells mediates the proposed ACh-melanogenesis signal-transduction pathway. To further confirm the inhibitory effect of ACh on melanogenesis, cultured B16F10 cells were transfected with pCRE-Luc, pTYR-Luc, and pMITF-Luc constructs before treatment of ACh, with or without BW284c51 (Fig. 6A). Correlating the response with melanin production, the luciferase activities downstream of CRE promoter, TYR promoter, and MITF promoter were significantly down-regulated by ACh, especially when BW284c51 was applied. The Western blotting result also indicated that the protein levels of MITF and TYR were reduced significantly in B16F10 cells, when the cultures were subjected to ACh administration and AChE inhibition (Fig. 6B). The level of pCRE-Luc activity did not change in either AChE-knockdown or AChE-overexpressing cells. Nonetheless, it was altered when the cells were cotreated with ACh and BW284c51 (Fig. 6C). In all cases, the applied AChE inhibitor did not fully reduce the expression of aforementioned markers, implying that part of these expressions might not be cholinergic dependent.
Figure 6.

ACh reduces melanin production when AChE-inhibited B16F10 cells. A, cultured B16F10 cells were treated with various concentrations of ACh with or without pretreatment of BW284c51 (BW) (20 μm) and the cell lysates were collected for determination of intracellular melanin content, pCRE-Luc, pMITF-Luc, and pTYR-Luc. Data are normalized and expressed as the percentage of control (sample treated with vehicle). B, cultured cells were treated with various concentrations of ACh with pretreatment of BW284c51 (20 μm), and cell lysates (40 μg) were collected for Western blot analysis. GAPDH was used as loading control. Data are normalized and expressed as the percentage of control. C, cultured B16F10 cells were transfected with ACHE shRNA or cotransfected with AChE and PRiMA cDNA, then treated with BW284c51 (20 μm) and ACh (1 mm). Cell lysates were collected for Ellman assay and luciferase assay. Data are normalized and expressed as the percentage of control (transfected with empty vector). All values are in mean ± S.D., n = 4, each with triplicate samples; *, p < 0.05; **, p < 0.01.
The immunostaining of mAChR M2 and M4 subtypes showed in B16F10 cells were positive (Fig. 7A). Over 50% AChE was colocalized with M2 and/or M4 mAChRs. To investigate whether M2/M4 mAChRs mediate inhibition on melanin synthesis, muscarine (an mAChR agonist) and oxotremorine sesquifumarate (an M2/M4 mAChR–specific agonist), AF-DX 116 (an M2 mAChR antagonist), and PD 102807 (an M4 mAChR antagonist) were applied in B16F10 cells. Muscarine or oxotremorine sesquifumarate caused down-regulation of pCRE-Luc and pMITF-Luc activities by ∼30%, but the effect was insensitive to BW284c51 treatment (Fig. 7B). The oxotremorine sesquifumarate–suppressed pCRE-Luc/pMITF-Luc activities were counteracted by AF-DX 116 and/or PD 102807 (Fig. 7C). These results suggest that ACh could mediate melanogenesis through M2/M4 mAChRs.
Figure 7.
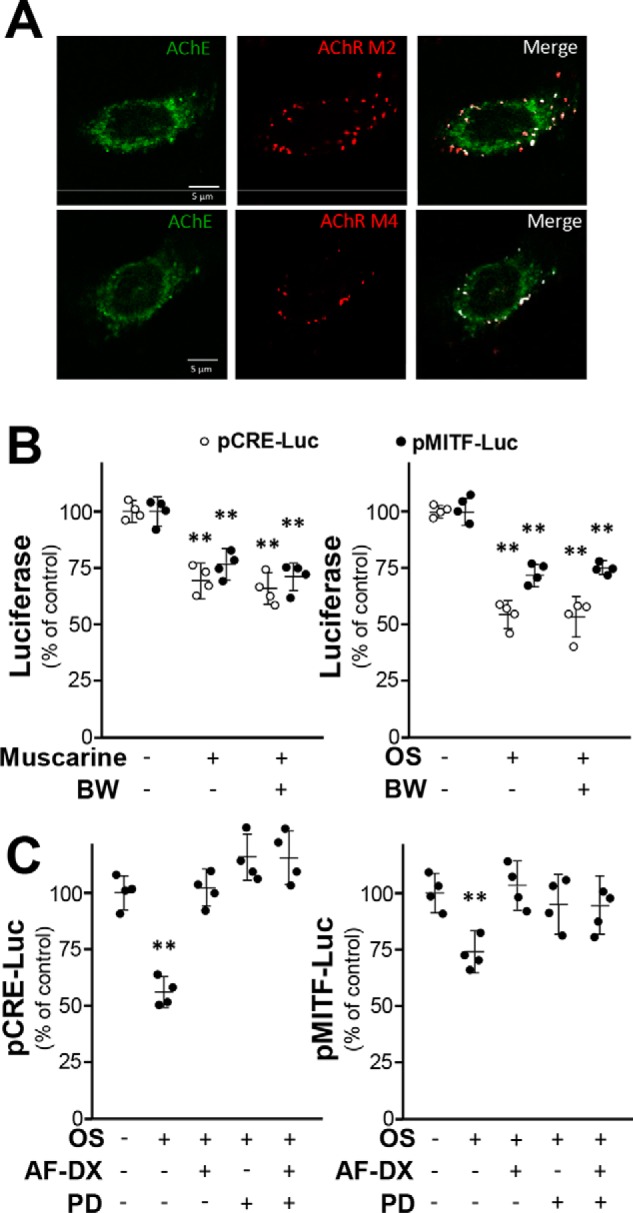
mAChR agonist inhibits melanogenesis pathway. A, B16F10 cells were fixed with 4% PFA for 15 min and stained with anti-AChE antibody and anti-M2/M4 mAChR antibody without Triton X-100. Confocal image showed AChE and M2/M4 mAChR localization. B, B16F10 cells were transfected with pCRE-Luc and pMITF-Luc and then treated with muscarine (1 mm) or oxotremorine sesquifumarate (OS) (50 μm) with or without pretreatment of BW284c51 (BW) (20 μm). Data are normalized and expressed as the percentage of control (sample treated with vehicle). C, in the transfected cells, OS (50 μm) with or without AF-DX 116 (AF-DX) (10 μm) and PD 102807 (PD) (10 μm) were applied. Cell lysates were collected for luciferase assay. Data are normalized and expressed as the percentage of control (sample treated with vehicle). All values are in mean ± S.D., n = 4, each with triplicate samples; *, p < 0.05; **, p < 0.01.
Discussion
Roles of AChE in cell development of different tissues were extensively studied for years. In muscle myogenesis, the regulation of AChE has been proposed to be mediated by a cAMP/PKA-dependent signaling (19, 26, 32). Similarly, in neuronal cells, the expression of AChE is activated by cAMP-signaling cascade (33). In cAMP-regulated AChE in muscle or in neuron, the transcriptional rate of ACHE gene was regulated by CREB (26). In erythrocyte, the glycosylation of AChE, as well as its transcript, was regulated during erythrogenesis (14), and during which GATA-1 was shown to be an activator of ACHE gene transcription (15). In osteoblastic differentiation, the expression of AChE was induced by Runx2, activated by Wnt/β-catenin signaling pathway, through the binding to ACHE promoter (21). This study serves to further expand our knowledge of the role of AChE during melanogenesis.
Our study identified AChE as one of the melanogenic markers. Melanogenesis is a mechanism to protect DNA from damage by UV radiation (34). The majority of melanin is produced by melanocyte, which resides in skin epidermis and hair follicles (11, 35). Regulation of melanin production is orchestrated by multiple signaling events, among which α-MSH–initiated cAMP-dependent signaling pathway is the most critical one (36). α-MSH produced by melanocytes or adjacent keratinocytes binds to MC1R, a Gs-protein–coupled receptor, subsequently increasing intracellular cAMP level. The intracellular cAMP activates PKA that catalyzes the phosphorylation of CREB, inducing the MITF transcription. MITF protein binds to E-box in genes for melanin synthesis including TYR, TRP1, and DCT, resulting in increased melanin production. In melanocytes and melanoma cells, the expression of AChE was down-regulated during melanogenesis; this regulation was mediated by CREB via cAMP signaling. Intriguingly, E-box is found between exon 1 and exon 2 of mammalian ACHE gene, as well as upstream of TYR gene, and the ACHE transcription was promoted by overexpression of MITF in melanoma cells. Although MITF-mediated AChE expression was in contrast to the role of cAMP, AChE levels decreased during melanogenesis in melanoma cells. This indicates that the suppressive effect from phosphorylated CREB is much stronger than the promotive effect, triggered by MITF. Thus, both CREB and MITF could be considered as AChE regulators during melanogenesis (see summary in Fig. 8).
Figure 8.

Schematic summary of AChE in melanogenesis. The α-MSH activates the adenylyl cyclase to induce melanin production through cAMP-dependent pathway in melanocytes. Although MITF slightly increases AChE expression though E-box site in the promoter of ACHE gene, the phosphorylated CREB eventually significantly down-regulates AChE expression through CRE-binding site in the promoter of ACHE gene. Meanwhile, the reduced AChE results in the increasing level of ACh that inhibits the adenylyl cyclase activity to decrease the intracellular cAMP, which alleviates the melanin production. This process is considered a negative feedback control for melanogenesis in melanocytes.
The CREB phosphorylation negatively regulates ACHE transcription, which seems contradictory to up-regulation of ACHE transcription in normal circumstances (19, 33), but coherent to our previous study of AChE expression during chick myogenesis (32). When expressed in melanoma cells, the pCRE-Luc construct showed inductive response to cAMP; whereas the pAChE-Luc construct containing CRE site showed suppressive effect. Additionally, the mutation on the CRE site in pAChE-Luc construct considerably blocked the suppressive response to cAMP. Moreover, it was suggested that cAMP also mediates the down-regulation of ACHE transcription via another regulatory element AP-2 in ACHE promoter in C2C12 cells (32). Alternatively, down-regulation of ACHE transcription in response to increased cAMP level might be caused by posttranscriptional regulation by microRNA (miRNA). It was reported that miR132 and miR212 expression in PC12 cells possibly could lead to degradation of ACHE transcripts (19). All of above indicates that the effect of CREB binding to CRE site may be loci- and cell-type dependent. In melanoma cells, this canonical positive regulation was altered because of other transcription machineries on ACHE promoter.
The role of cholinergic system in melanin production in skin has been proposed. The staining of AChE by using antibody in skin was negative during depigmentation but was positive in pigmentation of marginal dendritic melanocytes (37). ACh showed inhibitory effect on dopa oxidase activity, and thus low AChE amount was observed in skin with vitiligo (22). AChE controls the ACh level directly and suppresses ACh-induced events. In cultured melanoma cells, exogenous applied ACh has little effect on the levels of MITF, TYR, and melanin production when administered alone, but significantly inhibited the expressions when cotreated with BW284c51. This suggested the important role of AChE in cell surface as to control the amount of ACh. By LC-MS analysis, the amount of ACh level in culture medium was measured and was below the detection limit. Thus, the ACh-mediated inhibitory effect in intracellular cAMP should act as a negative feedback control of melanin production. AChE in melanocyte surface plays an indirect role in melanin production. Our preliminary results showed that MITF and TYR genes were down-regulated in skin of ACHE−/− mice, implying the important role of AChE in melanogenesis.4 On the other hand, the AChE inhibitor BW284c51 was reported as a blocker of nAChR in an expression system (38), and the activation of α7 nAChR increased intracellular cAMP levels in hippocampal neurons (39). The expression of nAChR has not been reported in melanocyte. The effect of this receptor activation, if any, should be minimal.
ACh was released from skin under sunlight, UV-A, and tactile stimulus (40). The release of ACh increased from 205 ± 58 to 349 ± 122 picomoles when the skin was exposed to sunlight (40). Keratinocytes, constituting the superficial layers of stratified skin epithelium, were shown to synthesize, degrade, and release ACh to their nearby cells including melanocyte (41). We therefore propose that ACh is mainly secreted by keratinocytes, and it is acting on mAChRs of melanocytes in proximity as a result to regulate the intracellular cAMP in these melanocytes. The underlying mechanisms behind the release of ACh from keratinocyte in response to sunlight are not fully clarified.
The cholinergic system is a primitive autocrine/paracrine signal transduction system evolved ∼2 billion years ago, before the emergence of central nervous system in higher eukaryotes (42). Melanocyte as major cell type in mammalian skin is derived from neural crest cells that later give rise to peripheral and enteric neurons, glia, bone, and cartilages (43). This cholinergic nature of melanocyte could be similar to that in the peripheral nervous system of vertebrates. Moreover, based on our findings, AChE may be a potential target in regulating the rate of melanogenesis, which therefore could pave a new direction in finding new drug candidates for diseases related to pigmentary disorder.
Experimental procedures
Chemicals
Bt2-cAMP, H89, and forskolin were purchased from Sigma-Aldrich. AF-DX 116 and PD 102807 were purchased from Tocris Bioscience (Bristol, U.K.). All cell culture reagents were from Thermo Fisher Scientific (Waltham, MA).
Cell cultures
B16F10 cell line was purchased from ATCC (Manassas, VA) and cultured in Dulbecco's modified Eagle's medium (DMEM). Human epidermal melanocytes were purchased from ScienCell Research Laboratories (Carlsbad, CA) and cultured in melanocyte medium (ScienCell). Melan-A cell line was a gift from Prof. Mingfu Wang at the University of Hong Kong. It was cultured in RPMI 1640 medium. RDF was collected from Sprague-Dawley male rats (2 days old) and prepared as described (44). All culture medium was supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin (10,000 units and 10,000 μg/ml, respectively) in a humidified atmosphere with 5% CO2 at 37 °C.
Animals
Animals were obtained for skin cryosection and primary culture preparation from Animal and Plant Care Facility of Hong Kong University of Science and Technology and performed according to the guidelines of Department of Health, The Government of Hong Kong SAR. The experimental procedures were reviewed and approved by Animal Ethics Committee at the University (Reference No.: (15–50) in DH/SHS/8/2/2 Pt.2). Housing was at a constant temperature (21 °C) and humidity (60%), under a fixed 12-h light/dark cycle and free access to food and water.
Measurement of melanin content
The melanin content from cell culture was determined as described previously, with modification (45). Briefly, B16F10 cells were seeded at a density of 2.0 × 105 cells in 6-well plates. After drug treatment, cells were washed with PBS and dissolved in 200 μl 1 m NaOH solution at 80 °C for 2 h. Then, 100 μl aliquots of the lysate containing dissolved melanin were transferred to 96-well plates, and the melanin contents were measured at 405 nm using Multiskan™ FC Microplate Photometer (Thermo Fisher Scientific) and normalized by protein concentration determined by Bradford protein assay (Bio-Rad Laboratories). The amounts of melanin were calculated according to the melanin standard curve (Fig. S2A).
Intracellular cAMP assay
B16F10 cells were seeded at 4 × 105 cells in 35-mm culture dishes. Collection of intracellular cAMP and determination of cAMP level was done using cAMP Enzyme Immunoassay kit (Sigma-Aldrich). To harvest the cultures, the cells were washed with sterile PBS twice and lysed with 0.1 mm HCl at room temperature. Cells were agitated at 200 rpm for 10 min at room temperature. The lysates were collected and centrifuged. After that 100 μl of supernatant was acetylated as the kit protocol instructed. Standards for calibration curves (Fig. S2C) were also freshly prepared and acetylated. The immunoassay assay procedure was performed according to the kit protocol. End point measurement of enzyme activity was adopted.
Protein kinase A activity assay
B16F10 cells were seeded at 4 × 105 cells until 80% confluence in 35-mm culture dishes. Extraction and measurement of intracellular PKA was done using PKA Colorimetric Activity Kit (Thermo Fisher Scientific). Cells were washed with sterile PBS twice prior to lysis with protease and phosphatase inhibitor cocktails. Cells were scraped from the confluence layers and subject to agitation at 200 rpm for 40 min at 4 °C. The cell lysates were then centrifuged for collection of supernatant, of which 20 μl were mixed with 20 μl of kinase reaction buffer. Standards for calibration were freshly prepared, and PKA activity assay was performed according to the protocol provided by the manufacturer (Fig. S2D).
Real-time quantitative PCR
Total RNA was extracted using RNAzol®RT reagent (Molecular Research Center, Cincinnati, OH). Briefly, cells were incubated in RNAzol at room temperature. Then, the total RNA was precipitated in 75% ethanol (v/v) by centrifugation at 12,000 × g for 10 min. The RNA pellet was washed by 75% ethanol and dissolved in RNase-free water. The RNA quality was determined according to the ratio (∼2.0) of absorbance at 260 nm and 280 nm by NanoDropTM (Thermo Fisher Scientific). Three μg RNA samples were applied for reverse transcription using First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) as the manufacturer's protocol. For absolute quantitative analysis, the sequences of specific primers for AChER, AChEH, and AChET are as follows: forward, 5′-CAG GGG ACC CCA ATG ACC CTC G-3′ and reverse, 5′-CCC ACT CCA TGC GCC TAC CGG T-3′ for mouse AChER; forward, 5′-CCG CGC AGC AAT ATG TGA GCC T-3′ and reverse, 5′-GCA GGT GCA AGG AGC CTC CGT-3′ for mouse AChEH; and forward, 5′-TAG AGG TGC GGC GGG GAC TG-3′ and reverse, 5′-TGA GCA GCG CTC CTG CTT GC-3′ for mouse AChET. Amplification was performed for 45 cycles. Each cycle consisted of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 20 s. For relative quantitative PCR, the sequences of specific primers are as follows: forward, 5′-CAC CGA TAC TCT GGA CGA GG-3′ and reverse, 5′-TCC TGC TTG CTA TAG TGG TCG-3′ for mouse AChE; forward, 5′-AGT CGT ATC TGG CCA TGG CTT G-3′ and reverse, 5′-GCA AGC TGT GGT AGT CGT CTT TGT C-3′ for mouse TYR; forward, 5′-AGA AGC TGG AGC ATG CGA ACC-3′ and reverse, 5′-GTT CCT GGC TGC AGT TCT CAA GAA C-3′ for mouse MITF; and forward, 5′-AAG GTC ATC CCA GAG CTG AA-3′ and reverse, 5′-CTG CTT CAC CTT GA-3′ for mouse GAPDH. Amplification was performed for 45 cycles. Each cycle consisted of denaturation at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 20 s, performed on Roche LightCycler 480 system (Roche).
Immunofluorescent staining
Cells were grown on glass coverslip for 48 h. After PBS wash, cells were fixed with 4% paraformaldehyde for 15 min. Cells were incubated with or without 0.1% Triton X-100 in PBS for 10 min and then blocked by 5% BSA for 1 h. Cultures were stained with anti-AChE antibody at 1:500 (sc-6432, Santa Cruz Biotechnology, Dallas, TX) for 16 h at 4 °C, followed with the Alexa Fluor 488 conjugated anti-goat antibodies (Sigma-Aldrich). Samples were mounted with ProLong™ Gold Antifade Mountant with or without DAPI (Thermo Fisher Scientific). Samples were then examined by a Zeiss Laser Scanning Confocal Microscope. The dorsal skin collected from C57BL/6 male mice (2 weeks old) was fixed with 4% PFA and embedded in OCT medium for section at 10 μm, followed by incubation with anti-AChE antibody, anti-cytokeratin (KRT) antibody (ab94894, Abcam), and anti-tyrosinase (TYR) antibody (sc-20035, Santa Cruz Biotechnology) at 4 °C overnight then with Alexa Fluor 488 conjugated antibody, Alexa Fluor 555 conjugated antibody, and Alexa Fluor 647 conjugated antibody (ab150117, ab150114, ab150115, Abcam).
DNA constructions and transfection
The promoter constructs of human pAChE-Luc and pPRiMA-Luc were described in Refs. 26 and 46. pBabe-MITF was a gift from Ivana de la Serna (Addgene plasmid no. 64888) (47). The DNA construct of CRE sequences tagged with a luciferase gene (pCRE-Luc) was from BD Biosciences. Transient transfection was performed using jetPRIME® reagent (Polyplus Transfection, New York, NY). In brief, the mixture of DNA constructs with reagents was added to cell culture and incubated for 4 h in incubator, followed by replacing medium.
Luciferase assay
Luciferase assay was performed using PierceTM Firefly Luciferase Glow Assay Kit (Thermo Fisher Scientific). Cells were lysed by 100 mm potassium phosphate buffer (pH 7.8), 0.2% Triton X-100, and 1 mm DTT and agitated for 30 min at °C. Afterward, cells were centrifuged at 16,000 × g for 10 min at 4 °C. Twenty μl of cell lysate were used for assay. The luminescent reaction was quantified in a GloMax® 96 Microplate Luminometer (Thermo Fisher Scientific), and the activity was expressed as percentage of untreated controls.
Sucrose density gradient analysis
Various AChE forms were separated by sucrose density gradient analysis according to previous method (48). In brief, continuous 5–20% sucrose gradients in lysis buffer containing 10 mm HEPES, pH 7.5, 1 mm EDTA, 1 mm EGTA, 0.2% Triton X-100, and 150 mm NaCl was prepared in 12-ml polyallomer ultracentrifugation tubes. Two hundred μl (1 μg/μl) cell lysates mixed with sedimentation markers, including alkaline phosphatase (6.1 s) and β-gal (16 s), were loaded onto the gradients followed by centrifugation at 38,000 rpm in SW 41 Ti Rotor (Beckman, Indianapolis, IN) at 4 °C for 16 h. Approximately 48 fractions were collected for determination of AChE activity by Ellman assay: 0.1 mm tetraisopropylpyrophosphoramide (iso-OMPA), 625 μm acetylthiocholine (ATCh), and 0.5 mm Ellman's Reagent were added to 30 μl of each fraction. The mixture was incubated at room temperature for 30 min and AChE activities were measured at 405 nm using Multiskan™ FC Microplate Photometer. AChE forms were determined by summation of the enzymatic activities corresponding to the peaks of sedimentation profile.
SDS-PAGE and Western blot analysis
Cells were lysed in whole cell lysis buffer and shaken for 30 min at 4 °C followed by centrifugation at 12,000 × g at 4 °C for 10 min. The supernatants were collected and protein concentrations were determined using Bradford protein assay (Bio-Rad). The aliquots normalized to 40 μg of protein were applied to 8% SDS-polyacrylamide gels and then transferred to nitrocellulose membranes. The membranes were blocked with 5% skim milk powder in TBS with 0.1% Tween 20 (TBST) for 2 h. After blocking, the membranes were incubated at 4 °C overnight with specific primary antibodies including anti-AChE at 1:500 (sc-6432, Santa Cruz Biotechnology), anti-tyrosinase at 1:200 (sc-20035, Santa Cruz Biotechnology), anti-MITF antibody at 1:100 (sc-52938, Santa Cruz Biotechnology), anti-t-CREB antibody at 1:1000 (4820S, Cell Signaling Technology, Danvers, MA), anti–phospho-CREB at 1:1000 (9198S, Cell Signaling Technology), and anti-GAPDH at 1:500,000 (G8795, Sigma-Aldrich), followed by incubation with horseradish peroxidase (HRP) secondary antibodies (Sigma-Aldrich) at 25 °C for 1 h. The immune-reactive proteins were detected using enhanced chemiluminescence (ECL) Western blotting detection kit (Thermo Fisher Scientific). The intensities of the bands were quantified using ChemiDoc Imaging System (Bio-Rad). The intensities of protein bands were in the nonsaturating range of calibration curves. AChE activity standard curve is shown in Fig. S2B.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed according to the protocol of ChIP kit (Abcam). In brief, cells were fixed with 4% paraformaldehyde and then lysed by sonication at 20% amplitude for 2.5 min using Q125 Sonicator (Qsonica, Melville, NY). The MITF-DNA complex was immunoprecipitated using anti-MITF antibody (ab12039, Abcam) at 2 μg/μl. Primers flanking the MITF-binding site of the AChE promoter were designed as follows: forward, 5′-AAC ATT GGC CGC CTC CAG-3′ and reverse, 5′-GCG GAT TGG TCC CGA CTC-3′. PCR was performed and the PCR products were run at 2% agarose gel electrophoresis. The intensities of the bands were measured using ChemiDoc Imaging System (Bio-Rad) and results were analyzed by fold enrichment method.
Statistics
Each result is represented as the mean ± S.D., calculated from independent replicate samples. Comparisons of the mean for untreated control cells and treated cells were analyzed using one-way analysis of variance (ANOVA) and Student's t test. Significant values were represented as *, p < 0.05, **, p < 0.01.
Author contributions
Q. W. and K. W. K. T. conceptualization; Q. W. formal analysis; Q. W. visualization; Q. W., A. H. Y. F., P. Y., and T. T. X. D. methodology; Q. W. writing-original draft; X. P. K. and T. T. X. D. resources; Q. P. X. investigation; K. W. K. T. supervision; K. W. K. T. funding acquisition; K. W. K. T. project administration; A. H. Y. F. qPCR experiments, skin section, and immunostaining; M. L. X. provided chemicals and tools; K. P. helped with luciferase assay; E. Y. L. L. provided chemicals and tools.
Supplementary Material
Acknowledgments
We thank Dr. Jacky Bonaventure (Institut Curie, PSL Research University, Orsay, France) for providing the pMITF-Luc plasmid and Dr. Jiada Li (State Key Laboratory of Medical Genetics, Central South University, Changsha, Hunan, China) for providing the pTYR-Luc plasmid.
This work was supported by the Hong Kong Research Grants Council Theme-based Research Scheme (T13–607/12R), Innovation Technology Fund (UIM/288, UIM/302, UIM/340, UIT/137, and ITS/022/16FP), TUYF15SC01, and Shenzhen Science and Technology Committee Research Grant (JCYJ 20,160,229,205,726,699, JCYJ 20,160,229,205,812,004, JCYJ 20,160,229,210,027,564, CKFW 2,016,082,916,015,476, JCYJ 20,170,413,173,747,440, and ZDSYS 201, 707,281,432,317 and 20,170, 326). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supporting Figs. S1 and S2.
Q. Wu, A. H. Y. Fung, M. L. Xu, E. Y. L. Liu, T. T. X. Dong, K. W. K. Tsim, unpublished results.
- α-MSH
- α-melanocyte stimulating hormone
- CRE
- cyclic AMP response elements
- CREB
- cyclic AMP-response element–binding protein
- MITF
- microphthalmia-associated transcription factor
- MC1R
- melanocortin 1 receptor
- TYR
- tyrosinase
- TRP1
- tyrosinase-related protein 1
- DCT
- DOPA-chrome tautomerase
- ACh
- acetylcholine
- mAChR
- muscarinic acetylcholine receptor
- AChE
- acetylcholinesterase
- AChER
- read through AChE
- AChEH
- hydrophobic AChE
- AChET
- tailed AChE
- PRiMA
- proline-rich membrane anchor
- qPCR
- quantitative polymerase chain reaction
- HEM
- human epidermal melanocytes
- RDF
- rat dermal fibroblast
- OCT
- optimal cutting temperature
- Bt2-cAMP
- dibutyryl-cyclic AMP
- nAChR
- nicotinic acetylcholine receptor.
References
- 1. Plonka P. M., Passeron T., Brenner M., Tobin D. J., Shibahara S., Thomas A., Slominski A., Kadekaro A. L., Hershkovitz D., Peters E., Nordlund J. J., Abdel-Malek Z., Takeda K., Paus R., Ortonne J.P., Hearing V.J., and Schallreuter K.U. (2009) What are melanocytes really doing all day long … ? Exp. Dermatol. 18, 799–819 10.1111/j.1600-0625.2009.00912.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yajima I., and Larue L. (2008) The location of heart melanocytes is specified and the level of pigmentation in the heart may correlate with coat color. Pigment Cell Melanoma Res. 21, 471–476 10.1111/j.1755-148X.2008.00483.x [DOI] [PubMed] [Google Scholar]
- 3. Goldgeier M. H., Klein L. E., Klein-Angerer S., Moellmann G., and Nordlund J. J. (1984) The distribution of melanocytes in the leptomeninges of the human brain. J. Invest. Dermatol. 82, 235–238 10.1111/1523-1747.ep12260111 [DOI] [PubMed] [Google Scholar]
- 4. Tachibana M. (1999) Sound needs sound melanocytes to be heard. Pigment Cell Res. 12, 344–354 [DOI] [PubMed] [Google Scholar]
- 5. Brenner M., and Hearing V. J. (2008) The protective role of melanin against UV damage in human skin. Photochem. Photobiol. 84, 539–549 10.1111/j.1751-1097.2007.00226.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rodriguez C., Mayo J. C., Sainz R. M., Antolín I., Herrera F., Martín V., and Reiter R. J. (2004) Regulation of antioxidant enzymes: A significant role for melatonin. J. Pineal Res. 36, 1–9 10.1046/j.1600-079X.2003.00092.x [DOI] [PubMed] [Google Scholar]
- 7. Hartman M. L., and Czyz M. (2015) MITF in melanoma: Mechanisms behind its expression and activity. Cell. Mol. Life Sci. 72, 1249–1260 10.1007/s00018-014-1791-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. del Marmol V., and Beermann F. (1996) Tyrosinase and related proteins in mammalian pigmentation. FEBS Lett. 381, 165–168 10.1016/0014-5793(96)00109-3 [DOI] [PubMed] [Google Scholar]
- 9. Grando S. A. (1997) Biological functions of keratinocyte cholinergic receptors. J. Investig. Dermatol. Symp. Proc. 2, 41–48 [DOI] [PubMed] [Google Scholar]
- 10. Buchli R., Ndoye A., Arredondo J., Webber R. J., and Grando S. A. (2001) Identification and characterization of muscarinic acetylcholine receptor subtypes expressed in human skin melanocytes. Mol. Cell. Biochem. 228, 57–72 [DOI] [PubMed] [Google Scholar]
- 11. Hirobe T. (2014) Keratinocytes regulate the function of melanocytes. Dermatologica Sinica 32, 200–204 10.1016/j.dsi.2014.05.002 [DOI] [Google Scholar]
- 12. Rachinsky T. L., Camp S., Li Y., Ekström T. J., Newton M., and Taylor P. (1990) Molecular cloning of mouse acetylcholinesterase: tissue distribution of alternatively spliced mRNA species. Neuron 5, 317–327 10.1016/0896-6273(90)90168-F [DOI] [PubMed] [Google Scholar]
- 13. Meshorer E., Erb C., Gazit R., Pavlovsky L., Kaufer D., Friedman A., Glick D., Ben-Arie N., and Soreq H. (2002) Alternative splicing and neuritic mRNA translocation under long-term neuronal hypersensitivity. Science 295, 508–512 10.1126/science.1066752 [DOI] [PubMed] [Google Scholar]
- 14. Luk W. K., Chen V. P., Choi R. C., and Tsim K. W. (2012) N-linked glycosylation of dimeric acetylcholinesterase in erythrocytes is essential for enzyme maturation and membrane targeting. FEBS J. 279, 3229–3239 10.1111/j.1742-4658.2012.08708.x [DOI] [PubMed] [Google Scholar]
- 15. Xu M. L., Luk W. K., Bi C. W., Liu E. Y., Wu K. Q., Yao P., Dong T. T., and Tsim K. W. (2018) Erythropoietin regulates the expression of dimeric form of acetylcholinesterase during differentiation of erythroblast. J. Neurochem. 10.1111/jnc.14448 [DOI] [PubMed] [Google Scholar]
- 16. Tsim K. W., Randall W. R., and Barnard E. A. (1988) An asymmetric form of muscle acetylcholinesterase contains three subunit types and two enzymic activities in one molecule. Proc. Natl. Acad. Sci. U.S.A. 85, 1262–1266 10.1073/pnas.85.4.1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Massoulié J., Pezzementi L., Bon S., Krejci E., and Vallette F. M. (1993) Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 41, 31–91 10.1016/0301-0082(93)90040-Y [DOI] [PubMed] [Google Scholar]
- 18. Chen V. P., Luk W. K., Chan W. K., Leung K. W., Guo A. J., Chan G. K., Xu S. L., Choi R. C., and Tsim K. W. (2011) Molecular assembly and biosynthesis of acetylcholinesterase in brain and muscle: The roles of t-peptide, FHB domain, and N-linked glycosylation. Front. Mol. Neurosci. 4, 36 10.3389/fnmol.2011.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu E. Y., Xu M. L., Jin Y., Wu Q., Dong T. T., and Tsim K. W. (2018) Genistein, a phytoestrogen in soybean, induces the expression of acetylcholinesterase via G protein-coupled receptor 30 in PC12 cells. Front. Mol. Neurosci. 11, 59 10.3389/fnmol.2018.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Soreq H., and Seidman S. (2001) Acetylcholinesterase—new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302 10.1038/35067589 [DOI] [PubMed] [Google Scholar]
- 21. Xu M. L., Bi C. W. C., Liu E. Y. L., Dong T. T. X., and Tsim K. W. K. (2017) Wnt3a induces the expression of acetylcholinesterase during osteoblast differentiation via the Runx2 transcription factor. J. Biol. Chem. 292, 12667–12678 10.1074/jbc.M117.777581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Iyengar B. (1989) Modulation of melanocytic activity by acetylcholine. Acta Anat. 136, 139–141 10.1159/000146813 [DOI] [PubMed] [Google Scholar]
- 23. Schallreuter K. U., Elwary S. M., Gibbons N. C., Rokos H., and Wood J. M. (2004) Activation/deactivation of acetylcholinesterase by H2O2: More evidence for oxidative stress in vitiligo. Biochem. Biophys. Res. Commun. 315, 502–508 10.1016/j.bbrc.2004.01.082 [DOI] [PubMed] [Google Scholar]
- 24. Schallreuter K. U., Elwary S., Gibbons N., Rokos H., and Wood J. M. (2005) Human epidermal acetylcholinesterase (AchE) is regulated by hydrogen peroxide (H2O2). Exp. Dermatol. 14, 155 10.1111/j.0906-6705.2005.0266j.x [DOI] [Google Scholar]
- 25. Schallreuter K. U., and Pittelkow M. P. (1988) Defective calcium uptake in keratinocyte cell cultures from vitiliginous skin. Arch. Dermatol. Res. 280, 137–139 10.1007/BF00456842 [DOI] [PubMed] [Google Scholar]
- 26. Choi R. C., Siow N. L., Zhu S. Q., and Tsim K. W. (2000) The cAMP-dependent protein kinase mediates the expression of AChE in chick myotubes. Neuroreport 11, 801–806 10.1097/00001756-200003200-00029 [DOI] [PubMed] [Google Scholar]
- 27. D'Mello S. A., Finlay G. J., Baguley B. C., and Askarian-Amiri M. E. (2016) Signaling pathways in melanogenesis. Int. J. Mol. Sci. 17, 1144 10.3390/ijms17071144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mutero A., Camp S., and Taylor P. (1995) Promoter elements of the mouse acetylcholinesterase gene transcriptional regulation during muscle differentiation. J. Biol. Chem. 270, 1866–1872 10.1074/jbc.270.4.1866 [DOI] [PubMed] [Google Scholar]
- 29. Hemesath T. J., Steingrímsson E., McGill G., Hansen M. J., Vaught J., Hodgkinson C. A., Arnheiter H., Copeland N. G., Jenkins N. A., and Fisher D. E. (1994) Microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev. 8, 2770–2780 10.1101/gad.8.22.2770 [DOI] [PubMed] [Google Scholar]
- 30. Tiwari P., Dwivedi S., Singh M. P., Mishra R., and Chandy A. (2013) Basic and modern concepts on cholinergic receptor: A review. Asian Pac. J. Trop. Dis. 3, 413–420 10.1016/S2222-1808(13)60094-8 [DOI] [Google Scholar]
- 31. Ashkenazi A., Winslow J. W., Peralta E. G., Peterson G. L., Schimerlik M. I., Capon D. J., and Ramachandran J. (1987) An M2 muscarinic receptor subtype coupled to both adenylyl cyclase and phosphoinositide turnover. Science 238, 672–675 10.1126/science.2823384 [DOI] [PubMed] [Google Scholar]
- 32. Siow N. L., Choi R. C., Cheng A. W., Jiang J. X., Wan D. C., Zhu S. Q., and Tsim K. W. (2002) A cyclic AMP-dependent pathway regulates the expression of acetylcholinesterase during myogenic differentiation of C2C12 cells. J. Biol. Chem. 277, 36129–36136 10.1074/jbc.M206498200 [DOI] [PubMed] [Google Scholar]
- 33. Wan D. C., Choi R. C., Siow N. L., and Tsim K. W. (2000) The promoter of human acetylcholinesterase is activated by a cyclic adenosine 3′, 5′-monophosphate-dependent pathway in cultured NG108—15 neuroblastoma cells. Neurosci. Lett. 288, 81–85 10.1016/S0304-3940(00)01200-3 [DOI] [PubMed] [Google Scholar]
- 34. Boukari F., Jourdan E., Fontas E., Montaudié H., Castela E., Lacour J. P., and Passeron T. (2015) Prevention of melasma relapses with sunscreen combining protection against UV and short wavelengths of visible light: A prospective randomized comparative trial. J. Am. Acad. Dermatol. 72, 189–190.e1 10.1016/j.jaad.2014.08.023 [DOI] [PubMed] [Google Scholar]
- 35. Cichorek M., Wachulska M., Stasiewicz A., and Tymińska A. (2013) Skin melanocytes: Biology and development. Postepy. Dermatol. Alergol. 30, 30–41 10.5114/pdia.2013.33376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Borovansky J., and Riley P. A. (eds) (2011) Melanins and Melanosomes: Biosynthesis, Structure, Physiological and Pathological Functions, John Wiley & Sons, Hoboken, NJ: 10.1002/9783527636150 [DOI] [Google Scholar]
- 37. Moellmann G., Lerner A. B., and Hendee J. R. Jr. (1974) The mechanism of frog skin lightening by acetylcholine. Gen. Comp. Endocrinol. 23, 45–51 10.1016/0016-6480(74)90051-3 [DOI] [PubMed] [Google Scholar]
- 38. Olivera-Bravo S., Ivorra I., and Morales A. (2005) The acetylcholinesterase inhibitor BW284c51 is a potent blocker of Torpedo nicotinic AchRs incorporated into the Xenopus oocyte membrane. Br. J. Pharmacol. 144, 88–97 10.1038/sj.bjp.0705965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng Q., and Yakel J. L. (2015) Activation of α7 nicotinic acetylcholine receptors increases intracellular cAMP levels via activation of AC1 in hippocampal neurons. Neuropharmacology 95, 405–414 10.1016/j.neuropharm.2015.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schlereth T., Birklein F., Haack K., Schiffmann S., Kilbinger H., Kirkpatrick C. J., and Wessler I. (2006) In vivo release of non-neuronal acetylcholine from the human skin as measured by dermal microdialysis: Effect of botulinum toxin. Br. J. Pharmacol. 147, 183–187 10.1038/sj.bjp.0706451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grando S. A., Kist D. A., Qi M., and Dahl M. V. (1993) Human keratinocytes synthesize, secrete, and degrade acetylcholine. J. Investig. Dermatol. 101, 32–36 10.1111/1523-1747.ep12358588 [DOI] [PubMed] [Google Scholar]
- 42. Baig A. M., Rana Z., Tariq S., Lalani S., and Ahmad H. R. (2018) Traced on the timeline: Discovery of acetylcholine and the components of the human cholinergic system in a primitive unicellular eukaryote Acanthamoeba spp. ACS Chem. Neurosci. 9, 494–504 10.1021/acschemneuro.7b00254 [DOI] [PubMed] [Google Scholar]
- 43. Sommer L. (2011) Generation of melanocytes from neural crest cells. Pigment Cell Melanoma Res. 24, 411–421 10.1111/j.1755-148X.2011.00834.x [DOI] [PubMed] [Google Scholar]
- 44. Seluanov A., Vaidya A., and Gorbunova V. (2010) Establishing primary adult fibroblast cultures from rodents. J. Vis. Exp. 44, e2033 10.3791/2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kim B., Lee S. H., Choi K. Y., and Kim H. S. (2015) N-nicotinoyl tyramine, a novel niacinamide derivative, inhibits melanogenesis by suppressing MITF gene expression. Eur. J. Pharmacol. 764, 1–8 10.1016/j.ejphar.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 46. Xie H. Q., Choi R. C., Leung K. W., Chen V. P., Chu G. K., and Tsim K. W. (2009) Transcriptional regulation of proline-rich membrane anchor (PRiMA) of globular form acetylcholinesterase in neuron: An inductive effect of neuron differentiation. Brain Res. 1265, 13–23 10.1016/j.brainres.2009.01.065 [DOI] [PubMed] [Google Scholar]
- 47. de la Serna I. L., Ohkawa Y., Higashi C., Dutta C., Osias J., Kommajosyula N., Tachibana T., and Imbalzano A. N. (2006) The microphthalmia-associated transcription factor requires SWI/SNF enzymes to activate melanocyte-specific genes. J. Biol. Chem. 281, 20233–20241 10.1074/jbc.M512052200 [DOI] [PubMed] [Google Scholar]
- 48. Choi R. C., Pun S., Dong T. T., Wan D. C., and Tsim K. W. (1997) NG108–15 cells induce the expression of muscular acetylcholinesterase when co-cultured with myotubes. Neurosci. Lett. 236, 167–170 10.1016/S0304-3940(97)00789-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.