

Abstract
In vivo barcoding using nuclease-induced mutations is a powerful approach for recording biological information, including developmental lineages; however, its application in mammalian systems has been limited. We present in vivo barcoding in the mouse with multiple homing guide RNAs that each generate hundreds of mutant alleles and combine for an exponential diversity of barcodes. Activation upon conception and continued mutagenesis through gestation result in developmentally barcoded mice wherein information is recorded in lineage-specific mutations. We use these recordings for reliable post hoc reconstruction of the earliest lineages and investigating axis development in the brain. Our results provide an enabling and versatile platform for in vivo barcoding and lineage tracing in a mammalian model system.
Keywords: hgRNA mouse, DNA barcodes, lineage tracing, Cas9, homing CRISPR, homing guide RNA, cell barcoding, in vivo barcoding, MARC1 mouse line, Cas9 mouse, NHEJ, brain development, brain A-P axis, brain L-R axis, visceral endoderm, parietal endoderm, yolk sac
One Sentence Summary:
In vivo barcoding is implemented in a mouse model using multiple independent barcoding loci to record and reconstruct lineages.
Introduction
In sexually reproducing multicellular eukaryotes, a single totipotent zygote remarkably develops into all cells of the full organism. This development occurs through a highly orchestrated series of differentiation events that take the zygote through many lineages as it divides to create all the different cell types (1). This path resembles a tree, with the zygote at the base of the trunk branching into stems of cell lineages that eventually end in the terminal cell types at the top of the tree (2, 3). The ability to map this tree of development will have a far ranging impact on our understanding of disease-causing developmental aberrations, our capacity to restore normal function in damaged or diseased tissues, and our capability to generate substitute tissues and organs from stem cells.
Tracing the lineage tree in non-eutelic higher eukaryotes with complex developmental pathways remains challenging. Clonal analysis, which entails cellular labeling and tracking with a distinguishable heritable marker, has been effective when evaluating a limited number of cells or lineages (4–7). Using more diverse pre-synthesized DNA sequences as markers, known as cellular barcoding, has allowed for analysis of larger cell numbers (7–9). What limits these approaches is the static nature of labeling that only allows analysis of a snapshot in time. Recent advances in genome engineering technologies, however, have enabled in vivo barcode generation (10, 11). In this approach, a locus is targeted for rearrangement or mutagenesis such that a diverse set of outcomes is generated in different cells (12). As these barcodes can be generated over a sustained period of time, they drastically expand the scope of cellular barcoding strategies, promising deep and precise lineage tracing from single-cell to whole-organism level (Fig. 1A) (13–15) and recording of cellular signals over time (16, 17). Multiple studies establish proof of this principle in recording and lineage tracing, with demonstrations in cultured cells (14–16, 18) and in lower vertebrates (13, 19–22). However, no demonstrations have yet been carried out in mice, a model organism more relevant to human health in many aspects such as development. The challenges associated with work in mice can account for this discrepancy. Gestation in mice takes place inside the mother’s womb, rendering genetic manipulation of individual zygotes or conceptuses difficult. Additionally, the longer gestation time in mice, together with the multitude of lineages that segregate throughout its development, demand sustained generation of highly diverse barcodes with minimal unwanted overwriting events to maximize the chance for successfully recording the events of interest.
Fig. 1. In vivo barcoding with hgRNAs and strategy to generate mouse with multiple hgRNA integrations.
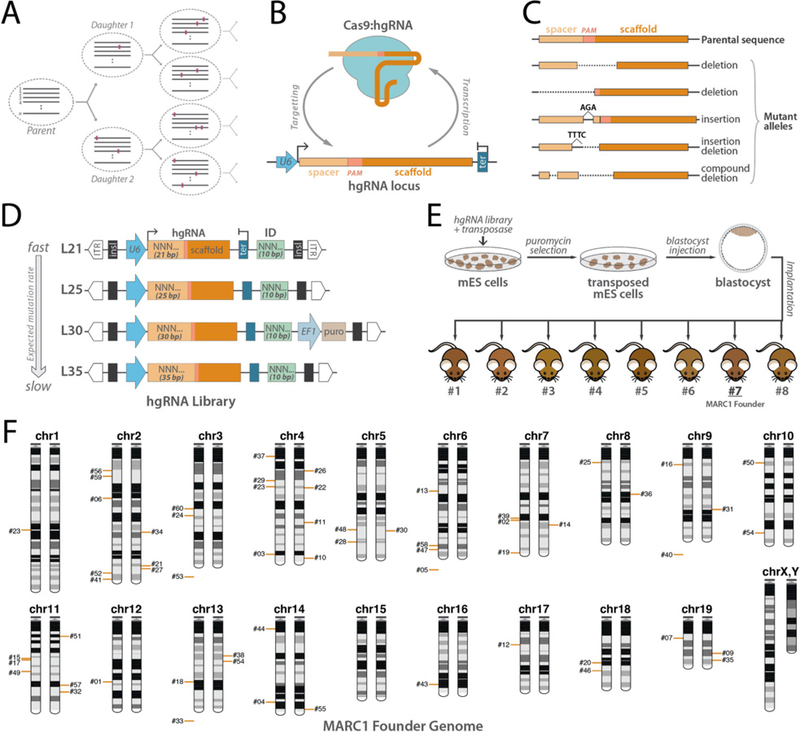
(A) Recording lineages using synthetically-induced mutations in the genome. A number of loci(n) gradually accumulate heritable mutations as cells divide, thereby recording the lineage relationship of the cells in an array of mutational barcodes. Dashed ovals represent cells, gray lines represent an array of n mutating loci, and colored rectangles represent mutations. (B) Homing CRISPR system, in which the Cas9:hgRNA complex cuts the locus encoding the hgRNA itself. As the NHEJ repair system repairs the cut (Lieber and Wilson 2010), it introduces mutations in the hgRNA locus. (C) Example of mutations that are created in the hgRNA locus that can effectively act as barcodes. (D) Design of PiggyBac hgRNA library for creating transgenic mouse. Four hgRNA sub-libraries with 21, 25, 30, and 35 bases of distance between transcription start site (TSS) and scaffold PAM were constructed and combined. The spacer sequence (light orange box) and the identifier sequence (green box) were composed of degenerate bases. (E) Blastocyst injection strategy for producing hgRNA mice. The hgRNA library was transposed into mES cells. Cells with a high number of transpositions were enriched using puromycin selection and injected in E3.5 mouse blastocysts to obtain chimeras. Chimera #7 was chosen as the MARC1 founder. (F) Chromosomal position of all 54 hgRNAs whose genomic position was deciphered in the MARC1 founder (red bars). Bars on left or right copy of the chromosome indicate the hgRNAs that are linked on the same homologous copy. hgRNAs whose exact genomic position is not known but whose chromosome can be determined based on linkage are shown below the chromosome. ITR: PiggyBac Inverted Terminal Repeats; insl: insulator; U6: U6 promoter; ter: U6 terminator; ID: Identifier sequence; EF1: Human elongation factor-1 promoter; puro: puromycin resistance.
Here, we deploy multiple independent barcoding loci in parallel for robust in vivo barcoding and lineage recording in mice. We create a mouse line that carries a scattered array of sixty genomically-integrated homing CRISPR guide RNA (hgRNA) loci. hgRNAs are modified versions of canonical single guide RNAs (sgRNAs) (23) that target their own loci (Fig. 1B) to create a substantially larger diversity of mutants than canonical sgRNAs (Fig. 1C) and thus act as expressed genetic barcodes (14). Crossing this hgRNA line with a Cas9 line results in developmentally barcoded offspring because hgRNAs stochastically accumulate mutations throughout gestation, generating unique mutations in each lineage and without deleting earlier mutations, in such a way that closely related cells have a more similar mutation profile, or barcode, than more distant ones. In developmentally barcoded mice, we extensively characterize the activity profile and mutant alleles of each hgRNA and carry out post hoc bottom-up reconstruction of the lineage tree in the early stage of development, starting with the first branches at its root and continuing through some of the germ layers. We also investigate lineage commitment with respect to the anterior-posterior and lateral axes in the brain.
Results
Founder mouse with multiple hgRNA loci
We created a library of hgRNAs with four different transcript lengths, variable spacer sequences, and 10-base identifiers downstream of the hgRNA scaffold in a transposon backbone (Fig. 1D, Materials and Methods). This library was transposed into mouse embryonic stem (mES) cells under conditions that would result in a high number of integrations per cell (Fig. 1E, Materials and Methods). Transfected mES cells were injected into blastocysts which were then implanted in surrogate females to generate chimeric mice. Twenty three chimeric mice resulted, of which eight males were more than 60% transgenic based on their coat color (Fig. 1E). Five of the eight showed more than 20 total hgRNA integrations in their somatic genomes and were crossed with wild-type mice to determine the number of hgRNAs in their germlines. The chimera with the highest average number of germline hgRNAs, which were transmitted to its progeny, was selected for further studies and starting a line. We refer to this mouse as the MARC1 (Mouse for Actively Recording Cells!) founder and its progeny as the MARC1 line. All results from here on focus on the MARC1 founder and its progeny.
Sequence, genomic position, and inheritance of hgRNA loci
By sequencing the hgRNA loci in the MARC1 founder, we identified 60 different hgRNAs (Table 1, Table S1). Each hgRNA has a unique 10-base identifier and a different spacer sequence (Table S1). We also sequenced the regions immediately flanking the transposed hgRNA elements (Materials and Methods), which allowed us to determine the genomic positions of 54 of the 60 hgRNAs (Fig. 1F, Table 1, Table S1), of which 26 are intergenic and 28 are located in an intron of a known gene (Materials and Methods, Table S3). Based on their locations, none are located in an exon or are expected to disrupt the gene. We then crossed the MARC1 founder with multiple females and analyzed germline transmission and the inheritance pattern of these hgRNAs in the more than a hundred resulting offspring. All 60 hgRNAs were transmitted through the germline and the offspring carrying them were fertile, had normal litter sizes, and presented no morphological abnormalities. 55 of the 60 showed a Mendelian inheritance pattern, appearing in about 50% of the offsprings (Table 1, Table S1). An additional 3 of the 60, all L30 hgRNAs, were detected in less than 20% of the offspring which we attribute to the low detection rate of L30 hgRNAs due to the performance of the PCR primer used for these and only these three hgRNAs (Materials and Methods). The remaining two were transmitted to almost 75% of the offspring, a result best explained by the duplication of the hgRNAs to loci more than 50 centimorgans away on the same chromosome or on different chromosomes and confirmed by the genomic location data (Table S1, Materials and Methods).
Table 1.
hgRNAs in the MARC1 founder male. For each hgRNA, its ID number, TSS to PAM length (L), observed inheritance probability (Inh%), its chromosome. See Tables S1, S2, and S3 for more details.
| ID (#) |
Length (bp) |
Inheritance (%) |
Location | Class | ID (#) |
Length (bp) |
Inheritance (%) |
Location | Class | ID (#) |
Length (bp) |
Inheritance (%) |
Location | Class | ID (#) |
Length (bp) |
Inheritance (%) |
Location | Class |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 21 | 49.6 | chr12 | slow | 16 | 35 | 47.2 | chr9 | inactive | 31 | 35 | 58.4 | chr9 | inactive | 46 | 25 | 47.2 | chr18 | slow |
| 2 | 35 | 55.2 | chr7 | inactive | 17 | 21 | 59.2 | chr11 | slow | 32 | 24 | 44 | chr11 | mid | 47 | 35 | 48.8 | chr6 | inactive |
| 3 | 35 | 55.2 | chr4 | inactive | 18 | 25 | 50.4 | chr13 | slow | 33 | 21 | 55.2 | chr13 | slow | 48 | 35 | 34.4 | chr5 | slow |
| 4 | 21 | 29.6 | chr14 | inactive | 19 | 21 | 39.2 | chr7 | fast | 34 | 35 | 40 | chr2 | mid | 49 | 35 | 58.4 | chr11 | slow |
| 5 | 35 | 41.6 | chr6 | inactive | 20 | 21 | 49.6 | chr18 | fast | 35 | 25 | 52 | chr19 | slow | 50 | 21 | 44 | chr10 | mid |
| 6 | 21 | 54.4 | chr2 | mid | 21 | 35 | 40 | chr2 | slow | 36 | 21 | 45.6 | chr8 | mid | 51 | 25 | 36.8 | chr11 | slow |
| 7 | 34 | 43.2 | chr19 | slow | 22 | 21 | 42.4 | chr4 | mid | 37 | 35 | 51.2 | chr4 | slow | 52 | 21 | 53.6 | chr2 | fast |
| 8 | 30 | 16.8 | slow | 23 | 21 | 80.8 | chr1&4 | mid | 38 | 21 | 38.4 | chr13 | slow | 53 | 35 | 47.2 | chr3 | inactive | |
| 9 | 35 | 51.2 | chr19 | slow | 24 | 21 | 51.2 | chr3 | inactive | 39 | 25 | 54.4 | chr7 | mid | 54 | 35 | 73.6 | chr10&13 | slow |
| 10 | 25 | 42.4 | chr4 | slow | 25 | 25 | 48 | chr8 | inactive | 40 | 35 | 45.6 | chr9 | inactive | 55 | 25 | 68.8 | chr14 | slow |
| 11 | 25 | 35.2 | chr4 | inactive | 26 | 35 | 46.4 | chr4 | inactive | 41 | 21 | 38.4 | chr2 | mid | 56 | 21 | 55.2 | chr2 | slow |
| 12 | 35 | 48.8 | chr17 | inactive | 27 | 21 | 39.2 | chr2 | slow | 42 | 30 | 15.2 | slow | 57 | 34 | 44 | chr11 | inactive | |
| 13 | 35 | 42.4 | chr6 | slow | 28 | 25 | 40 | chr5 | slow | 43 | 35 | 48.8 | chr16 | slow | 58 | 21 | 43.2 | chr6 | fast |
| 14 | 35 | 43.2 | chr7 | inactive | 29 | 35 | 52.8 | chr4 | slow | 44 | 21 | 51.2 | chr14 | inactive | 59 | 21 | 50.4 | chr2 | fast |
| 15 | 34 | 60.8 | chr11 | slow | 30 | 35 | 55.2 | chr5 | inactive | 45 | 30 | 7.2 | inactive | 60 | 25 | 51.2 | chr3 | slow |
We also compared the co-inheritance frequencies of these hgRNAs to those expected from Mendelian inheritance of independently segregating loci (fig. S1A). We found no mutually-exclusive cosegregating groups of hgRNAs (fig. S1A), indicating that the entire germline in the MARC1 founder was derived from only one of the injected stem cells and is thus genetically homogeneous. Considering that every hgRNA detected in the somatic tissue of the MARC1 founder was also transmitted to its offspring, these results further suggest that almost all transgenic cells within this chimera were derived from one of the stem cells that were injected into its blastocyst, an observation consistent with previous studies (24, 25). The co-inheritance analysis also revealed the groups of hgRNAs that deviate from an independent segregation pattern, suggesting that they are linked on a chromosome (fig. S1B). Close examination of this linkage disequilibrium allowed us to determine which linked hgRNAs were on different homologous copies of the same chromosome or were linked on the same copy of a chromosome (fig. S1C). Combined with the genomic location information that was obtained by sequencing, this co-inheritance analysis allowed us to decipher the cytogenetic location of most hgRNAs in the MARC1 founder with a high degree of confidence (Fig. 1F).
Activity of hgRNAs
We next studied the activity of MARC1’s hgRNAs upon activation with Cas9. For that, we crossed the MARC1 founder with Rosa26-Cas9 knockin females which constitutively express S. pyogenes Cas9 protein (26). Considering that major zygotic genome activation in the mouse occurs at the 2-cell stage (27), hgRNA activation is expected soon after conception. We sampled these Cas9-activated offspring at various stages after conception to measure the fraction of mutated spacers for each hgRNA. In all, we gathered 190 samples from 102 animals in seven embryonic stages and the adult stage (Table 2). The results confirm that hgRNAs start mutating their loci soon after the introduction of Cas9 (Fig. 2A). However, the rate at which these mutations accumulate range widely among the sixty MARC1 hgRNAs (Fig. 2A). Based on these activity levels, we classified hgRNAs into four categories with distinct activation profiles (Fig. 2B): five hgRNAs are “fast” as they mutate in at least 80% of the cells in each sample by E3.5 and in almost all cells by E8.5; twenty seven are “slow” as they mutate in only a minority of cells even in the adult stage; nine more are intermediate between “fast” and “slow” (“mid”) as they accumulate mutations throughout embryonic development and are mutated in almost all cells only in later embryonic or adult stages; the remaining hgRNAs appear to be inactive, at least with this level of Cas9 expression, mutating in less than 2% of sampled cells even in the adult stage (Table S2). Most mutations that are detected (about 80% for “fast” hgRNAs) are expected to render the hgRNA nonfunctional and thus prevent further changes (fig. S2).
Table 2.
Barcoded mice, their sample count, and their developmental stages of barcoded mice. Break down of the developmental stages (columns) of all mice used for hgRNA activity analysis and number of samples obtained per mouse (rows).
| Stage of development | Total mice |
Total samples |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E3.5 | E6.5 | E7.5 | E8.5 | E10.5 | E12.5 | E14.5 | E15.5 | E16.5 | adult | ||||
| Samples per mouse | one | 11 | 6 | 3 | 3 | 1 | 0 | 0 | 0 | 0 | 45 | 69 | 69 |
| two | 0 | 0 | 4 | 6 | 1 | 0 | 0 | 0 | 0 | 0 | 11 | 22 | |
| three | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 3 | |
| four | 0 | 0 | 0 | 0 | 7 | 6 | 0 | 0 | 2 | 0 | 15 | 60 | |
| five | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| six | 0 | 0 | 0 | 0 | 0 | 6 | 0 | 0 | 0 | 0 | 6 | 36 | |
| Total | 11 | 6 | 8 | 9 | 9 | 12 | 0 | 0 | 2 | 45 | 102 | 190 | |
Fig. 2. Activity of MARC1 hgRNAs.
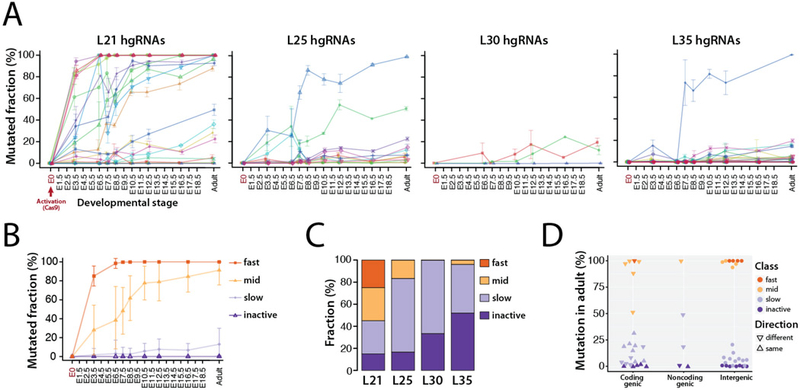
(A) Activity profiles of all 60 hgRNAs in embryonic and adult progenies of MARC1 founder crossed with Cas9-knockin females, broken down by hgRNA length. The fraction of mutant (non-parental) spacer sequences in each hgRNA is measured. Lines connect the observed average mutation rates of one hgRNA. Mean ±SEM is shown (N is different for each value, see Table 2). See Table S2 for numerical values of the plot. (B) Average activity profiles of each hgRNA class in embryonic and adult progenies of MARC1 founder crossed with Cas9-knockin females. Mean ±SEM is shown as a representation of range of activity (N is different for each value, see Table 2). (C) Functional categorization of hgRNAs based on their activity profile in panel A, broken down by length. (D) Position and transcription direction of hgRNAs with respect to all known coding and non-coding genes, annotated for their functional category. See Table S3 for the genes hgRNAs are located in and fig. S3 for breakdown of this plot by hgRNA length.
Transcript length clearly affects hgRNA activity: a far higher fraction of L21 hgRNAs, which have the shortest possible transcript length, are active compared to L25, L30, and L35 hgRNAs which are longer by 4, 9, and 14 bases, respectively (Fig. 2A,C). Furthermore, only L21 hgRNAs show “fast” activity while in longer hgRNAs the inactive proportion seems to grow (Fig. 2C). Beyond transcript length, the variation in activity among hgRNAs with an identical length (Fig. 2A) is far more than would be expected solely based on differences in their spacers (14), suggesting that genomic location may play a substantial role. Additionally, while we detected no significant difference between the activity of hgRNAs that are in intergenic regions compared to those within known genes (Wilcoxon p-value > 0.1), among hgRNAs that have landed within known coding and non-coding genes those that transcribe in the same direction as the gene have a lower activity than those that transcribe in the opposite direction (Wilcoxon p-value < 0.05, Fig. 2D, fig. S3, Table S3). These observations suggest that hgRNA activity is affected by both genomic location and interplay with endogenous elements.
Diversity and composition of hgRNA mutants
We next analyzed the diversity produced by MARC1 hgRNAs by considering all observed mutant spacer alleles in MARC1 x Cas9 offspring (Table S4). Only a handful of mutant spacer alleles were detected for each hgRNA in each sample (Fig. 3A, fig. S4A). However, when combining mutant spacers from all offspring, on average, more than 200 distinct mutant spacers for each “fast” hgRNA and more than 300 for each “mid” hgRNA were observed (Fig. 3B, fig. S4B). Furthermore, about 80% of all mutant spacer alleles were unique observations in a single offspring (Fig. 3C, fig. S5), suggesting that the mutant alleles observed with our sampling level constitute only a minority of all mutant spacers possible. These results indicate that each hgRNA can produce hundreds of mutant alleles.
Fig. 3. Diversity of mutant hgRNA alleles in offspring of MARC1 x Cas9 cross.
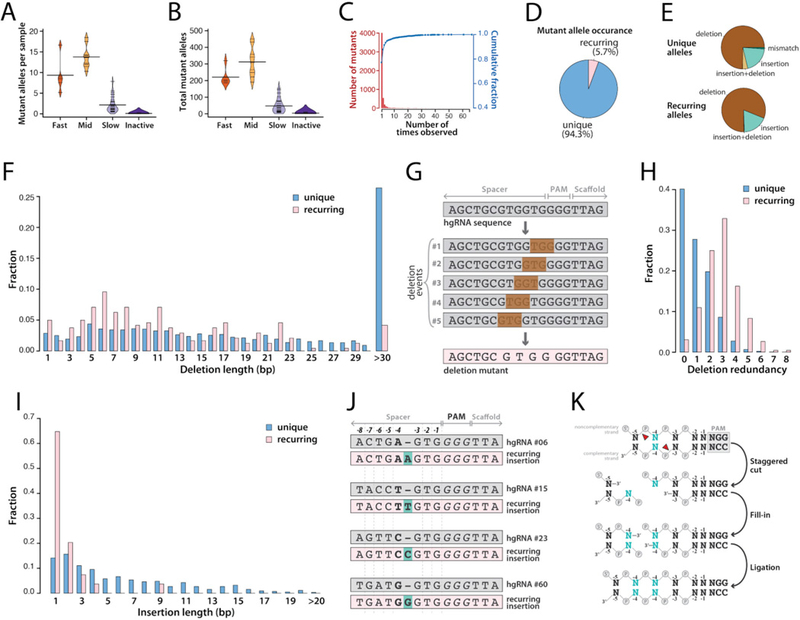
(A) For each hgRNA category, beanplot of the number of mutant spacer alleles observed in each mouse. Short horizontal lines mark the average for each hgRNA in the category, long horizontal lines mark the average of all the hgRNAs in the category. See fig. S4A for a separate plot for each hgRNA. (B) Beanplots of the total number of mutant spacer alleles observed for each hgRNA in all mice. See fig. S4B for a separate plot for each hgRNA. (C) Histogram (red bars) and cumulative fraction (blue connected dots) of the number of mice each mutant allele was observed in, combined for all hgRNAs. See fig. S5 for a separate plot for each hgRNA. (D) Relative ratio of recurring mutant spacer alleles (fig. S5, Materials and Methods) to the unique alleles. (E) Mutation types in unique (top) and recurring (bottom) spacer alleles. See Table S4 and Table S5 for the sequences and alignment of all mutants and recurring mutants, respectively, and fig. S6 for a separate plot for each hgRNA. (F) Distribution of deletion length for unique and recurring mutant spacer alleles. Deletions larger than 30 bp have been aggregated. (G) Schematic representation of how five distinct deletion events can lead to the same mutant spacer allele. (H) Distribution of deletion redundancy, that is, the number of independent simple deletion events in the parental spacer allele that would lead to the same observed deletion mutant, for unique and recurring spacer alleles. Simple deletion is defined as deletion of a contiguous stretch of bases without creating insertions or mismatches. Redundancy of “0” represents non-simple mutant alleles, which involve insertions, mismatches, or non-contiguous deletions. (I) Distribution of insertion length for unique and recurring mutant spacer alleles. Insertions of 20 bp or longer have been aggregated. (J) Four observed examples of recurring single-base insertions, involving duplication of the −4 position, for four different hgRNAs. (K) Schematic representation of how a single-base staggered overhang generated by Cas9 can lead to duplication of the −4 position.
Notably, while most mutant spacer alleles appeared in only a single sample, about 5% recurred in multiple MARC1 x Cas9 offspring (Fig. 3C,D, fig. S5). To understand this phenomenon, we compared the nature of unique and recurring mutant alleles (Table S4, Table S5). We observed that indels underlie the vast majority of alleles in both unique and recurring mutations (Fig. 3E, fig. S6A,B). The exact nature of these indel mutations, however, differ. First, short deletions of 23 bp or fewer are enriched in the recurring alleles (Fig. 3F). Interestingly, these mutations tend to be identical results of multiple distinct simple deletion events (Fig. 3G,H), suggesting that this group of recurring mutations can result from distinct mutagenesis events that lead to the same sequence. Second, single-base insertions are drastically enriched among recurring insertion mutants (Fig. 3I). A closer examination of these single-base insertions revealed that many follow the same pattern: duplication of the base at the −4 position of the PAM (Fig. 3J). In fact, this type of insertion was recurring in 34 of the 41 active hgRNAs. This observation can be best explained by Cas9 creating a staggered end by cutting at −4 position of the noncomplementary strand and at the −3 position of the complementary strand, thus creating a 5’ overhang which is then filled in on both ends and ligated (Fig. 3K). Therefore, our results suggest that Cas9 can produce staggered cuts, and that the nature of these cuts together with the sequence of the target site affect the eventual outcome of NHEJ.
Developmental hgRNA barcodes
The results thus far indicate that MARC1 hgRNAs accumulate mutations upon activation with Cas9 nuclease after conception. We next queried whether these mutations indeed reflect developmental events. For simplicity, we focused on “fast” and “mid” hgRNAs in eight post-E12 MARC1 x Cas9 offspring for which four different tissues had been sampled (Table 2). The sampled tissues were the placenta, the yolk sac, the head, and the tail. The barcode was defined for each hgRNA in each sample as the frequency vector of the relative abundances of all observed mutant alleles (Fig. 4A). For the 32 samples under consideration (8 conceptuses with 4 samples each), these barcodes showed diverse and complex patterns, with each sample having a unique barcode but with varying degrees of similarity to other samples (Fig. 4B, fig. S7). To compare the hgRNA barcodes between samples, we used a scaled Manhattan distance (L1) of their frequency vectors, such that a distance of 100 would indicate a completely non-overlapping set of mutant alleles and a distance of 0 would indicate a complete overlap of mutant alleles with identical relative frequencies (Materials and Methods). Pairwise comparison of all hgRNA barcodes among all samples (Fig. 4C) showed that more than 99% of barcode pairs have a scaled Manhattan distance of more than 5, indicating unique barcoding of each sample by each hgRNA. Furthermore, barcodes from different tissues of the same embryo were more similar to each other (median distance = 41) and more distinct from different embryos (median distance = 78) (Fig. 4C), suggesting that barcodes may record information about the history of samples relative to one another.
Fig. 4. In vivo barcoding in mouse embryos.
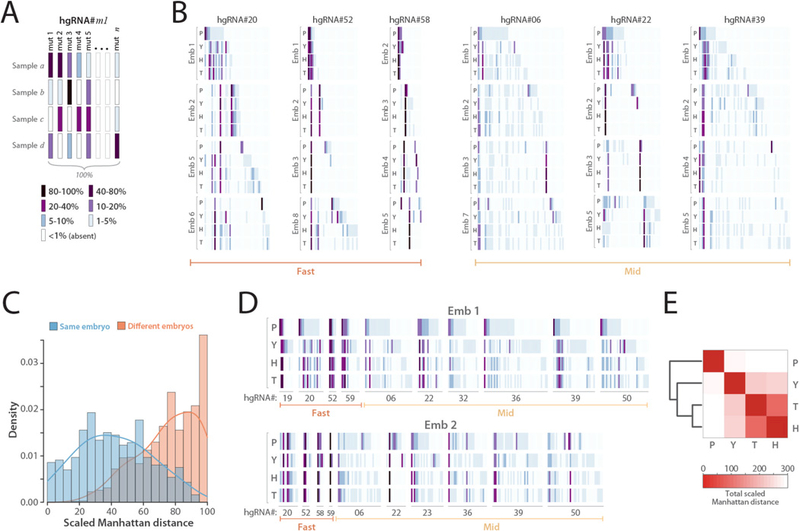
(A) Barcode depiction for each hgRNA in each sample. Each column corresponds to an observed mutant spacer and each row corresponds to a sample. The color of each block represents the observed frequency of the corresponding mutant spacer in the corresponding sample. (B) In vivo-generated barcodes of three “fast” and three “mid” hgRNAs in eight embryos from a MARC1 x Cas9 cross. Four tissues were sampled from each embryo: the placenta (P), the yolk sac (Y), the head (H), and the tail (T). Embryos 1 and 2 were obtained at E16.5 whereas embryos 3 to 8 were obtained at E12.5 (Table 2). For each hgRNA, the results for a maximum of four embryos is shown. Full barcodes for all hgRNAs in fig. S7. The color code is as annotated in panel A. Only mutant alleles with a maximum abundance of more than 1% are shown. (C) Histogram of the scaled Manhattan distances (L1) between the barcodes of all possible sample pairs for each hgRNA, broken down by sample pairs belonging to the same embryo (blue) and pairs belonging to different embryos (orange). (D) The complete barcode, composed of the concatenation of all hgRNA barcodes, for embryo 1 and embryo 2. (E) Heatmap of the average Manhattan distance between the “full” barcodes of placenta, yolk sac, head, and tail samples in all eight embryos. For a separate map for each embryo see fig. S8.
To further evaluate this recording of sample histories, we created a “full” barcode for each sample by combining the barcodes generated by each of its hgRNAs (Fig. 4D) and compared the distance between these barcodes in the four tissues obtained from each embryo (Fig. 4E, fig. S8). The results show higher similarity between the head and tail samples which together are the most different from placenta. The samples obtained here represent mixed and overlapping lineages. However, considering that the head and tail are derived from the inner cell mass (ICM) whereas the placenta is mostly derived from the trophectoderm (28–30), these results suggest that hgRNA barcodes of different tissues embody their lineage histories.
First lineage tree from barcode recordings
We next assessed if accurate lineage trees can be constructed de novo from developmentally barcoded mice. To assess this potential, we focused on the tree of the first lineages in development. The first lineage segregation events in mammals are the differentiation of blastomeres into trophectoderm and inner cell mass (ICM) before E3.5, followed by differentiation of ICM into primitive endoderm and epiblast by E4.5 (Fig. 5A) (28). To reconstruct this lineage tree, we used developmentally barcoded E12.5 conceptuses and sampled two distinct tissues from each of three lineages: the decidual zone (DZ) and the junctional zone (JZ) of the placenta, which are descendants of trophectoderm (29, 30), the parietal endoderm (PE) and visceral endoderm (VE) of the yolk sac, which are descendants of primitive endoderm, and the heart and a limb bud of the embryo proper which are descendants of the epiblast (28) (Fig. 5B). We then assembled the “full” barcode for each sample (Fig. 5C,D, Table S6) and using their Manhattan distances clustered them to form a tree for each embryo (Fig. 5E, Materials and Methods). Remarkably, despite the differences in the number and composition of hgRNAs inherited, the resulting tree perfectly matched the expected lineage in all four embryos, showing that the DZ and JZ form one clade of the tree while the other clade comprises two subclades, one with PE and VE and the other with the heart and limb bud (Fig. 5E). These results demonstrate that accurate lineage trees can be constructed from developmentally barcoded mice.
Fig. 5. Lineage derivation based on hgRNA-generated developmental barcodes.
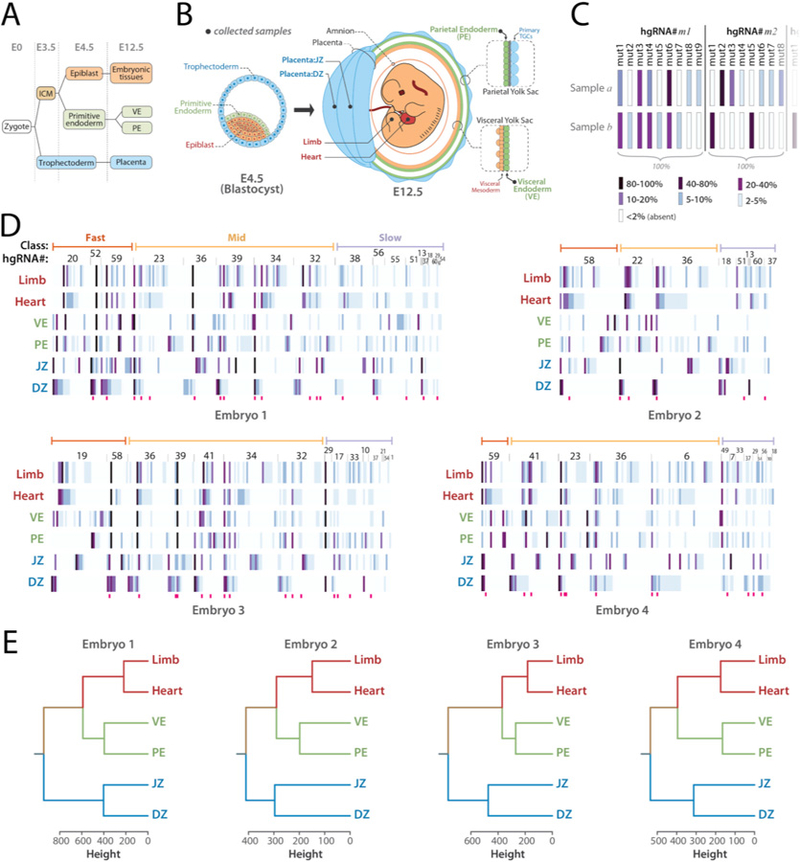
(A) Summary of the earliest lineages in mouse. (B) Schematic representation of a blastocyst and an E12.5 mouse conceptus, color-coded based on the origin of tissues in blastocyst. Black dots show the positions and tissues of the samples obtained from E12.5 conceptuses. (C) Summary of how hgRNA barcodes were compiled for each sample. Each bar represents a mutant spacer of an hgRNA and its color represents its abundance relative to other mutant spacers of the same hgRNA in the same sample. (D) “Full” hgRNA barcodes for all samples from the four mouse embryos analyzed. The barcode is annotated in panel C. Only mutant alleles with a maximum abundance of more than 2% are shown. Deep pink bars below each map mark highly recurring alleles which have been observed in more than 60% of all mice analyzed in Table 2. See Table S6 for a numerical version of each barcode map. (E) Lineage tree for each embryo calculated from the full barcodes in panel D.
We next evaluated the robustness of lineage tree derivation from hgRNA barcodes by calculating the tree topology with only parts of the full barcodes. For a bifurcating tree with six tips (Limb, Heart, VE, PE, JZ, and DZ, Fig. 6A) , 945 distinct rooted topologies are possible (31). Only a single one of these 945 tree topologies perfectly matches the expected lineage tree; we refer to this topology as “perfect” (Fig. 5E, Fig. 6B). Another eight topologies would be correct if unrooted: that is, if all four clades are correctly assigned, but the root is misplaced because a branch other than the one connecting the(DZ,JZ) clade to the((PE,VE),(Heart,Limb)) clade is the longest. We refer to these topologies as “correct” (Fig. 6B). If three, two, or less than two of the four clades have been assigned correctly, we consider the topologies as “incomplete”, “partial”, and “wrong”, respectively (Fig. 6B). With these distinctions, we evaluated the trees generated with all possible non-null subsets of the hgRNAs in each embryo. The results show that, depending on the embryo, 60% to 85% of all possible hgRNA subsets result in a correct topology (Fig. 6C) which compares favorably to the ~1% chance of finding a correct topology randomly. With only three hgRNAs, more than 50% of all derived trees have a correct topology for each embryo (Fig. 6D). Furthermore, calculated topologies improve with increasing number of hgRNAs (Fig. 6D). Combined, these results show that lineage tree derivation from in vivo-generated hgRNA barcodes is robust and that using a higher number of hgRNAs results in more reliable outcomes.
Fig. 6. Lineage tree derivation robustness and contribution of each hgRNA.
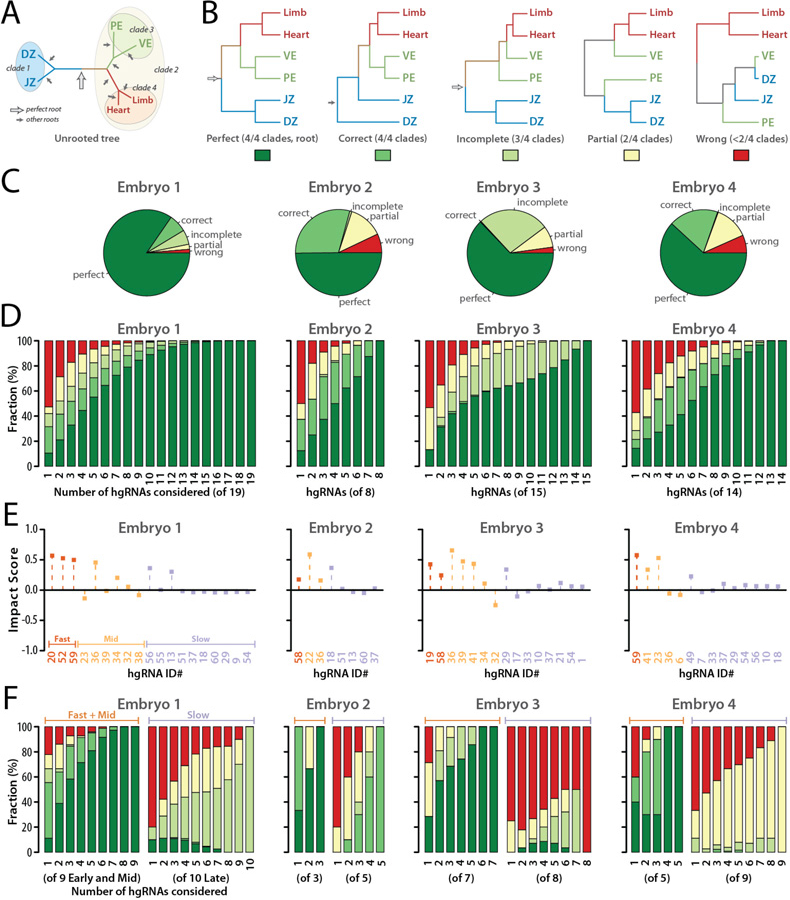
(A) The correct unrooted tree topology for the earliest lineages in mouse. Arrows indicate all possible roots. The empty arrow indicates the perfect root. (B) The perfect rooted topology and an example from each of the other topology classifications. The colored boxes below each topology comprise the color key for the following panels of the Fig.. (C) For each of the four embryos analyzed, distribution of tree calculation outcomes from all possible subsets of hgRNAs ( non-null subsets for an embryo with n hgRNAs). (D) Distribution of tree calculation outcomes when including only m of the n hgRNAs in each embryo ( combinations,). Color code is as described in panel B. See also fig. S9 and fig. S10 for all combinations included and excluding each hgRNA. (E) Impact Score of each hgRNA in the early lineage tree of each embryo. (F) Distribution of tree calculation outcomes when only including k of the fast and mid hgRNAs (left side of each panel, combinations) or k of the slow hgRNAs (right side of each panel, combinations). Color code is as described in panel B.
We then examined the contribution of each hgRNA to deriving the correct tree topology for each embryo. We defined the “Impact Score” of an hgRNA in each embryo’s early lineage tree as the difference between the fraction of all “correct” and “perfect” topologies in which the hgRNA was considered and the fraction of all “wrong” and “partial” topologies in which the hgRNA was considered (Fig. 6E, fig. S9, fig. S10, Materials and Methods). As such, an impact score of +1 would indicate that whenever the hgRNA was included in tree derivation, a “correct” or a “perfect” topology was obtained and no such topologies were obtained without that hgRNA. An impact score of −1 would indicate that when the hgRNA was included in tree derivation only “partial” or “wrong” topologies were obtained. Values between +1 and −1 define the range between those entirely constructive or destructive outcomes with an impact score of 0 indicating that the likelihood of obtaining a correct topology is the same with or without the hgRNA. Impact scores for hgRNAs in our four embryos show positive average contribution by all three active hgRNA classes with “slow” hgRNAs, which are largely unmutated early in development (Fig. 2B), having an average impact close to 0, and “mid” and “fast” hgRNAs, which are active early in development (Fig. 2B), having increasingly positive impacts on the derivation of the correct tree (Fig. 6E). In fact, only three “fast” and “mid” hgRNAs are enough to obtain a correct topology in more than 90% of all derived trees (Fig. 6F). By contrast, exclusive use of “slow” hgRNAs does not recover the early lineage tree as reliably (Fig. 6F). Combined, these results suggest that active mutagenesis during a differentiation event allows it to be recorded. They also suggest that when the developmental stage in which a lineage differentiates is known, hgRNA activity profiles (Fig. 2A) can aid in choosing the appropriate hgRNAs such that correct trees can be reliably obtained with just a few hgRNAs.
Interestingly, when only “slow” hgRNAs are considered in tree construction for early lineages, increasing the number of hgRNAs still results in improved outcomes (Fig. 6F). This observation suggests that even when hgRNAs have low activity levels at the time an event is being recorded, partial recordings from multiple hgRNAs can be combined to obtain a more complete recording. As another example, four different hgRNAs from Embryo 3 predict a “partial” tree when considered on their own yet the “perfect” tree is derived when all four are considered together (fig. S11), further supporting that hgRNA recordings are integrable.
In two of our lineage-analyzed embryo samples (Fig. 5), we noted several hgRNAs in which all ICM-derived tissue samples (PE, VE, Heart, Limb) were dominated by a single mutant allele, while the corresponding trophectoderm-derived tissue samples (DZ, JZ) displayed a more uniform distribution of multiple mutant alleles (Fig. 7). These profiles suggest that, in these embryos, these hgRNAs mutated as trophectoderm and ICM lineages differentiated and that fewer blastomeres led to the ICM compared to trophectoderm. These observations are consistent with previously reported observations (32, 33) and suggest that hgRNA mutation profiles could be used to measure both the relationship between lineages and the relative number of cells that seed lineages.
Fig. 7. Trophectoderm and ICM barcodes show differences in the number of mutant hgRNA alleles.
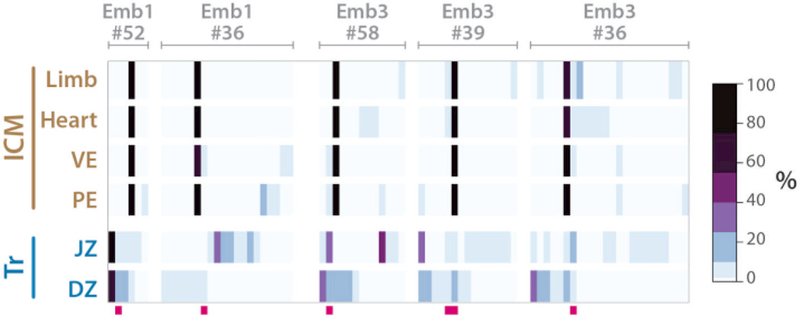
Five barcodes from two embryos in Fig. 5D that distinguish trophectoderm-derived and ICM-derived samples. Deep pink bars below each map mark highly recurring alleles which have been observed in more than 60% of all mice analyzed in Table 2. See Table S6 for a numerical version of each barcode map. Only mutant alleles with a maximum abundance of more than 2% are shown.
Axis development in the brain
We next used developmentally barcoded mice to address lineages above the first lineages in the tree with a focus on the establishment of the anterior-posterior (A-P) and the lateral (L-R) axes with respect to each other in the brain. Patterning of the nervous system and its progenitors starts in gastrulation (E6.5) when the embryo has radial symmetry (34, 35). By E8.5, both A-P and L-R axes are established in the neural tube (Fig. 8A); however, it remains unclear which axis is established first (36, 37). At a morphological level they appear concurrently (38) and previous single-cell labelling and tracing experiments carried out ex vivo do not adequately address the issue (39). We analyzed two developmentally barcoded adult mice. In one we dissected the left and right cortex and cerebellum while in the other we additionally dissected the tectum. The cortex, tectum, and cerebellum respectively originate from embryonic forebrain (prosencephalon), midbrain (mesencephalon), and hindbrain (rhombencephalon) vesicles in the neural tube (Fig. 8A). From each region, two samples of neuronal nuclei were sorted (s1 and s2, Materials and Methods). We also obtained a sample of the blood and one of a muscle from each mouse, both mesoderm-derived, to serve as outgroups. We then assembled the full barcode for each sample and applied clustering as before (Fig. 8B,C, fig. S12). In addition to segregating the mesoderm- and ectoderm-derived cells, the results clearly show that neurons from the left side of each brain region are more closely related to neurons from the right side of the same region than they are to neurons from either of the other two regions. Considering that no extensive migration of neuronal cell bodies between the regions sampled here has been reported (40), these results suggest that commitment to the A-P axis is established before commitment to the L-R axis in development of the central nervous system.
Fig. 8. Anterior-posterior axis is established before the left-right axis in the development of the brain.
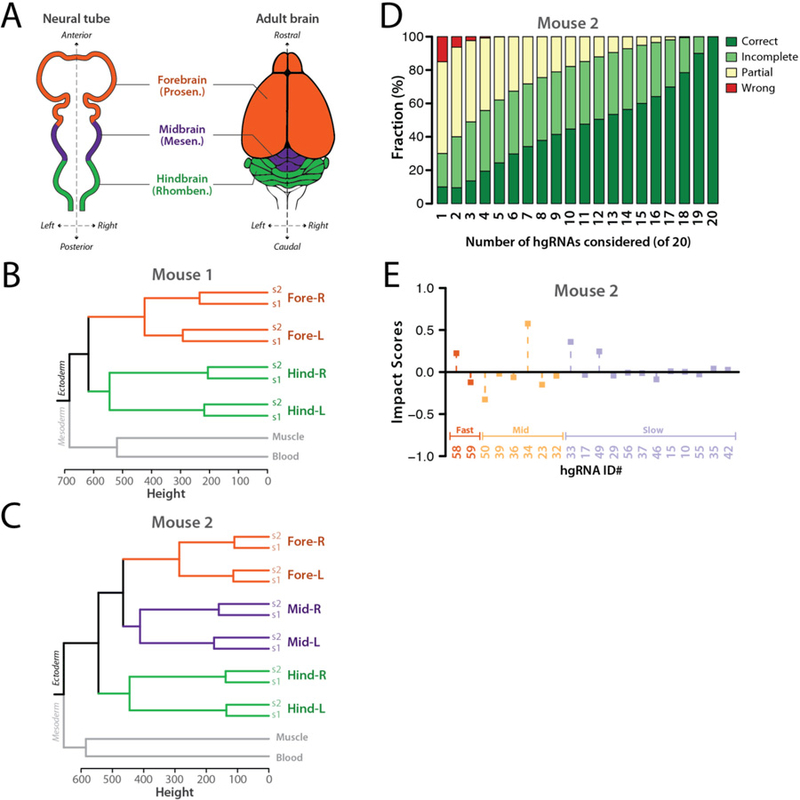
(A) Dorsal view of the neural tube and superior view of the adult brain in mouse. The primary brain vesicles in the neural tube and their corresponding structures in adult brain are shown. (B,C) Calculated trees based on hgRNA barcodes in two adult mice. See fig. S12 for the full barcodes. (D) Distribution of tree calculation outcomes for mouse 2 when only including of the n hgRNAs in each mouse ( combinations). Only hgRNAs with at least 7% mutation rate in one of the samples were considered. (E) Impact Score of each hgRNA in the early lineage tree of mouse 2.
Similar to the first lineage tree analysis above (Fig. 6), we evaluated the robustness of the brain axis tree derivation as well as the contribution of each hgRNA in Mouse 2 (Fig. 8D,E, Materials and Methods). We assigned topologies with all three left and right sample pairs placed closest to one another as “correct”, and those with two, one or zero pairs placed as “incomplete”, “partial”, and “wrong”, respectively. We then calculated the distribution of tree derivation outcomes with all possible subsets of hgRNAs that were active in Mouse 2 (Fig. 8D). The results show that half of the combinations with only three hgRNAs derive a correct or partially correct topology, a ratio that only improves when including more hgRNAs. We also calculated the Impact Score of each hgRNA (Fig. 8E, Materials and Methods). Compared to Impact Scores for the first lineage tree (Fig. 6E), we found lower relative contribution by “fast” hgRNAs, which would be expected for lineages that segregate much later in development. Taken together, these results demonstrate that lineages across diverse developmental times are recorded in our developmentally barcoded mice and can be extracted.
Discussion
In this study, we created an hgRNA mouse line for in vivo barcoding and used it to generate developmentally barcoded mice in which lineage information is recorded in cell genomes and can be extracted and reconstructed.
Our strategy to create the MARC1 line was designed to address challenges associated with in vivo barcoding in a mouse model. First, genetic manipulation of individual mouse embryos is more challenging than that of lower vertebrates. Therefore, a line with genomically integrated, stable, and heritable barcoding elements that can be activated by simply crossing with other lines is powerful, versatile, and shareable (supplementary text). Second, tracking development in mice demands that the system be capable of generating a great many barcodes with little overwriting or deletion (14). As such, we scattered hgRNAs throughout the genome instead of using a contiguous array, circumventing large deletion events that can occur with multiple adjacent cut sites (41–43) and remove prior recordings. In fact, we estimate that less than 1% of all mutations resulted in unidentifiable alleles by removing a amplification primer binding sites or all unique sequences (supplementary text). The scarcity of these unwanted deletion events led to a great success rate in analyzing barcoded mice (4/4 in Fig. 5, 2/2 in Fig. 8). Furthermore, as hgRNA loci in this scattered array accumulate mutations independently, their mutant alleles combine exponentially to create a large diversity of barcodes. Consequently, the 41 active MARC1 hgRNAs can in theory combine to create more than 1074 different barcodes ( where ni is the total observed mutant alleles for hgRNA# i in this study which is likely an underestimation (see Results)). Even only five “fast” and five “mid” hgRNAs can combine for roughly1023 different barcodes(2005×3005), where 200 and 300 are the observed average number of mutant alleles for “fast” and “mid” hgRNAs respectively). This remarkable diversity is adequate for uniquely barcoding every one of the ~1010 cells in a mouse. Furthermore, assuming a perfect binary developmental tree as a first order approximation, this diversity is adequate for uniquely marking all the ~2×1010 internal and terminal nodes of the mouse developmental tree.
Close analysis of the nature of mutant alleles in hgRNA barcodes showed the interplay between target site sequence and Cas9-induced double-strand breaks that determines the possible NHEJ outcomes (Fig. 3). Specifically, short indels underlie recurring NHEJ outcomes. Notable among these is a recurring duplication of the base at the −4 position of the cut site in a majority of active hgRNAs. The most likely explanation for this observation is Cas9 creating staggered cuts that produce a single-base 5’ overhang (Fig. 3K), since a terminal transferase activity would not duplicate the base adjacent to the cut site and RuvC exonuclease activity on the noncomplementary strand would not result in an insertion at all. Whether Cas9 creates blunt or staggered overhangs in vivo has been a subject of debate. Our observation in mice combined with a recent report in yeast (44) and previous in vitro and in vivo evidence (43, 45–49) clarify that Cas9 can create both staggered ends as well as blunt ends, though the ratio of the two is unknown as of yet.
By crossing the MARC1 line with a line that constitutively expresses Cas9, we generated developmentally barcoded mice in which lineage information is recorded in the hgRNA barcodes. We were able to reconstruct parts of the lineage tree using these mice, with the first branches that emerge after the zygote, on to some of the germ layer, neuroectoderm, and the neural tube branches (Fig. 5, Fig. 8). We find remarkable robustness and flexibility in these recordings (Fig. 6, Fig. 8D). Specifically, there is overlap in recordings made by various hgRNAs and therefore the derived lineage tree is robust to removing any part of the barcode. Furthermore, partial non-overlapping recordings from different hgRNAs can be integrated to reconstruct a complete tree. Combined with evidence of sustained hgRNA mutagenesis throughout gestation, these results suggest that developmentally barcoded mice embody information from various stages of development in embryonic and extraembryonic tissues. Extracting such information will be a matter of the type of question being investigated and an ability to isolate cells from relevant lineages. Another interesting possibility is creating other types of barcoded mice by crossing the MARC1 line with other S. pyogenes Cas9 lines. Among these are inducible Cas9s (50, 51), ones with different activity levels (52), tissue or lineage-specific versions based on Cre drivers (26, 52), or base-editing Cas9s (53, 54). Such barcoded mice may enhance the capabilities of the system, overcome its shortcomings (supplementary text), or better focus its potential on specific problems.
In conclusion, the results presented here provide a platform for in vivo barcoding and lineage tracing in the mouse. While we have focused here on the recording aspect of in vivo developmental barcoding, more effective readout strategies, in particular those with transcriptome-coupled single-cell readouts (19, 21, 22), or with in situ readouts (55) will be necessary. Finally, in addition to lineage tracing applications, this platform may also be applied to recording cellular signals over time (16, 17, 56–58) and uniquely barcoding each cell in a tissue or an organism for identification purposes, particularly for connectome mapping in the brain (59–61).
Methods summary
All animal procedures were approved by the Harvard University Institutional Animal Care and Use Committee (IACUC). For embryonic samples, MARC1 founder was crossed with a Cas9-knockin female. Pregnant females were then dissected at the desired embryonic timepoints, designating noon of the day of vaginal plug detection as E0.5. For isolating neurons from adult barcoded female mice, brains were dissected into the regions of interest and homogenized. Nuclei were isolated from the homogenize by gradient ultracentrifugation, labeling with a NeuN antibody, and sorting the NeuN positive fraction in flow cytometry. From all obtained samples, DNA was extracted and amplified with specific primers for hgRNA loci. The resulting amplicons were sequenced with paired ends and analyzed to identify the hgRNA itself based on the identifier sequence, and the mutant allele based on the spacer sequence. The sequencing results were processed and filtered to obtain a list of high-confidence unique spacer-identifier pairs observed in each sample and their respective abundances. For obtaining lineage trees, these lists were converted into frequency matrices and clustered hierarchically using Ward’s criterion. All procedures for the experiments and data analyses are described in detail in the supplementary materials.
Supplementary Material
Acknowledgements
The authors would like to acknowledge Drs. Rudolf Jaenisch for generously sharing reagents, Dr. John Aach for critical reading of the manuscript and helpful comments, Andyna Vernet for assistance with animal husbandry, Garry Cuneo for assistance in FACS, Dr. Lin Wu and the Genome Modification Facility for blastocyst injections as well as helpful discussions, Dr. Scott Kennedy, Dr. John Young, Brandon Fields, and Angela Reslow for generously sharing equipment and providing technical advice, and Drs. Matthew Warman, Clifford Tabin, Connie Cepko, Yonatan Stelzer, Seth Shipman, Jeffrey Macklis, Babak Khalaj, Noah Davidsohn, Alex Ng, and Richie Kohman for helpful discussions.
Funding: This work has been supported by funding from NIH grants MH103910 and HG005550 (G.M.C.), the Intelligence Advanced Research Projects Activity (IARPA) via Department of Interior/Interior Business Center (DoI/IBC) contract number D16PC00008 (G.M.C.), Burroughs Wellcome Fund 1013926 (P.M.) and NIH grants R01HG009285, RO1CA222826, and RO1GM123313 (P.M.).
Footnotes
Competing interests: The authors declare no competing financial interest. GMC’s competing financial interests can be found at v.ht/PHNc.
Data and material availability: All sequencing data are available in the Sequence Read Archive (SRP155997), all other data are available in supplementary materials, and other materials are available upon request.
Final published version: http://science.sciencemag.org/content/early/2018/08/08/science.aat9804.
References and notes
- 1.Kaufman MH, The Atlas of Mouse Development (1992).
- 2.Sulston JE, Schierenberg E, White JG, Thomson JN, The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol 100, 64–119 (1983). [DOI] [PubMed] [Google Scholar]
- 3.Riddle DL, Blumenthal T, Meyer BJ, Priess JR, in C. elegans II 2nd edition (Cold Spring Harbor Laboratory Press, 1997). [PubMed] [Google Scholar]
- 4.Weisblat DA, Sawyer RT, Stent GS, Cell lineage analysis by intracellular injection of a tracer enzyme. Science 202, 1295–1298 (1978). [DOI] [PubMed] [Google Scholar]
- 5.Walsh C, Cepko CL, Widespread dispersion of neuronal clones across functional regions of the cerebral cortex. Science 255, 434–440 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Dymecki SM, Tomasiewicz H, Using Flp-recombinase to characterize expansion of Wnt1-expressing neural progenitors in the mouse. Dev. Biol 201, 57–65 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Kretzschmar K, Watt FM, Lineage tracing. Cell 148, 33–45 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Naik SH, Schumacher TN, Perié L, Cellular barcoding: a technical appraisal. Exp. Hematol 42, 598–608 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Gerrits A et al. , Cellular barcoding tool for clonal analysis in the hematopoietic system. Blood 115, 2610–2618 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Woodworth MB, Girskis KM, Walsh CA, Building a lineage from single cells: genetic techniques for cell lineage tracking. Nat. Rev. Genet 18, 230–244 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma J, Shen Z, Yu Y-C, Shi S-H, Neural lineage tracing in the mammalian brain. Curr. Opin. Neurobiol 50, 7–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peikon ID, Gizatullina DI, Zador AM, In vivo generation of DNA sequence diversity for cellular barcoding. Nucleic Acids Res 42, e127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McKenna A et al. , Whole-organism lineage tracing by combinatorial and cumulative genome editing. Science 353, aaf7907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalhor R, Mali P, Church GM, Rapidly evolving homing CRISPR barcodes. Nat. Methods 14, 195–200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frieda KL et al. , Synthetic recording and in situ readout of lineage information in single cells. Nature 541, 107–111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perli SD, Cui CH, Lu TK, Continuous genetic recording with self-targeting CRISPR-Cas in human cells. Science 353 (2016), doi: 10.1126/science.aag0511. [DOI] [PubMed] [Google Scholar]
- 17.Sheth RU, Yim SS, Wu FL, Wang HH, Multiplex recording of cellular events over time on CRISPR biological tape. Science 358, 1457–1461 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt ST, Zimmerman SM, Wang J, Kim SK, Quake SR, Quantitative Analysis of Synthetic Cell Lineage Tracing Using Nuclease Barcoding. ACS Synth. Biol 6, 936–942 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alemany A, Florescu M, Baron CS, Peterson-Maduro J, van Oudenaarden A, Whole-organism clone tracing using single-cell sequencing. Nature 556, 108–112 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Flowers GP, Sanor LD, Crews CM, Lineage tracing of genome-edited alleles reveals high fidelity axolotl limb regeneration. Elife 6 (2017), doi: 10.7554/eLife.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spanjaard B et al. , Simultaneous lineage tracing and cell-type identification using CRISPR–Cas9-induced genetic scars. Nat. Biotechnol 36, 469–473 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raj B et al. , Simultaneous single-cell profiling of lineages and cell types in the vertebrate brain. Nat. Biotechnol (2018), doi: 10.1038/nbt.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mali P et al. , RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Markert CL, Petters RM, Manufactured hexaparental mice show that adults are derived from three embyronic cells. Science 202, 56–58 (1978). [DOI] [PubMed] [Google Scholar]
- 25.Wang Z, Jaenisch R, At most three ES cells contribute to the somatic lineages of chimeric mice and of mice produced by ES-tetraploid complementation. Dev. Biol 275, 192–201 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Platt RJ et al. , CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee MT, Bonneau AR, Giraldez AJ, Zygotic genome activation during the maternal-to-zygotic transition. Annu. Rev. Cell Dev. Biol 30, 581–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu CC, Brennan J, Robertson EJ, From fertilization to gastrulation: axis formation in the mouse embryo. Curr. Opin. Genet. Dev 11, 384–392 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Rossant J, Cross JC, in Mouse Development (2002), pp. 155–180.11782409
- 30.Simmons DG, Cross JC, Determinants of trophoblast lineage and cell subtype specification in the ouse placenta. Dev. Biol 284, 12–24 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Felsenstein J, Inferring Phylogenies (Sinauer Associates Incorporated, 2004). [Google Scholar]
- 32.Leung CY, Zhu M, Zernicka-Goetz M, Polarity in Cell-Fate Acquisition in the Early Mouse Embryo. Curr. Top. Dev. Biol 120, 203–234 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Anani S, Bhat S, Honma-Yamanaka N, Krawchuk D, Yamanaka Y, Initiation of Hippo signaling is linked to polarity rather than to cell position in the pre-implantation mouse embryo. Development 141, 2813–2824 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Beddington RS, Robertson EJ, Axis development and early asymmetry in mammals. Cell 96, 195–209 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Molnár Z, Price DJ, in Kaufman’s Atlas of Mouse Development Supplement (Elsevier, 2016), pp. 239–252. [Google Scholar]
- 36.Arnold SJ, Robertson EJ, Making a commitment: cell lineage allocation and axis patterning in the early mouse embryo. Nat. Rev. Mol. Cell Biol 10, 91–103 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Kiecker C, Lumsden A, The role of organizers in patterning the nervous system. Annu. Rev. Neurosci 35, 347–367 (2012). [DOI] [PubMed] [Google Scholar]
- 38.Altmann CR, Brivanlou AH, Neural patterning in the vertebrate embryo. Int. Rev. Cytol 203, 447–482 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Lawson KA, Meneses JJ, Pedersen RA, Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development 113, 891–911 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Ayala R, Shu T, Tsai L-H, Trekking across the brain: the journey of neuronal migration. Cell 128, 29–43 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Lee HJ, Kim E, Kim J-S, Targeted chromosomal deletions in human cells using zinc finger nucleases. Genome Res 20, 81–89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canver MC et al. , Characterization of Genomic Deletion Efficiency Mediated by Clustered Regularly Interspaced Palindromic Repeats (CRISPR)/Cas9 Nuclease System in Mammalian Cells. J. Biol. Chem 289, 21312–21324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Byrne SM, Ortiz L, Mali P, Aach J, Church GM, Multi-kilobase homozygous targeted gene replacement in human induced pluripotent stem cells. Nucleic Acids Res 43, e21 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lemos BR et al. , CRISPR/Cas9 cleavages in budding yeast reveal templated insertions and strand-specific insertion/deletion profiles. Proc. Natl. Acad. Sci. U. S. A 115, E2040–E2047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gasiunas G, Barrangou R, Horvath P, Siksnys V, Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U. S. A 109, E2579–86 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jinek M et al. , A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jinek M et al. , Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343, 1247997 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zuo Z, Liu J, Cas9-catalyzed DNA Cleavage Generates Staggered Ends: Evidence from Molecular Dynamics Simulations. Sci. Rep 5, 37584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsai SQ et al. , GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dow LE et al. , Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol 33, 390–394 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Polstein LR, Gersbach CA, A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol 11, 198–200 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chiou S-H et al. , Pancreatic cancer modeling using retrograde viral vector delivery and in vivo CRISPR/Cas9-mediated somatic genome editing. Genes Dev 29, 1576–1585 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR, Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gaudelli NM et al. , Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JH et al. , Highly Multiplexed Subcellular RNA Sequencing in Situ. Science 343, 1360–1363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marblestone AH et al. , Physical principles for scalable neural recording. Front. Comput. Neurosci 7, 137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shipman SL, Nivala J, Macklis JD, Church GM, CRISPR-Cas encoding of a digital movie into the genomes of a population of living bacteria. Nature 547, 345–349 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Punthambaker S, Hume RI, Potent and long-lasting inhibition of human P2X2 receptors by copper. Neuropharmacology 77, 167–176 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cai D, Cohen KB, Luo T, Lichtman JW, Sanes JR, Improved tools for the Brainbow toolbox. Nat. Methods 10, 540–547 (2013). [PubMed] [Google Scholar]
- 60.Kebschull JM et al. , High-Throughput Mapping of Single-Neuron Projections by Sequencing of Barcoded RNA. Neuron 91, 975–987 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Church G, Marblestone A, Kalhor R, in The Future of the Brain, Marcus G, Freeman J, Eds. (Princeton University Press, 2014), pp. 50–64. [Google Scholar]
- 62.Lieber MR, Wilson TE, SnapShot: Nonhomologous DNA End Joining (NHEJ). Cell 142, 496–496.e1 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.