

Summary
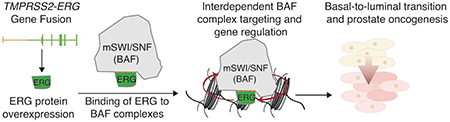
Chromosomal rearrangements resulting in the fusion of TMPRSS2, an androgen-regulated gene, and the ETS family transcription factor ERG occur in over half of prostate cancers. However, the mechanism by which ERG promotes oncogenic gene expression and proliferation remains incompletely understood. Here, we identify a binding interaction between ERG and the mammalian SWI/SNF (BAF) ATP-dependent chromatin remodeling complex, which is conserved among other oncogenic ETS factors, including ETV1, ETV4 and ETV5. We find that ERG drives genome-wide retargeting of BAF complexes in a manner dependent on binding of ERG to the ETS DNA motif. Moreover, ERG chromatin occupancy and target gene regulation are dependent on BAF complex ATPase activity. In a prostate organoid model, BAF complexes are required for ERG-mediated basal-to-luminal transition, a hallmark of ERG activity in prostate cancer. These observations suggest a fundamental interdependency between ETS transcription factors and BAF chromatin remodeling complexes in cancer.
Graphical Abstract
Introduction
Translocations involving ETS family transcription factors occur in 60% of prostate cancers, with the majority (~50%) of cases bearing the TMPRSS2-ERG fusion (Cancer Genome Atlas Research, 2015; Clark and Cooper, 2009; Helgeson et al., 2008; Paulo et al., 2012; Tomlins et al., 2007; Tomlins et al., 2006; Tomlins et al., 2005). Recent studies have identified disease-associated loss-of-function mutations in factors that either target ERG for degradation (SPOP) (Gan et al., 2015) or function as repressors of ERG activity (ERF) (Bose et al., 2017), further converging on ERG activity as a critical mediator of prostate oncogenesis. Despite the high incidence of translocation-induced overexpression of ETS factors such as ERG and ETV1 in prostate cancers, the mechanisms by which ETS family oncogenes induce cell transformation remain incompletely defined.
As a member of the ETS transcription family, ERG binds specifically to the conserved ETS GGA(A/T) DNA-binding motif via the ETS DNA-binding domain (Donaldson et al., 1996; Wei et al., 2010). ERG is a ~55kDa protein containing additional structured domains such as the pointed (PNT) domain, which enables homodimerization of the ERG protein and has been suggested to mediate heterodimerization with other ETS factors (Carrere et al., 1998). Structural domains within the ERG protein have also been shown to facilitate interactions with other proteins, including AR and the AP-1 complex (Adamo and Ladomery, 2016; Clark and Cooper, 2009; Verger et al., 2001; Yu et al., 2010). ERG is not expressed in normal prostate tissue, and its expression pattern is restricted to a limited set of adult tissues, including vascular, adipose, mammary, and splenic tissue (Consortium, 2015; Mohamed et al., 2010). Moreover, the association of ERG with other transcription factors can influence ERG binding specificity, and can hence impact transcriptional activation or repression of target genes (Adamo and Ladomery, 2016; Basuyaux et al., 1997; Verger et al., 2001). Thus, the repertoire of ERG-bound protein interactions can greatly influence the specific roles of ERG in different cellular contexts, highlighting the importance of understanding the mechanism of action for TMPRSS2-ERG in prostate cancer.
Transcription factors may act, at least in part, by recruiting chromatin regulatory or modifying complexes to their target sites across the genome. Interrogation of chromatin landscape features across primary prostate tumors suggests that ERG mediates changes in cis-regulatory elements and chromatin topology, suggesting a broader function of ERG across the genome (Adamo and Ladomery, 2016; Kron et al., 2017; Rickman et al., 2012; Yu et al., 2010). Several recent studies have implicated transcription factor-mediated shifts in the epigenetic landscape as a critical driver in prostate cancer, however the mechanisms by which these factors induce such changes is not well understood (Kron et al., 2017; Pomerantz et al., 2015; Shukla et al., 2017). Understanding the mechanism by which oncogenic ETS factors drive prostate oncogenesis has broad implications to transcription factor biology and oncogenesis, suggesting principles by which these factors alter the chromatin landscape to drive disease.
Here using mass-spectrometry coupled with biochemical approaches, we identified a robust interaction between ERG with mSWI/SNF (BAF) chromatin remodeling complexes. We found that overexpression of ERG results in global re-targeting of BAF complexes in a dose-dependent manner, with gain of BAF complex targeting over ETS sites coupled with concomitant loss of BAF complex targeting to AR sites. Notably, we demonstrated that BAF complex perturbation via suppression of specific subunits displaces the ERG transcription factor on chromatin, and that ERG-mediated target gene regulation is dependent on the ATPase activity of the BRG1 subunit. Using a prostate organoid model, we found that BAF complexes are required to facilitate basalto-luminal transition, a hallmark of prostate oncogenesis. Taken together, these studies reveal a fundamental interdependence between a transcription factor and a chromatin remodeler and suggest a mechanism by which transcription factors alter the chromatin landscape through competitive binding and targeting of BAF chromatin remodeling complexes.
Results Identification of endogenous TMPRSS2-ERG protein interactions
To understand how ERG drives oncogenic gene expression programs, we sought to identify ERG interacting proteins in prostate cancer cells which contain the TMPRSS2-ERG fusion (VCaP) and hence, high levels of ERG mRNA and protein (Fig. 1a) (Tomlins et al., 2005). Immunoprecipitation of endogenous ERG from VCaP cells followed by SILAC-based proteomic mass spectrometry revealed peptides corresponding to components of the mammalian SWI/SNF (BAF) complex within the top 50 differentially enriched proteins between ERG and control IgG conditions (Fig. 1b-d, Fig. S1, Table S1). Specifically, we identified peptides corresponding to 12 canonical BAF complex subunits, including ARID1A (BAF250A), SMARCA4 (BRG1) and SMARCC1 (BAF155), as well as more recently identified BAF components such as BCL7B (Kadoch et al., 2013). Moreover, we found that most BAF complex subunit proteins were more substantially enriched than previously-identified ERG interacting proteins such as SPOP (Gan et al., 2015) and EWSR1 (Kedage et al., 2016) (Fig. S1c). Peptides corresponding to subunits specific to PBAF (polybromo-associated BAF) complexes were substantially less enriched than those corresponding to canonical BAF complexes, suggesting that this interaction is specific for a subset of mSWI/SNF family complex assemblies (Fig. 1d). Previous studies have implicated BAF complexes in prostate cancer, suggesting its roles in proliferative control (Shen et al., 2008) and in antagonizing the tumor suppressor functions of BAF complexes (Prensner et al., 2013), however the precise contribution of BAF complexes, or ATP-dependent chromatin remodeling, to prostate oncogenesis remains to be defined. These results demonstrate that subunits of BAF complexes are significantly enriched among ERG interacting proteins as detected in the unbiased SILAC mass spectrometry.
Figure 1.

SILAC mass spectromety for ERG-interacting proteins reveals subunits of BAF complexes.
(A) Schematic of TMPRSS2-ERG gene fusion that results in aberrant ERG expression levels.
(B) Schematic of SILAC mass spectrometry experiment in TMPRSS2-ERG-containing VCaP prostate cancer cells.
(C) SILAC mass-spectrometry screen for ERG interactors reveals significant enrichment of BAF complex subunits. Anti-ERG SILAC hits are plotted as log2-fold change for each experimental replicate. Highlighted are ERG (green) and BAF complex subunit components (red).
(D) Table of mammalian SWI/SNF protein subunits identified in proteomic mass-spectrometry, indicating number of unique peptides, sequence coverage (%), associated p-values, and rank among the full set of enriched proteins.
Binding of oncogenic ETS factors to BAF complexes
To validate the SILAC mass spectrometry findings, we performed reciprocal immunoprecipitation studies using two anti-ERG antibodies specific for distinct epitopes and an antibody specific for the BRG1 ATPase subunit of BAF complexes (anti-BRG1) and confirmed the interaction of endogenous ERG with BAF complexes (Fig. 2a). Treatment of nuclear extracts with ethidium bromide and benzonase failed to disrupt ERG-BAF complex binding, indicating that the ERG-BAF interaction is not DNA-dependent (Fig. 2b). In addition, immunoprecipitation of BAF complexes from VCaP cells (using antibodies specific to complex subunits, BRG1 and BAF47) followed by mass spectrometric analyses resolved ERG peptides, further confirming this interaction (Fig. S2 a,b, Table S2). These results affirm the ERG-BAF complex interaction as identified in the SILAC mass spectrometry results.
Figure 2.

In-solution binding of ERG to BAF complexes in prostate cancer.
(A) Immunoprecipitation using anti-ERG (mouse monoclonal) and anti-BRG1 antibodies (left); IP with an alternate anti-ERG (rabbit polyclonal) antibody (right), in VCaP cell nuclear extracts.
(B) Immunoprecipitation using IgG and anti-ERG antibodies, with or without ethidium bromide (EtBr) treatment (left). Immunoprecipitation using IgG and anti-ERG antibodies with or without treatment of benzonase, presence of plasmid DNA indicated for benzonase treatment control (right).
(C) Immunodepletion studies performed on VCaP cell nuclear extracts using an anti-BRG1 antibody.
(D) Urea denaturation analysis performed on anti-ERG IPs from VCaP nuclear extracts treated with [urea]= 0–2.5M.
(E) Nuclear protein input and anti-ERG IP on nuclear extracts from LNCaP cells in empty and ERG conditions.
(F) Input (left) and anti-V5 IP (right) in HEK-293-T cells transfected with V5-ETV1, V5-ETV4, V5-ETV5 or empty vector.
We sought to determine the properties and specificity of the ERG-BAF complex interaction. To determine the fraction of nuclear ERG that was associated with BAF complexes, we performed sequential depletion studies using an anti-BRG1 antibody, which depleted ERG from the nuclear extract (~45% depletion, average) to similar levels as other BAF complex subunits (~45–65% depletion, average) (Fig. 2c). Immunodepletion of ERG from VCaP nuclear extracts did not substantially deplete BAF complex subunit proteins, suggesting that while the majority of nuclear ERG protein interacts with BAF complexes, a smaller percentage of total BAF complexes interact with ERG in solution (Fig. S2c). Silver stain analyses of ERG and BAF complex immunoprecipitations revealed similar banding patterns in ERG-expressing cell lines (Fig. S2d). We found that ERG remains bound to BAF complexes in up to ~0.5M urea and sediments in monomeric fractions of a 10–30% glycerol gradient (Fig. 2d, Fig. S2e), indicative of a robust yet transient transcription factor interaction [as compared to subunits, which are stable at ≥2.5M urea and co-migrate in high-density fractions (Kadoch and Crabtree, 2015)]. To determine whether introduction of ERG into cells lacking ERG expression drives the interaction between ERG and BAF complexes, we introduced ERG (corresponding to the most common T1:E4 TMPRSS2-ERG fusion) into LNCaP cells (Fig. 2e) or HEK-293-T cells (Fig. S2f) and found that ERG co-immunoprecipitated BAF complex subunits. In addition to ERG, other ETS family transcription factors, including ETV1, ETV4 and ETV5 are overexpressed in an additional ~10% of prostate cancer cases. To determine if these oncogenic ETS factors also bind to BAF complexes, we overexpressed ETV1, ETV4 and ETV5 in HEK-293-T cells and found that all three transcription factors comparably tethered to BAF complexes (Fig. 2f). We also observed interaction between endogenous ETV1 and ETV5 with BRG1 in the ETV1/5-overexpressing MDA-PCa-2b prostate cancer cell line (Fig. S2g). Collectively, these observations indicate that overexpression of ERG (by the TMPRSS2-ERG gene fusion or by exogenous expression) and oncogenic ETS factors results in an interaction with BAF complexes.
ERG retargets BAF complexes genome-wide to ETS target sites
To determine the consequences of this interaction on genome-wide chromatin binding of both BAF complexes and ERG, we performed chromatin immunoprecipitation (ChIP)-sequencing studies in VCaP cells using antibodies specific for ERG and the core BAF complex subunit BAF155. We found widespread co-occupancy of ERG and BAF155 in VCaP cells, with corresponding enrichment levels of ERG and BAF155 observed across all ERG target sites in VCaP cells (Fig. 3a-b). Overlap between ERG and BAF complexes is observed at ERG target genes such as PLAT and AR (Fig. 3c). Notably, we found the ERG sequence motif exhibited significant central enrichment across all BAF complex (BAF155) sites, suggesting a potentially instructive role for ERG in targeting BAF complexes to these sites rather than incidental co-localization (Fig. 3d). These results demonstrate the genome-wide co-occupancy of ERG and BAF complexes in prostate cancer cells.
Figure 3.

ERG directs genome-wide retargeting of BAF complexes in a dose-dependent manner.
(A) Heatmap of ERG and BAF155 occupancy in VCaP cells over all VCaP ERG sites (87220) ranked by ERG occupancy.
(B) Venn diagram of ERG and BAF155 peaks in VCaP cells.
(C) Example tracks of ERG and BAF155 co-occupancy at the PLAT (top) and AR (bottom) loci in VCaP cells.
(D) Centrimo motif enrichment plot of the ERG motif over BAF155 sites in VCaP cells.
(E) Nuclear protein immunoblot from VCaP cells in shCt and shERG conditions.
(F) Metagene plots of ERG and BAF155 at ERG-BAF155 shared sites in shCt and shERG conditions in VCaP cells.
(G) Correlation plot of log2(fold change) in ERG and BAF155 occupancy (shERG/shCt) over all ERG–BAF155 sites in VCaP shCt cells.
(H) Venn diagram of BAF155 peaks in shCt and shERG conditions and ERG peaks in shCt condition in VCaP cells.
(I) Centrimo motif enrichment plots as well as significance of enrichment for ERG and AR sequence motifs at condition-specific BAF155 sites in VCaP cells as defined by log2FC in BAF155 occupancy. See also Figure S3E.
(J) Example ERG and BAF155 ChIP-seq and RNA-seq tracks at the HOXB13 locus in VCaP cells in shCt and shERG conditions.
As transcription factors and chromatin regulators exhibit varying degrees of similarity in genome-wide occupancy patterns (Hnisz et al., 2013), we sought to determine whether ERG has a direct, instructive role in dictating BAF complex localization. We found that shRNA-mediated suppression of ERG levels in VCaP cells resulted in substantial loss of both ERG and BAF155 occupancy at BAF155-ERG sites across the genome (Fig. 3e-f, Fig. S3a-c). Furthermore, we found that the degree to which BAF155 occupancy was lost directly corresponded to the degree of ERG depletion (Fig. 3g, Fig. S3d), suggesting ERG levels dictate BAF complex occupancy and eviction upon ERG suppression. Notably, we found that ERG suppression resulted in both gain and loss of BAF complex targeting, and that peaks called specifically in the shCt condition exhibited marked overlap with ERG sites (64.6%) whereas peaks called only in the shERG condition exhibited more modest overlap with ERG occupancy (27.9%) (Fig. 3h). To further dissect this, we determined the fold change in BAF155 occupancy at all BAF155 sites in either condition, and categorized BAF155 sites as shCt-specific (26798), shCt-shERG (40350), or shERG-specific (1479) based on the degree of BAF155 retargeting (Fig. S3e-f). We found that the ERG sequence motif was centrally enriched at BAF complex sites that were lost upon ERG suppression as well as sites unaffected, but not at sites gained upon ERG suppression (Fig. 3i). By contrast, the AR sequence motif exhibited central enrichment at unchanged and gained sites, but minimal enrichment at lost sites (Fig. 3i).
As the antagonism between ERG and AR has been suggested to play an important role in prostate cancer (Yu et al., 2010), we characterized the effects of ERG expression on androgen receptor (AR) regulation genome-wide. We found that while ERG occupancy was biased to sites lost upon ERG suppression, AR occupancy increased globally upon ERG suppression, with a greater percent occupancy at gained sites and shared sites than lost sites (Fig. S3g-h). Previous studies have reported the interaction between AR and BAF complex subunits (Link et al., 2008), and we validated binding between AR and BRG1 in both VCaP and LNCaP cells (Fig. S3i). Together, these observations suggest that the antagonism between ERG and AR is likely the consequence of competitive binding and regulation of BAF complexes, with ERG expression regulating the AR cistrome in a manner similar to that observed for critical transcription factors such as FOXA1 and HOXB13 (Pomerantz et al., 2015). For example, at the HOXB13 locus, ERG suppression leads to corresponding depletion of BAF155 occupancy at promoter-distal sites and decreased gene expression (Fig. 3j, Fig. S3j). These observations demonstrate that BAF complexes are targeted by ERG genome-wide, identifying a critical role for BAF chromatin remodeling complexes in ERG-mediated oncogenic gene expression.
ERG requires DNA binding activity to recruit BAF complexes but not for genome-wide chromatin occupancy
We next sought to determine how overexpression of oncogenic ETS factors induces target gene regulation in prostate cancer. To do this, we overexpressed V5-ERG, V5-ETV1, or an empty vector control in LNCaP cells and performed RNA-seq experiments (Fig S4a-c). We found that gene expression changes induced by ERG and ETV1 expression are highly similar between these oncogenic ETS factors, with significant concordance in target gene regulation (Fig. S4d-e). This includes concordant activation of ERG target genes such as FZD4 and GRPR (Fig. S4f). These results suggest a convergent regulatory mechanism for oncogenic ETS factors that bind BAF complexes.
To further confirm these findings and to determine how DNA binding activity of these transcription factors influences de novo genome-wide retargeting of BAF complexes, we overexpressed either wild-type ERG or an ERG point mutation (R367K) described to disrupt its DNA binding ability (Verger et al., 2001) (Fig. 4a). We confirmed that this mutant could not bind to DNA as assessed by electrophoretic mobility shift assays (EMSA) performed on purified recombinant ERG proteins (Fig. S4g-h). We found that the ERG-R367K DNA-binding mutant maintained the ability to interact with BAF complexes (Fig. 4b), further affirming that the binding of ERG to BAF complexes is not chromatin- or DNA-dependent (Fig. 2b). We next performed ChIP-seq for ERG and BAF155 in LNCaP cells in empty vector, ERG, and ERG-R367K conditions in LNCaP cells (Fig. S4i). We found that ERG expression also resulted in both gain and loss of BAF complex targeting, and that peaks called specifically in the ERG condition exhibited marked overlap with ERG sites (70.7%) whereas peaks called only in the empty condition exhibited more modest overlap with ERG occupancy (18.6%) (Fig. 4c). Similar to VCaP, we categorized BAF155 sites by the fold changes in BAF155 occupancy, and designated sites as empty-specific (1666), empty-ERG (69451), or sERG-specific (10837) based on the degree of BAF155 retargeting (Fig. S4j). We also performed ChIP-seq for H3K27ac and H3K27me3 in empty and ERG conditions in LNCaP cells and found moderate increases in H3K27ac at ERG-specific BAF155 sites, indicative of increased activity but not de novo accessibility (Fig. S4k). We found that the ERG motif was the top enriched motif at ERG-specific BAF155 sites (Fig. 4d), whereas the AR motif was significantly enriched at empty-specific and empty-ERG sites (Fig. S4l). ChIP-seq of AR in LNCaP cells in empty and ERG conditions demonstrated that AR binding is altered by ERG expression, suggesting a role for ERG in altering the AR cistrome genome-wide in prostate cancer (Fig. S4m-n).
Figure 4.

Distinct roles of DNA binding and BAF complex binding in genome-wide ERG targeting and regulation
(A) Nuclear protein immunoblot of LNCaP cells in empty vector, ERG, and ERG-R367K (DNA-binding mutant).
(B) Nuclear protein input, IgG, and anti-ERG IPs on nuclear extracts from LNCaP cells in empty, ERG, and ERG-R367K conditions.
(C) Venn diagram of BAF155 peaks in empty and ERG conditions with ERG peaks in the ERG condition in LNCaP cells.
(D) Centrimo motif enrichment plot of the ERG motif over ERG-specific BAF155 sites (10837) in LNCaP cells as defined by the fold change in BAF155 occupancy. See also Figure S4J.
(E) Metagene plots of ERG and BAF155 occupancy over ERG-specific BAF155 sites (10837) in LNCaP cells in empty, ERG, and ERG-R367K conditions.
(F) Heatmaps of ERG and BAF155 occupancy in LNCaP cells in empty, ERG, and ERG-R367K conditions, over all ERG-specific BAF155 sites (10837) ranked by ERG occupancy in the ERG condition.
(G) Metagene plots of ERG and BAF155 occupancy over Empty-ERG BAF155 sites (69451) in LNCaP cells in empty, ERG, and ERG-R367K conditions.
(H-I) Example tracks of ERG and BAF155 in LNCaP cells in empty, ERG, and ERG-R367K conditions at the (H) GRPR and (I) MYC loci.
(J) RT-qPCR experiments indicating RNA expression of ERG target genes in LNCaP cells. Two biological replicates are presented for each condition, Error bars = Mean ± SD for n=3 technical replicates.
We next determined whether a DNA binding defect in the ERG transcription factor affected genome-wide occupancy of ERG and targeting of BAF complexes. We analyzed ERG and BAF155 occupancy in empty, ERG, and ERG-R367K conditions in LNCaP cells over sites to which BAF155 targets in the presence of wild-type ERG. As expected, ERG R367K exhibited substantially less chromatin binding. Importantly, we found that ERG-R367K failed to increase BAF complex occupancy at ERG-specific BAF155 sites. Specifically, we found that ERGR367K exhibited a low level of occupancy at these sites and BAF155 showed a similar occupancy to that observed in cells expressing an empty vector (Fig. 4e-f). Surprisingly, at shared empty-ERG BAF155 sites, we found that ERG-R367K bound at levels similar to wildtype ERG, and that the observed occupancy levels corresponded to the endogenous and unchanged BAF complex occupancy at these sites (Fig. 4g, S4o). These distinct classes of ERG binding sites were seen at the GRPR and MYC loci, where DNA-independent and DNA-dependent ERG targeting occurred at promoter-distal sites (Fig. 4h-i, S4p). Finally, using RNA-qPCR, we found that ERG-R367K was unable to induce expression of several ERG target genes, suggesting the requirement for wild-type ERG-mediated BAF complex targeting for gene activation (Fig. 4j). Understanding the mechanisms by which transcription factors are bound at sites lacking their target sequence is an important question in gene regulation. These findings suggest that protein-protein interactions with chromatin regulators may be a critical mediator of transcription factor positioning at these sites.
BAF complex activity is required for global ERG chromatin occupancy and target gene regulation
To ascertain whether the ERG-BAF complex interaction was required for ERG-mediated cell proliferation in VcaP cells, we introduced shRNAs specific to core members of the BAF complex (ARID1A, BRG1, and BAF155) (Fig. 5a). Protein-level depletion of any of these subunits resulted in attenuated VCaP proliferation (Fig. 5b). These observations extend recent results from an shRNA-based screen that revealed several BAF complex components were required for the proliferation of TMPRSS2-ERG-containing VCaP cells but not 22rv1 prostate cells lacking ERG overexpression (Mounir et al., 2016). Finally, as further validation of VCaP cell dependence on BAF complexes, we evaluated the relationship between ERG expression and dependency on BAF complex subunits by analyzing genome-scale shRNA synthetic lethal screens performed in hundreds of cancer cell lines across many lineages (Project Achilles) (Tsherniak et al., 2017). We found that VCaP cells exhibited a strong dependence on BRG1 (SMARCA4) and ARID1A (Fig S5a-b). These findings suggest that BAF complexes are required for VCaP cell proliferation.
Figure 5.

ERG is dependent on BAF complex catalytic activity for chromatin occupancy and gene regulation.
(A) Immunoblot performed on VCaP nuclear extracts in shCt, shBRG1, shARID1A, and shBAF155 conditions.
(B) Proliferation analyses of VCaP cells in shCt, shBRG1, shARID1A, and shBAF155 conditions. Error bars = Mean ± SEM for n=3 biological replicates. (*** p < 0.001, ** p < 0.01, two-tailed t-test vs. shCt)
(C) Metagene plots of ERG at ERG-BAF155 shared sites in shCt and shARID1A conditions in VCaP cells.
(D) Correlation plot of log2(fold change) for ERG and BAF155 in shCt and shARID1A conditions over all ERG–BAF155 sites in VCaP shCt cells.
(E) Example ERG, BAF155, and RNA-seq tracks in shCt, shERG, and shARID1A conditions at the GRPR locus in VCaP cells
(F) (left) Overlap of significantly changed genes in VCaP shERG and shARID1A conditions (right) Heatmap of log2FC in expression for VCaP shERG and shARID1A RNA-seq over shared set of 537 significantly-changed genes. Gene changes are significantly concordant between both conditions (P = 1.53e-26, Fisher’s exact test).
(G) Input and anti-ERG IP in SW13 (SMARCA2/4-dual deficient) nuclear extracts in empty, ERG and GFP control, ERG and BRG1, and ERG and BRG1-K785R (ATPase catalytically-inactive) conditions.
(H) Plot of RNA-seq changes in SW13 cells in empty and ERG conditions, in the absence (empty condition, left) or presence (BRG1 condition, right) of BRG1 ATPase, with significantly changed genes upon ERG expression indicated in orange.
(I) Proportion of genes upregulated (left) or downregulated (right) by ERG in the presence of BRG1, that are also able to be regulated by ERG in the GFP control or BRG1-K785R conditions.
To define the consequences of BAF complex subunit perturbation on ERG targeting on chromatin and gene regulation, we performed ERG and BAF155 ChIP-seq as well as RNA-seq in VCaP cells expressing an shRNA targeting ARID1A (Fig. S5c-f). These experiments were performed at the same time as shCt and shERG experiments (Fig. S3a-c), to allow direct comparison of these perturbations. We found that ARID1A suppression led to genome-wide attenuation of both ERG and BAF155 binding at BAF155-ERG shared sites, to a comparable degree as ERG suppression, despite only modest changes in ERG levels at the RNA and protein level (Fig. 5c, S5d, S5g). In addition, we found that the attenuation in ERG binding upon ARID1A suppression directly corresponded to the degree to which BAF155 occupancy was lost (Fig. 5d, S5g). Concordant attenuation of ERG-BAF complex co-targeting was seen at target loci such as GRPR (Fig. 5e, S5h). By RNA-seq, we found significant concordance in resulting gene expression changes from shERG and shARID1A (p = 1.53e-26, Fisher’s exact test, Fig. 5f), including interdependent regulation of key ERG target genes such as KLK3 and PLA1A (Fig. 5f, S5i). These observations demonstrate that binding of ERG to chromatin and regulation of gene expression is dependent on the presence and recruitment of the BAF complex chromatin remodeler.
Finally, to determine whether ERG requires functional BAF complexes for de novo ERG target gene regulation, we used the SW13 adrenal carcinoma cell line, which lacks both ATPase subunits of the BAF complex (BRG1- and BRM- dual deficient). We performed dual introduction of either a control vector or ERG, alongside which we introduced wild-type BRG1, an ATPase-dead BRG1 mutant (K785R), or a GFP control, into SW13 cells (Fig. 5g). We found that the interaction of ERG with BAF complexes was dependent on the presence of BRG1, suggesting that ERG binding requires the presence of the core ATPase and its associated subunits within BAF complexes (Fig. 5g). We performed RNA-seq on these engineered SW13 cells (Fig. S5j-k), and found that ERG-mediated gene regulation is dependent on a catalytically active BAF complex (Fig. 5h). Specifically, we found that in the absence of an ATPase subunit within the BAF complex, ERG expression only altered the expression of 52 genes, whereas in the presence of BRG1, ERG expression resulted in changed expression of 652 genes (Fig. 5h). The presence of a catalytically active BRG1 ATPase was required for the regulation of the majority of ERG target genes, as most genes that were upregulated (310/367, 84.4%) or downregulated (243/285, 85.2%) by ERG in the presence of BRG1 failed to change in the GFP or BRG1-K785R mutant conditions, including ERG target genes such as PLA1A and FZD4 (Fig. 5i, Fig. S5l-n). These observations demonstrate that ERG target gene regulation is dependent on the catalytic activity of the BRG1 ATPase, such that BAF complex binding alone is not sufficient to induce ERG target gene regulation in the absence of BAF complex catalytic activity.
BAF complexes are required for ERG-mediated basal-to-luminal transition in prostate organoids
We next sought to characterize the physiologic relevance of the interdependence between ERG and the BAF complex. Previous studies have shown that ERG activity is responsible for the basal to luminal transition of prostate cells, a hallmark of prostate oncogenesis (Bose et al., 2017; Klezovitch et al., 2008). To test whether loss of BAF complex components affects ERG-driven basal to luminal transition in prostate epithelia, we introduced either an shRNA targeting Brg1 or a non-targeting shRNA control into prostate cells derived from Pten+/− mice expressing the Tmprss2-ERG fusion (Baena et al., 2013) and then derived 3D organoids in matrigel and DHT (Fig. 6a). We found that suppression of Smarca4 (encoding the BRG1 ATPase) levels by ~40–45% resulted in a substantial increase in basal organoid morphology and subsequent loss of luminal morphology as compared to control Tmprss2-ERG+ prostate cells (Fig. 6b, Fig. S6a,b). Moreover, suppression of BRG1 in Pten+/− ,Tmprss2-ERG+ organoids resulted in morphology resembling that of Pten+/− mice that lack Tmprss2-ERG entirely (Fig. 6b, Fig. S6a,b). As a control, we performed knockdown of ERG in Pten+/− ,Tmprss2-ERG+ organoids and found that this reverted the organoids to a predominantly basil morphology, affirming the driving role for the ERG transgene in luminal phenotype specification(Fig. S6c-d). We verified these morphological observations by performing hematoxylin and eosin (H&E) staining coupled with immunohistochemistry for the luminal marker CK8 and the basal marker TRP63. We observed robust CK8 staining and modest TRP63 staining in the shCt organoids indicating a luminal phenotype induced by ERG. In contrast, shBRG1 organoids exhibited faint CK8 and strong TRP63 staining, hallmarks of a basal cell phenotype (Fig. 6c, Fig. S6e). These observations provide evidence that functional BAF complexes play a critical role in facilitating ERG-mediated basal to luminal transition under physiological conditions.
Figure 6.

BAF complexes are required for ERG-driven basal-to-luminal transition in prostate organoids.
(A) Schematic representation of the prostate organoid workflow to assay ERG-driven basal to luminal morphology.
(B) Proportion of Pten+/− ERG+ prostate organoids infected with shCt or shBRG1, or Pten+/− prostate organoids exhibiting basal, luminal, or intermediate phenotypes. Error bars = Mean ± SEM for n=2 indicated, each replicate is derived from an independent mouse. (*** = p < 1e-10 by Fisher exact test).
(C) H&E and immunohistochemical analyses for CK8 (luminal marker) and TRP63 (basal marker) performed on mouse prostate organoids infected with shCt or shBRG1. Scale bars (red) indicate a distance of 50mm.
(D) Model for interdependence of ERG and BAF complexes in prostate oncogenesis.
Discussion
Here we report the discovery and characterization of an unexpected physical interaction between TMPRSS2-ERG and the mSWI/SNF (BAF) complex, which is necessary for ERG-directed BAF complex targeting, gene expression, prostate cancer cell proliferation and ERG-driven basal to luminal transition. We find that ERG retargets BAF complexes genome-wide to ERG target sites, and that ERG requires DNA binding to retarget BAF complexes to new, de novo sites, but not for genome-wide occupancy at BAF complex sites. We also find that ERG requires BAF complexes and their catalytic activity for genome-wide chromatin occupancy as well as target gene regulation. Our results suggest an interdependent mechanism of recruitment by which a transcription factor both recruits and requires a chromatin remodeler for chromatin occupancy and regulation (Fig. 6d).
Understanding the mechanism by which oncogenic ETS factors contribute to prostate oncogenesis is important for the broader function of ETS factors in oncogenesis and disease. The interaction of ERG with BAF is conserved among oncogenic ETS family transcription factors, ETV1, ETV4 and ETV5, which are overexpressed in a range of malignancies, including breast, colorectal and gastric tumors (Oh et al., 2012), gastrointestinal stromal tumor (Chi et al., 2010), as well as acute lymphoblastic leukemia (Zhang et al., 2016) and metastatic lesions in the lung (Okimoto et al., 2016). We found that the interaction between ERG and BAF is required for ERG-mediated gene expression and oncogenic function which provides a mechanistic explanation for recent genome-scale synthetic lethal screening efforts which have indicated that TMPRSS2-ERG-expressing cells require the expression of several BAF complex subunits (Mounir et al., 2016). Although many cell lines depend on BAF components, analysis of genome-scale loss-of-function genetic screens (Project Achilles) further confirmed that ERGexpressing prostate cancer cell lines exhibit dependence on the expression of BAF components. As such, the interplay between BAF complexes and ETS factors is likely required for tumor maintenance. The observations described herein suggest a gain-of-function mechanism by which BAF complexes contribute to oncogenesis via tethering to an oncogenic transcription factor.
Studies examining the enhancer landscape in prostate cancer have also implicated the competition of transcription factors as an important contributor to oncogenesis (Kron et al., 2017; Pomerantz et al., 2015; Shukla et al., 2017). Our studies describe altered genomic targeting of BAF complexes by ERG binding to ETS motif sites, and that ERG is also dependent on BAF complexes for genome-wide occupancy and target gene regulation. Our results align with ETV1 overexpression in gastrointestinal stromal tumor (GIST), in which ETV1 cooperates with a pioneer factor (FOXK1) to regulate target genes, but ETV1 alone does not pioneer accessibility (Chi et al., 2010; Ran et al., 2018). This is distinct from the de novo gain or loss of enhancer activation by BAF complexes observed in other cancers (Boulay et al., 2017; Nakayama et al., 2017), suggesting that BAF complex recruitment can modulate target gene regulation absent major changes in histone marks or accessibility. In addition, we demonstrated that in the absence of DNA-binding activity, ERG bound to the genomic sites occupied by BAF complexes that were not altered by wild-type ERG expression, but failed to actively direct BAF complexes to de novo sites as with wild-type ERG. These observations identify two distinct classes of ERG binding sites and suggest that transcription factor occupancy at non-motif sites may be facilitated by protein-protein interactions with chromatin regulators.
Exome sequencing studies have demonstrated that the genes encoding BAF complex subunits are mutated in >20% of human cancer, generating both gain- and loss-of-function phenotypes (Kadoch and Crabtree, 2013; Kadoch et al., 2013). A recent study has also identified mutations in BAF complex subunits in a prostate cancer subtype lacking ETS factor overexpression, suggesting that BAF complexes can function as both tumor suppressors and oncogenes in distinct mechanisms of prostate oncogenesis (Armenia et al., 2018). Understanding the relationship between transcription factors and chromatin remodelers has been a fundamental question in gene regulation (Burns and Peterson, 1997; Fryer and Archer, 1998), with recent studies demonstrating both loss- and gain-of-function perturbations by which BAF complexes can be regulated by transcription factors and fusion oncoproteins (Boulay et al., 2017; McBride et al., 2018; Pulice and Kadoch, 2016; Takaku et al., 2016). These findings suggest a fundamental interdependence between chromatin remodeling complexes and transcription factors that has implications for the large family of ETS factors involved in development and cancer. Together, these findings demonstrate the importance of the ERG-BAF complex interaction for prostate oncogenesis and suggest that targeting this interaction may disrupt ERG-driven oncogenesis.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Cigall Kadoch (cigall_kadoch@dfci.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Cell Culture
VCaP, MDA-PCa-2b, HEK-293-T and SW13 cells were used in this study. All cell lines were cultured in DMEM medium (Gibco, Grand Island, NY, USA), supplemented with 10% fetal bovine serum, 1% Glutamax (Gibco), 1% Sodium Pyruvate (Gibco) and 1% Penicillin-Streptomycin (Gibco). LNCaP cells were cultured in RPMI medium containing 10% FBS and 1% Penicillin-Streptomycin and maintained in a humidified incubator at 37°C with 5% CO2.
Prostate Organoid Culture
Tmprss2-ERG knockin mice were generated previously (Baena et al., 2013). Pten+/− mice were generated by crossing PtenL/+ mice to Gata1-Cre mice. All mice were maintained on a mixed genetic background and housed in pathogen-free barrier environment. All mouse studies were approved by the Institutional Animal Care and Use Committee (IACUC).
METHOD DETAILS
SILAC media preparation and cell culture conditions
Standard SILAC media preparation and labeling steps were followed as previously described(Ong and Mann, 2006) with the addition of light proline to prevent the conversion of arginine to proline (Bendall et al., 2008). Briefly, L-methionine and 200mg/L of L-Proline were added to base media according to standard formulations for DMEM (Caisson Labs). This base media was divided into three parts and to each was added either l-arginine (Arg0) and l-lysine (Lys0) (light), 13C614N4-l-arginine (Arg6) and 4,4,5,5-D4-l-lysine (Lys4) (medium), or 13C615N4-l-arginine (Arg10) and 13C615N2-l-Lysine (Lys8) (heavy) to generate the three SILAC labeling mediums. Each medium with the full complement of amino acids at the standard concentration for each media, was sterile filtered through a 0.22μ filter (Milipore, Bedford MA). VCaP cell line was grown in the corresponding labeling media, supplemented with 2 mM L-glutamine (Gibco), 10% dialyzed fetal bovine serum (Sigma) and antibiotics (Gibco), in a humidified atmosphere with 5% CO2. Cells were grown for at least eight cell divisions in labeling media.
ERG-protein interaction studies
VCaP cells were grown for 3 weeks (8 cell doublings) in DMEM depleted of L-arginine and L-lysine (Caisson Labs Inc.) and supplemented with 10% dialyzed FBS (Sigma) and amino acids as described above to generate light- and heavy-labeled cells. Cells were lysed in low volume of IP lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.1% sodium deoxycholate, 1 mM EDTA) supplemented with complete protease inhibitor cocktail (Roche) generating highly concentrated lysates (~10mg/ml). For the immunoprecipitation reactions, lysates were diluted ten-fold into mild IP buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, protease inhibitors cocktail) to a concentration of 1mg/ml. 5 mg of heavy-labeled protein lysate was incubated over night with 4μg anti ERG (C-20) antibody (Santa Cruz). 5mg of light-labeled lysates were incubated with 4μg isotype-matched IgG antibody (Santa Cruz) as a non-specific control for binding to antibody and/or to Protein A/G sepharose beads. The reactions were incubated with 50μl of 50% beads slurry (pre-washed 3 times in PBS) for 2 hours at room temperature. Finally, the reactions were washed 3 times in IP buffer and one time in the same buffer lacking the NP40. The beads/antibody/ERG complex were eluted in 25μl of 0.1% trifluoroacetic acid for 1min at room temperature followed by immediate neutralization with 25μl of 1M Tris HCl pH 8.0. The supernatants were subjected to mass spectrometric analysis, as described below. For a second replicate, labels were swapped such that heavy labeled lysates were incubated with control and light labeled lysates with anti-ERG antibody.
1D-SDS-PAGE and MS analysis for ERG-protein interaction studies
The beads from immunopurification samples were washed once with IP lysis buffer (Pierce), then the two different lysates of each replicate were combined, washed again and reduced and alkylated, on bead, in 2 mM DTT and 10 mM iodoacetamide respectively. One part LDS buffer (Invitrogen) was added to three parts sample (including beads) and tubes heated to 70°C for 10 minutes. Proteins were resolved on a 4–12% gradient 1.5 mm thick Bis-Tris gel with MES running buffer (Nupage, Invitrogen) and Coomassie stained (Simply Blue, Invitrogen). Gel lanes were excised into eight pieces and then further cut into 1.5 mm cubes. The gel pieces were further destained in a solution containing 50% EtOH and 50% 50 mM ammonium bicarbonate, then dehydrated in 100% EtOH before addition of sufficient trypsin (12.5 ng/μL) to swell the gel pieces completely. An additional 100 μL of 50 mM ammonium bicarbonate was added before incubating at 37°C overnight on a thermomixer (Eppendorf). Enzymatic digestion was stopped by the addition of 100 μL of 1% TFA to tubes. A second extraction with 300 μL of 0.1% TFA was combined with the first extract and the peptides from each gel slice cleaned up on C18 StageTips (Rappsilber et al., 2007). Peptides were eluted in 50 μL of 80% acetonitrile/0.1% TFA and dried down in an evaporative centrifuge to remove organic solvents. The peptides were then reconstituted with 3% ACN in 0.1% formic acid. Reconstituted peptides were separated on an online nanoflow EASY-nLC 1000 UHPLC system (Thermo Fisher Scientific) and analyzed on a benchtop Orbitrap Q Exactive mass spectrometer (Thermo Fisher Scientific). The peptide samples were injected onto a capillary column (Picofrit with 10 μm tip opening / 75 μm diameter, New Objective, PF360–75-10-N-5) packed in-house with 20 cm C18 silica material (1.9 μm ReproSil-Pur C18-AQ medium, Dr. Maisch GmbH, r119.aq). The UHPLC setup was connected with a custom-fit microadapting tee (360 μm, IDEX Health & Science, UH-753), and capillary columns were heated to 50 °C in column heater sleeves (Phoenix-ST) to reduce backpressure during UHPLC separation. Injected peptides were separated at a flow rate of 200 nL/min with a linear 80 min gradient from 100% solvent A (3% acetonitrile, 0.1% formic acid) to 30% solvent B (90% acetonitrile, 0.1% formic acid), followed by a linear 6 min gradient from 30% solvent B to 90% solvent B. Each sample was run for 150 min, including sample loading and column equilibration times. Data-dependent acquisition was obtained using Xcalibur 2.2 software in positive ion mode at a spray voltage of 2.00 kV. MS1 Spectra were measured with a resolution of 70,000, an AGC target of 3e6 and a mass range from 300 to 1800 m/z. Up to 12 MS2 spectra per duty cycle were triggered at a resolution of 17,500, an AGC target of 5e4, an isolation window of 2.5 m/z and a normalized collision energy of 25. Peptides that triggered MS2 scans were dynamically excluded from further MS2 scans for 20 s.
Identification and quantification of proteins for ERG-protein interaction studies
All mass spectra were analyzed with MaxQuant software version 1.3.0.5(Cox and Mann, 2008) using a human Uniprot database. MS/MS searches for the proteome data sets were performed with the following parameters: Oxidation of methionine and protein N-terminal acetylation as variable modifications; carbamidomethylation as fixed modification. Trypsin/P was selected as the digestion enzyme, and a maximum of 3 labeled amino acids and 2 missed cleavages per peptide were allowed. The mass tolerance for precursor ions was set to 20 p.p.m. for the first search (used for nonlinear mass re-calibration) and 6 p.p.m. for the main search. Fragment ion mass tolerance was set to 20 p.p.m. For identification we applied a maximum FDR of 1% separately on protein, peptide and PTM-site level. We required 2 or more unique/razor peptides for protein identification and a ratio count of 2 or more for protein quantification per replicate measurement. To assign interacting proteins we used the Limma package in the R environment to calculate moderated t-test p, as described previously(Udeshi et al., 2013).
Mass Spectrometry
VCaP nuclear extracts were immunoprecipitated with cross-linked antibodies against IgG (Cell Signaling Technology), ERG (C-17, Santa-Cruz), BAF47 (A-5, Santa-Cruz) or BRG1 (EPNCIR111A, Abcam). Samples were then run on a 4%−12% Bis-Tris Gel (Thermo Scientific) and subjected to Coomassie staining. Bands were then cut from each IP from the 45–65KDa and 140–250KDa regions and submitted to the Taplin Biological Mass Spectrometry Facility (Harvard Medical School) for analysis.
Silver Stains
VCaP or HEK-293-T nuclear extracts were immunoprecipitated with cross-linked antibodies. Samples were then run on a 4%−12% Bis-Tris Gel (Thermo Scientific) and stained using the SilverQuest silver stain kit (Invitrogen).
Nuclear Extract Preparation
Cells were homogenized in Buffer A (25 mM HEPES (pH 7.6), 25 mM KCL, 0.05 mM EDTA, 10% glycerol, 5 mM MgCl2, 0.1% NP-40 supplemented with fresh 1 mM DTT, protease inhibitors [Roche], and 1 mM PMSF) on ice. Nuclei were sedimented by centrifugation (1,200 rpm), resuspended in Buffer C (10 mM HEPES (pH 7.6), 3mM MgCl2, 100 mM KCL, 0.1 mM EDTA, 10% glycerol, 1 mM DTT and protease inhibitors), and lysed by the addition of ammonium sulfate to a final concentration of 300 mg/mL. Soluble nuclear proteins were separated by ultracentrifugation (100,000 × g) and precipitated with 0.3 mg/ml ammonium sulfate for 20 min on ice. Protein precipitate was isolated by ultracentrifugation (100,000 × g) and resuspended in IP buffer 1 (200–300 mM NaCl, 50 mM Tris-HCl [pH 8.0], 1% NP-40, 1mM EDTA, 1mM DTT, 1 mM PMSF with protease inhibitors) or IP buffer 2 (150 mM NaCl, 50 mM Tris-HCl, 1mM EDTA, 1% Triton X-100, 1mM DTT, 1mM PMSF with protease inhibitors) for immunoprecipitation analyses or HEMG-0 buffer (25 mM HEPES [pH 7.9], 0.1 mM EDTA, 12.5 mM MgCl2, 100 mM KCl, supplemented with DTT and PMSF) for analyses on glycerol gradient.
Immunoprecipitation
Nuclear extracts were resuspended in IP buffer 1 and placed in protein lo-bind tubes (Eppendorf). Protein concentration was determined using Bradford assay and adjusted to the final volume of 200μl at a final concentration of 1 mg/ml with IP buffer. Each IP was incubated with 1.5–2.5μg of antibody, (Antibody specifications are found in Table S3) overnight at 4°C and then for 2h with 20μl Protein G Dynabeads (Themo-Fisher) or Protein G Sepharose beads (GE Healthcare). The beads were then washed five times at 4°C with IP buffer and resuspended in 20μl 2× gel loading buffer: (4× LDS buffer; Invitrogen) + DTT and water. For experiments treated with Ethidium Bromide, nuclear extracts were incubated at 4°C for 30 min with 50μg/ml EtBr prior to immunoprecipitation. In Benzonase (Sigma-Aldrich) treated experiments, Benzonase was added at 1/1000 to each sample prior to overnight incubation with antibody.
Alternatively, immunoprecipitations were performed by washing cells with cold PBS and resuspended in EB0 hypotonic buffer containing 50mM Tris pH 7.5, 0.1% NP-40, 1mM EDTA, 1mM MgCl2 supplemented with protease inhibitors. Lysates were pelleted at 5,000rpm for 5min at 4°C. Supernatants were discarded and nuclei were resuspended in EB300 high salt buffer containing 50mM Tris pH 7.5, 300mM NaCl, 1% NP-40, 1mM EDTA, 1mM MgCl2 supplemented with protease inhibitors. Lysates were incubated on ice for 10 min with occasional vortexing. Lysate was pelleted at 21000g for 10 min at 4°C. Supernatants were quantified and supplemented with 1 mM DTT. 200μg of protein was used for immunoprecipitation with 1–2ug of antibodies over night at 4°C. Protein-G Dynabeads were added for 2 hours and washed with EB150 (EB300 with 150mM NaCl). Beads were eluted with loading LDS and loaded onto SDS-PAGE.
Depletion Studies
Nuclear extracts were prepared to a final concentration of 2.5 mg/ml with IP buffer 2. For each IP, 75ug (30 mL) of nuclear extract was incubated with 2.5 mg of antibody overnight at 4°C and then for 1h with 15 mL pre-washed Protein G Sepharose beads. After centrifugation (10,000 rpm for 1min) 45 mL of the supernatant was either saved or used for another round of IP. In total 2–3 rounds of IP were performed. Quantitative densitometry analyses were performed with the Li-Cor Oddessy Imaging System (Li-COR Biosciences, Lincoln, NE, USA).
Urea Denaturation Studies
Nuclear extracts (150 μg) were subjected to partial urea denaturation, ranging from 0.125 to 2.5 M urea (in IP buffer), for 30 min at room temperature (RT) prior to anti-ERG IP. The co-precipitated proteins were analyzed by immunoblot.
Density Sedimentation Analyses
Nuclear extract (500 μg) was resuspended in 200 ml of 0% glycerol HEMG buffer and carefully overlaid onto a 10 ml 10%–30% glycerol (in HEMG buffer) gradient prepared in a 14 × 89 mm polyallomer centrifuge tube (331327, Beckman Coulter, Brea, CA, USA). Tubes were centrifuged in an SW40 rotor at 4°C for 16 hr at 40,000 rpm. Fractions (0.5 ml) were collected and used in analyses.
Transient Transfection Studies
Briefly, HEK-293-T cells were plated in 6-well plates to 80% confluence prior to transfection using polyethylenimine (PEI) in a 3:1 PEI:DNA ratio and were harvested after 48h.
Lentiviral Generation
Lentivirus was produced by PEI (Polysciences Inc.) transfection of HEK-293-T LentiX cells (Clontech) with gene delivery vector co-transfected with packaging vectors pspax2 and pMD2.G as previously described(Kadoch and Crabtree, 2013). Supernatants were harvested 72h post-transfection and centrifuged at 20,000 rpm for 2h at 4°C. Virus containing pellets were resuspended in PBS and placed on cells dropwise. Selection of lentivirally-infected cells was achieved with either blasticydin or puromycin, both used at 2μg/ml. Overexpression or knock-down (KD) efficiency was determined by Western blot analyses or RT-qPCR. shRNA constructs used are found in Table S3.
Expression and Purification of Recombinant ERG in Escherichia coli
ERG (33–479) and ERG (33–479) R367K a were cloned into a Pet28PP bacterial expression vector with an N-terminus 6X histidine affinity tag followed by a PreScission protease cleavage recognition site. ERG constructs were transformed in Rosetta DE3 competitive cells from Millipore, grown to optical density of 0.6 in Luria-Bertani (LB) broth and subjected to 1mM IPTG overnight at 16°C for induction of ERG protein. Briefly, bacterial cell pellets were homogenized in lysis buffer containing 50mM NaPi pH 7.4, 500mM NaCl, 10% glycerol, supplemented with 1mM DTT, 10mM Imidazole, 0.1% Igepal and protease inhibitors. Following sonication, soluble fraction was incubated with high capacity Ni-NTA affinity chromatography matrix, washed twice with 50mM NaPi pH 7.4, 500mM NaCl, 10% glycerol, supplemented with 1mM DTT, 50mM Imidazole and finally eluted with 50mM NaPi pH 7.4, 500mM NaCl, 10% glycerol, supplemented with 1mM DTT, 150mM Imidazole and protease inhibitors. Eluted proteins were concentrated, assess for purity prior to quantification.
Electrophoretic Mobility Shift Assay
The protein-DNA binding reaction was carried out according to Verger et al. with modifications(Verger et al., 2001). We used the polyomavirus enhancer (Py) probe 5’/IRD800/-GATCTTTAAGCAGGAAGTGACTAACTGACCGCAGGTGGATC-3’ modified at the 5’ end with the infrared fluorescent dye IRD800 (IRD800-Py) for facile mobility shift detection on the Odyssey CLx imaging system from LI-COR. Duplex DNA formation was carried out on a thermocycler with the complement of IRD800-Py. The DNA binding assay was assessed in a total volume of 10uL of binding buffer at room temperature for 20 min. ERG protein and duplex IRD800-Py DNA complex was analyzed on a ThermoFisher Scientific 6% DNA Retardation Gel in 0.5X TBE buffer at 100V for 50 min.
Chromatin Immunoprecipitation
ChIP assays were carried out on VCaP or LNCaP cultures of approximately 2–5 million cells per sample and per epitope. ChIP experiments were performed following the procedures described previously (Boulay et al., 2017). Briefly, cells were cross-linked for 10 min in 1% formaldehyde at 37 °C. This reaction was subsequently quenched in 125 mM glycine for 5 min. Chromatin from formaldehyde-fixed cells was fragmented to a size range of 200–700 bases with a Covaris E220 focused-ultrasonicator (Covaris, Inc). Solubilized chromatin was immunoprecipitated with the indicated antibodies overnight at 4°C. Antibody-chromatin complexes were pulled down with protein G-Dynabeads (Life Technologies), washed, and then eluted. After crosslink reversal, RNase A, and proteinase K treatment, immunoprecipitated DNA was extracted with AMP Pure beads (Beckman Coulter).
RNA collection/preparation
Cells were harvested following 48h exposure to lentivirus and either 2 days (SW13), 4 days (LNCaP) or 8 days (VCaP) of selection. The overexpression constructs used in LNCaP cell experiments were selected with blasticidin. The knockdown constructs used in VCaP cell experiments were selected with puromycin. SW13 cells were selected with both blasticidin and puromycin as the empty and V5-ERG vectors were blasticidin resistant, while the V5-GFP, V5-BRG1 and V5-BRG1 K785R constructs were puromycin resistant. Of note, the WT SMARCA4 gene was subcloned from MGC Human SMARCA4 Sequence-Verified cDNA purchased from GE Dharmacon (Accession: BC136644, Clone ID: 9020634). The K785R SMARCA4 mutant was subcloned from pBJ5 BRG1 DN, a gift from Gerald Crabtree (Addgene plasmid # 17874). RNA-seq samples were prepared in duplicate with independent infection, selection and cell culture. All RNA was produced using the RNeasy Mini Kit (Qiagen).
Library Prep and Sequencing for ChIP-seq and RNA-seq
Library prep and sequencing (75bp single end on Illuminia Nextseq 500) was performed by the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute.
Sequence Data Processing
ChIP-seq reads were mapped to the human reference genome (hg19) using Bowtie2(Langmead and Salzberg, 2012) version 2.1.0 with parameters –k 1. RNA-seq reads were mapped to the human reference genome (hg19) using STAR(Dobin et al., 2013) version 2.3.1 with default parameters. All sequence data is deposited in the Sequence Read Archive under GSE110657. See Table S4 for summary of statistics on sequencing experiments.
ChIP-seq Data Analysis
Peaks were called against input reads using MACS2 (Zhang et al., 2008) version 2.1.0 at q=1e-3. Broad peak calls were used for all marks in this study. Peaks were filtered to remove peaks that overlap with ENCODE blacklisted regions, as well as peaks mapped to unmappable chromosomes (only chr1–22,X,Y included). Duplicate reads were removed using samtools rmdup for all downstream analyses. ChIP-seq track densities were generated per million mapped reads with MACS2 2.1.0 using parameters –B –SPMR.
Metagene read densities were generated using HTSeq (Anders et al., 2015), with fragment length extended to 200bp to account for the average 200bp fragment size selected in sonication, using the center of each peak set used. Total read counts for each region were normalized the number of mapped reads to give reads per million mapped reads. Metagene plots were generated using average read densities across all sites indicated for each condition, with narrow metagene plots were generated around the center of the peak. Heatmaps were generated using the same HTSeq read densities as in metagene plots, sites were then ranked by mean ChIP-seq signal for the epitope and condition indicated in each figure. Heatmaps were visualized using Python matplotlib with a midpoint of 0.5 reads per million for the heatmap color scale to set the threshold for visualization. For quartile changes in ERG and BAF155 occupancy, metagene read densities were generated as above over the ERG-BAF155 peak set. From this, average RPM values over each peak window were used to calculate log2FC values between conditions, and ranking all sites by the log2FC in ERG occupancy to bin the sites by ERG displacement, and then generate metagene plots as above. For motif enrichment analysis, 500bp core sequences centered on peak centers were submitted to MEME-ChIP analysis(Machanick and Bailey, 2011). Motifs were selected based on rank in Centrimo significance determination, with transcription factor families collapsed for motif similarity.
To generate plots of log2 fold change for ChIP-seq reads, the peak sets for BAF155 and ERG were intersected to generate ERG-BAF complex sites in VCaP. ChIP-seq read counts for each BAF complex site were generated using Rsubread featureCounts, and read counts in each peak region were normalized per million mapped reads. Input RPM values for each region in each condition were subtracted from each ChIP epitope in that condition, values with higher input enrichment than ChIP enrichment were set to 0. Log2 fold change values were determined for each ChIP epitope using the normalized RPM values above, with a pseudocount of 0.1. Pairwise correlation was determined using a Pearson correlation coefficient between normalized fold change values for each pair of ChIP experiments. For condition-specific BAF155 sites, BAF155 sites in both conditions (VCaP: shCt and shERG; LNCaP: empty and ERG) were merged, then processed as above. Condition-specific BAF155 sites were determined as those that had a 1.5FC in BAF155 occupancy between conditions.
RNA-seq Data Analysis
RPKM values for samples were generated using GFold (Feng et al., 2012) version 1.1.0. All error bars represent Mean±SEM. Significance was assessed using the R package DESeq2 (Love et al., 2014) using raw read counts generated with Rsubread featureCounts against the hg19 refFlat annotation. Significantly changing genes were assessed with a Bonferri-corrected p-value of less than 1e-3 and a two-fold gene expression change (|log2FC|>1) to determine set of significantly changing genes. DESeq2 files were filtered for genes that were expressed (RPKM ≥ 1 in at least one condition), as well as to remove small RNA genes (MIR and SNO) remove. Significance on RPKM bar graphs is derived from the Bonferroniadjusted p-value assessed by DESeq2. For waterfall analysis of RNA-seq, log2 fold change values were generated with filtering as above for expressed and short RNA genes. RPKMs for biological duplicate RNA-seq in each cell line were combined using the average replicates of each condition, then doing a log2 fold change comparison with a pseudocount of 1 in each condition, i.e. log2( ( RPKMCOND2 + 1)/(RPKMCOND1 + 1) ). RNA-seq tracks were generated using bedtools genomecov –split –scale with the mapped read count to generate tracks normalized per million mapped reads. For pearson correlation coefficients, RPKM values were log2 normalized for determination of replicate concordance. Concordance of perturbations at RNA-seq level was determined using a fisher exact test for the concordance of significantly-changed genes in both conditions.
VCaP proliferation experiments
VCaP cells were plated in 12 well plates at 200K cells per well, next, each well was separately infected with shRNAs targeted against a BAF complex member or luciferase (control). Puromycin selection began after 48h, and media with puromycin was changed every 48h. 3 separate wells of cells for each condition were counted on days 7, 10, 14 and 17 using a Vi-CELL Cell Counter (Beckman).
RNAi dependency waterfall plots
RNAi dependency data (DEMETER scores) was downloaded from the Project Achilles data portal (http://portals.broadinstitute.org/achilles/datasets/all), using the 2.20.2 release. Dependency] data for SMARCA4 and ARID1A were gathered for the 362 cell lines that lack BAF complex mutations included in the dataset. ERG gene expression was gathered from the CCLE data portal (https://portals.broadinstitute.org/ccle/data) from the latest release (CCLE_RNAseq_081117.rpkm.gct). Log2 RPKM of the ERG gene was scored for each cell line. Cutoffs were chosen for high expression (RPKM >= 8) and low expression (0 < RPKM < 8).
Organoids
Murine prostates were isolated from Pten+/− or Pten+/−; Tmprss2-ERG mice. Single cell suspension from murine prostate was spinfected with lentiviruses with slight modifications as described in Xin et al. (Xin et al., 2003). Infected prostatic single cell suspensions were cultured then in organoid culture conditions, consisting of embedding cells within a Matrigel™ matrix and incubating with modified ENR media as described in Karthaus et. al (Karthaus et al., 2014). Growth of organoids was strictly controlled by selection with 0.3–0.6 μg/ml puromycin 2 days post spinfection. Organoids were passaged either via trituration with a glass Pasteur pipet or trypsinization with TrypLE for 5 min at 37C. Passage was performed every week with a 1:5 ratio. Scored organoid colonies are represented as the proportion of colonies scored as basal, luminal, or intermediate in each biological replicate derived from an independent mouse. Error bars are Mean±SEM for n=2 biological replicates. Significance is determined using a Fisher exact test between the two conditions compared, using the sum total of both biological replicates for statistical comparison.
Quantitative RT-PCR analysis
RNA from cell lines or infected mouse organoids was obtained using the RNeasy Mini Kit (Qiagen). cDNA preparation was performed by the Molecular Biology Core Facilities at the Dana-Farber Cancer Institute or by using the SuperScript VILO cDNA Synthesis Kit (Thermo Fisher). The primers for quantitative RT-PCR analysis are listed in Table S3. Quantitative real-time PCR (qRT-PCR) was performed with the Power SYBR Green PCR Master Mix (Applied Biosystems), and performed according to standard PCR conditions in a CFX384 Touch™ Real-Time PCR Detection System (Biorad).
Paraffin sectioning
Prostate organoids were processed in an automatic tissue processer (Tissue-TEK VIP, Sakura) and embedded into paraffin blocks. The blocks were cut using microtome (Leica RM 2145) to 5 μm thickness. The sections were placed on Apex Superior Adhasive glass slides (Leica) and stored at room temperature until further use.
Haematoxylin and eosin (H&E) staining
H&E staining was performed using standard procedures. After staining, sections were dehydrated and mounted using Vectamount and visualized under a light microscope (Olympus BX50) and analyzed by Cell Sens Standard ver. 1.15 software.
Immunohistochemistry
Deparaffinized 5μm-thick tissue sections were immunostained using the Vectastain R.T.U Elite ABC KIT according to manufacturer’s instructions. Briefly, antigen retrieval was performed by heating slides either in TRIS-EDTA buffer (pH 9) (Abcam) or with citrate buffer (pH 6) (Thermo Scientific) in a microwave and sections were sequentially blocked for 10 minutes with BLOXALL Blocking Solution (SP-6000), wash in PBS and blocked for 1hr with 10% horse blocking serum. Slides were then incubated for overnight in 4°C with primary antibody diluted in SignalStain Ab Diluent (Cell Signaling) Antibodies were used at the following concentrations: anti–pan-p63 (clone 4A4) 1/400, and anti-CK8/18 antibody (ab53280) 1/75. For all antibodies appropriate secondary Vectastain ABC Elite Kit HRP-conjugated antibodies and chromogenic 3,3-diaminobenzidine substrate were then successively added. Nuclei were counterstained with Hematoxylin. After staining, sections were dehydrated and mounted in the Permaslip medium (Alban Scientific Inc). Samples were visualized under a light microscope (Olympus BX50) and analyzed by Cell Sens Standard ver. 1.15 software.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical comparisons between two groups for proliferation analyses were performed with GraphPad Prism software 7.0 using a two-tailed unpaired t-test. The sample size (n) is indicated in the figure legends and represents biological replicates. Details for sequence data analyses and statistical significance are described in the specific Method Details section.
DATA AND SOFTWARE AVAILABILITY
Sequence data for all cell line experiments are deposited under GSE110657.
Supplementary Material
Highlights.
ERG binds to BAF complexes in solution and in a DNA-independent manner
ERG targets BAF complexes to ETS motif sites genome-wide
BAF complex ATPase activity is required for ERG target gene regulation
BAF complexes are required for ERG-mediated basal-to-luminal transition
Acknowledgments
We thank members of the Molecular Biology Core Facility at the Dana-Farber Cancer Institute, especially Zach Herbert, for expertise and technical assistance with ChIP-seq and RNA-seq datasets. We thank members of the Taplin Mass-Spec Facility (HMS) for assistance with mass spec experiments and data analysis. We thank G.R. Crabtree and A. Kuo for the anti-BAF155 rabbit polyclonal antibody used in ChIP-seq experiments. We thank W.G. Kaelin, D.R. Liu, L. Ellis and E.S. Lander for manuscript review and critical feedback. This work was supported in part by awards from the Prostate Cancer Foundation (W.C.H., L.A.G., D.A.T.), NIH/NCI U01 CA176058 (W.C.H.), the Department of Defense Prostate Cancer Research Program Postdoctoral Training Award W81XWH-15–1-0659 (G.J.S.), the NIH/NCI Tumor Cell Biology Training Program Grant T32-CA009361 (G.J.S.), the Department of Defense Prostate Cancer Research Program Idea Development Award (W81XWH-15–1-0546) and the NIH award UH2 CA213392 (Z.L.), the NIH DP2 New Innovator Award 1DP2CA195762–01 (C.K.), the American Cancer Society Research Scholar Award RSG-14–051-01-DMC (C.K.) and the Pew-Stewart Scholars in Cancer Research Grant (C.K.).
Footnotes
Declaration of Interests
C.K. is a Scientific Founder, shareholder, and consultant of Foghorn Therapeutics (Cambridge, MA).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamo P, and Ladomery MR (2016). The oncogene ERG: a key factor in prostate cancer. Oncogene 35, 403–414. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics (Oxford, England) 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenia J, Wankowicz SAM, Liu D, Gao J, Kundra R, Reznik E, Chatila WK, Chakravarty D, Han GC, Coleman I, et al. (2018). The long tail of oncogenic drivers in prostate cancer. Nat Genet 50, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baena E, Shao Z, Linn DE, Glass K, Hamblen MJ, Fujiwara Y, Kim J, Nguyen M, Zhang X, Godinho FJ, et al. (2013). ETV1 directs androgen metabolism and confers aggressive prostate cancer in targeted mice and patients. Genes Dev 27, 683–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuyaux JP, Ferreira E, Stehelin D, and Buttice G (1997). The Ets transcription factors interact with each other and with the c-Fos/c-Jun complex via distinct protein domains in a DNA-dependent and -independent manner. J Biol Chem 272, 26188–26195. [DOI] [PubMed] [Google Scholar]
- Bendall SC, Hughes C, Stewart MH, Doble B, Bhatia M, and Lajoie GA (2008). Prevention of amino acid conversion in SILAC experiments with embryonic stem cells. Mol Cell Proteomics 7, 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose R, Karthaus WR, Armenia J, Abida W, Iaquinta PJ, Zhang Z, Wongvipat J, Wasmuth EV, Shah N, Sullivan PS, et al. (2017). ERF mutations reveal a balance of ETS factors controlling prostate oncogenesis. Nature 546, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, et al. (2017). Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178 e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns LG, and Peterson CL (1997). The yeast SWI-SNF complex facilitates binding of a transcriptional activator to nucleosomal sites in vivo. Mol Cell Biol 17, 4811–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2015). The Molecular Taxonomy of Primary Prostate Cancer. Cell 163, 1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrere S, Verger A, Flourens A, Stehelin D, and Duterque-Coquillaud M (1998). Erg proteins, transcription factors of the Ets family, form homo, heterodimers and ternary complexes via two distinct domains. Oncogene 16, 3261–3268. [DOI] [PubMed] [Google Scholar]
- Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, et al. (2010). ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 467, 849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark JP, and Cooper CS (2009). ETS gene fusions in prostate cancer. Nat Rev Urol 6, 429–439. [DOI] [PubMed] [Google Scholar]
- Consortium GT (2015). Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348, 648–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson LW, Petersen JM, Graves BJ, and McIntosh LP (1996). Solution structure of the ETS domain from murine Ets-1: a winged helix-turn-helix DNA binding motif. EMBO J 15, 125–134. [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Feng J, Meyer CA, Wang Q, Liu JS, Shirley Liu X, and Zhang Y (2012). GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics (Oxford, England) 28, 2782–2788. [DOI] [PubMed] [Google Scholar]
- Fryer CJ, and Archer TK (1998). Chromatin remodelling by the glucocorticoid receptor requires the BRG1 complex. Nature 393, 88–91. [DOI] [PubMed] [Google Scholar]
- Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, Varmeh S, Zhang J, Cheng L, Sun Y, et al. (2015). SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol Cell 59, 917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgeson BE, Tomlins SA, Shah N, Laxman B, Cao Q, Prensner JR, Cao X, Singla N, Montie JE, Varambally S, et al. (2008). Characterization of TMPRSS2:ETV5 and SLC45A3:ETV5 gene fusions in prostate cancer. Cancer Res 68, 73–80. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2013). Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2015). Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1, e1500447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, and Crabtree GR (2013). Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45, 592–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karthaus WR, Iaquinta PJ, Drost J, Gracanin A, van Boxtel R, Wongvipat J, Dowling CM, Gao D, Begthel H, Sachs N, et al. (2014). Identification of multipotent luminal progenitor cells in human prostate organoid cultures. Cell 159, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedage V, Selvaraj N, Nicholas TR, Budka JA, Plotnik JP, Jerde TJ, and Hollenhorst PC (2016). An Interaction with Ewing’s Sarcoma Breakpoint Protein EWS Defines a Specific Oncogenic Mechanism of ETS Factors Rearranged in Prostate Cancer. Cell Rep 17, 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klezovitch O, Risk M, Coleman I, Lucas JM, Null M, True LD, Nelson PS, and Vasioukhin V (2008). A causal role for ERG in neoplastic transformation of prostate epithelium. Proc Natl Acad Sci U S A 105, 2105–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kron KJ, Murison A, Zhou S, Huang V, Yamaguchi TN, Shiah YJ, Fraser M, van der Kwast T, Boutros PC, Bristow RG, et al. (2017). TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat Genet 49, 1336–1345. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link KA, Balasubramaniam S, Sharma A, Comstock CE, Godoy-Tundidor S, Powers N, Cao KH, Haelens A, Claessens F, Revelo MP, et al. (2008). Targeting the BAF57 SWI/SNF subunit in prostate cancer: a novel platform to control androgen receptor activity. Cancer Res 68, 4551–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machanick P, and Bailey TL (2011). MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics (Oxford, England) 27, 1696–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MJ, Pulice JL, Beird HC, Ingram DR, D’Avino AR, Shern JF, Charville GW, Hornick JL, Nakayama RT, Garcia-Rivera EM, et al. (2018). The SS18-SSX Fusion Oncoprotein Hijacks BAF Complex Targeting and Function to Drive Synovial Sarcoma. Cancer Cell 33, 1128–1141.e1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed AA, Tan SH, Mikhalkevich N, Ponniah S, Vasioukhin V, Bieberich CJ, Sesterhenn IA, Dobi A, Srivastava S, and Sreenath TL (2010). Ets family protein, erg expression in developing and adult mouse tissues by a highly specific monoclonal antibody. J Cancer 1, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounir Z, Korn JM, Westerling T, Lin F, Kirby CA, Schirle M, McAllister G, Hoffman G, Ramadan N, Hartung A, et al. (2016). ERG signaling in prostate cancer is driven through PRMT5-dependent methylation of the androgen receptor. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama RT, Pulice JL, Valencia AM, McBride MJ, McKenzie ZM, Gillespie MA, Ku WL, Teng M, Cui K, Williams RT, et al. (2017). SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet 49, 1613–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Shin S, and Janknecht R (2012). ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim Biophys Acta 1826, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okimoto RA, Breitenbuecher F, Olivas VR, Wu W, Gini B, Hofree M, Asthana S, Hrustanovic G, Flanagan J, Tulpule A, et al. (2016). Inactivation of Capicua drives cancer metastasis. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, and Mann M (2006). A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc 1, 2650–2660. [DOI] [PubMed] [Google Scholar]
- Paulo P, Barros-Silva JD, Ribeiro FR, Ramalho-Carvalho J, Jeronimo C, Henrique R, Lind GE, Skotheim RI, Lothe RA, and Teixeira MR (2012). FLI1 is a novel ETS transcription factor involved in gene fusions in prostate cancer. Genes Chromosomes Cancer 51, 240–249. [DOI] [PubMed] [Google Scholar]
- Pomerantz MM, Li F, Takeda DY, Lenci R, Chonkar A, Chabot M, Cejas P, Vazquez F, Cook J, Shivdasani RA, et al. (2015). The androgen receptor cistrome is extensively reprogrammed in human prostate tumorigenesis. Nat Genet 47, 1346–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prensner JR, Iyer MK, Sahu A, Asangani IA, Cao Q, Patel L, Vergara IA, Davicioni E, Erho N, Ghadessi M, et al. (2013). The long noncoding RNA SChLAP1 promotes aggressive prostate cancer and antagonizes the SWI/SNF complex. Nat Genet 45, 1392–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulice JL, and Kadoch C (2016). Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb Symp Quant Biol 81, 53–60. [DOI] [PubMed] [Google Scholar]
- Ran L, Chen Y, Sher J, Wong EWP, Murphy D, Zhang JQ, Li D, Deniz K, Sirota I, Cao Z, et al. (2018). FOXF1 Defines the Core-Regulatory Circuitry in Gastrointestinal Stromal Tumor. Cancer discovery 8, 234–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, and Ishihama Y (2007). Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Rickman DS, Soong TD, Moss B, Mosquera JM, Dlabal J, Terry S, MacDonald TY, Tripodi J, Bunting K, Najfeld V, et al. (2012). Oncogene-mediated alterations in chromatin conformation. Proc Natl Acad Sci U S A 109, 9083–9088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Powers N, Saini N, Comstock CE, Sharma A, Weaver K, Revelo MP, Gerald W, Williams E, Jessen WJ, et al. (2008). The SWI/SNF ATPase Brm is a gatekeeper of proliferative control in prostate cancer. Cancer Res 68, 10154–10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla S, Cyrta J, Murphy DA, Walczak EG, Ran L, Agrawal P, Xie Y, Chen Y, Wang S, Zhan Y, et al. (2017). Aberrant Activation of a Gastrointestinal Transcriptional Circuit in Prostate Cancer Mediates Castration Resistance. Cancer Cell 32, 792–806 e797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaku M, Grimm SA, Shimbo T, Perera L, Menafra R, Stunnenberg HG, Archer TK, Machida S, Kurumizaka H, and Wade PA (2016). GATA3-dependent cellular reprogramming requires activation-domain dependent recruitment of a chromatin remodeler. Genome biology 17, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, Menon A, Jing X, Cao Q, Han B, et al. (2007). Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 448, 595–599. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Mehra R, Rhodes DR, Smith LR, Roulston D, Helgeson BE, Cao X, Wei JT, Rubin MA, Shah RB, et al. (2006). TMPRSS2:ETV4 gene fusions define a third molecular subtype of prostate cancer. Cancer Res 66, 3396–3400. [DOI] [PubMed] [Google Scholar]
- Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. (2005). Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 310, 644–648. [DOI] [PubMed] [Google Scholar]
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. (2017). Defining a Cancer Dependency Map. Cell 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, and Carr SA (2013). Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics 12, 825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verger A, Buisine E, Carrere S, Wintjens R, Flourens A, Coll J, Stehelin D, and Duterque-Coquillaud M (2001). Identification of amino acid residues in the ETS transcription factor Erg that mediate Erg-Jun/Fos-DNA ternary complex formation. J Biol Chem 276, 17181–17189. [DOI] [PubMed] [Google Scholar]
- Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. (2010). Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 29, 2147–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin L, Ide H, Kim Y, Dubey P, and Witte ON (2003). In vivo regeneration of murine prostate from dissociated cell populations of postnatal epithelia and urogenital sinus mesenchyme. Proc Natl Acad Sci U S A 100 Suppl 1, 11896–11903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Yu J, Mani RS, Cao Q, Brenner CJ, Cao X, Wang X, Wu L, Li J, Hu M, et al. (2010). An integrated network of androgen receptor, polycomb, and TMPRSS2-ERG gene fusions in prostate cancer progression. Cancer Cell 17, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, McCastlain K, Yoshihara H, Xu B, Chang Y, Churchman ML, Wu G, Li Y, Wei L, Iacobucci I, et al. (2016). Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Nat Genet 48, 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome biology 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.