

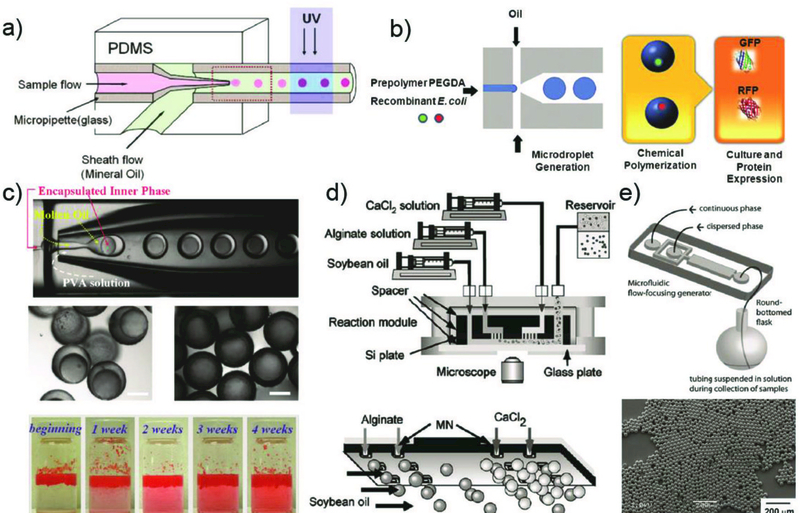
Abstract
Droplet microfluidics offers exquisite control over the flows of multiple fluids in microscale, enabling fabrication of advanced microparticles with precisely tunable structures and compositions in a high throughput manner. The combination of these remarkable features with proper materials and fabrication methods has enabled high efficiency, direct encapsulation of actives in microparticles whose features and functionalities can be well controlled. These microparticles have great potential in a wide range of bio-related applications including drug delivery, cell-laden matrices, biosensors and even as artificial cells. In this review, we briefly summarize the materials, fabrication methods, and microparticle structures produced with droplet microfluidics. We also provide a comprehensive overview of their recent uses in biomedical applications. Finally, we discuss the existing challenges and perspectives to promote the future development of these engineered microparticles.
1. Introduction
Microparticles with sizes ranging from 1 μm to 1000 μm have emerged as advanced functional materials for a wide range of biomedical applications, such as drug delivery, tissue engineering, biosensing, and cellular life science.1–3 These applications of microparticles depend on their properties which correlate with their size, structure, composition and configuration. Therefore, it is essential to fabricate microparticles in a controlled manner to improve their pharmaceutical capability and reliability for biological studies.4–6 However, it has long been a challenge to produce microparticles with such desired properties through conventional methods including emulsion polymerization, dispersion polymerization and spray drying.7 These methods normally result in microparticles with large polydispersity, poor reproducibility, limited functionality, and less tunable morphology. To overcome these limitations, various technologies, including droplet microfluidics, flow lithography microfluidics, electrohydrodynamic co-jetting, photolithography, and soft lithography-based imprinting and micromolding have recently been explored for tailored fabrication of microparticles.8–10
Among these, droplet microfluidics is one of the most effective techniques, as it offers exquisite control over multiple fluids at the microscale. Therefore, it allows precise tuning of the compositions and geometrical characteristics of microparticles.11,12 Exploiting these advantages, engineered microparticles with controlled sizes, monodispersity, diverse morphologies, and specific functions can be generated, and are playing an increasingly important role in biomedical fields.5,13 For instance, as drug delivery vehicles,6,14,15 microcapsules or multi-core microparticles can be prepared with well-defined structures and compositions that allow for high encapsulation efficiency and well-controlled release of the encapsulants. As cell carriers,16 hydrogel microparticles can be produced to act as extracellular matrix (ECM) to protect cells from the surrounding environment and maintain efficient nutrient and metabolic exchanges for long term cell culture. As a result, these cell-laden microparticles have direct applications in tissue engineering,17 stem cell therapy,18 and single cell studies.19 In addition, liposomes or polymersomes with multicompartment structures can be generated by droplet microfluidics in an exquisite and facile manner, making them ideal candidates for artificial cells.20, 21 Furthermore, tremendous effort has been expended on exploring new droplet microfluidic system as well as materials chemistry to produce microparticles with good biocompatibility, rich functionalities, and high production rates. This leads to new and exciting opportunities for further development in their use for advanced diagnostics and therapeutics.
In this review, we provide an overview of microparticles fabricated by droplet microfluidics, and highlight the most recent progress in biomedical fields. We introduce the droplet formation mechanism and describe devices used to generate various types of droplets. We summarize methods to prepare microparticles templated from these droplets and emphasize the unique and complex structures enabled by microfluidic techniques. We then describe the biomedical applications of these microparticles, focusing on recent advancements in their use as drug delivery vehicles and cell-laden matrices. Other applications including biosensors and artificial cells are also briefly described. Lastly, we discuss the existing challenges that can potentially impact the practical use of these microparticles and conclude with perspectives and potential implications.
2. Droplet generation
In droplet microfluidics, properties of immiscible fluids are exploited at a microscale to generate and manipulate droplets.22 To produce droplets that meet the sophisticated requirements in biomedical applications, microfluidic chips that allow precise manipulation of fluidic elements on a small length scale are required. In this section, we first discuss the mechanisms of droplet formation and various device geometries used for droplet generation. Then, we describe two of the most widely used microfluidic devices including glass capillary devices and lithographically fabricated poly(dimethylsiloxane)(PDMS) devices for generating various types of emulsion droplets from single emulsions to double emulsions and to even more complex emulsions. Lastly, other devices made from materials that have high stability and tolerate harsh operating conditions, as well as technologies for large scale production are discussed.
2.1. Droplet generation mechanism
An emulsion is a mixture of two immiscible liquids where one liquid is dispersed in another immiscible liquid. Most conventional methods for generating emulsions involve droplet breakup using shear or impact stresses generated by agitation. However, due to the nonuniform shear stresses applied, the resulting emulsions are highly polydisperse in size. In contrast, microfluidic devices offer an alternate and versatile route to produce emulsions.11,23 An emulsion is produced in a microfluidic device by precisely fabricating one drop at a time. This process is an outcome of a well-controlled balance between various forces acting on the fluid flow. These forces include inertial force, viscous force, interfacial tension, and buoyancy. In some cases, external forces such as electric,24,25 magnetic,26,27 and centrifugal forces28 are also utilized. The balance of these forces determines the fluid behaviour and thus the mechanism of droplet formation. Typically, in microfluidic systems, buoyancy is small compared to interfacial and viscous forces during droplet formation due to the relatively small channel size, flow velocity and droplet volume. Therefore, the complex phenomenon of droplet breakup is determined by various dimensionless numbers, which are related to the fluid properties, channel geometries, and the flow conditions. Capillary number (Ca) and Weber number (We) are two main dimensionless numbers that determine the flow behavior in the channel. The capillary number represents the relative effect of viscous forces and surface tension, while the Weber number reflects the balance between inertial and surface tension forces. For instance, in a single emulsion system comprising of a dispersed phase and a continuous phase, Ca of the continuous phase and We of the dispersed phase are typically low, yielding formation of droplets one by one, which is the dripping mode.
While there are various modes of droplet breakup including squeezing, dripping, jetting, tip-streaming,29 and tip-multi-breaking,30 the dripping mode is most widely utilized due to the high monodispersity of the droplets generated. In this mode, the size of droplets can be tuned by changing the tip size and the flow rates of the fluid phases. Moreover, droplets within droplets dispersed in a third continuous phase, or double emulsions, and even higher order multi-phase (multiple) emulsions with tunable size, morphology, and the number of inner drops, can be produced. More detailed descriptions about droplet formation mechanism and their relationships to various dimensionless numbers can be found in other reviews.31,32
The droplet breakup modes can be applied to various channel geometries including cross-flow, co-flow, and flow-focusing. These three device geometries rely mainly on the viscous shear force for droplet breakup, as shown in Fig. 1a. In a cross-flow geometry, also called T-junction, the dispersed phase is sheared in a T-shaped junction which has an angle θ (0° < θ ≤ 180°) between the dispersed and the continuous phase channels. This device geometry is widely used in the production of single emulsions due to the simplicity in device fabrication, which requires no additional alignment. The droplets generated have high monodispersity with a coefficient of variation (CV) typically less than 2%. The size of the droplets generated in a T-junction is generally larger than 10 μm due to the limitation of the channel dimension. In a co-flow geometry, also called coaxial junction, dispersed phase channel is inserted into and aligned with the continuous phase channel, and dispersed phase and continuous phase fluids flow in parallel through the channels. In most cases, droplets are formed in a dripping mode, and have low CV (less than 3%). However, the droplet sizes are larger than the tip diameter, thus, are typically larger than 10 μm. Flow-focusing has a channel geometry that is similar to that of co-flow but has a focus unit that suddenly shrinks the fluid passageway. Fluid phases form a hydrodynamic flow that contracts through the focus unit resulting in high fluid flow rate. This enhances the viscous shear force and allows formation of droplets with sizes down to a few hundred nanometers.
Fig. 1.
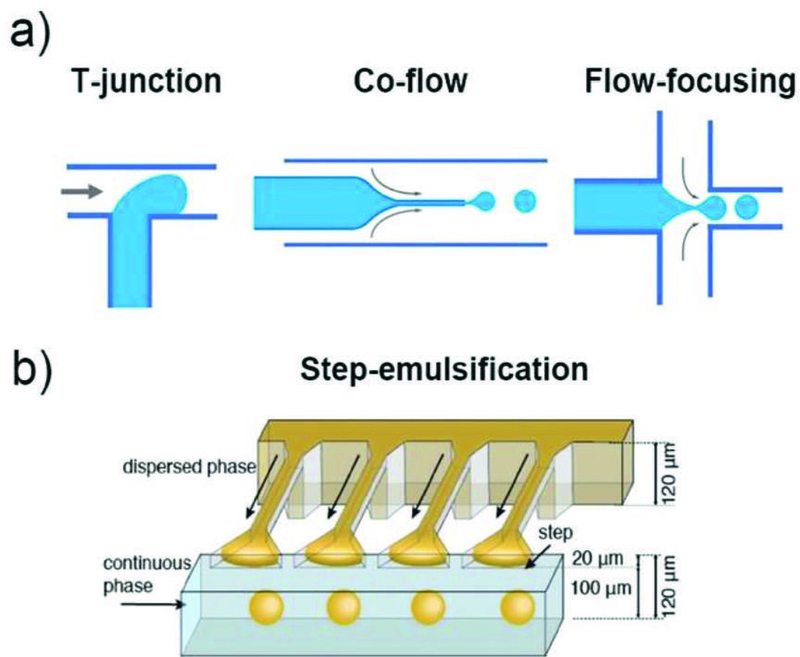
a) Schematic illustration of various channel geometries including cross-flow, co-flow, and flow-focusing. b) Schematic illustration of a step-emulsification channel arranged with four parallelized drop markers.33 The drop breakup occurs at the step between the nozzle and the continuous phase channel. Reprinted with permission from ref. 33. Copyright 2016, John Wiley & Sons, Inc.
There are also geometries that allow massive parallelization of multiple nozzles to achieve high throughput production of droplets. For example, step-emulsification devices can produce droplets with higher uniformity with high throughput.33,34 In such a device, droplet formation is governed by the large difference in the capillary pressure caused by the step change in channel height as shown in Fig. 1b.33,35,36 Since the droplet formation is mainly driven by interfacial tension, without shear forces, the droplet size is independent of the flow rates of both the dispersed and the continuous phases.37,38 As long as the dispersed phase flow rate is less than the droplet formation rate at the step, the droplet size is determined only by the height of the inlet channel. This spontaneous droplet formation mechanism often limits the production rate from each nozzle, which is typically much slower than conventional flow-focusing device. However, these devices are much suitable for parallelization as droplet formation does not rely on the shear force from the adjacent immiscible phase. Examples of these devices include, microchannel devices,37 edge-based droplet generation devices,39 millipede devices (Fig. 2a),36 and step emulsification devices. In addition, nozzle geometry36 as well as device materials other than PDMS33 have been investigated to further expand the utility of these devices. Moreover, external forces such as magnetic,40 and centrifugal forces41 can also be utilized in combination with the step-emulsification device to achieve droplet formation without use of any pumps. Buoyancy can effectively remove droplets from the parallelized step-emulsifier nozzle exits allowing operation with high production rates without impairing monodispersity as shown in Fig. 2b.42 Furthermore, tandem emulsification,43 a two-step process in which both step-emulsification and flow-focusing geometry with distinctive surface wettability are utilized, can also produce double emulsion droplets with high-throughput. Here, single emulsions generated from the first device are re-injected directly into the second device to circumvent the spatial control of wettability in a single device as shown in Fig. 2c.
Fig. 2.
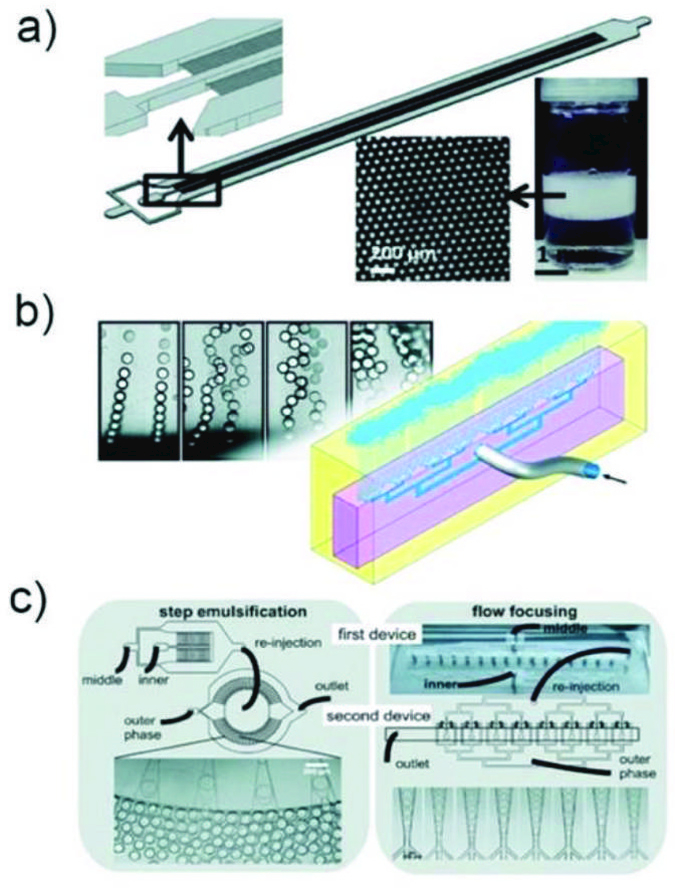
a) Schematic illustration of the millipede device36 and the drops produced. Reprinted with permission from ref. 36. Copyright 2016, The Royal Society of Chemistry. b) Droplet clearance from the nozzle exits in PDMS volcano device.42 Reprinted with permission from ref. 42. Copyright 2018, The Royal Society of Chemistry. c) Schematic illustration of tandem-emulsification.43 Reprinted with permission from ref. 43. Copyright 2017, The Royal Society of Chemistry.
2.2. Droplet microfluidic device
Fabrication of microfluidic devices with well-defined geometry and material compatibility is important for generating and manipulating droplets. Various microfluidic devices have been explored to fulfil the following requirements: material-inherent properties and compatibility with the operating fluid combinations, flexibility in tuning the channel morphologies, level of automation in the fabrication process, as well as the feasibility for large-scale commercialization. Two dominant types of droplet microfluidic devices used in generating various types of emulsion droplets are discussed. One is glass capillary microfluidics that have the advantage of high chemical resistance and ideal coaxial flow-focusing which enables preparation of droplets with wide range of material compositions and structures. The other is lithographically fabricated poly(dimethylsiloxane) (PDMS) devices that have the advantage of preparing a large number of identical devices, making them attractive for large-scale production of droplets.
2.2.1. Capillary microfluidic devices.
Capillary microfluidic devices have been widely used to form monodisperse emulsions since 2005.44 Capillary microfluidic devices do not require complicated fabrication process and are simple and highly robust, as they involve simply assembling basic modules including injection tube, transition tube, and collection tube.45 Typically, a capillary microfluidic device consists of coaxial assemblies of glass capillaries. For example, a tapered cylindrical glass capillary is carefully inserted into a square glass capillary to form water-in-oil (W/O) (Fig. 3a) or oil-in-water (O/W) single emulsion droplets. Prior to insertion, the injection cylindrical capillary surface is rendered to have a higher affinity to the continuous phase than to the dispersed phase; this prevents the adhesion or wetting of droplets on the capillary surface. The surface is rendered hydrophobic for W/O single emulsion, whereas it is rendered hydrophilic for O/W single emulsion. Flowing one fluid inside the cylindrical capillary and flowing a second fluid through the interstices between the square and the cylindrical capillary from the same direction results in a three-dimensional (3D) coaxial flow of the two fluids; this is known as the co-flow geometry. When both fluid flow rates are low, monodisperse droplets are formed at the tip of the capillary orifice in the dripping mode. If the flow rate of either fluid reaches beyond a certain critical limit, the result is a jet, which is a long stream of the inner fluid with droplets forming downstream. This typically results in droplets with a broader size distribution than that of droplets formed from dripping. Another type of geometry for droplet formation in capillary devices is the flow-focusing geometry. In contrast to co-flow capillary devices, the two fluids are introduced from the two ends of the same square capillary, from opposite directions. The inner fluid is hydrodynamically focused by the outer fluid through the narrow orifice of the tapered cylindrical capillary, enabling formation of monodisperse droplets with sizes smaller than that of the orifice.
Fig. 3.
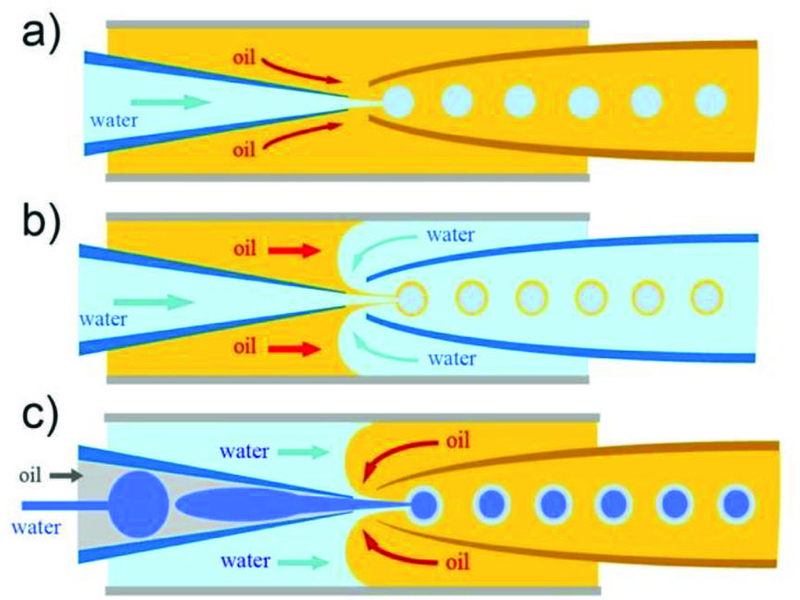
Schematic illustration of the coaxial capillary microfluidic devices for generation of a) single (W/O), b) double (W/O/W) and c) triple (W/O/W/O) emulsions.
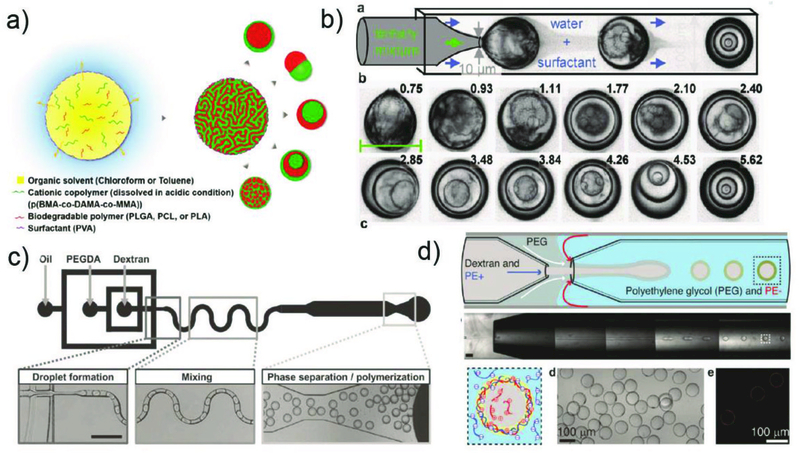
A double emulsion is one droplet within another droplet dispersed in a third immiscible phase. The high degree of control offered by glass capillary microfluidic devices enables fabrication of these droplets with a core-shell structure. The most common design combines both co-flow and flow-focusing in which the device consists of two tapered cylindrical capillaries that are inserted into opposite ends of a square capillary from the opposite direction. Similar to the single emulsion device, the glass capillaries are treated to have the desired surface wettability prior to assembly. The inner phase fluid flows through the tapered injection cylindrical capillary while the middle phase fluid, which is immiscible with the inner and outer fluids, flows through the interstices between the injection cylindrical capillary and the square capillary in the same direction, as shown in Fig. 3b. Simultaneously, the outermost phase fluid flows through the interstices between the collection cylindrical capillary and the square capillary in the opposite direction, and hydrodynamically focuses the coaxially flowing stream of the inner and middle fluids. When the three fluids enter the collection tube, double emulsions are formed. By selectively tuning the surface wettability of the cylindrical capillaries and properly choosing fluid types for these three phases, stable formation of either water-in-oil-in-water (W/O/W) (Fig. 3b) or oil-in-water-in-oil (O/W/O) double emulsions can be achieved. Furthermore, by using a modified glass capillary device, which has an injection capillary with two separate internal channels, multi-core double emulsion droplets can also be prepared.46 This structure has great potential for co-encapsulation of more than two incompatible actives or reactants without cross-contamination.
Even higher-order multiple emulsions (triple-, quadruple-), can be made through sequential co-flow emulsification by adding additional co-flow junctions composed of injection and transition tubes,45 or by combining basic building blocks (a drop maker, a connector and a liquid extractor) to form a scalable device.47 Here, surface treatment of the glass capillaries is also required to enhance controllability in droplet formation and promote droplet stability. The high droplet stability ensures accurate and independent control of droplet size and the number of the inner droplets in multiple emulsions. Furthermore, triple emulsion droplets can also be prepared by simultaneous breakup of a core-sheath stream in a single step without sequential emulsification. One major advantage of this approach is that it allows formation of ultra-thin inner shells in triple emulsion droplets,48 such as water-in-oil-in-water-in-oil (W/O/W/O) (Fig. 3c). This strategy is also used to form ultra-thin shells in double emulsion droplets (Fig. 4).49 For both cases, the shell thickness can be reduced to less than 1 µm, which is difficult to achieve through sequential emulsification. Capillary microfluidic devices can also be parallelized to some extent. A parallelized device comprising of four drop makers produces monodisperse double emulsion droplets from each device simultaneously.50
Fig. 4.
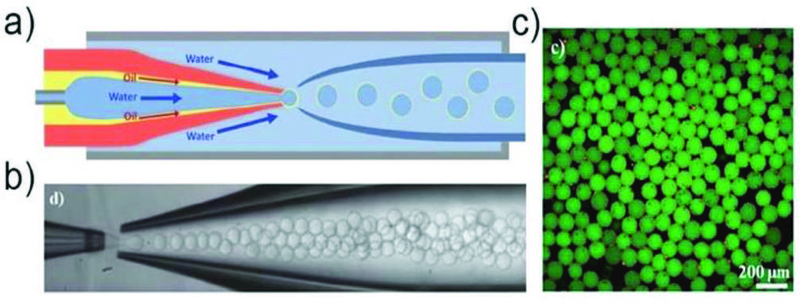
a) Schematic illustration of the microfluidic device for preparation of double-emulsion droplets with an ultra-thin shell.49 b) Optical microscope image showing the continuous generation of double emulsion in dripping mode. c) Confocal microscope images of microcapsules with a poly(lactic acid) membrane. Reprinted with permission from ref. 49. Copyright 2011, The Royal Society of Chemistry.
2.2.2. PDMS microfluidic devices.
While capillary microfluidic devices are easy to assemble and enable high precision flow control, the manual fabrication process makes it difficult to prepare more than a few devices at a time and the devices prepared often suffer from a lack of reproducibility. Thus, producing large numbers of devices with similar geometry is challenging. A widely used alternative is PDMS microfluidic devices due to the simple and reproducible device fabrication procedures. Such PDMS devices are prepared using soft lithography which enables the formation of identical devices through replica molding, fabrication of devices with micron-scale resolution, and greater flexibility in the channel design compared to capillary devices. Moreover, PDMS is optically transparent, biologically inert, permeable to gases, and low in cost of fabrication51,52
A PDMS device fabrication process involves preparing master from photomask, forming a PDMS replica, and sealing. Briefly, to fabricate three-dimensional microfluidic devices, the photomasks are firstly prepared. To create a device from the photomask, a silicon wafer is coated with a photoresist to the desired height of the microfluidic channels. The coated wafer is then heated to evaporate the solvent, and cooled to solidify the coating. Then, the photomask is placed on top of the coated wafer and the two are exposed to ultra-violet (UV) light. The light that passes through the transparent regions of the photomask is crosslinked while the uncrosslinked parts are removed by using a solvent. To mold a microfluidic device from this “master,” another polymer, PDMS is poured to form a clear, rubbery layer. Then, the imprinted side is bonded to either another block of PDMS or a glass substrate for sealing with an oxygen plasma treatment. There are also methods such as the membrane sandwich method53 and solid-object printing54 that enable preparation of PDMS device with 3D microchannels. Moreover, geometries including “T-junction”55 and “flow-focusing”56 have been developed in PDMS microfluidic devices for droplet formation. While T-junction and flow-focusing droplet makers behave quite similarly, T-junctions tend to yield more monodisperse droplets at low flow rates, due to the enhanced flow stability with only a single continuous phase inlet.57 Flow-focusing junctions yield emulsions at higher flow rates since the centered position of the dispersed phase enables dripping at higher flow speeds.
Regardless of the geometry used for droplet formation, the wetting properties of the channels are of critical importance in determining the types of emulsions that can be formed. The inherent hydrophobic nature of PDMS often limits the types of these emulsion droplets. For example, to form O/W single emulsions, the microchannel surface needs to be modified to be hydrophilic to ensure an effective wetting of the channel surface by the continuous aqueous phase. To resolve these issues, various surface modification techniques have been developed to make the PDMS surface hydrophilic. These methods include oxidation, silane treatment, acid treatment, chemical vapor deposition (CVD), Layer-by-Layer (LBL) deposition, sol-gel method and graft polymer coating.58–60 However, to form double or higher order emulsion droplets in PDMS microfluidic devices, the surface wettability needs to be spatially patterned to have subsequent continuous fluids favourably wet the microchannel to form double or even multiple emulsions using multiple flow junctions in microfluidic devices; this allows the inner droplets to be formed in one part of the device and the outer droplets in another part. The need to spatially control the wettability in PDMS devices has stimulated development of various techniques to spatially modify device surface properties.61,62 For example, a simple and rapid method to spatially pattern the surface wetting properties of PDMS microfluidic devices is through layer-by-layer deposition of polyelectrolytes using syringe-vacuum-induced segmented flow in 3D microchannels.63 This technique offers selective surface modification in microfluidic chips with multiple flow-focusing junctions, enabling production of monodisperse double- and triple- emulsion droplets.
2.2.3. Other microfluidic devices.
Other microfluidic devices based on various materials have also been explored to fulfil specific requirements for droplet formation. For example, fluoropolymer-based devices are commonly used in systems that involve chemical reactions with long-residence-time due to excellent chemical stability and moderate heat resistance. Organic reactions under strongly acidic and basic conditions can be carried out in perfluoropolyether (PFPE) devices even at elevated temperatures.64 Quantum dots can be synthesized using the polytetrafluoroethylene (PTFE)-based microfluidic device.65 However, PTFE tube is not resistant to high pressure, and the upper limit in operating temperature is only about 200 oC.
With the recent advances in 3D printing technology, 3D printed chips are gaining more attention in microsystems. The basic concept of 3D printing is additive manufacturing of materials. Specifically, 3D digital models from the computer designed by CAD software or 3D scanners are used to create a series of layers in the Z direction that is solidified layer-by-layer to form a 3D entity. Various techniques utilized in 3D printing process mainly differ by the form of the source material (liquids/powders), various sequences to achieve multiple layers (bottom-up or top-down), solidification methods (UV or heat), and the feature resolution. Among these source materials, photocurable resins are widely used due to their biocompatibility. These resins include PEG diacrylate, gelatin methacrylate, hyaluronic acid and PEGylated/functionalized alginates. Acrylonitrile has also been used to form T-junction microfluidic device for droplet production. Monodisperse aqueous droplets are produced with polydispersity index similar to those produced in analogous PDMS chips.66 Microfluidic flow-focusing devices are also made to generate aqueous droplets around 500 μm in diameter.67 Double emulsions are also made using a 3D-printed microcapillary assembly (Fig. 5a).68 These examples reveal that 3D printing allows formation of a true 3D design of channels such as tapered geometry in XYZ in a cost-effective manner. Another unique advantage of 3D printing is the automation of microfluidic kit. Microfluidic channels as well as pumps, mixers, and valves can be printed simultaneously.69 Even non-expert users can produce functional modules with 3D printing and can combine them to create their specific integrated microfluidic devices.70 However, 3D printing techniques have many remaining challenges, such as relatively low resolution, lack of suitable materials, and immature methods to modify the surface wetting properties.
Fig. 5.

a) A 3D-printed assembly of capillary device ready for use.68 Reprinted with permission from ref. 68. Copyright 2014, The Royal Society of Chemistry. b) Roll-to-roll hot embossing of microfluidic chips.75 Reprinted with permission from ref. 75. Copyright 2016, The Royal Society of Chemistry.
Microfluidic devices can also be prepared through hot-embossing,71 which is a very versatile replication method that uses high pressure and elevated temperature to transfer structures in the master to the polymer. Many thermoplastic polymers, such as polycarbonate, polymethylmethacrylate, polyethylene, copolyester, cyclic olefin copolymers, polystyrene, polythiophene, can be processed using hot-embossing to fabricate microfluidic devices.72 Advantages of hot-embossing for microfluidics include the ability to achieve very clean and precise features quickly and cost-effectively with materials that otherwise cannot be prepared using other technologies. Three modes of hot-embossing have been developed, including plate-to-plate, roll-to-plate and roll-to-roll. Among these, the roll-to-roll (R2R) process allows large-scale production of microfluidic devices in the most cost-effective manner.73,74 In a typical R2R process, a thermoplastic foil is continuously fed through a pressurized area between a heated embossing cylinder and a blank counter cylinder to generate microscale geometries with high throughput.73 Although R2R hot embossing (Fig. 5b) offers high throughput, high automation, and decreased cost compared to conventional soft-lithography, high aspect ratio structures are challenging to fabricate using this method.75
3. Fabrication of microparticles
Droplet microfluidics provides a powerful strategy for generating versatile monodisperse droplets, which can be used as templates for fabricating various microparticles with tailored physical and chemical properties. With the recent growing recognition of the necessity of producing microparticles for biomedical applications, a variety of materials and fabrication methods are being explored in microfluidically produced emulsions with complex structures. Each immiscible phase or compartment within these emulsions serves as a template that bears physical and chemical reactions either in the entirety or between the interfaces, enabling flexible synthesis of microparticles with tunable sizes and morphologies, as well as distinctive bio-related functionalities. Moreover, recent advances in utilizing aqueous two-phase systems (ATPs) in droplet microfluidics76 as well as high-order multiple emulsion drops48 enable the generation of engineered microparticles that fulfil sophisticated requirements in biomedical applications such as biocompatibility and multicomponents. A host of materials can be selected for microparticle fabrication, which is essential for achieving desired properties to meet different application purposes. These materials can be categorized into two types:16,77 first, natural macromolecules including natural polysaccharides (alginate, agarose, dextran, chitosan), proteins (gelatin, collagen, peptides) and hyaluronic acid, and second, synthetic polymers including poly(ethylene glycol), polyglycerol, poly(acrylic acid) and poly(acrylamide).
In this section, we introduce the basic principles on how microparticles are fabricated directly using monodisperse droplets as templates. These produced droplets are converted into solid microscale particles by various methods including polymerization, ionic crosslinking, solvent evaporation, etc. Each fabrication method is described in detail, followed by approaches in fabricating microparticle with various structures from simple microspheres to multi-compartmental microcapsules. Importantly, each fabrication method is not exclusive and by combining these methods, microparticles with unique properties can be achieved.
3.1. Methods for microparticle fabrication
3.1.1. Polymerization.
Polymerization is the most popular method for converting emulsion droplets to solid particles due to the versatile and efficient protocols, as well as the variety in the choice of polymerizable materials. By addition of monomers or oligomers in the dispersed phase along with a small amount of suitable initiator such as thermal or photoinitiator, emulsion droplets can be polymerized upon heating or UV irradiation. For thermally triggered polymerization, thermal initiator produces radicals which initiate and propagate the polymerization above a certain temperature. However, this strategy is less preferred as high temperature can perturb the activity of biomolecules and also cause unstable emulsion interfaces, leading to coalescence before solidification.
Photopolymerization is one of the most prevalent ways to initiate polymerization due to the fast response time, which can induce polymerization within seconds, enabling less consideration of emulsion stability. For instance, UV-polymerizable hydrogel precursor droplets dispersed in an oil carrier phase can be polymerized upon exposure to UV, resulting in biocatalyst immobilized microparticles (Fig. 6a).78 The size of the microparticles can be adjusted from tens to hundreds of micrometers by changing the flow rate of the constituent fluids. The same principle has been extensively applied to a large number of monomers and oligomers for the synthesis of hydrogel microparticles. By using W/O droplets with water-soluble monomers, hydrogel microparticles such as polyacrylamide (PAAm),79 poly(ethylene glycol) diacrylate (PEGDA)80 and gelatin methacrylate (GelMA)81 were fabricated, which possess great potential as substrates or carriers in biochemical analysis and building units for tissue engineering. In addition to photoinitiated polymerization, microparticles can also be synthesized via redox-initiated polymerization.82,83 For example, E-coli loaded PEG microparticles were fabricated without usage of UV irradiation, as the irradiation can potentially damage the encapsulated microorganisms and cells even under a short period of exposure. Thus, enhanced cell viability in microparticles was achieved (Fig. 6b).82 Also, organic polymer resin microparticles such as ethoxylated trimethylolpropane triacrylate (ETPTA), 1,6-hexanediol diacrylate (HDDA), and tripropylene glycol diacrylate (TPGDA) were prepared through use of oil droplets as templates.84–86 Furthermore, thiol–ene photopolymerization was recently exploited to fabricate advanced microcapsules with tunable encapsulation, degradation, and thermal properties.87
Fig. 6.
Various methods utilized in converting emulsion droplets to solid microparticles. a) Fabrication of microparticles via photopolymerization.78 Reprinted with permission from ref. 78. Copyright 2005, American Chemical Society. b) Synthesis of microparticles encapsulating cells via redox-initiated chemical polymerization.82 Reprinted with permission from ref. 82. Copyright 2010, John Wiley & Sons, Inc. c) Fabrication of microcapsules via freezing. Series of photographs below show the leakage of Allura Red AC food dye from the microcapsules over time.88 Reprinted with permission from ref. 88. Copyright 2011, American Chemical Society. d) Synthesis of alginate microparticles through coalescence of two separately prepared drops and subsequent physical crosslinking.90 Reprinted with permission from ref. 90. Copyright 2005, Elsevier. e) Synthesis of PLGA microparticles through solvent evaporation.93 Reprinted with permission from ref. 93. Copyright 2009, John Wiley & Sons, Inc.
3.1.2. Temperature induced gelation and freezing.
Microparticles prepared by polymerization method result in a polymeric matrix with covalent linkages which make them chemically resistant as well as mechanically durable as compared to other methods. However, radicals generated during polymerization can cause serious problems considering their biotoxicity. Solidification of droplets through temperature induced physical gelation and freezing can alleviate some of these issues. The gelation method is applicable to droplets comprising of natural biological resources including collagen, agarose, and gelatin, which can be transformed into hydrogel by simply changing the temperature.16 The freezing method relies on the fact that some hydrocarbon molecules and lipid molecules including glycerides possess melting point at or above room temperature. By operating above the melting point and collecting the generated droplets at either room temperature or in an ice bath, temperature triggered phase transition results in solid microparticles or a solid shell in microcapsules. These types of microparticles are particularly appealing as delivery vehicles due to their biocompatibility and thermoresponsive behavior. However, microcapsules prepared by freezing exhibit large pores in the shell, inducing leakage of encapsulants, thus additional treatments are required to block these pores (Fig. 6c).88,89
3.1.3. Ionic crosslinking.
One of the most studied ionic crosslinking material in droplet microfluidics is alginate hydrogel microparticles where gel precursor droplets are cross-linked by divalent ions. This is typically triggered by ionic reactions among charged polysaccharide residues and divalent cations such as calcium ions (Ca2+). However, unlike photopolymerization, incorporation of both polymer precursors and cross-linking agents in the same compartment can result in instantaneous consolidation and clogging of the microfluidic channel. Thus, delicate control over the onset of solidification is required. One method utilizes coalescence or fusing of two separately prepared droplets, each comprising precursor polymers and cross-linking agents, which mix and crosslink downstream of the microfluidic device (Fig. 6d).90 Other methods rely on diffusion of Ca2+ from the continuous phase into the dispersed aqueous phase solely containing alginate polymers.91 Nanoparticles which release divalent cations by triggered ionization can also be incorporated within the samedroplet but in an inactive state to control the onset of gelation reaction.92
3.1.4. Solvent evaporation/diffusion.
Polymers approved by the U.S. Food and Drug Administration (FDA) such as poly(lactic acid) (PLA) and poly(lactic-co-glycolic acid) (PLGA) are biocompatible and biodegradable, which make them one of the most appealing materials for drug delivery systems (DDS). However, these polymers are not applicable for polymerization or ionic cross-linking, and thus solvent evaporation or diffusion method is applied to produce microparticles. This fabrication method typically utilizes dissolution and assembly of organic compound in a volatile solvent, allowing the use of almost unlimited sets of materials in microparticles; any polymers or active materials dissolvable in a volatile solvent can be incorporated. For example, monodisperse biodegradable drug-loaded PLGA microparticles were prepared by dissolving both PLGA and drug in dichloromethane (DCM) droplets which rapidly evaporates to form solid particles. The size of the particles can be tuned by controlling the flow rate. Also, the resulting monodisperse microparticles exhibit significantly reduced burst release and slower overall rates of drug release than conventional, polydisperse microparticles with similar characters (Fig. 6e).93 In a separate work,94 PLA microparticles were generated by dissolving the polymers in ethyl acetate, an organic solvent with low toxicity. Furthermore, use of a volatile organic solvent in the middle phases of multi-phase emulsion droplets enables formation of solid shells by removal of the solvent through evaporation or diffusion into the adjacent phases. Due to the relatively slow consolidation process, this method demands a long lifetime or stability of the multiple-emulsion droplets; otherwise, undesired products can be obtained. With the stability of the emulsion droplets secured, functional materials can be readily incorporated within the shell membrane to impart stimuli-responsiveness as well as microcapsules with even sub-micron shell thickness.49
3.1.5. Phase separation/dewetting.
One remarkable feature of solvent evaporation/diffusion method is that the co-solvent or two organic compounds with different solubilities may undergo internal physical processes, such as phase separation and dewetting, as the composition changes due to solvent evaporation or mass transfer with the surroundings through the interfaces. This process often yields at least two immiscible phases, which can remodel the droplets into more complex configurations. Here, we highlight some representative examples including generation of Janus microparticles and higher-order microparticles from a homogeneous single emulsion via phase separation,95 and production of liposomes/polymersomes via dewetting.96,97
Janus particles composed of biodegradable and pH-responsive compartments have also been prepared using solvent evaporation induced phase separation (Fig. 7a).98 Briefly, emulsion droplets containing two polymers are homogeneous at the beginning, and then transform into microparticles with multiple compartments as solvent is depleted from the droplets. By tuning the polymer pairs, pH value of the continuous phase, and type of organic solvent, configuration of the microparticles can be altered into core-shell, Janus, and core-double shell structures. In addition, phase separation of two biodegradable polymers, PCL and PLGA, has also been studied in double emulsion droplets using the similar principles to result in a Janus shell, where one component forms the shell and the other component dewets on the surface.99 Phase separation can even result in higher-order multiple emulsions with onion-like configuration from a homogeneous single emulsion droplet.95,100 It was showed that a ternary mixture of ethanol, water, and an oil, diethyl phthalate (DEP), emulsified in an aqueous solution can result in droplets with multiple layers by internal phase separation (Fig. 7b). In a separate material system, higher-order emulsion droplets can be prepared from single-emulsion droplets.100
Fig. 7.
Other methods utilized in converting emulsion droplets to solid microparticles. a) Formation of microparticles with various configuration through phase separation in emulsion drops.98 Reprinted with permission from ref. 98. Copyright 2016, American Chemical Society. b) Formation of quintuple emulsion drops from single emulsion drops via phase separation.95 Reprinted with permission from ref. 95. Copyright 2014, John Wiley & Sons, Inc. c) Synthesis of dextran/PEG microparticles by utilizing ATPs.109 Reprinted with permission from ref. 109. Copyright 2012, John Wiley & Sons, Inc. d) Fabrication of microparticles through interpolymer complexation between polyelectrolytes in ATPs.113 Reprinted with permission from ref. 113. Copyright 2016, John Wiley & Sons, Inc.
Phase separation can also be used to fabricate vesicles, which are microcapsules with a membrane composed of a molecular bilayer. Depending on the composition of the bilayer membrane, they can be categorized into liposome for lipid molecules, and polymersomes for amphiphilic block-copolymers. Vesicles are promising for delivery vehicles, as well as for investigating cellular physiological activities in response to various stimuli due to their structure which resembles natural biomembrane systems. While various methods have been developed101 to prepare both liposomes and polymersomes, droplet microfluidic techniques using multi-phase emulsion droplets as templates96,97 allow fabrication of vesicles in a highly controlled manner. Briefly, W/O/W double emulsion droplets are generated with an oil shell containing either lipids or amphiphilic polymers in two solvent mixture; one is a good solvent with high volatility and the other is a poor solvent with low volatility. Evaporation and diffusion of the good solvent into the continuous phase result in the amphiphiles aligned at both inner and outer interfaces to attract and the poor solvent with low volatility to dewet. After dewetting of oil on the surface of the inner droplet, this either remains to form a local multilayer or becomes completely separated from the water core, resulting in a single bilayer vesicle. By tuning the composition of each phase of the emulsion droplets, unilamellar and multicompartmental liposomes can be prepared. In addition, multiple polymersomes, or polymersomes-in-polymersomes, can be prepared for programmed release of different ingredients.102 Furthermore, multi-functional vesicles have been fabricated by embedding hydrophobic functional materials such as quantum dots, gold, and magnetic nanoparticles in the shell for triggered release.103
3.1.6. Aqueous two-phase system (ATPs).
Typical droplet formation in microfluidic devices involves the use of an oil phase and an aqueous phase. However, droplets can also be generated in aqueous two-phase systems (ATPs) consisting of two aqueous solutions that are immiscible with each other. ATPs comprises of hydrophilic materials that are each dissolved in an aqueous phase that demix into two water-rich phases when their concentrations exceed a certain value. The phase separation occurs when the entropic driving force that favours mixing becomes less than the enthalpic penalty that opposes it.76 All aqueous environment offered by ATPs provides a mild surrounding environment compared to typical oils, enabling incorporation of active biomolecules such as proteins without potential denaturation. This makes them well suited for biomedical applications. However, the interfacial tension between the two phases in ATPs is substantially low compared to that between oil and water. Therefore, droplets are often formed at jetting regime as the viscous and inertial forces dominate during drop formation, resulting in polydisperse droplets. To overcome this challenge, various techniques have been developed, such as operation at low droplet generation rate with weak viscous and inertial forces,104 all-aqueous electrospray technique,105 applying external forces including pulse,106 mechanical vibration,107 and piezo-electricity.108 The most established ATPs system comprising of PEG and dextran has been employed in synthesizing hydrogel microparticles through mechanical piezoelectric induced generation of W/W droplets.108 The same system has also been used to synthesize microparticles with complex shape via polymerization in single-emulsion droplets (Fig. 7c).109
3.1.7. Interfacial reaction and complexation.
Many of the methods described in the previous sections rely on converting a selected compartment within an emulsion droplets to a solid. However, one of the major advantages of having more than one immiscible fluid is that one can induce reactions at the fluid interfaces. This is generally achieved by using either two reactive reagents or complementary interacting polymer pairs that are each separately dissolved in the dispersed phase and the continuous phase. This facilitates reactions or complexation that occur on droplet interfaces, enabling fabrication of microcapsules directly from single emulsions. For example, hollow metal-organic framework (MOF) microcapsules were synthesized by the interfacial reaction among the organic and inorganic precursors that were initially dissolved in two immiscible solvents.110 Guest species were also encapsulated in microcapsules under the mild reaction conditions. Similarly, core-shell chitosan microcapsules were fabricated by cross-linking with glutaraldehyde that was initially present in the inner and outer fluids in O/W/O double emulsions.111 Polyelectrolyte (PE) microcapsules have been prepared through interfacial complexation of polymers in emulsions followed by spontaneous droplet hatching112 Briefly, two complementary polyelectrolytes were each initially dissolved in the inner and middle phase of W/O/W double emulsion drops. Then, the two polymers formed a complex at the inner W/O interface of the W/O/W double emulsion followed by dewetting of the oil drop resulting in polyelectrolyte microcapsules. Polyelectrolyte microcapsules can also be synthesized by combining ATPs and interfacial complexation in W/W/W double emulsion droplets (Fig. 7d).113 These double emulsion droplets serve as a transient template while interpolymer complexation occurs between two oppositely charged polyelectrolytes.
3.1.8. Colloidal Assembly.
Emulsion droplets prepared via droplet microfluidics serve as an excellent template to geometrically confine and induce organization of colloidal particles into ordered superstructures, including colloidal clusters and 3D spherical colloidal crystals. By utilizing colloidal nanoparticles with tunable size, uniformity, and surface functionality as the building block in droplets, sacrificial templates, interfacial stabilizers, colloidal crystal beads, inverse opals, and particles with surface patterns can be synthesized in a highly controlled way for various applications. For example, magneto-responsive Janus photonic crystal particles for bioassay were synthesized by combining phase separation and self-assembly of silica and magnetic nanoparticles.114 Moreover, multi-color encoded microspheres with silica nanoparticles arrays were synthesized by combining photopolymerization and self-assembly of functional silica nanoparticles on the surface of the microparticle for multiplex immunoassays.115
3.2. Microparticle Structures
In this section, we describe how to prepare microparticles with various structures, as summarized in Fig. 8. We also focus on how these microparticles fulfill the different requisites in diverse fields of biomedical applications.
Fig. 8.
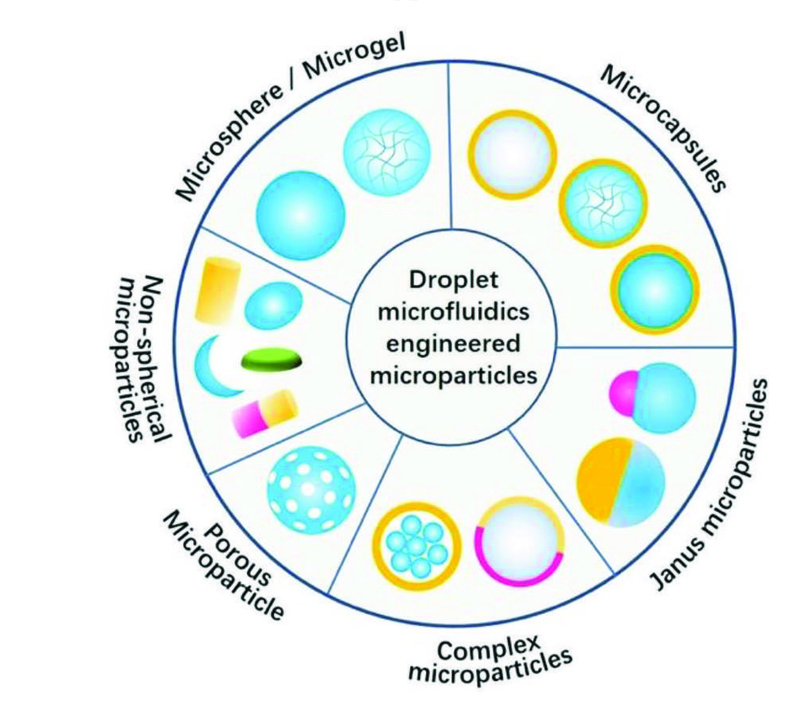
Microfluidically engineered microparticles with various structures. The gray color represents the liquid phase, while all other colors represent either solid or hydrogel phase.
3.2.1. Microspheres and microgels.
Monodisperse emulsion droplets produced via droplet microfluidics in their natural state maintain a spherical shape due to the minimization of their surface energy. In the simplest case, polymeric microspheres or microgels composed of either polymer chains or crosslinked polymer networks can be obtained by solidification of single emulsion droplets. Production of monodisperse microparticles with well-defined sizes, mechanical properties, and functionalities have an enormous impact on biomedical applications such as drug delivery and cell encapsulation. For instance, polymeric microspheres or microgels prepared via droplet microfluidics enable flexible delivery of drugs as particle size and material composition strongly affects their release profile, biodistribution, and administration route. Moreover, encapsulation of cells in microgels provide a biocompatible 3D microenvironment for living cells by protecting the cells from the surroundings while simultaneously supplying adequate amount of water, oxygen, and nutrients required to sustain the cells. Molecular architecture within the microgel has also been tuned to vary the mechanical properties of the microgels encapsulating the cells; these factors govern cell migration, proliferation, and differentiation.116,117 Cell-encapsulating microgels also serve as scaffolds for tissue engineering and implantable carriers for cell therapy.
3.2.2. Non-spherical microparticles.
Nonspherical microparticles can be prepared by adjusting the reaction conditions during the fabrication process. This includes, for example, an aqueous solution of sodium alginate flowing through an oil layer and into a reservoir containing Ca2+. Due to relatively slow sedimentation process and reaction rate, which can be tuned by changing the viscosity and Ca2+ concentration, tear droplet or tail shaped particles can be prepared (Fig. 9a).118 Other examples utilize polymerization of the deformed droplet during spatial confinement to result in microparticles with various shapes including spheres, disks, and rods.119 Furthermore, by selectively solidifying a compartment in a Janus type emulsion droplet, microparticles with complex shape can be prepared. For instance, dumbbell, acornlike, sharp-edged, or moon-crescent type microparticles were produced by tuning the relative values of the interfacial tension (Fig. 9b).120 A similar conceptual idea was applied to create microlenses with shapes from convex to planar and even concave.121
Fig. 9.
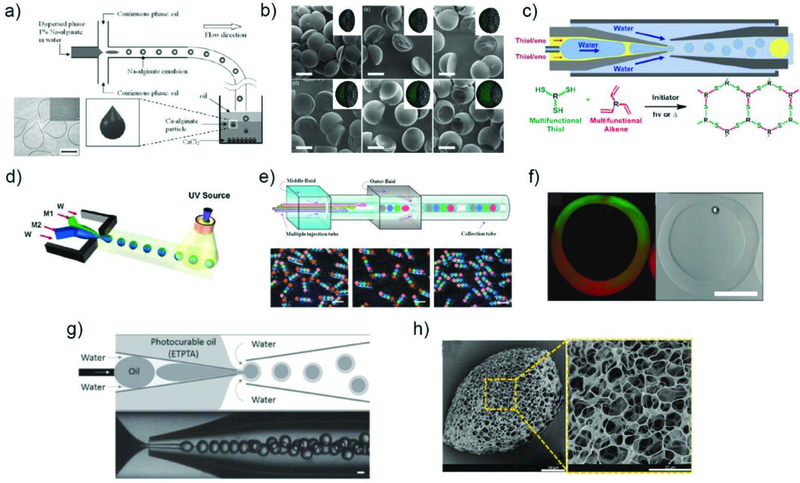
Fabrication of microparticles with various structures. Synthesis of a) teardrop or tail shaped alginate microparticles,118 Reprinted with permission from ref. 118. Copyright 2013, John Wiley & Sons, Inc. b) microparticles with complex shapes by selective solidification of a Janus type emulsion drop,120Reprinted with permission from ref. 120. Copyright 2011, American Chemical Society. c) microcapsules with tunable encapsulation, degradation, and thermal properties by exploiting thiol-ene chemistry,87 Reprinted with permission from ref. 87. Copyright 2017, American Chemical Society. d) amphiphilic Janus particles.124 Reprinted with permission from ref. 124. Copyright 2006, American Chemical Society. e) microcapsules with multiple core components by using multiple inner flows during emulsification,128 Reprinted with permission from ref. 128. Copyright 2010, Nature Publishing Group. f) microcapsules with Janus shells,129 Reprinted with permission from ref. 129. Copyright 2010, American Chemical Society. g) triple emulsion drops with an ultra-thin intermediate layer for encapsulation of hydrophobic cargo in polymeric microcapsules48(Reprinted with permission from ref. 48. Copyright 2016, John Wiley & Sons, Inc.) and h) porous microparticles by addition of tiny oil drops as porogens.132 Reprinted with permission from ref. 132. Copyright 2014, American Chemical Society.
3.2.3. Microcapsules with core-shell structure.
Core-shell microparticles, or microcapsules, are typically composed of solid, liquid, or gas bubbles surrounded by a protective shell. Due to the unique core−shell structure, such microcapsules enable highly efficient encapsulation and protection of valuable and delicate cargoes from the surrounding environment. Moreover, the high flexibility of material selection for the shell enables production of microcapsules with diverse properties and functionalities such as enhanced retention, controlled release, and stimuli-responsiveness. Therefore, they have been widely applied in the delivery of therapeutics. The ability to fabricate uniform emulsion droplets with precisely controlled size and morphology offers powerful advantages as it allows generation of microcapsules with adjustable release profiles as well as minimal possibilities of burst release, which can cause serious issues in delivery of therapeutics. While microcapsules can be prepared from homogeneous single emulsion droplets by phase separation95,100 or by merging of single emulsion droplets by wettability control,122 they are most commonly derived from double emulsion droplets using different shell solidification processes. As long as the stability of the double emulsion droplets is secured during the solidification of the shell phase, almost unlimited variety of substances can be employed as the core and the shell materials. Moreover, by creating a biphasic flow within the injection capillary and forming a sheath flow consisting of a thin layer of the middle phase fluid with high affinity to the capillary wall, double emulsion droplets with even sub-micron thick shells can be prepared.49 All these provide enormous opportunities for tailoring microcapsules for specific applications. For example, advanced microcapsules with tunable encapsulation, degradation, and thermal properties can be prepared by exploiting thiol-ene chemistry in the middle phase of double emulsion droplets (Fig. 9c).87 The details of many other examples can be found in several other papers.11,123
3.2.4. Janus microparticles.
Janus microparticles have two separate compartments with distinctive physicochemical properties which make them appealing for drug delivery, micromotors, and self-assembly. They are synthesized from either Janus droplets or homogeneous droplets. When Janus droplets serve as templates, the particles are directly synthesized through either polymerization or ionic crosslinking of the constituent phases. Various types of Janus particles have been synthesized using droplet microfluidics. Amphiphilic Janus particles with different volume fractions of the constituent phases form clusters with different aggregation numbers (Fig. 9d).124 Also, selective modification of the surface of Janus particles was employed to impart additional functionality. In another example, Janus hydrogel particles with superparamagnetic properties and chemical anisotropy were self-assembled into two-dimensional chain-like structures under an external magnetic field.125 Homogeneous emulsion droplets can also be employed to synthesize Janus particles through phase separation. For example, a pH-responsive cationic copolymer and another biodegradable polymer were dissolved in an organic solvent which went through phase separation upon evaporation.98 Thermoresponsive volume-phase transition can also induce particles with Janus structure when poly(N-isopropylacrylamide) (PNiPAM) nanoparticle suspensions containing acrylamide are heated above its transition temperature.126
3.2.5. Microparticles with complex structures (multi-core, higher-order).
The structure of microcapsules can be much more diversified by incorporating an additional compartment in the emulsion droplet, either in the core or the shell. For example, microcapsules with multiple core components were synthesized by using several separate inner flows during emulsification. These inner cores can be either separated by a solid shell or have a single Janus core within the shell after UV irradiation.127 By adding more inner flows, microcapsules with even more number of cores can be produced (Fig. 9e).128 Furthermore, microcapsules with Janus shells can also be prepared by using parallel middle phase flows during emulsification (Fig. 9f).129
Triple emulsion droplets offer even more flexibility in the selection of core and shell material as well as an effective route to achieve high encapsulation efficiency of incompatible cargos. Triple emulsion droplets with an ultra-thin intermediate layer enable highly efficient encapsulation of hydrophobic cargoes in polymeric microcapsules, directly dispersed in water (Fig. 9g).48 In addition, by replacing the ultra-thin water layer with a fluorocarbon oil layer, high loading efficiency of a broad range of polar and non-polar cargoes in a single platform was achieved.130
3.2.6. Porous microparticles.
Porous microparticles can be prepared by introducing sacrificial templates into the precursor droplet and subsequently removing them after solidification. For example, PNiPAM microparticles with tunable structures and temperature-sensitive properties were prepared by addition of PEG with different concentration and molecular weight131 or by addition of tiny oil droplets that act as porogens (Fig. 9h).132 Polystyrene (PS) microbeads were also used as a sacrificial template and selectively removed with organic solvent to create porous hydrogel microparticles.133 Other examples include using gas bubbles, either initially included134 or post-generated,135 self-assembling dendrimer-dye complex136 as porogens to synthesize monodisperse porous microspheres. In addition, phase-separation can also be employed to prepare porous microparticles.137
4. Biomedical applications
Droplet microfluidics provides a unique method for fabrication of monodisperse microparticles with control over the size, morphology, and functionality, in a high throughput manner. This technique also allows on-chip encapsulation of various bioactives, such as drugs, cells and other bio-reagents in microparticles with high efficiency (Fig. 10), and even direct co-encapsulation of multi-components.138 Moreover, enhanced retention as well as controlled release of bioactives can be achieved by adjusting the particle size, structure and composition. These advantages make microfluidically engineered microparticles beneficial for various biomedical applications including development of advanced delivery vehicles and fundamental studies of cell biology.
Fig. 10.
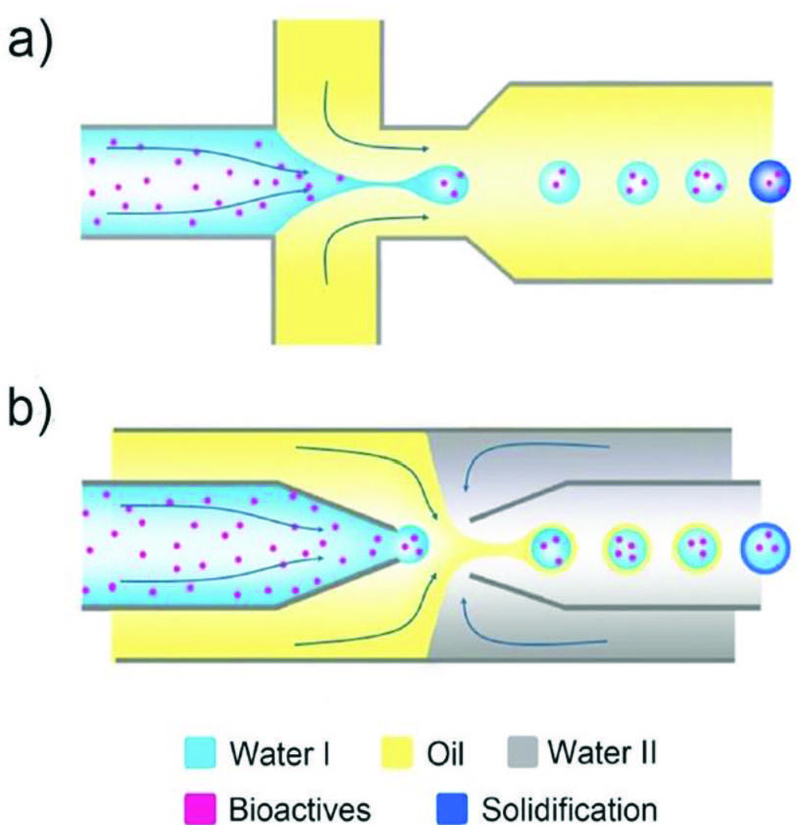
Encapsulation of bioactives, such as drugs, proteins, and cells, a) in microspheres/microgels templated with single emulsions generated from a PDMS device and b) in the liquid core of microcapsules templated with double emulsions generated from a glass capillary device.
4.1. Drug delivery
An advanced drug delivery carrier requires excellent biocompatibility, biodegradability, and high drug loading efficiency. This drug carrier also need to deliver drugs to the target site with desired release rate upon demand. The microparticles generated by droplet microfluidics allow high drug loading efficiency (>96%) through on-chip encapsulation.139, 140 Their uniformity in size, structure, and composition also guarantee a consistent drug release profile. Various biocompatible materials and fabrication methods discussed in Section 3 are applicable in producing microparticles with desired properties and functions as advanced microcarriers.141 The size of the microparticle is essential for choosing the appropriate drug administration route. Microparticles produced by microfluidics with sizes ranging from a few microns to hundreds of microns are most suitable for oral drug delivery. Particles with sizes of 10 to 250 μm can also be used for subcutaneous or intramuscular injection.2
4.1.1. Drug encapsulation.
A typical drug encapsulation process using droplet microfluidic technique is shown in Fig. 10. Briefly, a precursor solution containing drugs, such as antibodies, proteins, anticancer drugs or other therapeutics, is first emulsified to form discrete droplets. Drugs directly encapsulated inside these droplets are then solidified into microparticles. To achieve effective therapeutic delivery, drugs should be well-encapsulated to avoid side effects due to premature leakage. Besides, a delicate selection of materials and emulsion templates based on the physicochemical properties of drugs including molecular weight, polarity, and bioactivity, is necessary.
Single emulsion templated microparticles solidified from W/O or O/W emulsions can directly encapsulate either hydrophilic or hydrophobic drugs within these particles. To efficiently encapsulate drugs, it is essential to use materials that have good compatibility with the drugs. For instance, chitosan-based microspheres have been fabricated by droplet microfluidics to encapsulate hydrophilic ampicillin142 or Bovine serum albumin (BSA) drugs.143 Amphiphilic microgels fabricated from random copolymers comprising of hydrophilic acrylic acid (AA) and hydrophobic n-butyl acrylate (BuA) enable encapsulation of both hydrophobic and hydrophilic moieties.144
Microparticles with core-shell structure allow encapsulation of drugs inside the core with high loading efficiency, while the shell performs as a diffusion barrier to control the release profile by enhancing the retention of the drugs. The drug release period can be adjusted by tuning the shell thickness and the mesh size of the polymeric shell. For example, microcapsules with a semi-permeable PLGA shell were prepared.145 The shell thickness can be controlled in the range of 70–150 nm by adjusting the initial PLGA concentration to achieve sustained release of the payloads up to 3–5 months. The enhanced encapsulation can also be achieved through modification of the shell or the core of the microcapsules. By generating solid precipitates within the shell of microcapsules,88 small pores of the shell can be blocked, enabling a significant reduction of the dye leakage from 16% to 3% in 4 weeks (Fig. 11a). Microcapsules with a hydrogel core were fabricated for enhanced fragrance encapsulation and retention.146 The hydrogel core serves as a physical barrier to immobilize the individual fragrance droplets and prevents them from interacting with the shell, resulting in a substantial reduction in leakage (Fig. 11b). This is especially beneficial for long-term retention of perfume in cosmetics.
Fig. 11.
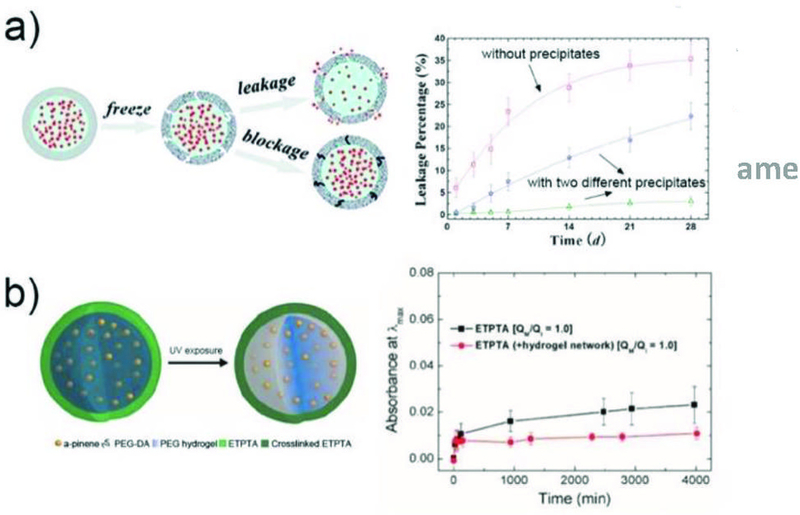
Drug encapsulation in microparticles for long term retention. a) The enhanced encapsulation of actives in self-sealing microcapsules by formation of precipitates in the capsule shells.88 Reprinted with permission from ref. 88. Copyright 2011, American Chemical Society. b) The microcapsules with a hydrogel core enhance retention of fragrant molecules.146 Reprinted with permission from ref. 146. Copyright 2016, American Chemical Society.
Microparticles with multicompartment structure enable separate coencapsulation and synergistic release of multiple drugs. This approach reduces the risk of cross-contamination between the drugs and keeps independent control over drug release, which is advantageous for combination therapy. It also ensures the correct local concentrations of both drugs, which can be essential for synergistic release. For example, microcapsules with one or multiple GelMA hydrogel cores and PLGA solid shell were prepared (Fig. 12a).147 Hydrophilic and hydrophobic drugs, doxorubicin hydrochloride (DOX) and camptothecin (CPT) were loaded into the core and the shell, respectively. These drugs can be released sequentially upon gradual degradation of the biopolymer shell. Multicompartment microcapsules with heterogeneous Ca-Alginate hydrogel shells were fabricated by using combined coextrusion microfluidic devices (Fig. 12b).148 Each compartment is covered with a distinctive shell, allowing incorporation of multiple components and independent control over their release. In dual compartmental capsules with thermoresponsive nanogels introduced into one-half of the Janus shell, the release of actives in the two compartments can be triggered by temperature and concentration gradient, respectively.
Fig. 12.
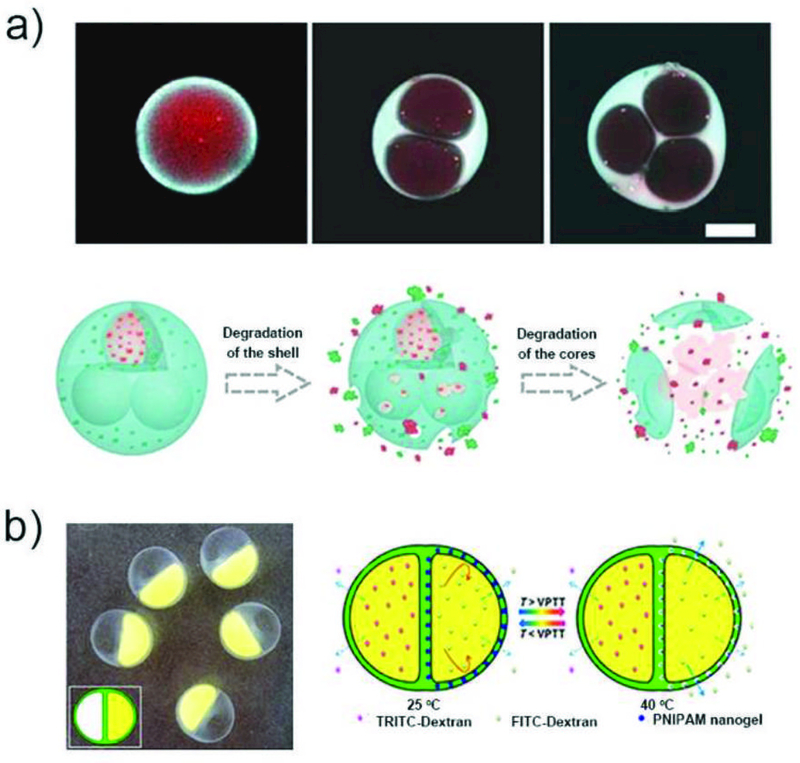
Multicompartment microparticles for coencapsulation of multidrugs. a) CLSM images of DOX (red) and CPT (blue) loaded microparticles with single or multiple cores, and illustration of sequenced fracture of the shell and the core to release the drugs.147 Reprinted with permission from ref. 147. Copyright 2017, Science China Press. b) Optical micrograph of microcapsules with dual compartments and heterogeneous shell, and illustration of microcapsules exhibiting both temperature triggered release and sustained release.148 Reprinted with permission from ref. 148. Copyright 2016, American Chemical Society.
4.1.2. Drug release.
Drugs loaded in the microparticles can be released either by diffusion through the pores of the particle matrix or by degradation of the particle matrix.141 The release of drugs can also be realized from microparticles comprised of stimuli-responsive materials, which change their physical or chemical properties upon external stimuli such as temperature,149, 150 pH,151–153 ultrasound,154, 155 or light.156–158 Different drug release profiles, including sustained release and burst release, can be achieved by tuning the properties of the microparticles. Sustained release refers to gradual release of drugs over an extended period of time. It normally entails an initial fast release followed by a slow and steady release. This release profile is desirable for many therapeutic treatments which require minimal drug dosage for maximum patient compliance to achieve long-term safe and effective drug delivery.159, 160 By contrast, burst release refers to a quick and abrupt release of drugs, resulting in a rapid increase in local drug concentration within a short period. Microparticles with stimuli-responsive properties can exhibit burst release profile, which are desirable for release of drugs at the designated site to achieve fast relief.161 It is important to develop microparticles with the desired release profile to fulfil various therapeutic requirements.
4.1.2.1. Sustained Release.
Release of drugs in microparticles through diffusion or degradation of the polymer matrix159 can be regulated by several parameters, including particle size,147 shell thickness,145 particle structure,143 matrix mesh size,162,163 or affinity of the drugs to the polymer matrix in the microparticles.132 By tuning these parameters, drugs can be gradually released from the microparticles for a predetermined period of time. In general, increase of particle size and shell thickness results in decreased release rate and prolonged duration.145,147
The mesh size of the polymer matrix in microparticles is one of the key parameters controlling the diffusion and release of drugs.164 Drugs with hydrodynamic diameter that are smaller than the mesh size can migrate freely through the polymer matrix. As the size of the drug increases, steric hindrance by the matrix becomes stronger, resulting in a slower release rate. For instance, fluorescein sodium salt (Mw = 376 Da) encapsulated inside a protein-based microcapsules is released 60% in 1 h, because of its smaller size compared to the mesh size of the microcapsule membrane.165 By contrast, large FITC-bovine serum albumin (BSA) (Mw = 68,000 Da) almost does not leak within the same period. For a microcapsule membrane composed of polyelectrolytes,113 the release time is extended significantly, from 10 to 1000 min, when the molecular weight of neutral dextran molecules increases from 5 to 40 kDa. The interaction between the drug and polymer matrix also influences the release rate, apart from the mesh size. When a negatively charged protein with a size similar to the 40-kDa dextran is encapsulated in the polyelectrolyte microparticles, only ~20 % is released after 600 min. This large suppression in release is a result of the enhanced electrostatic interaction between the protein and the polyelectrolyte membrane. A similar phenomenon is also observed when using nanofibril microgels composed of amyloid proteins as microcarriers:166 dyes which have a strong affinity to amyloid gels are not fully released after 1 week, whereas other dyes or drugs reach their maximum release rate after 1 hr.
The rate of sustained release can also be tuned by altering the microparticle structure. Chitosan microspheres with three different types of structures, including solid, porous, and core-shell structures, were fabricated from W/O emulsions by controlling the solidification time. Different release profiles of BSA were demonstrated due to the difference in mesh size and structure,143 as shown in Fig. 13a. Introduction of a shell on a microparticle has proven to be an efficient strategy for suppressing drug release. Four model drugs with different solubility and lipophilicity were encapsulated in porous silicon microparticles covered with a solid lipid membrane shell.167 The release rate of these drugs from these particles was always slower than that of the bare silica microparticles in all pH conditions tested (1.2, 5.0 and 7.4). In another example, polyacrylamide (PAM)/poly(ethylene glycol) (PEG) microcapsules with multicore structures were prepared from hydrogel/water/oil (H/W/O) emulsions, which exhibit different protein release rate.79 The release of BSA in the first hour from a single-core microparticle is slower than that from a quadruple-core microparticle.
Fig. 13.
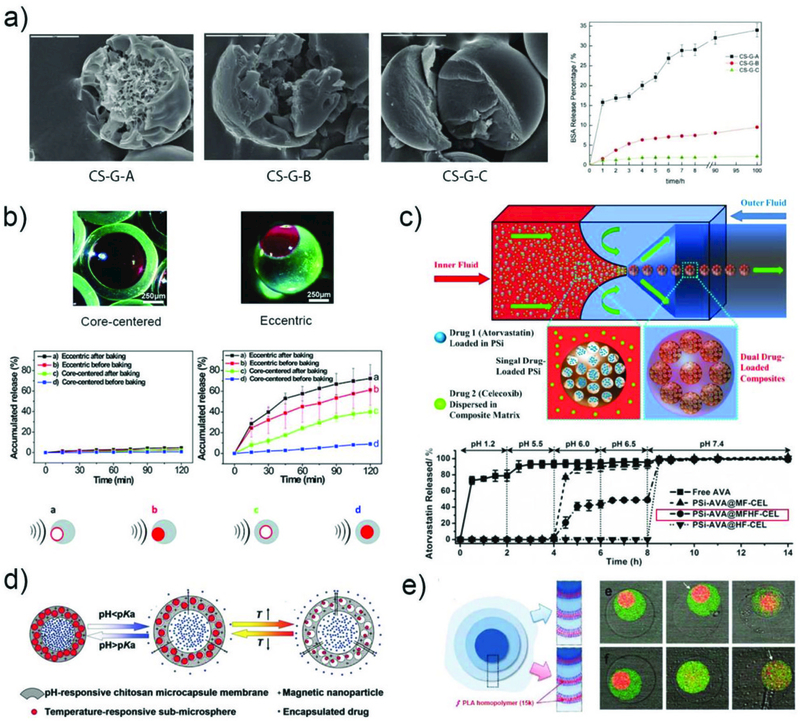
The sustained release of drugs. a) SEM images of chitosan microspheres with three different structures and their in vitro BSA release profiles.143 Reprinted with permission from ref. 143. Copyright 2012, John Wiley & Sons, Inc. b) Optical images of the eccentric and core-centered internal structures, and release profiles of rhodamine 6G from the four types of microcapsules with and without ultrasound.168 Reprinted with permission from ref. 168. Copyright 2014, The Royal Society of Chemistry. c) The fabrication of multi-drugs loaded polymer/porous silicon (PSi) composite microparticles for multi-stage release of AVA drugs in different pH conditions.169 Reprinted with permission from ref. 169. Copyright 2014, John Wiley & Sons, Inc. d) Schematic illustration of the multi-stimuli-responsive microcapsules with adjustable controlled-release.173 Reprinted with permission from ref. 173. Copyright 2014, John Wiley & Sons, Inc. e) Schematic illustration of a triple polymersome showing bilayers with no internal homopolymer on the top right and two of the bilayers containing homopolymer on the bottom right. Series of confocal images showing the sequential dissociation of these two kinds of membranes in the mixture of water and ethanol.102 Reprinted with permission from ref. 102. Copyright 2011, American Chemical Society.
Sustained release can also be achieved in microparticles composed of responsive materials by applying external stimuli. For example, the release time scale can be adjusted by introducing ultrasound as an external stimulus which can change the mesh size or destroy the particle matrix. Microcapsules of PDMS with eccentric and core-centered internal hollow structures were produced from W/O/W droplets, and their release profile was regulated by ultrasound. For both microcapsules, rhodamine 6G was only very slightly released in the absence of ultrasound whereas it was continuously released from the microcapsules in the presence of ultrasound. The eccentric microcapsules with thinner walls exhibited higher release rate than core-centered microcapsules, as shown in Fig. 13b.168
Microparticles with pH responsive property show great potential for targeted drug delivery due to the variation of pH in the human body, such as the acidic condition in the stomach and the basic condition in parts of the intestine and colon. A series of composite microparticles were microfluidically engineered and exhibited pH triggered sustained release profile. The utility of these particles as advanced oral drug co-delivery vehicles for combination anticancer therapy was demonstrated.169, 170 For example, multi-drug loaded polymer/porous silicon (PSi) composite microparticles were fabricated from O/W droplets in a flow-focusing capillary device (Fig. 13c).169 Two drugs atorvastatin (AVA) and celecoxib (CEL) were selected due to their synergetic effect for cancer therapy. The AVA was loaded inside PSi, which were then encapsulated together with the CEL into microparticles composed of two pH-responsive polymers. These composite microparticles showed multi-stage pH-responsive behavior. They maintained their structural integrity at pH from 1.2 to 5.5, whereas they started to dissolve between pH 6.0 and 6.5 and released 50% of the drugs in 2 hrs. Above pH 6.5, the structure completely collapsed to release all of the remaining drugs in half an hour. Upon incubation of these microparticles with colorectal cancer cells, cell viability decreases to less than 50% under pH 6.5 while 90% cell viability is observed under pH 5.5 after 24 hrs. By encapsulating multi-drug loaded silicon nanoparticles and magnetic nanowires into the same polymeric microspheres, pH-responsive microspheres with long term sustained release profile and magnetic targeting properties was achieved.171 These microspheres can protect the payloads under acidic conditions and release them gradually under basic conditions, making them superior carriers for intestinal delivery of therapeutics. Core-shell alginate microcapsules encapsulating Vitamin A and gelatin were also prepared for intestine-targeted drug delivery. The alginate microcapsule remains intact in an acidic environment while it degrades in alkaline environment to release the encapsulated gelatin.172
Microparticles with multi-stimuli responsive properties have been prepared to achieve enhanced control over drug release. For instance, responsive microcapsules with adjustable mesh size in the shell were fabricated from W/O/W emulsions.173 The shell of these microcapsules is composed of crosslinked pH-responsive chitosan membrane with embedded magnetic nanoparticles and thermoresponsive nanospheres as shown in Fig. 13d. When the local pH is lower than the pKa of chitosan, the membrane swells, resulting in a high drug release rate. The release rate can be further tuned by varying the interspace distance between the nanosphere in the capsule membrane. This is achieved by the temperature regulated volume change of the nanospheres. In addition, due to the magnetic nanoparticles, microcapsules can be directed to the desired site by an external magnetic field.
The programmable release of multiple components can be achieved through preparation of microparticles with complex structures; this includes microparticles with several shell layers and additional compartments. For instance, polymersome-in-polymersomes were fabricated from multiple-stage W/O/W double emulsions, providing convenient strategies for encapsulating multiple actives into different membrane layers.102 The programmed rupture of the membrane triggered by mechanical strain or osmotic pressure enables sequential release of the payloads (Fig. 13e).
Microparticles prepared from biodegradable materials have also been demonstrated to show sustained drug release in vivo. Microcapsules with biodegradable thin shells of PLGA exhibit continuous release of encapsulated bioactives in times ranging from two to five months, at different pH conditions (Fig. 14).145 When these microparticles are introduced into mouse dorsum through subcutaneous injection, the encapsulated drugs are observable even after one month, while the free drugs dissolved in the tissue are barely detected after one day. Acetalated dextran (AcDX)-based biodegradable microspheres were prepared by droplet microfluidics for spinal cord injury treatment.174 These monodisperse microspheres have a diameter of 7.2 μm and exhibit gradual degradation at pH 7.4 during a period of one month. After intrathecally injecting into injured rats, they not only effectively suppress the astrogliosis and inflammation in the injured spinal cord, but also protect the neurons from glutamate-induced excitotoxicity. This reveals the potential application of these AcDX microspheres for severe neurological diseases treatment.
Fig. 14.
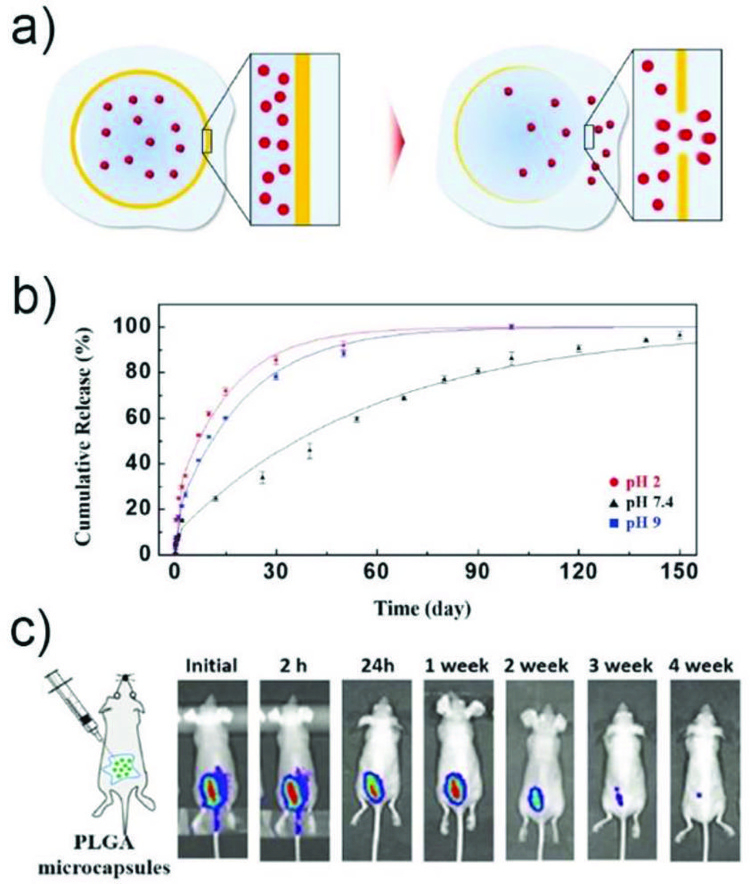
a) Schemetic illustration of release of encapsulants through a hole formed at the thinnest part of a PLGA membrane by degradation.145 b) Cumulative release of sulforhodamine B from the microcapsules at different pH. c) In vivo fluorescence images for ICG-loaded PLGA microcapsules that are subcutaneously injected into dorsum of mice. Reprinted with permission from ref. 145. Copyright 2017, John Wiley & Sons, Inc.
4.1.2.2. Burst Release.
Microparticles for burst release are typically composed of stimuli-responsive materials, which can respond to external stimuli and abruptly change or rupture their structures followed by ejection of the drugs.
Temperature is one of the most widely used stimuli for triggering a fast response. The temperature alteration can induce the microparticles to either melt or change the volume to achieve burst release. For microcapsules with a frozen shell, increasing the ambient temperature above the melting point of the shell material can trigger the solid shell to melt in few seconds and abruptly release the payloads. For instance, microcapsules with fatty acid glyceride shell remain stable at room temperature for at least six months with no observable leakage. However, when heated above 37 °C, the solid shell turns into liquid in 30 sec and encapsulated actives are completely released after 5 mins.59 Microparticles composed of PNiPAM, a thermoresponsive polymer with a lower critical solution temperature (LCST) about 32oC, can collapse or swell by tuning the temperature above or below the LCST. Thermoresponsive microcapsules made from PNiPAM have been fabricated using double-emulsion templates.175,176 They release actives two ways: 1) The actives are encapsulated at low temperature, and when the temperature is raised above the LCST, the particles quickly shrink and rupture due to the sudden increase in internal pressure,175 2) The actives are sealed in the core at high temperature and when the temperature is lowered below the LCST, the shell sharply swells to enlarge the mesh size, which releases the actives.176 Block copolymers of PNiPAM122 and nanogels177 have also been used as composites in the microcapsule shells. Increasing the temperature results in enlarged mesh size due to shrinkage of the PNiPAM nanogels, and therefore release of the encapsulated actives.
Microparticles composed of pH responsive polymers have also been explored for burst release. A large pH variation dramatically changes the polymer solubility, resulting in change of mesh size or even decomposition of the particle matrix. Monodisperse microcapsules composed of a crosslinked chitosan shell and a core containing fluorescent dyes are sealed at neutral pH. At acidic conditions (pH~3), the chitosan shell decomposes rapidly and releases the dyes completely within a few minutes.111 To achieve more effective acute gastrosis therapy, different chitosan based pH-responsive microcapsules can be fabricated containing both free drugs and drug-loaded PLGA nanoparticles for programmed sequential release of drugs.178 In an acidic environment, microcapsules release both the free drugs and the PLGA nanoparticles rapidly within 60 sec due to the decomposition of the chitosan shell. This results in sufficient drug dose for immediate relief of patient symptoms after drug administration (Fig. 15a). Then the drugs inside the PLGA nanoparticles are slowly released over 2 days to maintain the therapeutic effect and reduce complications.
Fig. 15.
The burst release of drugs. a) The CLSM snapshots showing the decomposition of chitosan shell to release the encapsulated free RhB drugs and the RhB-PLGA nanoparticles rapidly in an acidic environment.178 Reprinted with permission from ref. 178. Copyright 2016, American Chemical Society. b) The schematic illustration of osmotic pressure triggered release of encapsulated enzymes.179 Reprinted with permission from ref. 179. Copyright 2017, John Wiley & Sons, Inc. c) The images of PFC-alginate microcapsules before and after ultrasound exposure.180 Reprinted with permission from ref. 180. Copyright 2014, American Chemical Society. d) The fluorescent micrographs showing the triggered release of FITC-dextran cargo from hydrated microcapsules under ultraviolet light.185 The release profile of FITC-dextran from the microcapsules as a function of the rehydration time. Reprinted with permission from ref. 185. Copyright 2016, American Chemical Society. e) The enzyme-triggered release of protein-based microcapsules and the release profile of the FITC-BSA.165 Reprinted with permission from ref. 165. Copyright 2014, John Wiley & Sons, Inc. f) The microcapsules showing both pH and ionic strength triggered release. The fluorescent images of PAA/bPEI microcapsules containing FITC-dextran showing pH-triggered release, the optical images showing the salt-triggered deformation of the microcapsules and the encapsulated FITC-dextran molecules release profile in NaCl solutions.112 Reprinted with permission from ref. 112. Copyright 2015, American Chemical Society. g) Temperature triggered release of the upper-oriented inner core when the magnet is in the bottom.187 Reprinted with permission from ref. 187. Copyright 2014, The Royal Society of Chemistry.
Osmotic shock is induced by a sudden change in solute concentration around the microparticles, causing a rapid change in the movement of solvent across its shell membrane. This particular stimulus has been widely applied for the burst release of actives. Polymersomes are prepared from self-assembly of amphiphilic diblock copolymers (PEG-b-PLA) by using multiple-stage W/O/W droplets. Their shells are semi-permeable membranes with relatively low mechanical properties and high-water diffusivity, enabling easy rupture of the shell under osmotic pressure to release the payloads.96, 103 Microcapsules with ultra-thin polymeric shell have also been utilized to achieve osmotic pressure triggered burst release of enzymes. These microparticles fabricated from W/O/W double emulsions have a shell with thickness ranging from tens to hundreds of nanometers. Enzymes protected by these thin shells maintain a much prolonged activity compared to free enzymes, while over 90% of the encapsulated enzymes can be released when triggered by osmotic shock (Fig. 15b).179
Ultrasound and light are attractive stimuli for triggering drug release from microparticles due to their remote manipulation capability and non-invasiveness. For example, alginate core-perfluorocarbon-oil-shell microcapsules180 can maintain their integrity after injection into a polyacrylamide gel that mimics the tissue matrix. However, upon exposure to ultrasound for 15 min, the particles can be disrupted, demonstrating their potential for ultrasound-based therapies (Fig. 15c). Light is another easily applied stimulus. In particular, near-infrared (NIR) can penetrate deeply into tissue and is barely absorbed by the hemoglobin and body water, making it an attractive external stimulus for in vivo drug release.181, 182 Microcapsules exhibiting NIR-sensitive properties are normally comprised of temperature responsive polymer materials183 and NIR-absorbing materials such as gold nanorods or nanoparticles. For example, microcapsules with an ethyl cellulose shell containing PNiPAM nanogels and gold nanorods were fabricated.177 After irradiation with NIR laser, the gold nanomaterials absorb energy and generate localized heat. The increase of the local temperature induces shrinkage of the PNiPAM nanogels and formation of macrogaps, resulting in enhanced release of encapsulants. Microcapsules composed of PLGA and gold nanomaterials can also be ruptured under NIR illumination since PLGA melts at temperature above its Tg (40–60 oC).184 In addition, UV-responsive supramolecular microcapsules were formed through interface host-guest interaction between amphiphilic polymer micelles containing azobenzene groups and cucurbit[8]uril (CB[8]).185 Upon exposure to UV for 3 min, the trans-to-cis photoisomerization of azobenzene groups induces a facile increase of the microcapsule porosity, releasing 84% of the payloads after 18 min (Fig. 15d).
Other stimuli such as enzymes and ions are attractive in achieving fast responses in biological environments. For example, protein based microcapsules can be degraded when a digestive enzyme is introduced. This induces the degradation of shell layer into small pieces due to the cleavage of amide bonds, leading to a total release of the encapsulants in 40 min (Fig. 15e).165 Microcapsules with a shell membrane composed of 15-crown-5, NiPAM, and AAm-based copolymer can recognize K+ ion to form a sandwich complex and induce phase transition of PNiPAM. The capsules shrink and rupture rapidly, leading to the ejection of all the encapsulated lipophilic actives in the cores.186
Microparticles with multi-responsive properties have also been developed for burst release. Polyelectrolyte microcapsules were fabricated through interfacial complexation of two oppositely charged polymers at the inner (W/O) interface of W/O/W double emulsions.112 These microcapsules show both pH and ionic strength responsive behavior (Fig. 15f). The change of pH from 5 to 7 reduces the interaction between the two polymers causing drastic swelling of the microcapsules in less than 13 min. Increasing the ionic strength of the solution from 1 to 100 mM also induces sudden deformation of the particles and subsequent release of the encapsulants. Through incorporation of magnetic nanoparticles in the shell, microcapsules can respond to a magnetic field for targeted drug release. In particular, microparticles containing magnetic particles with an asymmetric shape can burst release drugs at the target site. Microcapsules with a magnetically eccentric core-shell structure were fabricated from O/W/O double emulsions through a combination of gravitational and magnetic forces.187 The microparticles are composed of a PNiPAM shell with an off-center core and magnetic nanoparticles dispersed in the shell, on the opposite side of the core from the nanoparticles. The off-centered core induces heterogeneity in the shell thickness: the shell near the core is thinner while the side far from the core is thicker, as shown in Fig. 15g. When the temperature is raised above the LCST of PNiPAM, the shell breaks at the thinner side and eject the core. With the presence of magnetic nanoparticles, drugs can be released at a desired location with a specific orientation by applying an external magnetic field.
4.2. Cell-laden matrix
Cells in the body exist in a 3D microenvironment comprising of proteins and polysaccharides, which defined as the extracellular matrix (ECM).188 The properties of ECM strongly influence cellular phenotype and function. Biocompatible hydrogel microparticles with sizes ranging from 60 to 200 μm are promising platforms for in vitro cell culture, as they perform as 3D matrices that can mimic various aspects of the ECM.16, 96 Their properties such as stiffness and porosity can be tuned by altering the gel components. In addition, the intrinsic porous structure and the high surface-to-volume ratio of hydrogel microparticles facilitate exchange of oxygen, nutrients and waste, while protecting the cells from the surrounding environment and retaining cell viability. These advantages lead to emergence of a large number of biomedical applications related to cell-laden hydrogel microparticles including cell life study,77,189 drug screening and discovery,190,191 and tissue engineering.17,192
Although several techniques have been exploited for generating cell-laden microparticles, such as bioprinting,193 micromolding,10 and soft-lithography,194 droplet microfluidics is often the most suitable choice as it allows continuous production of monodisperse hydrogel microparticles in a high-throughput manner. More importantly, microfluidics allows control over both the number of cells encapsulated in each gel particle, and the overall encapsulation efficiency. In this section, we introduce recent advances in cell encapsulation and culture in hydrogel microparticles generated by droplet microfluidics. We also discuss the application of cell-laden microparticles in cell-fate studies, tissue engineering, and single cell analysis.
4.2.1. Cell encapsulation and culture.
In a typical cell-encapsulation process, a hydrogel precursor solution containing cells is emulsified in a continuous oil phase to form discrete droplets on-chip, followed by crosslinking to result in cell-embedded hydrogel microparticles as shown in Fig. 10. The cells can also adhere to the surface of microparticles after their formation with the advantages of fast cell proliferation and easy collection. However, this strategy only allows cells cultured on a curved surface instead of a 3D matrix inside the microparticles. The most common examples of engineered cell-laden micromatrix include microgels with uniform spherical structure, and core-shell microcapsules with either a liquid or a hydrogel core. Only a few examples of cell-laden microparticles with Janus structure have been addressed.195,196 Overall, the number, type, and distribution of cells can be custom designed, leading to versatile 3D compartments within these hydrogel microparticles as shown in Fig. 16.
Fig. 16.
Schematic illustration of cells distributed in different positions of the hydrogel microparticles.
4.2.1.1. Cell encapsulation and culture in microgels.
In the simplest case, cell-laden microgels can be prepared by encapsulating cells in W/O emulsions, followed by gelation. The size, shape, porosity, and mechanical properties of the microgels can be tuned by varying the channel size, flow rate, polymer concentration, and crosslinking density. All of these factors significantly affect the cell fate. Therefore, deliberate consideration of materials selection, crosslinking method, and fabrication conditions is required for production of cell-laden microgels. In general, high biocompatibility of precursors, low precursor concentration, and fast gelation kinetics under mild condition are key requirements for the formation of microgels that resemble ECM. Moreover, they need to be immediately transferred into an aqueous solution such as cell culture medium after microgel formation to retain high cell viability.197
Many natural polymers including alginate,198 gelatin,199 collagen,200 agarose,116 and peptide201 have been investigated for cell-laden microgels due to their biocompatibility and degradability. By virtue of their fast solidification kinetics, alginate-calcium microgels have been extensively studied for cell encapsulation and culture. For instance, encapsulated breast tumor cells can form multicellular spheroids in alginate microbeads.52 However, the rapid ionic crosslinking between alginate and calcium ions often results in heterogeneous microgels. To resolve this issue, a method that utilizes water-soluble calcium-ethylenediaminetetraacetic acid (Ca-EDTA) complex as a crosslinking precursor has been developed (Fig. 17a).202 This method allows homogenous distribution of calcium ions within the generated alginate droplets. The dissociation of this complex is triggered by pH reduction, leading to gelation of the droplets with high homogeneity. Human mesenchymal stem cells (MSCs) were encapsulated and cultured inside these homogenous microgels for 15 days and stable cell growth and proliferation were observed. It was also reported that different shapes of cell aggregates can be produced by adjusting the concentration of alginate and CaCl2.203 Using this approach, human cervical carcinoma, human hepatocellular liver carcinoma and human umbilical vein endothelial cell aggregates with spherical, spindle- and branch-like shapes are successfully obtained in a controllable manner. Cell viability assay shows that more than 95% of the retrieved cells retain their proliferation ability. In addition, different types of cells can be embedded in alginate microbeads for co-culture. For example, equal amounts of HeLa and HEK293 cells were embedded in the alginate microbeads, and formed heterologous 3D spheroids after 3 days culture.204
Fig. 17.
Cell encapsulation and culture in microgels. a) Formation of homogeneously crosslinked alginate microparticles by on-demand release of calcium ions from a water-soluble calcium–EDTA complex. Cell viability is determined to be 70% after 15 days culture. Microscopic images showing the stable growth and proliferation of cells.202 Reprinted with permission from ref. 202. Copyright 2015, John Wiley & Sons, Inc. b) Encapsulation of cells and proteins in PEG-4MAL microgels by using a flow-focusing microfluidic chip through a cytocompatible crosslinking reaction. Viability of cells was imaged and quantified, indicating human islets maintain high viability after culture for 8 days in microgels.206 Reprinted with permission from ref. 206. Copyright 2014, John Wiley & Sons, Inc.
Synthetic polymers can be chemically modified, unlike natural polymers, and thus have advantages for producing microgels with tunable properties by utilizing cytocompatible reactions. These polymeric materials are often incorporated in cell-laden microgels by free radical photoinitiated polymerization, among which PEG-DA has been most widely used. However, as this reaction involves use of free radicals which have cytotoxic effects on cells, different methods have been developed to achieve cytocompatibility. By combining the droplet microfluidic technique with bio-orthogonal thiol−ene click reactions, cell-laden microgels were fabricated without involvement of free radicals in the system. Cell-laden microgels were prepared by Michael addition of dithiolated PEG macro-cross-linkers to acrylated hyperbranched polyglycerol (hPG) building blocks.205 By varying the PEG molecular weight, microgels with different properties can be achieved, which affects the viability of encapsulated cells. Yeast cells in the PEG6.0 kDa matrix show higher viability compared to those in the PEG1.5 kDa matrix, due to the high diffusivity of PEG6.0 kDa microgel that facilitates the metabolism of encapsulated cells. Moreover, yeast cells encapsulated in PEG6.0 kDa microgels are able to proliferate extensively, forming huge colonies of cells. Cell-laden 4-arm PEG maleimide microgels, functionalized with cell adhesive peptides were reported, and are also crosslinked by Michael addition reaction in the presence of small molecule dithiothreitol (DTT) (Fig. 17b).206 Human pancreatic islets encapsulated in microgels retained viability of about 90% after 8 days of culture. This approach was shown to have almost no detrimental effect on islet cells function. PEG norbornene (PEGNB)-based droplets encapsulating cells were generated in a flow focusing microfluidic device. These cell-laden droplets were collected and then exposed to UV light in bulk solution to induce step-growth photopolymerization which is not inhibited by oxygen.207 The higher cytocompatible of PEGNB over PEG-DA has been demonstrated by higher viability of cells in PEGNB microgels compared to PEG-DA microgels after long-term encapsulation.
Uniform microgels formed through reversible crosslinking have also been developed. This allows the cells to not only be encapsulated but also to be released afterwards, which benefits subsequent operations and examination for deeper understanding of the cells. Importantly, all these advantages can be achieved without any detrimental effect on their properties and functions. Cell-laden microgels with pH-cleavable properties are fabricated through azide–alkyne cycloaddition using droplet microfluidics.208 Here, PEG- dicyclooctyne and dendritic poly(glycerol azide) serve as bioinert hydrogel precursors. Azide conjugation was performed using different substituted acid-labile benzacetal linkers that allowed precise control of the microgel degradation kinetics in the pH range from 4.5 to 7.4; pH-controlled release of the encapsulated cells was achieved upon demand with minimum effect on cell viability and spreading. Reversibly cross-linked microgels from linear PEG precursor polymers that carry bipyridine moieties on both chain ends were also prepared.209
The gelation was induced by complexation of iron(II) ions with the polymers. By using this method, the encapsulated mammalian cells retained 90% cell viability. The microgels were degradable by addition of competitive ligands under very mild conditions with negligible effect on cell viability of the encapsulated as well as the released cells.
Besides using a single component, hybrid microgels can also be prepared by combination of multiple components to achieve tunable and optimized properties for cell encapsulation and culture. For example, collagen-gelatin composite microbeads were fabricated. By varying the composition of the precursor droplets, microgels exhibited tailored mechanical properties in the range of 1–10 kPa, comparable to human tissue.210 Enzymatically crosslinked dextran-tyramine conjugates have also been explored for on-chip microgel formation.211 When encapsulated in these microgels, MSCs demonstrated higher viability (95%) compared to alginate (81%) or PEG-DA (69%) microgels. Hydrogel microbeads embedded with Human neonatal dermal fibroblasts (HDFn) were formed from peptide-star PEG conjugates and oligosaccharides through noncovalent assembly. Cell viability as high as 98% was maintained even after 7 days of continuous culture.201 Additional functionalities can be further afforded through both covalent and non-covalent interactions by using cell attachment sequence or biotinylated peptide.
4.2.1.2. Cell encapsulation and culture in microcapsules.
Although microgels with uniform structure are suitable carriers for cell behavior study in vitro, their simple structure limits their ability to mimic the 3D physical microenvironment. More sophisticated structures are thus required for complex cell culture study, such as cell–cell interaction study with controllable spatial distribution of different cells in a 3D cellular microenvironment.
Microcapsules templated from W/O/W double emulsions have also been generated for cell encapsulation, and can be categorized into two types: matrix-core/shell and liquid-core/shell microcapsules. Incorporating cells in the core of the microcapsules enhances their resistance against external effects, such as enzymatic attack and UV irradiation, and prevents them from egressing. It is also critical to rapidly transfer the crosslinked microcapsules into liquid phase to avoid any detrimental effects on the cells. Cell-laden PEG-DA and alginate/Ca2+ microcapsules were fabricated by utilizing droplets with an ultra-thin shell as templates in a capillary microfluidic device. Due to its large surface area, the thin oil shell spontaneously dewets upon crosslinking of the innermost precursor droplet, allowing direct dispersion of the microgels in the aqueous phase.212 Moreover, this thin-shell rupture approach prevents long-term exposure of cells to oil and surfactants, leading to biocompatible cell encapsulation.
Microcapsules with hydrogel-core/shell can better restrict the cells from egressing, and thus allow long-term cell culture studies. Encapsulating cell-laden PEG microbeads within a non-degradable gel shell can prevent cell escape, allowing the study of cell behavior in different microenvironments during 2 weeks of culture.213 For instance, the proliferation of mouse embryonic stem cells (ESC) in microcapsules with different elasticities was studied. Cells in stiff microgels showed a lower proliferation rate and lower cell viability than in soft microgels after 4 days of culture (Fig. 18a). Microcapsules were prepared in a T-junction microfluidic device by electrostatic self-assembly of peptide amphiphiles (E3-PA and K3-PA).214 The stiffness of these capsules with a fibrillar structure can be tuned by changing the peptide concentration. Human dermal fibroblasts were encapsulated within these capsules and their morphologies were studied. In 0.5 wt% E3-PA capsules, fibroblasts exhibited extended protrusions that interacted with the fibrillar matrix, and adopted a spindle shape with fine filopodia after 7 days. After 14 days, the spread cells elongated and formed a 3D network. On the contrary, cells cultured in 1 and 2 wt% E3-PA capsules always maintained a round shape. Further study also proved that keratinocytes can adhere to the peptide capsule surface and be cocultured with fibroblasts within the capsules.
Fig. 18.
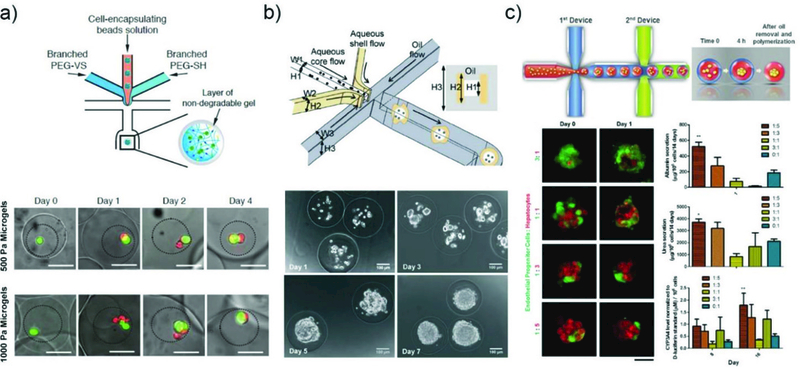
Cell encapsulation and culture in microcapsules.a) Microfluidic approach used for coencapsulation of cell containing bead-in-a-bead. Images showing the growth of ESCs encapsulated in soft and stiff microgels at different time points.213 GFP marks Oct4 expression of the ESC colonies, and the dead cells exhibit red fluorescence. Reprinted with permission from ref. 213. Copyright 2015, John Wiley & Sons, Inc. b) Generation of microcapsules by using a non-planar (3D) microfluidic flow-focusing device.219 Phase contrast images of ES cells encapsulated in the pre-hatching embryo-like microcapsules after different number of days, showing proliferation of the cells to form a single aggregate. Reprinted with permission from ref. 219. Copyright 2013, The Royal Society of Chemistry. c) The process of generating microencapsulated hepatocyte spheroid using double emulsion droplet generated by two connected microfluidic devices.220 Tracking of cell organization in the composite spheroids at different co-culture ratios. Functional assessments of hepatocyte with different ratio of EPC to hepatocyte. Reprinted with permission from ref. 220. Copyright 2016, John Wiley & Sons, Inc.
Microcapsules with solid barcode particle core and GelMA hydrogel shell which are applicable for cell culture and drug screening were also prepared.215 The microgel shell layer serves as a 3D ECM microenvironment for enhanced cell adhesion and culture. The photonic crystal cores of the barcode particles provide stable diffraction peaks that can encode different cell aggregates during culture and distinguish their biological response during drug testing. Different kinds of cells (HepG2, HCT-116 and NIH-3T3) were loaded on the barcode particles and cocultured as liver and tumor cell spheroids to test the cytotoxic effect of tegafur (TF). The cytotoxicity of TF against the HCT-116 tumor cell spheroids was enhanced in the multiple cell coculture system, revealing the effectiveness of this cell spheroids and barcodes platform for drug screening.
Microcapsules with liquid-core/shell structure allow encapsulated cells to form cell aggregates in the liquid core due to enhanced cell-cell interactions. This strategy is of special value for stem cell studies. For example, the comparison between P19 EC cells cultured in alginate microbeads and liquid-core/shell microcapsules shows that the latter allows formation of single spherical embryoid body (EB) cells within 2 days.216 In contrast, cells in the alginate microbeads only form bumpy shapes from several clusters of cells. Liquid-core/alginate shell microcapsules were also adopted for human MSCs culture. The cells can rapidly form stem cell spheroids with controllable size (30–80 μm) in 150 mins and can be retrieved via a droplet-releasing agent.217 Further study proved that additional RGD-modification of the microcapsules enhanced the hMSCs osteogenic differentiation. Furthermore, liquid-core/shell microcapsules are also beneficial for pluripotent stem cells with high differentiation and self-renewal ability; culturing pluripotent stem cells in an aqueous environment with enhanced cell-cell interaction is helpful in forming 3D cell aggregates and better maintaining their stemness.218 Similar liquid-core/alginate shell structure was also applied to encapsulate mouse embryonic stem (ES) cells, which mimics miniaturized 3D architecture of pre-hatching embryos (Fig. 18b).219 In each microcapsule, the cells proliferated and formed multiple small aggregates within 3 days, and further merged into a single aggregate on day 5–7 depending on the number of cells initially encapsulated in the core.
Hepatocytes are liver epithelial cells which dedifferentiate rapidly in vitro, resulting in low level of hepatic function. They require 3D culture systems with enhanced cell-cell interaction or co-culturing with feeder cells to rescue or enhance the liver-specific function. Microcapsules with a liquid core and polymer shell have also been applied for hepatocytes co-culture. For example, alginate or alginate/collagen double emulsion droplets encapsulating primary hepatocytes were produced using two connected microfluidic flow-focusing PDMS devices (Fig. 18c).220 These droplets promote cell assembly within the liquid core in 4 hrs to form the spheroids. Microgels with single spheroids were subsequently generated upon oil removal and polymerization of the inner alginate phase. Hepatocyte functions can be further enhanced when mixed with endothelial progenitor cells at an optimal ratio of 5:1 to form co-cultured spheroids in the microgel. Microcapsules with a liquid core and PEG hydrogel shell were also used for trapping primary hepatocytes and assemble the cells into spheroids.221 The hydrogel shell protects cells from shear associated with suspension cultures. High levels of hepatic function were maintained for two weeks. Furthermore, microencapsulated hepatocytes were cocultured within a 3T3 fibroblast monolayer shell, without mixing the two kinds of cells to further enhance hepatic function. This microencapsulated-spheroid formation method has high yield, versatility, and uniformity, showing great potential in liver-tissue engineering.
4.2.2. Tissue engineering and regenerative medicine.
Tissue engineering and regenerative medicine are related to the generation of biological tissue replacements for transplant and tissue culture.17, 222 These can be used for patients who suffer from tissue damage or disease. Tissues are integrated 3D structures of multiple types of cells and ECMs. The function of a tissue is typically governed by multiple cues, such as intercellular signaling and cell interactions with the surrounding ECMs. Cell-laden microgel “modules” carrying different types of cells can be combined or reconfigured to mimic various types of tissues. These microgels generated from microfluidics serve as building blocks for construction of tissue engineering scaffolds. Their applications in tissue engineering, including organ-on-a-chip, bone/cartilage regeneration, stem cell culture and therapy have been extensively studied.17
4.2.2.1. 3D organ/tumor models.
Organs such as the liver consist of multiple cells, which are arranged in a 3D scaffold. To mimic organs or tissues with improved functionality, 3D tissue models that have a spatial distribution of multiple different types of cells in the ECMs are required. Microgels encapsulating different types of cells are thus promising for building in vitro organ models for artificial tissue applications.
A 3D liver model was produced through controlled assembly of heterotypic cells in a 3D core-shell hydrogel microparticle.223 Microcapsules with a liquid core and an alginate hydrogel shell were generated by using a flow-focusing microfluidic device. Different types of cells were spatially distributed within the microcapsules (Fig. 19a). The hierarchical assembly of hepatocytes in the core and fibroblasts in the shell resulted in the formation of hetero-cellular spheroids. The high permeability of the alginate shell allows long-term culture (>10 days) of the spheroids to form microtissues with dense cell aggregates in the microcapsules. The core-shell structure allows co-culture of hepatocytes and fibroblasts spatially separated, which is beneficial for the expression of liver-specific functions. A large number of monodisperse microtissues, each in every microcapsule, were achieved.
Fig. 19.
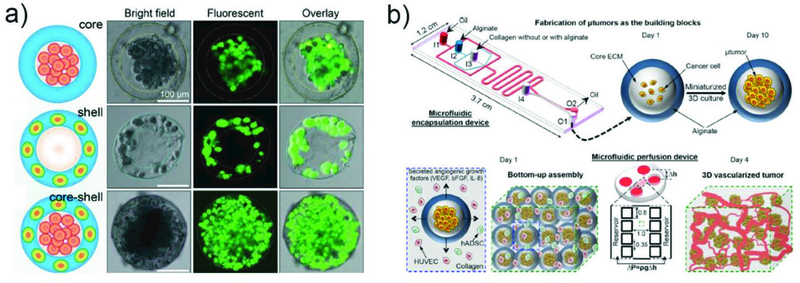
a) Spatial assembly of different cells in the 3D core–shell scaffold, including HepG2 cells confined in the core by the hydrogel shell, NIH-3T3 fibroblasts immobilized by the crosslinked alginate network in the shell, and simultaneous assembly of hepatocytes in the core and fibroblasts in the shell, forming an artificial liver in a droplet.223 Reprinted with permission from ref. 223. Copyright 2016, The Royal Society of Chemistry. b) A non-planar microfluidic device is used for encapsulating cancer cells in microcapsules, and cells cultured in the microcapsules for 10 days to form microtumors.191 The microtumors in microcapsules are assembled together with human umbilical vein endothelial cells (HUVECs) and human adipose-derived stem cells (hADSCs) in collagen hydrogel by using microfluidic perfusion device. Reprinted with permission from ref.191. Copyright 2017, American Chemical Society.
Development of high-fidelity 3D models to recapitulate the tumor microenvironment is essential for high-throughput cancer drug screening and drug discovery assays. A 3D vascularized human tumor with controlled formation of a complex 3D vascular network is produced through a “bottom-up” approach (Fig. 19b).191 Microcapsules consisting of a fibrous collagen core enclosed in a semipermeable alginate hydrogel shell were generated. MCF-7 human mammary cancer cells were encapsulated in the core for miniaturized 3D culture, which can gradually develop into avascular microtumors after prolonged culture. The microtumors were then used as building blocks and assembled with endothelial cells and other stromal cells to form macroscale 3D vascularized tumor. Moreover, in vitro study illustrated that such 3D tumors are more resistant to free anticancer drugs compared to conventional 3D avascular microtumors and 2D-cultured cells, but can be effectively killed by nanoparticle encapsulated drugs, demonstrating its value in anticancer drug discovery.
4.2.2.2. Injectable Scaffolds.
The small size of the hydrogel microparticles is particularly attractive for injectable cell delivery systems in regenerative medicine, as it allows direct delivery of cells through needles to the damaged tissue area.224 This minimizes surgical invasiveness and thus is beneficial in practical clinical applications. Gelatin-silica monodisperse microparticles were fabricated for in vitro cell culture scaffolds and injectable tissue constructs.81 The gelatin microgels were first prepared and cardiac side population (CSP) cells adhered on the surface of these microgels, which showed high proliferation, migration and spreading onto their cell-conductive surrounding. A thin layer of degradable silica hydrogel was then coated on the surface of these cell-laden microgels. This layer serves as an effective protection layer against oxidative stress which is detrimental to the cells during and after injection and implantation. Simultaneously, it keeps the migratory and proliferative capability of the CSP cells. In a separate work, bacterial cellulose (BC) microspheres with a hollow structure were fabricated by using agarose microgels as the template to form an injectable scaffold (Fig. 20a).225 Through assembling these hollow microspheres, a robust scaffold with high porosity was achieved. Cells loaded in this porous scaffold exhibit enhanced proliferation after culturing for 2 or 4 days. Furthermore, these hollow microspheres with cells have been used as an injectable scaffold in vivo for wound healing; this was tested by using a male Sprague Dawley rat skin wound-healing model. These scaffolds promoted significant and rapid wound closure due to high porosity and the ability to retain culture medium (Fig. 20b).
Fig. 20.
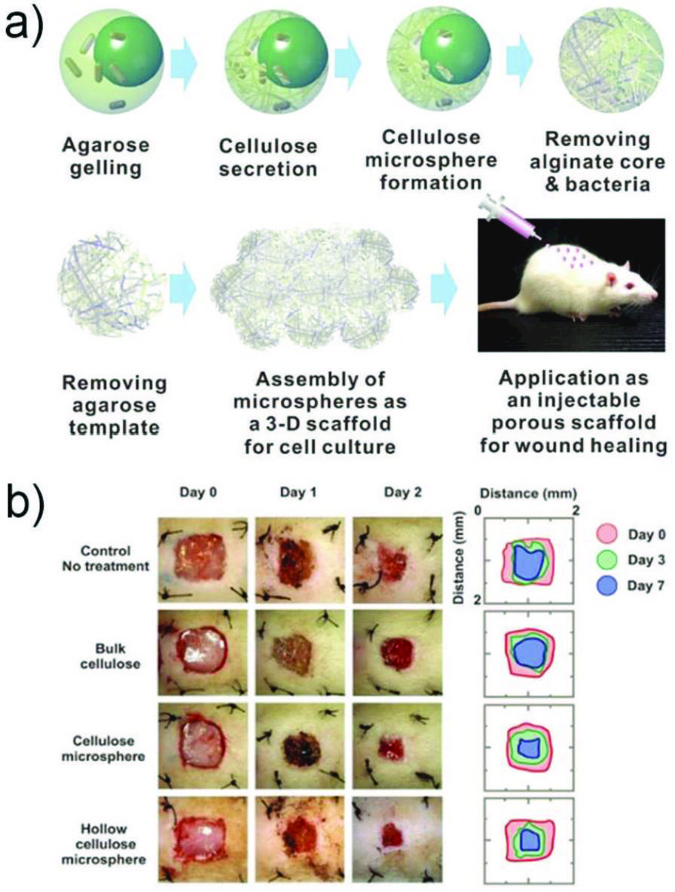
a) Schematic illustration of the steps to produce hollow bacterial cellulose microspheres.225 This includes gelling, cellulose secretion, purification, and application of the microsphere as a cell culture scaffold in vitro and an injectable scaffold for wound healing in vivo. b) Representative images of wound closure in an in vivo epidermal wound-healing model in male Sprague Dawley rats and the traces of wound-bed closure for the different treatments. Reprinted with permission from ref. 225. Copyright 2016, John Wiley & Sons, Inc.
4.2.2.3. Stem cell therapy.
Stem cells have gained considerable attention in tissue restoration and regenerative medicine due to their inherent capability of renewing and differentiating into specialized cell types.226 Microgels prepared via microfluidics show great promise for stem cell research,18 as stem cells can be spatially paired with suitable environmental factors to modulate their viability, growth, proliferation and differentiation.
Among stem cells, bone marrow-derived mesenchymal stem cells (BMSCs) have been widely used for bone regeneration owing to their osteogenic differentiation capability, low immunogenicity, and high proliferation ability.227 GelMA microgels encapsulating BMSCs and growth factors were produced as injectable osteogenic tissue constructs (Fig. 21a).228 GelMA microgels were able to sustain stem cell viability, support cell spreading inside the microspheres, and enhance cell proliferation (Fig. 21b). Moreover, an osteogenic growth factor, bone morphogenic protein-2 (BMP-2), was co-encapsulated within these microgels. BMSCs laden GelMA microgels, with or without BMP-2, were implanted into a rabbit femoral defect for in vivo bone formation evaluation. It was found that microgels with both BMSCs and BMP-2 showed synergistic therapeutic efficacy on bone formation (Fig. 21c). This work reveals that BMSCs laden GelMA microgels exhibit enhanced osteogenesis both in vitro and in vivo, making them a promising engineered injectable tissue construct for regenerative medicine. Instead of GelMA microgels that are generated under UV exposure, gelatin-based visible light curable microgels were produced by thiol-ene reaction between gelatin norbornene (GelNB) and a PEG-dithiol cross-linker.199 This reaction enables rapid in situ microencapsulation of human BMSCs under biocompatible conditions and the cells can maintain high viability (91±2%) after 7 days of culture. Moreover, the encapsulated human BMSCs exhibit rapid cell migration and considerable improvement in chondrogenic differentiation. Thus, these injectable microgels hold great promise as therapeutic candidates for articular cartilage regeneration.
Fig. 21.
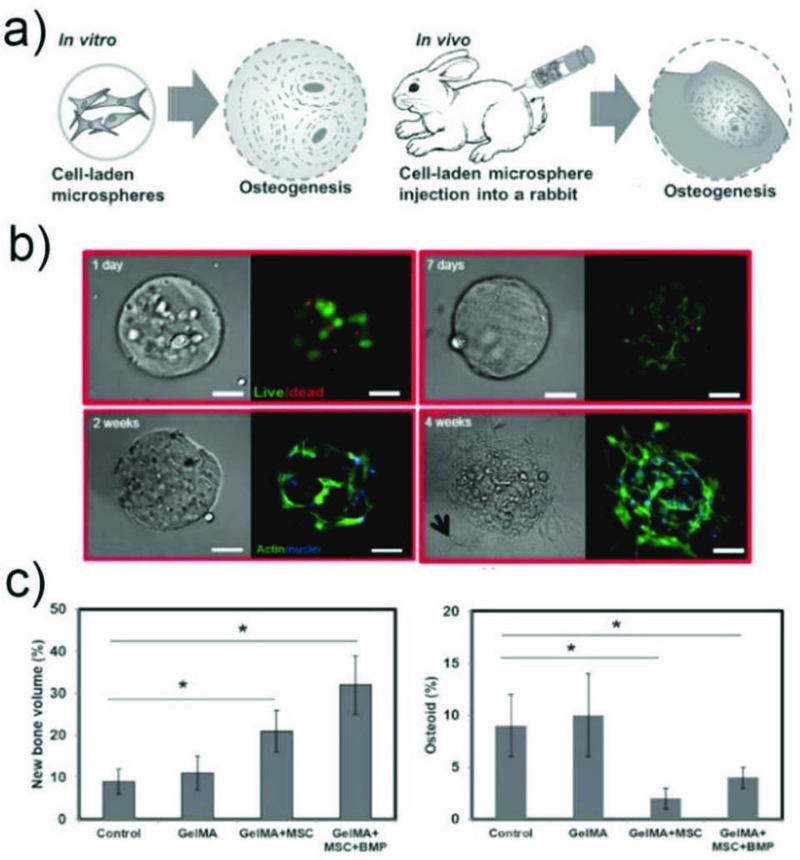
a) Schematic illustration of the application of BMSC-laden GelMA microspheres for osteogenesis and regeneration of injured bones in vitro and in vivo.228 b) Viability of BMSCs encapsulated in GelMA after 1 and 7 d of culture. Phalloidin/DAPI images of BMSCs cultured in GelMA after 2 and 4 weeks. c) Bone defect repair in vivo. Histomorphometrical analysis (%) of new bone formation and (E) osteoid (arrows) formation and total area in the defect zone (* p < 0.05). Reprinted with permission from ref. 228. Copyright 2016, John Wiley & Sons, Inc.
4.2.3. Single cell study.
Single cell study enables detection of cellular heterogeneity between large cell populations at the genomic and proteomic levels.229–231 The results have a significant impact on basic biology, cancer, immunology, and stem cell research. Droplet microfluidics provides an efficient method to encapsulate single cells in monodisperse microdroplets, allowing high-throughput analysis and manipulation of single cells, and thus highly sensitive assays.232,233 Currently, most of the microfluidic single cell studies have focused on studying a cell in a droplet. More sophisticated studies are enabled by encapsulation of cells in microgels. This allows transfer of microgels into an aqueous culture medium for prolonged cell culture. Here, we focus on the strategy of encapsulating and culturing single cells in microgels. We will also introduce the most recent progress on single-cell-laden microgels for analysis.
4.2.3.1. Single cell encapsulation and culture.
Ensuring that single cells are encapsulated in each microgel can be accomplished by using the Poisson distribution. A highly diluted cell solution containing polymer is injected into a microfluidic device to create monodisperse droplets that are then solidified to obtain single cell-laden microniches. Encapsulation of single cells in a microgel with controllable properties enable studying the influence of local microenvironment on single cells. For instance, single prostate cells encapsulated in a Matrigel microbead, without any interaction with neighbouring cells, were able to proliferate and differentiate into an acinus within 6 days.234 Hyaluronic acid microbeads containing cell binding site-fibrinogen were prepared through mild biorthogonal thiol-ene click chemistry for culture of single human MSCs.235 This was the first example for long term culture of human MSCs up to 4 weeks and the cells displayed round morphology independent of mechanical property. Also, these microbeads supported the preferential differentiation of human MSCs into adipocytes after 14 days of culture.
These examples have demonstrated the potential of using microfluidics for generating single-cell-laden microgels. However, since they all follow Poisson encapsulation statistics, the majority of the microniches generated are empty. To purify the cell-laden microniches, post-encapsulation sorting is required. To overcome this problem, single cells can be encapsulated in a thin layer of alginate-calcium microgel by using a cross-junction microfluidic device (Fig. 22a).236 Cells pre-coated with calcium carbonate were delivered together with an aqueous solution of alginate polymer. Acetic acid dissolved in the oil phase mediated calcium release and resulted in the formation of microgels. The fraction of microgels containing mMSCs and pre-adipocyte cells (OP9s) dramatically increased by a factor of ten in comparison to direct injection without pre-coating. The encapsulation efficiencies were over 90% and the cells exhibited high in vitro cell viability over 3 days. Furthermore, increased differentiation of the cells was observed in singly encapsulated mMSCs compared to larger microgels encapsulating multiple mMSCs due to the higher ratio of cell to hydrogel matrix in singly encapsulated cells. In addition, in vivo study proved that intravenous injection of singly encapsulated mMSCs into mice delayed clearance kinetics and sustained donor-derived soluble factors.
Fig. 22.
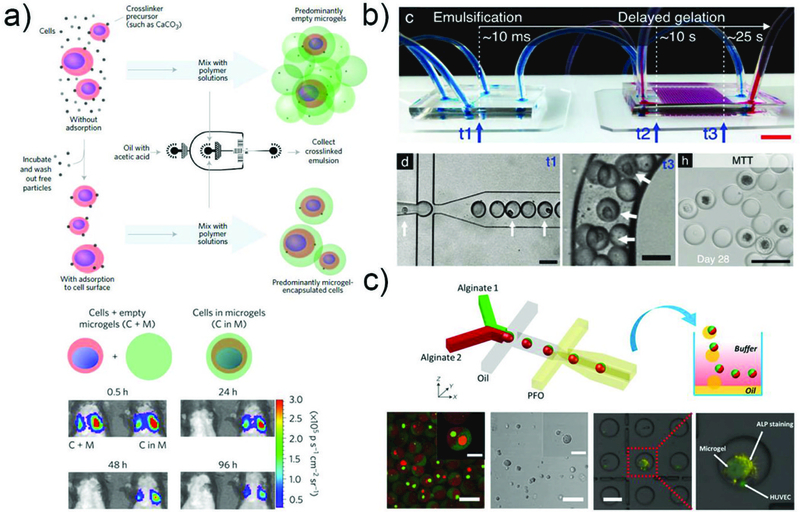
Single cell encapsulation and culture. a) Schematic showing the steps in encapsulation of single cells in thin layers of alginate gel. Representative bioluminescence images showing the biodistribution of mMSCs overexpressing Firefly luciferase with or without microgel encapsulation after in vivo injection.236 Reprinted with permission from ref 236. Copyright 2017, Nature Publishing Group. b) A standard microfluidic droplet generator was connected to the H2O2 diffusion-based crosslinking chip.238 The position of cells in microgel precursor droplets was analyzed immediately after droplet generation (t1), and at the end of the crosslinking chip (t3). MSCs encapsulated in delayed enzymatically crosslinked microgels remained viable and metabolically active throughout 28 d of in vitro culture. Reprinted with permission from ref. 238. Copyright 2017, John Wiley & Sons, Inc. c) Schematic illustration of PDMS microfluidic device for the production of Janus microgels.195 Each microgel contains two different cells labelled using red and green cell trackers, respectively, in adjacent compartments. The positive ALP staining assay results indicate that the presence of HUVEC favour the differentiation of MSC towards osteogenesis. Reprinted with permission from ref. 195. Copyright 2018, John Wiley & Sons, Inc.
Another problem for single cell study is that cells egress from the microniches during subsequent cell culture. Strategies to center cells inside the microgels have been developed to retain long term cell culture. .Orbital shaking at 1000 rpm during the gelation step at 37°C allows positioning of the cells in the center of the microniches after creation in a microfluidic device with two consecutive cross junctions.237 This strategy almost fully prevents the encapsulated cells from escape, and 79 ± 17% and 83 ± 2% of cells are viable after 7 days culture within 5% and 7.5% (w/v) polymer microniches, respectively. Another strategy for centering cells was designed by using a PDMS microfluidic chip consisting of three equally-long and high parallel channels. This enables delayed enzymatic crosslinking of tyramine-conjugated hydrogel precursor microdroplets and resulted in almost perfectly centered single cells in the microgels (Fig. 22b).238 Only 4 ± 1% cells escaped after 7 days in these centered microconstructs. The prevention of cell escape enabled long-term (28 d) culture of the MSCs and differentiation of MSCs in less than 5-µm-thick 3D hydrogel coatings. Single cell analysis revealed high cell viability (>90%).
Apart from encapsulating single cells in microgels, multiple single cells can be encapsulated inside microgels in distinct compartments for the cell-cell interaction study at a single cell level. The interactions between these cells are important in regulating cell behavior by biochemical signaling cues from neighbouring cells. A one-step microfluidic approach for preparing multi-compartment microgels was developed, and a cell-cell interaction study was performed using a Janus microgel.195 By encapsulating both single human MSCs and single HUVEC cells in a Janus microgel, positive ALP activity, which represents the osteogenesis of MSCs, was only observed when co-cultured with HUVECs (Fig.22c). This biocompatible microfluidic approach can be applied for controlled cell delivery in cell immobilization and targeted reorganization, and for directed bottom-up assembly of hierarchical tissue-like structures.
4.2.3.2. Single cell modular bioinks.
Single-cell-laden microgels have great potential as modular bioinks for 3D biomaterial fabrication. Engineering of multifunctional tissues by a modular approach using bioink that comprises of single-cell-laden microgels in an injectable macrogel was reported.239 The single-cell-laden PEG-DA microgels (35 µm) were first produced by using a microfluidic flow-focusing device with a small nozzle size. After purification with fluorescence-activated cell sorting (FACS), the single-cell-laden microgels were incorporated into multiple distinct injectable macrogel precursors to effectively create several modular bioinks. These include macroconstructs of PEGDA created using photolithography and dextran-tyramine conjugates created using emulsification. They are used for the biofabrication of various 3D constructs with an independently controllable micro- and macroenvironment.
4.2.3.3. Single cell screening.
Single cell screening is very important in cell biology and therapeutic applications. To perform a single cell immunoassay on a heterogeneous population of cells, alginate microgels were used as permeable cell culture chambers for screening single cells secreting antigen-specific antibodies (Fig. 23a).240 A co-flow microfluidic device was used to generate alginate droplets, which encapsulated single antibody-secreting cells and high-molecular-weight capturing complexes. These droplets were used to capture the secreted antibodies, followed by solidification to form microgels. The alginate microgel had a nanometer-sized porous structure which allows easy removal of the unbound fluorescent antigens, enhancing the detection efficiency, while the cells and macromolecules were retained within the particle. This approach was employed to screen antibodies against TNFα antigen produced by single cells.
Fig. 23.
a) Schematic illustration of the heterogeneous immunoassay in alginate microparticles.240 Reprinted with permission from ref. 240. Copyright 2014, The Royal Society of Chemistry. b) Analytical procedure for single-cell forensic STR typing, including encapsulation of single cells and DNA isolation, STR target amplification and STR products analysis.243 Reprinted with permission from ref. 243. Copyright 2014, American Chemical Society. c) Platform for DNA barcoding thousands of Cells.244 Cells are encapsulated into droplets with lysis buffer, reverse-transcription mix, and hydrogel microspheres carrying barcoded primers. After encapsulation, primers are released. cDNA in each droplet is tagged with a barcode during reverse transcription. Droplets are then broken and material from all cells is linearly amplified before sequencing. Reprinted with permission from ref. 244. Copyright 2015, Elsevier.
More complex structures have also been used for high throughput molecular screening. For example, microparticles composed of a polyelectrolyte shell embedded with an agarose bead have been developed.241 The core contains transformed enzyme-producing E.Coli, cell lysate and an enzyme-activity-sensing mixture for enzyme screening. The active enzyme triggers fluorescent signals, enabling it to be distinguished from the other, non-active enzymes. Since the inner core is gelled after the enzyme reaction, when it is cooled to 4oC, the enzyme, the encoding plasmid, and the florescence signals, can all be analysed well. After FACS, the polymeric shell was removed by increasing the pH above 12. Then, cells with the highest fluorescent intensity were separated and subsequently, plasmids from fluorescent cells were collected for further evolution or genomic analysis. Several rounds of selection can be carried out sequentially to obtain the best enzymatic activity.
4.2.3.4. Single cell sequencing.
Mapping the gene expression at the single-cell level is very important for observing heterogeneity within cell populations. Droplet microfluidics is a powerful technology for quantitative in-depth single cell sequencing, as it allows simultaneous processing of a large number of cells.
In most of the studies, droplets containing cells and hydrogel beads are used to prepare single cells for sequencing. Only a few examples use hydrogel microparticles for single cell sequencing. An agarose gel platform was developed by using droplet microfluidics for high-throughput single-cell genetic detection and sequencing in 2011.242 The same platform was used for multiplex short tandem repeat (STR) typing at the single-cell level.243 The procedures are shown in Fig. 23b. Briefly, single cells are first encapsulated with primer-functionalized beads in nanoliter agarose droplets using a microfluidic droplet generator. Then the gelled droplets are incubated in cell lysis buffer to release genomic DNA. Small reagents can diffuse into the interior of the porous microgels, while large DNA remains within the microgels; thus, multiplex STR amplification can be realized within the nanoliter reactors. The microgels also allow mechanical manipulation and long-term storage of the microdroplets.
Polymer hydrogel microspheres generated by droplet microfluidics are also used as barcode beads for single cell sequencing. For example, a high-throughput inDrop (indexing droplets) droplet microfluidic approach was developed for indexing thousands of individual cells for RNA sequencing (Fig. 23c).244 A library of barcoded polyacrylamide hydrogel microspheres containing acrydite-modified DNA primer is prepared by droplet microfluidics. Then, the barcoded beads are prepared by split-pool synthesis for combinatorial addition of the barcodes. Each bead carries 109 covalently coupled, photo-releasable primers encoding one of 147,456 barcodes. Because the beads are deformable, they can be loaded into drops with high precision, with over 90% of the droplets containing exactly one bead. Cells are added at high dilution, ensuring that only very few cells are drops with more than a single cell. This allows a barcoded library of the genomic material from individual cells to be prepared. InDrop sequencing was evaluated by analyzing mMSCs, revealing in detail the structure of the population and the heterogeneous onset of differentiation after a leukemia inhibitory factor was withdrawn. This method has become widely used for other sequencing-based assays.
4.3. Biosensors
Biosensors are devices comprised of biological recognition elements and signal transduction elements for quantitative and semi-quantitative analysis.245, 246 An ideal biosensor can detect analytes, such as glucose, enzymes, DNA, and antibodies in a rapid, efficient, and convenient manner. They are increasingly in demand for many biomedical applications from fundamental biological studies to clinical diagnostics. Microparticles containing sensing components prepared from droplet microfluidics have been exploited for sensing applications.247
One of the most widely used microparticles for biosensors are PEG-based hydrogel microparticles by virtue of their biocompatibility and antifouling properties. Single emulsion droplets coupled with UV polymerization were used to produce PEG-based microgels for sensing glucose concentrations (Fig. 24a).248 The functional TRITC-CoA-dextran-FITC complex was directly integrated into the microgels. TRITC-ConA is a sugar binding protein that can bind to dextran thereby quenching the fluorescent signal of FITC-dextran. In the presence of glucose, higher binding affinity of glucose to ConA than dextran results in the release of FITC-dextran from TRITC-ConA, and increases the signal proportional to the amount of glucose. Alternatively, functional sensing elements can also be introduced into microparticles through post-functionalization to further enrich their applicability for biosensing. For example, PEG microspheres containing either carboxylates or primary amines were fabricated by droplet microfluidics (Fig. 24b).249 These chemically functionalized microspheres can conjugate with various molecules, such as antibodies or peptide probes, which allow binding of biospecific targets for sensing or medical diagnostic applications.
Fig. 24.

Microparticle-based biosensors. a) PEG-based microparticles produced in PDMS device for measuring the concentration of glucose in vitro.248 Reprinted with permission from ref. 248. Copyright 2012, AIP Publishing. b) Microcapsule-based biosensor encapsulating quantum dots or gold nanorods for the detection of glucose and heparin.249 Reprinted with permission from ref. 249. Copyright 2018, The Royal Society of Chemistry. c) Hydrogel microspheres with tunable chemical functionalities for biomolecular conjugation reactions.250 Reprinted with permission from ref. 250. Copyright 2017, American Chemical Society. d) Microparticles encoded with colored core droplets and functionalized silica nanoparticles for multiplex immunoassay.115 Reprinted with permission from ref. 115. Copyright 2011, John Wiley & Sons, Inc. (e) Microparticle biosensors for monitoring the glucose concentration in vivo.253 Reprinted with permission from ref. 253. Copyright 2010, National Academy of Sciences.
The generation of microparticles with complex structures enabled by droplet microfluidics enables the production of biosensors with extended properties. For instance, hollow PEG-based microcapsules were fabricated to encapsulate nanosensors such as glucose-responsive quantum dots and heparin-responsive gold nanorods in the liquid core (Fig. 24c).250 The microcapsules not only protect the nanosensors from the external matrix, but also allow free diffusion of biomolecules into the microcapsules. The biomolecules interact with the encapsulated nanosensors, generating detectable optical signals. These microcapsules were further immobilized into alginate calcium hydrogels for implantable detection devices. The multiplex immunoassay was realized by utilizing multicolor encoded multicompartment microcapsules (Fig. 24d).115 The microparticles were encoded with multiple core droplets of three distinct colors for optical coding. The surface of the microparticles was decorated with silicon particle arrays, enabling integration of functional groups for immobilizing target biomolecules.
Other materials based hydrogel microbeads including polyacrylamide251 and poly(acrylamide-co-acrylic acid)252 can also serve as the matrix to encapsulate colloidal nanoparticles for DNA or glucose detection. Fluorescent polyacrylamide hydrogel beads were fabricated using droplet microfluidics. These monodispersed microbeads behave as highly sensitive, bio-stable, long-lasting, and injectable sensors that can continuously monitor the glucose level in vivo (Fig. 24e).253
Microparticle sensors are not limited to the detection of biomolecules; other microsensors which can detect microenvironment conditions, such as temperature, osmolyte concentration, pH, etc, have also been prepared by droplet microfluidics. Microcapsules with a semipermeable membrane have been reported to directly measure the osmolyte concentration in the media.254 By having an ultra-thin shell with known osmolarity solution inside, highly sensitive sensors that detect a small amount of sample have been demonstrated. When injected to the target area, microcapsules either shrink or swell depending on the difference in the osmolyte concentration across the shell membrane. This sensor does not require any delicate equipment. Microparticle-based microsensors that sense the temperature variation can be produced by encapsulating temperature sensitive materials, such as PNIPAM-based gels,255 or temperature dependent photonic liquid crystals.256 The temperature sensitive materials were encapsulated either inside the core or in the shell of the microparticles, endowing them with reversible color and spectral shifts upon temperature changes.255
4.4. Artificial cells
Artificial cells are micrometer-sized systems that are able to mimic or substitute some of the functional characteristics of living cells.20 They typically have a semi-permeable membrane, similar to living cells, which encloses cell constituents. Artificial cells enable a deeper understanding of the cellular functions and the origin of life. They can also accelerate the development of biomedical applications, including cell substitution, gene therapy, and targeted drug delivery. However, it is an immense challenge to generate artificial cells that can mimic real cellular structure and function.
Droplet microfluidics can be used to produce multicompartment microparticles to serve as artificial cells with well-designed functions. Liposomes, polymersomes and polymer microcapsules are three typical structures which can be generated by droplet microfluidics to resemble natural cells. Since natural cell membranes are composed of lipid bilayers, most research on artificial cells uses liposomes as basic models (Fig. 25a).257 Although the first unilamellar liposome templated by the W/O/W emulsion was published as early as 2006,258 their application as artificial cells was long bottlenecked by the existence of trace oil between the lipid bilayers. This may change the natural state of membrane proteins or the way they are integrated onto the membrane.101 Fortunately, this problem was partly resolved by utilizing surfactant assisted dewetting processes.259, 260 Both octanol259 and Pluronic F-68260 have a strong tendency to lower the surface tension of lipid membranes, leading to complete dewetting and thus formation of oil-free liposomes. Liposomes with a more complex structure, named vesosome have also been generated to better mimic cell functions. These include two-compartment liposomes,261 multi-lamellar262 and multivesicular liposomes.260, 263 For example, a two-step approach was used to form vesosomes, including concentric, pericentric and multicompartment structures (Fig. 25b).264 Hierarchical vesosomes were generated by sequential dewetting of lipid-bilayer-templated liposomes. To mimic the biofunctions, the inner liposome modelled the nucleus, and was filled with an in vitro transcription mixture that allowed for RNA generation.
Fig. 25.
Microfluidic microparticle based artificial cells. a) Demonstration of the typical structure of eukaryotic cells and corresponding artificial cells.257 Reprinted with permission from ref. 257. Copyright 2017, The Royal Society of Chemistry. b) Schematics and snapshots of the microfluidic preparation of vesosomes from emulsion dewetting and their application for molecular recognition reaction, membrane protein expression and integration.264 Reprinted with permission from ref. 264. Copyright 2017, American Chemical Society. c) dsGUV cell-like compartments encapsulated in water-in-oil copolymer-stabilized droplets.270 Representative combined images of green fluorescence from lipids (ATTO 488-labelled DOPE) and bright-field microscopy of the encapsulated LUVs. Schematic representation of the process for incorporating transmembrane and cytoskeletal proteins into dsGUVs using high-throughput droplet-based pico-injection technology. Reprinted with permission from ref. 270. Copyright 2018, Nature Publishing Group.
Transportation of small ions from “nucleus” to cytoplasm was also achieved by integration of transmembrane nanopores which should enable RNA transfer. Membrane composition is another important factor for designing artificial cells. Natural cell membranes are asymmetric and have varied regional microdomains,265 which can be generated by droplet microfluidic techniques.97, 266, 206 Asymmetric phospholipids based liposomes have been generated by a droplet emulsion-transfer method.266 A tetra-layer system was constructed, where a single-layer emulsion can move through the different layers by gravity induced sedimentation; it is subsequently coated with a second lipid layer with different compositions. Vesicles with controlled micro-domains on lipid bilayers were successfully generated by using double emulsions as templates. A solution of lipids dissolved in a mixture of a highly volatile good solvent and a less volatile poor solvent is used as the middle oil phase in double emulsion droplets. The evaporation of the good solvent triggered the dewetting of the less volatile poor solvent and subsequently formed micro-domains.
Polymersomes, composed of amphiphilic block copolymers, have many features that make them superior structures as artificial cells by comparison to liposomes. They are more stable than liposomes due to the molecular size and structure of the polymers, and they can be custom-tailored to have diverse properties. For example, their permeability can be tuned by varying the polymer compositions.267 Biocompatible and biodegradable polymersomes with multicompartments,268 and double bilayers269 have been developed to mimic the properties of cells. For instance, polymersome-based artificial cells for protein expression, aggregation and triggered release have been reported.267 These polymersomes were produced from double-emulsion templates by glass capillary devices, where PEG-b-PLA copolymers and an in vitro protein expression mixture were used as the middle phase and the inner phase, respectively. The stable structure of these polymersomes provides an isolated environment to encapsulate different cell contents and allows monitoring of long-term reactions. The cytoskeletal actin-like protein MreB was expressed using in vitro transcription and translation. These proteins inside the polymersomes can be trigger-released by osmolarity difference across the semi-permeable membrane, and the polymersomes can self-heal afterwards.
Multicompartment polymer microcapsules were fabricated using only aqueous media to mimic eukaryotic cells.257 Briefly, anionic alginate was infused from a capillary device into a container filled with cationic chitosan and CaCl2 to drive the crosslinking. Cell-mimicking membrane was formed immediately upon contact between anionic and cationic biopolymers. The resulting small capsules were then mixed with alginate and extruded as the aqueous phase to another capillary device to form multicompartment structures. Their applicability as artificial cells was confirmed by embedding two groups of E. coli as sensors: one for generating target molecules and one for responding to the target molecules.
The stability of the artificial cells can be further enhanced by combining the merits of both liposomes and polymersomes (Fig. 25c).270 An AuNPs@PEG@perfluorinated polyether multilayer structure was used as a shell to stabilize liposomes. Small and giant unilamellar liposomes were produced by accurately tuning the concentration of encapsulated lipids. In addition, both membrane and cytoskeleton proteins have been effectively loaded into the structures by pico-injection. Subsequently, in vitro expression of integrin and its integration to the cell membrane were demonstrated. Moreover, integrin mediated cellular interactions with both BSA and Fibrinogen-coated glasses were characterized. The results revealed that the artificial cells can effectively mimic the behavior of cell attachment as do living cells.
The advance of artificial cells constructed by droplet microfluidics has also promoted mimicry of the cellular functions. Liposome-based artificial cells are especially advantageous for research on cell membrane-based functions, where structuring of the membranes,97, 271 integration of membrane proteins259, 261 and transmembrane reactions,261, 272–274 have been explored. Encapsulation of protein expression systems in artificial cells and subsequent integration of membrane protein are of special value for “bottom-up” functioning of artificial cells.275 Despite these advances, there are still challenges for future development. For example, segmented transmembrane reactions inside artificial cells are still not well developed because of the difficulty in generating functional vesosome structures. Although this limitation has been partially resolved,263, 264 it remains a challenge to achieve RNA transport from the nucleus to the cytoplasm, or transport of the Krebs cycle product through the mitochondria membrane. The instability of artificial liposomes also precludes the possibility of carrying out cellular reactions under harsh conditions. In contrast, polymersomes can be more stable, and are easier to fabricate as complex structures such as multi-segment structures. However, applications of polymersomes besides protein expression and controlled release are still in the early stage of development.
5. Challenges and Perspective
Recent advances in droplet microfluidic technology have enabled production of uniform microparticles with well-defined structures, and multiple functionalities. Diverse microfluidic devices have been developed to generate complex droplet templates. Moreover, various materials and solidification methods have been exploited to produce microparticles encapsulating bioactives for different applications. Despite these encouraging achievements, there remain challenges to be resolved to realize the broader applicability of these microparticles in biomedical fields.
5.1. Microparticles production
Although droplet microfluidic technology is easy to use in the lab for microparticle production, it is still not feasible to employ them outside an academic lab.276 The lack of robustness and high cost of devices are two main concerns. For instance, capillary-based devices are resistance to organic solvent, and the particles can be produced by various chemical reactions. However, they have to be delicately made one by one by hand, especially when producing particles with complex structures by using assembled devices. Alternatively, PDMS or other polymer-based devices fabricated by lithographic techniques can alleviate this limitation. These devices, even with complicated geometries, can be precisely designed and replicated to produce microparticles on demand. However, the cost of production of the particles or other structures remains relatively high.
Another concern is the scale-up and mass production of microparticles. With droplet microfluidics, microparticles are fabricated droplet-by-droplet, which is far below the production rate of traditional bulk emulsification methods. A typical single droplet generator can produce droplets with throughput in the range of 0.1–10 mL/hr, while typically rates above L/hr, are required for commercial-scale manufacturing. Some strategies have been reported to scale-up generation of droplets,36,277 among which parallelization of multiple droplet generators has been developed sufficiently to attain commercially viable flow rates. Three-dimensional (3D) microchannels are designed to implement the parallelization of droplet generators. Nevertheless, there are still several critical concerns for commercial-scale manufacturing, including 1) device design to achieve an even distribution of fluids in each droplet generator, 2) device materials that are mechanically robust enough to withstand high pressure fluid injection, 3) surface wettability control of the microchannels for stable generation of droplets. Moreover, most scale-up studies are focused on production of single emulsions. High-throughput production of complex emulsions by droplet microfluidics has been less addressed.277 Improvement in the mass production of complex structures like multicompartment droplets are expected to meet more sophisticated applications.
5.2. Biomedical applications
Microparticles fabricated by droplet microfluidics have great potential in the fields of drug delivery, cell biology and biosensors. Materials with good biocompatibility and controlled biodegradability are required to form such microparticles. Currently, only a few types of materials from synthetic polymers to natural macromolecules have been exploited to fulfil this requirement. More biocompatible materials with optimized functions and properties suitable for microparticle fabrication need to be further investigated.
Microparticles with sizes ranging from a few microns to hundreds of microns can be generated by droplet microfluidics. They are ideal vehicles for drug delivery through oral or subcutaneous administration. However, the ideal size of the drug-loaded particles should be at sub-micron scales to avoid occlusion of blood vessels. Although nanoscale particles can be fabricated by droplet microfluidics,278, 279 low concentration of precursor solutions are used, which reduces the production rate. Thus, development of efficient methods for producing particles with nanoscale size is crucial for effective drug delivery.297
Hydrogel microparticles produced by droplet microfluidics can serve as ideal ECM-like materials for 3D cell culture. Cytocompatible materials and reactions have been developed to generate microparticles for effective cell encapsulation and long-term 3D cell culture. However, encapsulation of cells with controlled number of cells per particle is still a big challenge, especially for single cell study. Currently, encapsulation of single cells can be achieved from precursor solutions with low cell concentration, and the results follow a Poisson distribution with only a few single-cell-laden microgels being obtained. Enhancing the efficiency of single cell encapsulation remains an obstacle that must be overcome.
Apart from the intensive research on cell encapsulation and culture, there is a growing interest in the application of these cell-laden hydrogel microparticles as therapeutics or artificial organ/tissue building blocks for tissue engineering. A very important area is the development of functional tissues with assembled multiple cells in vitro for tissue repair. While microparticles prepared by droplet microfluidics can be used for controlled assembly and miniaturized 3D culture of cells to form a microtissue, it is not easy to build a macrotissue by assembling these cell-laden microparticles. 3D printing technology can form tissues using cell-laden microgels as the ink. However, it usually takes several hours to do the printing, which reduces the cell viability during the operation. Moreover, a properly organized vascular system must be incorporated into the artificial tissues, which is essential for the metabolism of embedded cells to maintain the tissues viability. Different methods have been developed to engineer the vascular networks, including generation of microchannels by using sacrificial materials, such as gelatin, alginate or LCST polymers.193, 280 However, the size of the channels formed is typically hundreds of microns, which is far beyond that of the human vascular system. These issues need to be resolved before cell-laden microparticles can be practically used for in vivo tissue engineering.
Droplet microfluidics also offers great opportunity to generate cargo-loaded microparticles for cell mimicry.281 The outcome of this research can provide insights in the understanding of the basic principles of life, and holds promise for various cell-based biomedical applications. This appealing research area is still at an early development stage, with major challenges remaining in the fabrication of microparticles with high stability, enhanced functionality, and environmental sensitivity for mimicking natural cells.
6. Conclusion
In this paper, we summarize the mechanism of droplet formation, microfluidic devices, and solidification methods to convert these droplets into microparticles with various structures. These microparticles with controlled properties and integrated functions have solved many major problems in biomedical fields. Considerable progress has been made in controlled drug delivery, 3D cell culture, artificial organ building, biomolecules sensing, and cell mimicking. More research still needs to be done to overcome the remaining challenges, and bring such engineered microparticles out of academic research and into practical applications. This paper provides a compendium of the work in droplet microfluidics that will be a valuable resource for the scientific and industrial communities.
Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21474060, 21574078 and 21327902), the Shanghai Rising-Star Program (No. 16QA1401800), the China Scholarship Council (201606895013 and 201606210262) and the National Key R&D Program of China (2016YFA0201200) is acknowledged. This work was also supported by Basic Science Research Program through the National Research Foundation of Korea(NRF) funded by the Ministry of Education (NRF-2018R1D1A1B07041102), POSTECH Basic Science Research Institute, the National Science Foundation (DMR-1708729, DMR-1420570), and by the National Institutes of Health (P01GM096971, R01EB023287).
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Hernandez RM, Orive G, Murua A and Pedraz JL, Adv. Drug Deliv. Rev, 2010, 62, 711–730. [DOI] [PubMed] [Google Scholar]
- 2.Choi A, Seo KD, Kim DW, Kim BC and Kim DS, Lab Chip, 2017, 17, 591–613. [DOI] [PubMed] [Google Scholar]
- 3.Shang L, Cheng Y and Zhao Y, Chem. Rev, 2017, 117, 7964–8040. [DOI] [PubMed] [Google Scholar]
- 4.Tran VT, Benoit JP and Venier-Julienne MC, Int. J. Pharm, 2011, 407, 1–11. [DOI] [PubMed] [Google Scholar]
- 5.Thiele J, Macromol. Chem. Phys, 2017, 218, 1600429. [Google Scholar]
- 6.Duncanson WJ, Lin T, Abate AR, Seiffert S, Shah RK and Weitz DA, Lab Chip, 2012, 12, 2135–2145. [DOI] [PubMed] [Google Scholar]
- 7.Saralidze K, Koole LH and Knetsch MLW, Materials, 2010, 3, 3537–3564. [Google Scholar]
- 8.Seo KD, Kim DS and Sanchez S, Lab Chip, 2015, 15, 3622–3626. [DOI] [PubMed] [Google Scholar]
- 9.Dendukuri D and Doyle PS, Adv. Mater, 2009, 21, 4071–4086. [Google Scholar]
- 10.Yeh J, Ling Y, Karp JM, Gantz J, Chandawarkar A, Eng G, Blumling Iii J, Langer R and Khademhosseini A, Biomaterials, 2006, 27, 5391–5398. [DOI] [PubMed] [Google Scholar]
- 11.Lee TY, Choi TM, Shim TS, Frijns RA and Kim SH, Lab Chip, 2016, 16, 3415–3440. [DOI] [PubMed] [Google Scholar]
- 12.Kim JH, Jeon TY, Choi TM, Shim TS, Kim SH and Yang SM, Langmuir, 2014, 30, 1473–1488. [DOI] [PubMed] [Google Scholar]
- 13.Heida T, Neubauer JW, Seuss M, Hauck N, Thiele J and Fery A, Macromol. Chem. Phys, 2017, 218, 1600418. [Google Scholar]
- 14.Wang W, Zhang M-J and Chu L-Y, Acc. Chem. Res, 2014, 47, 373–384. [DOI] [PubMed] [Google Scholar]
- 15.Liu DF, Zhang HB, Fontana F, Hirvonen JT and Santos HA, Lab Chip, 2017, 17, 1856–1883. [DOI] [PubMed] [Google Scholar]
- 16.Velasco D, Tumarkin E and Kumacheva E, Small, 2012, 8, 1633–1642. [DOI] [PubMed] [Google Scholar]
- 17.Jiang W, Li M, Chen Z and Leong KW, Lab Chip, 2016, 16, 4482–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allazetta S and Lutolf MP, Curr. Opin. Biotechnol, 2015, 35, 86–93. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Z and Yang CYJ, Acc. Chem. Res, 2017, 50, 22–31. [DOI] [PubMed] [Google Scholar]
- 20.Trantidou T, Friddin M, Elani Y, Brooks NJ, Law RV, Seddon JM and Ces O, ACS Nano, 2017, 11, 6549–6565. [DOI] [PubMed] [Google Scholar]
- 21.Kamiya K and Takeuchi S, J. Mater. Chem. B, 2017, 5, 5911–5923. [DOI] [PubMed] [Google Scholar]
- 22.Shah RK, Shum HC, Rowat AC, Lee D, Agresti JJ, Utada AS, Chu L-Y, Kim J-W, Fernandez-Nieves A, Martinez CJ and Weitz DA, Mater. Today, 2008, 11, 18–27. [Google Scholar]
- 23.Seiffert S, Macromol. Rapid. Comm, 2011, 32, 1600–1609. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Jain M, Ma Y and Nandakumar K, Soft Matter, 2015, 11, 3884–3899. [DOI] [PubMed] [Google Scholar]
- 25.Link DR, Grasland-Mongrain E, Duri A, Sarrazin F, Cheng Z, Cristobal G, Marquez M and Weitz DA, Angew. Chem. Int. Ed, 2006, 45, 2556–2560. [DOI] [PubMed] [Google Scholar]
- 26.Huang J-P, Ge X-H, Xu J-H and Luo G-S, Chem. Eng. Sci, 2016, 152, 293–300. [Google Scholar]
- 27.Yan Q, Xuan S, Ruan X, Wu J and Gong X, Microfluid. Nanofluid, 2015, 19, 1377–1384. [Google Scholar]
- 28.Maeda K, Onoe H, Takinoue M and Takeuchi S, Adv. Mater, 2012, 24, 1340–1346. [DOI] [PubMed] [Google Scholar]
- 29.Suryo R and Basaran OA, Phys. Fluids, 2006, 18, 082102. [DOI] [PubMed] [Google Scholar]
- 30.Zhu PG, Kong TT, Kang ZX, Tian XW and Wang LQ, Sci. Rep, 2015, 5, 11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu P and Wang L, Lab Chip, 2017, 17, 34–75. [DOI] [PubMed] [Google Scholar]
- 32.Chong ZZ, Tan SH, Ganan-Calvo AM, Tor SB, Loh NH and Nguyen NT, Lab Chip, 2016, 16, 35–58. [DOI] [PubMed] [Google Scholar]
- 33.Ofner A, Moore DG, Rühs PA, Schwendimann P, Eggersdorfer M, Amstad E, Weitz DA and Studart AR, Macromol. Chem. Phys, 2016, 218, 1600472. [Google Scholar]
- 34.Li Z, Leshansky AM, Pismen LM and Tabeling P, Lab Chip, 2015, 15, 1023–1031. [DOI] [PubMed] [Google Scholar]
- 35.Sahin S, Bliznyuk O, Rovalino Cordova A and Schroën K, Sci. Rep, 2016, 6, 26407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amstad E, Chemama M, Eggersdorfer M, Arriaga LR, Brenner MP and Weitz DA, Lab Chip, 2016, 16, 4163–4172. [DOI] [PubMed] [Google Scholar]
- 37.Sugiura S, Nakajima M, Tong J, Nabetani H and Seki M, J. Colloid Interface Sci, 2000, 227, 95–103. [DOI] [PubMed] [Google Scholar]
- 38.Sugiura S, Nakajima M, Kumazawa N, Iwamoto S and Seki M, J. Phys. Chem. B, 2002, 106, 9405–9409. [Google Scholar]
- 39.Sahin S and Schroen K, Lab Chip, 2015, 15, 2486–2495. [DOI] [PubMed] [Google Scholar]
- 40.Kahkeshani S and Di Carlo D, Lab Chip, 2016, 16, 2474–2480. [DOI] [PubMed] [Google Scholar]
- 41.Schuler F, Schwemmer F, Trotter M, Wadle S, Zengerle R, von Stetten F and Paust N, Lab Chip, 2015, 15, 2759–2766. [DOI] [PubMed] [Google Scholar]
- 42.Stolovicki E, Ziblat R and Weitz DA, Lab Chip, 2018, 18, 132–138. [DOI] [PubMed] [Google Scholar]
- 43.Eggersdorfer ML, Zheng W, Nawar S, Mercandetti C, Ofner A, Leibacher I, Koehler S and Weitz DA, Lab Chip, 2017, 17, 936–942. [DOI] [PubMed] [Google Scholar]
- 44.Utada AS, Lorenceau E, Link DR, Kaplan PD, Stone HA and Weitz DA, Science, 2005, 308, 537–541. [DOI] [PubMed] [Google Scholar]
- 45.Chu L-Y, Utada AS, Shah RK, Kim J-W and Weitz DA, Angew. Chem, 2007, 119, 9128–9132. [Google Scholar]
- 46.Sun BJ, Shum HC, Holtze C and Weitz DA, ACS Appl. Mater. Interfaces, 2010, 2, 3411–3416. [DOI] [PubMed] [Google Scholar]
- 47.Wang W, Xie R, Ju XJ, Luo T, Liu L, Weitz DA and Chu LY, Lab Chip, 2011, 11, 1587–1592. [DOI] [PubMed] [Google Scholar]
- 48.Choi C-H, Lee H, Abbaspourrad A, Kim JH, Fan J, Caggioni M, Wesner C, Zhu T and Weitz DA, Adv. Mater, 2016, 28, 3340–3344. [DOI] [PubMed] [Google Scholar]
- 49.Kim S-H, Kim JW, Cho J-C and Weitz DA, Lab Chip, 2011, 11, 3162–3166. [DOI] [PubMed] [Google Scholar]
- 50.Kim S-H, Kim JW, Kim D-H, Han S-H and Weitz DA, Microfluid. Nanofluid, 2013, 14, 509–514. [Google Scholar]
- 51.Clausell-Tormos J, Lieber D, Baret J-C, El-Harrak A, Miller OJ, Frenz L, Blouwolff J, Humphry KJ, Köster S, Duan H, Holtze C, Weitz DA, Griffiths AD and Merten CA, Chem. Biol, 2008, 15, 427–437. [DOI] [PubMed] [Google Scholar]
- 52.Yu L, Chen MC and Cheung KC, Lab Chip, 2010, 10, 2424–2432. [DOI] [PubMed] [Google Scholar]
- 53.Anderson JR, Chiu DT, Jackman RJ, Cherniavskaya O, McDonald JC, Wu H, Whitesides SH and Whitesides GM, Anal. Chem, 2000, 72, 3158–3164. [DOI] [PubMed] [Google Scholar]
- 54.McDonald JC, Chabinyc ML, Metallo SJ, Anderson JR, Stroock AD and Whitesides GM, Anal. Chem, 2002, 74, 1537–1545. [DOI] [PubMed] [Google Scholar]
- 55.Garstecki P, Fuerstman MJ, Stone HA and Whitesides GM, Lab Chip, 2006, 6, 437–446. [DOI] [PubMed] [Google Scholar]
- 56.Ward T, Faivre M, Abkarian M and Stone HA, Electrophoresis, 2005, 26, 3716–3724. [DOI] [PubMed] [Google Scholar]
- 57.Abate AR, Poitzsch A, Hwang Y, Lee J, Czerwinska J and Weitz DA, Phys. Rev. E, 2009, 80, 026310. [DOI] [PubMed] [Google Scholar]
- 58.Vickers JA, Caulum MM and Henry CS, Anal. Chem, 2006, 78, 7446–7452. [DOI] [PubMed] [Google Scholar]
- 59.Zhou J, Khodakov DA, Ellis AV and Voelcker NH, Electrophoresis, 2012, 33, 89–104. [DOI] [PubMed] [Google Scholar]
- 60.Roman GT, Hlaus T, Bass KJ, Seelhammer TG and Culbertson CT, Anal. Chem, 2005, 77, 1414–1422. [DOI] [PubMed] [Google Scholar]
- 61.Abate AR, Krummel AT, Lee D, Marquez M, Holtze C and Weitz DA, Lab Chip, 2008, 8, 2157–2160. [DOI] [PubMed] [Google Scholar]
- 62.Abate AR, Thiele J, Weinhart M and Weitz DA, Lab Chip, 2010, 10, 1774–1776. [DOI] [PubMed] [Google Scholar]
- 63.Choi C-H, Lee H and Weitz DA, ACS Appl. Mater. Interfaces, 2018, 10, 3170–3174. [DOI] [PubMed] [Google Scholar]
- 64.Kim J-O, Kim H, Ko D-H, Min K-I, Im DJ, Park S-Y and Kim D-P, Lab Chip, 2014, 14, 4270–4276. [DOI] [PubMed] [Google Scholar]
- 65.Nightingale AM, Krishnadasan SH, Berhanu D, Niu X, Drury C, McIntyre R, Valsami-Jones E and deMello JC, Lab Chip, 2011, 11, 1221–1227. [DOI] [PubMed] [Google Scholar]
- 66.Lidia D, Laura G, Alfio L, Giacomo M, Alfio N and Marco R, J. Micromech. Microeng, 2015, 25, 035013. [Google Scholar]
- 67.Femmer T, Jans A, Eswein R, Anwar N, Moeller M, Wessling M and Kuehne AJC, ACS Appl. Mater. Interfaces, 2015, 7, 12635–12638. [DOI] [PubMed] [Google Scholar]
- 68.Martino C, Berger S, Wootton RCR and deMello AJ, Lab Chip, 2014, 14, 4178–4182. [DOI] [PubMed] [Google Scholar]
- 69.Waheed S, Cabot JM, Macdonald NP, Lewis T, Guijt RM, Paull B and Breadmore MC, Lab Chip, 2016, 16, 1993–2013. [DOI] [PubMed] [Google Scholar]
- 70.Lee KG, Park KJ, Seok S, Shin S, Kim DH, Park JY, Heo YS, Lee SJ and Lee TJ, RSC Adv, 2014, 4, 32876–32880. [Google Scholar]
- 71.Yang S and DeVoe DL, in Microfluidic Diagnostics: Methods and Protocols, eds. Jenkins G and Mansfield CD, Humana Press, Totowa, NJ, 2013, pp. 115–123. [Google Scholar]
- 72.Nge PN, Rogers CI, and Woolley AT, Chem. Rev 2013, 113, 2550–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liedert R, Amundsen LK, Hokkanen A, Maki M, Aittakorpi A, Pakanen M, Scherer JR, Mathies RA, Kurkinen M, Uusitalo S, Hakalahti L, Nevanen TK, Siitari H and Soderlund H, Lab Chip, 2012, 12, 333–339. [DOI] [PubMed] [Google Scholar]
- 74.Wang X, Liedert C, Liedert R and Papautsky I, Lab Chip, 2016, 16, 1821–1830. [DOI] [PubMed] [Google Scholar]
- 75.Wang X, Liedert C, Liedert R and Papautsky I, Lab Chip, 2016, 16, 1821–1830. [DOI] [PubMed] [Google Scholar]
- 76.Hann SD, Stebe KJ and Lee D, Langmuir, 2017, 33, 10107–10117. [DOI] [PubMed] [Google Scholar]
- 77.Rossow T, Lienemann PS and Mooney DJ, Macromol. Chem. Phys, 2017, 218, 1600380. [Google Scholar]
- 78.Jeong WJ, Kim JY, Choo J, Lee EK, Han CS, Beebe DJ, Seong GH and Lee SH, Langmuir, 2005, 21, 3738–3741. [DOI] [PubMed] [Google Scholar]
- 79.Guo S, Yao T, Ji X, Zeng C, Wang C and Zhang L, Angew. Chem. Int. Ed, 2014, 53, 7504–7509. [DOI] [PubMed] [Google Scholar]
- 80.Liu H, Qian X, Wu Z, Yang R, Sun S and Ma H, J. Mater. Chem. B, 2016, 4, 482–488. [DOI] [PubMed] [Google Scholar]
- 81.Cha C, Oh J, Kim K, Qiu Y, Joh M, Shin SR, Wang X, Camci-Unal G, Wan K.-t., Liao R and Khademhosseini A, Biomacromolecules, 2014, 15, 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee KG, Park TJ, Soo SY, Wang KW, Kim BII, Park JH, Lee C-S, Kim DH and Lee SJ, Biotechnol. Bioeng, 2010, 107, 747–751. [DOI] [PubMed] [Google Scholar]
- 83.Kim J-W, Utada AS, Fernández-Nieves A, Hu Z and Weitz DA, Angew. Chem. Inter. Ed, 2007, 46, 1819–1822. [DOI] [PubMed] [Google Scholar]
- 84.Gu H, Ye B, Ding H, Liu C, Zhao Y and Gu Z, J. Mater. Chem. C, 2015, 3, 6607–6612. [Google Scholar]
- 85.Visaveliya N and Kohler JM, J. Mater. Chem. C, 2015, 3, 844–853. [Google Scholar]
- 86.Nisisako T and Hatsuzawa T, Microfluid. Nanofluid, 2010, 9, 427–437. [Google Scholar]
- 87.Amato DV, Lee H, Werner JG, Weitz DA and Patton DL, ACS Appl Mater Interfaces, 2017, 9, 3288–3293. [DOI] [PubMed] [Google Scholar]
- 88.Zhao Y, Shum HC, Adams LLA, Sun B, Holtze C, Gu Z and Weitz DA, Langmuir, 2011, 27, 13988–13991. [DOI] [PubMed] [Google Scholar]
- 89.Comunian TA, Abbaspourrad A, Favaro-Trindade CS and Weitz DA, Food Chem, 2014, 152, 271–275. [DOI] [PubMed] [Google Scholar]
- 90.Sugiura S, Oda T, Izumida Y, Aoyagi Y, Satake M, Ochiai A, Ohkohchi N and Nakajima M, Biomaterials, 2005, 26, 3327–3331. [DOI] [PubMed] [Google Scholar]
- 91.Zhang H, Tumarkin E, Peerani R, Nie Z, Sullan RMA, Walker GC and Kumacheva E, J. Am. Chem. Soc, 2006, 128, 12205–12210. [DOI] [PubMed] [Google Scholar]
- 92.Tan WH and Takeuchi S, Adv. Mater, 2007, 19, 2696–2701. [Google Scholar]
- 93.Xu Q, Hashimoto M, Dang TT, Hoare T, Kohane DS, Whitesides GM, Langer R and Anderson DG, Small, 2009, 5, 1575–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Watanabe T, Ono T and Kimura Y, Soft Matter, 2011, 7, 9894–9897. [Google Scholar]
- 95.Haase MF and Brujic J, Angew. Chem. Int. Ed, 2014, 53, 11793–11797. [DOI] [PubMed] [Google Scholar]
- 96.Shum HC, Kim J-W and Weitz DA, J. Am. Chem. Soc, 2008, 130, 9543–9549. [DOI] [PubMed] [Google Scholar]
- 97.Arriaga LR, Datta SS, Kim S-H, Amstad E, Kodger TE, Monroy F and Weitz DA, Small, 2014, 10, 950–956. [DOI] [PubMed] [Google Scholar]
- 98.Min NG, Ku M, Yang J and Kim S-H, Chem. Mater, 2016, 28, 1430–1438. [Google Scholar]
- 99.Shi W and Weitz DA, Macromolecules, 2017, 50, 7681–7686. [Google Scholar]
- 100.Choi C-H, Weitz DA and Lee C-S, Adv. Mater, 2013, 25, 2536–2541. [DOI] [PubMed] [Google Scholar]
- 101.van Swaay D and deMello A, Lab Chip, 2013, 13, 752–767. [DOI] [PubMed] [Google Scholar]
- 102.Kim S-H, Shum HC, Kim JW, Cho J-C and Weitz DA, J. Am. Chem. Soc, 2011, 133, 15165–15171. [DOI] [PubMed] [Google Scholar]
- 103.Amstad E, Kim S-H and Weitz DA, Angew. Chem. Int. Ed, 2012, 51, 12499–12503. [DOI] [PubMed] [Google Scholar]
- 104.Moon B-U, Abbasi N, Jones SG, Hwang DK and Tsai SSH, Anal. Chem, 2016, 88, 3982–3989. [DOI] [PubMed] [Google Scholar]
- 105.Song Y, Sauret A and Cheung Shum H, Biomicrofluidics, 2013, 7, 061301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Moon B-U, Jones SG, Hwang DK and Tsai SSH, Lab Chip, 2015, 15, 2437–2444. [DOI] [PubMed] [Google Scholar]
- 107.Sauret A, Appl. Phys. Lett, 2012, 100, 154106. [Google Scholar]
- 108.Ziemecka I, van Steijn V, Koper GJ, Rosso M, Brizard AM, van Esch JH and Kreutzer MT, Lab Chip, 2011, 11, 620–624. [DOI] [PubMed] [Google Scholar]
- 109.Ma S, Thiele J, Liu X, Bai Y, Abell C and Huck WT, Small, 2012, 8, 2356–2360. [DOI] [PubMed] [Google Scholar]
- 110.Ameloot R, Vermoortele F, Vanhove W, Roeffaers MBJ, Sels BF and De Vos DE, Nat. Chem, 2011, 3, 382–387. [DOI] [PubMed] [Google Scholar]
- 111.Liu L, Yang J-P, Ju X-J, Xie R, Liu Y-M, Wang W, Zhang J-J, Niu CH and Chu L-Y, Soft Matter, 2011, 7, 4821–4827. [Google Scholar]
- 112.Kim M, Yeo SJ, Highley CB, Burdick JA, Yoo PJ, Doh J and Lee D, ACS Nano, 2015, 9, 8269–8278. [DOI] [PubMed] [Google Scholar]
- 113.Zhang LY, Cai LH, Lienemann PS, Rossow T, Polenz I, Vallmajo-Martin Q, Ehrbar M, Na H, Mooney DJ and Weitz DA, Angew. Chem. Int. Ed, 2016, 55, 13470–13474. [DOI] [PubMed] [Google Scholar]
- 114.Shang L, Shangguan F, Cheng Y, Lu J, Xie Z, Zhao Y and Gu Z, Nanoscale, 2013, 5, 9553–9557. [DOI] [PubMed] [Google Scholar]
- 115.Kim SH, Shim JW and Yang SM, Angew. Chem. Int. Ed, 2011, 50, 1171–1174. [DOI] [PubMed] [Google Scholar]
- 116.Kumachev A, Greener J, Tumarkin E, Eiser E, Zandstra PW and Kumacheva E, Biomaterials, 2011, 32, 1477–1483. [DOI] [PubMed] [Google Scholar]
- 117.Raz N, Li JK, Fiddes LK, Tumarkin E, Walker GC and Kumacheva E, Macromolecules, 2010, 43, 7277–7281. [Google Scholar]
- 118.Lin Y-S, Yang C-H, Hsu Y-Y and Hsieh C-L, Electrophoresis, 2013, 34, 425–431. [DOI] [PubMed] [Google Scholar]
- 119.Xu S, Nie Z, Seo M, Lewis P, Kumacheva E, Stone HA, Garstecki P, Weibel DB, Gitlin I and Whitesides GM, Angew. Chem. Int. Ed, 2005, 44, 724–728. [DOI] [PubMed] [Google Scholar]
- 120.Kim S-H, Abbaspourrad A and Weitz DA, J. Am. Chem. Soc, 2011, 133, 5516–5524. [DOI] [PubMed] [Google Scholar]
- 121.Nisisako T and Torii T, Adv. Mater, 2007, 19, 1489–1493. [Google Scholar]
- 122.Deng N-N, Wang W, Ju X-J, Xie R, Weitz DA and Chu L-Y, Lab Chip, 2013, 13, 4047–4052. [DOI] [PubMed] [Google Scholar]
- 123.Datta SS, Abbaspourrad A, Amstad E, Fan J, Kim S-H, Romanowsky M, Shum HC, Sun B, Utada AS, Windbergs M, Zhou S and Weitz DA, Adv. Mater, 2014, 26, 2205–2218. [DOI] [PubMed] [Google Scholar]
- 124.Nie Z, Li W, Seo M, Xu S and Kumacheva E, J. Am. Chem. Soc, 2006, 128, 9408–9412. [DOI] [PubMed] [Google Scholar]
- 125.Yuet KP, Hwang DK, Haghgooie R and Doyle PS, Langmuir, 2010, 26, 4281–4287. [DOI] [PubMed] [Google Scholar]
- 126.Shah RK, Kim J-W and Weitz DA, Adv. Mater, 2009, 21, 1949–1953 [Google Scholar]
- 127.Zhao Y, Shum HC, Chen H, Adams LLA, Gu Z and Weitz DA, J. Am. Chem. Soc, 2011, 133, 8790–8793. [DOI] [PubMed] [Google Scholar]
- 128.Zhao Y, Xie Z, Gu H, Jin L, Zhao X, Wang B and Gu Z, Npg Asia Mater, 2012, 4, e25. [Google Scholar]
- 129.Seiffert S, Romanowsky MB and Weitz DA, Langmuir, 2010, 26, 14842–14847. [DOI] [PubMed] [Google Scholar]
- 130.Lee H, Choi C-H, Abbaspourrad A, Wesner C, Caggioni M, Zhu T, Nawar S and Weitz D, Adv. Mater, 2016, 28, 8425–8430. [DOI] [PubMed] [Google Scholar]
- 131.Huang S, Lin B and Qin J, Electrophoresis, 2011, 32, 3364–3370. [DOI] [PubMed] [Google Scholar]
- 132.Mou C-L, Ju X-J, Zhang L, Xie R, Wang W, Deng N-N, Wei J, Chen Q and Chu L-Y, Langmuir, 2014, 30, 1455–1464. [DOI] [PubMed] [Google Scholar]
- 133.Chu LY, Kim JW, Shah RK and Weitz DA, Adv. Funct. Mater, 2007, 17, 3499–3504. [Google Scholar]
- 134.Wan J, Bick A, Sullivan M and Stone HA, Adv. Mater, 2008, 20, 3314–3318. [Google Scholar]
- 135.Gong X, Wen W and Sheng P, Langmuir, 2009, 25, 7072–7077. [DOI] [PubMed] [Google Scholar]
- 136.Duncanson WJ, Zieringer M, Wagner O, Wilking JN, Abbaspourrad A, Haag R and Weitz DA, Soft Matter, 2012, 8, 10636–10640. [Google Scholar]
- 137.Watanabe T, Lopez CG, Douglas JF, Ono T and Cabral JT, Langmuir, 2014, 30, 2470–2479. [DOI] [PubMed] [Google Scholar]
- 138.Shim TS, Kim SH and Yang SM, Part. Part. Syst. Char, 2013, 30, 9–45. [Google Scholar]
- 139.Vasiliauskas R, Liu DF, Cito S, Zhang HB, Shahbazi MA, Sikanen T, Mazutis L and Santos HA, ACS Appl. Mater. Interfaces, 2015, 7, 14822–14832. [DOI] [PubMed] [Google Scholar]
- 140.Araujo F, Shrestha N, Shahbazi MA, Liu DF, Herranz-Blanco B, Makila EM, Salonen JJ, Hirvonen JT, Granja PL, Sarmento B and Santos HA, ACS Nano, 2015, 9, 8291–8302. [DOI] [PubMed] [Google Scholar]
- 141.Zhao CX, Adv. Drug Deliv. Rev, 2013, 65, 1420–1446. [DOI] [PubMed] [Google Scholar]
- 142.Yang CH, Huang KS and Chang JY, Biomed. Microdevices, 2007, 9, 253–259. [DOI] [PubMed] [Google Scholar]
- 143.Xu JH, Zhao H, Lan WJ and Luo GS, Adv. Healthc. Mater, 2012, 1, 106–111. [DOI] [PubMed] [Google Scholar]
- 144.Lu B, Tarn MD, Pamme N and Georgiou TK, J. Mater. Chem. B, 2016, 4, 3086–3093. [DOI] [PubMed] [Google Scholar]
- 145.Lee TY, Ku M, Kim B, Lee S, Yang J and Kim SH, Small, 2017, 13, 11. [DOI] [PubMed] [Google Scholar]
- 146.Lee H, Choi C-H, Abbaspourrad A, Wesner C, Caggioni M, Zhu T and Weitz D, ACS Appl. Mater. Interfaces, 2016, 8, 4007–4013. [DOI] [PubMed] [Google Scholar]
- 147.Li YN, Yan D, Fu FF, Liu YX, Zhang B, Wang J, Shang LR, Gu Z and Zhao Y, Sci. China-Mater, 2017, 60, 543–553. [Google Scholar]
- 148.He F, Wang W, He XH, Yang XL, Li M, Xie R, Ju XJ, Liu Z and Chu LY, ACS Appl. Mater. Interfaces, 2016, 8, 8743–8754. [DOI] [PubMed] [Google Scholar]
- 149.Pradhan P, Giri J, Rieken F, Koch C, Mykhaylyk O, Döblinger M, Banerjee R, Bahadur D and Plank C, J. Control. Release, 2010, 142, 108–121. [DOI] [PubMed] [Google Scholar]
- 150.Zhang K, Wu W, Guo K, Chen J and Zhang P, Langmuir, 2010, 26, 7971–7980. [DOI] [PubMed] [Google Scholar]
- 151.San Miguel A, Scrimgeour J, Curtis JE and Behrens SH, Soft Matter, 2010, 6, 3163–3166. [Google Scholar]
- 152.Broaders KE, Pastine SJ, Grandhe S and Frechet JMJ, Chem. Comm, 2011, 47, 665–667. [DOI] [PubMed] [Google Scholar]
- 153.Cui L, Cohen JL, Chu CK, Wich PR, Kierstead PH and Fréchet JMJ, J. Am. Chem. Soc , 2012, 134, 15840–15848. [DOI] [PubMed] [Google Scholar]
- 154.Kooiman K, Böhmer MR, Emmer M, Vos HJ, Chlon C, Shi WT, Hall CS, de Winter SHPM, Schroën K, Versluis M, de Jong N and van Wamel A, J. Control. Release, 2009, 133, 109–118. [DOI] [PubMed] [Google Scholar]
- 155.Pavlov AM, Saez V, Cobley A, Graves J, Sukhorukov GB and Mason TJ, Soft Matter, 2011, 7, 4341–4347. [Google Scholar]
- 156.Ochs M, Carregal-Romero S, Rejman J, Braeckmans K, De SC Smedt and W. J. Parak, Angew. Chem, 2013, 125, 723–727. [DOI] [PubMed] [Google Scholar]
- 157.Volodkin D, Skirtach A and Möhwald H, Polym. Int, 2012, 61, 673–679. [Google Scholar]
- 158.Pastine SJ, Okawa D, Zettl A and Fréchet JMJ, J. Am. Chem. Soc , 2009, 131, 13586–13587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Giteau A, Venier-Julienne MC, Aubert-Pouessel A and Benoit JP, Int. J. Pharm, 2008, 350, 14–26. [DOI] [PubMed] [Google Scholar]
- 160.Loira-Pastoriza C, Todoroff J and Vanbever R, Adv. Drug. Deliv. Rev, 2014, 75, 81–91. [DOI] [PubMed] [Google Scholar]
- 161.Huang X, Brazel CS, Control J. Release, 2001, 73, 121–136.. [DOI] [PubMed] [Google Scholar]
- 162.Deveza L, Ashoken J, Castaneda G, Tong X, Keeney M, Han L-H and Yang F, ACS Biomateri. Sci. Eng, 2015, 1, 157–165. [DOI] [PubMed] [Google Scholar]
- 163.Kim B, Lee TY, Abbaspourrad A and Kim S-H, Chem. Mater, 2014, 26, 7166–7171. [Google Scholar]
- 164.Li J and Mooney DJ, Nat. Rev. Mater, 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhou JH, Hyun DC, Liu H, Wu HK and Xia YN, Macromol. Rapid Comm, 2014, 35, 1436–1442. [DOI] [PubMed] [Google Scholar]
- 166.Shimanovich U, Efimov I, Mason TO, Flagmeier P, Buell AK, Gedanken A, Linse S, Åkerfeldt KS, Dobson CM, Weitz DA and Knowles TPJ, ACS Nano, 2015, 9, 43–51. [DOI] [PubMed] [Google Scholar]
- 167.Liu DF, Herranz-Blanco B, Makila E, Arriaga LR, Mirza S, Weitz DA, Sandler N, Salonen J, Hirvonen J and Santos HA, ACS Appl. Mater. Interfaces, 2013, 5, 12127–12134. [DOI] [PubMed] [Google Scholar]
- 168.Huang J, Li W, Li Y, Luo C, Zeng Y, Xu Y and Zhou J, J. Mater. Chem. B, 2014, 2, 6848–6854. [DOI] [PubMed] [Google Scholar]
- 169.Liu D, Zhang H, Herranz-Blanco B, Makila E, Lehto VP, Salonen J, Hirvonen J and Santos HA, Small, 2014, 10, 2029–2038. [DOI] [PubMed] [Google Scholar]
- 170.Zhang HB, Liu DF, Shahbazi MA, Makila E, Herranz-Blanco B, Salonen J, Hirvonen J and Santos HA, Adv. Mater, 2014, 26, 4497–4503. [DOI] [PubMed] [Google Scholar]
- 171.Maher S, Santos A, Kumeria T, Kaur G, Lambert M, Forward P, Evdokiou A and Losic D, J. Mater. Chem. B, 2017, 5, 4097–4109. [DOI] [PubMed] [Google Scholar]
- 172.Huang KS, Yang CH, Kung CP, Grumezescu AM, Ker MD, Lin YS and Wang CY, Electrophoresis, 2014, 35, 330–336. [DOI] [PubMed] [Google Scholar]
- 173.Wei J, Ju XJ, Zou XY, Xie R, Wang W, Liu YM and Chu LY, Adv. Funct. Mater, 2014, 24, 3312–3323. [Google Scholar]
- 174.Liu D, Chen J, Jiang T, Li W, Huang Y, Lu X, Liu Z, Zhang W, Zhou Z, Ding Q, Santos HA, Yin G and Fan J, Adv Mater, 2018, 10.1002/adma.201706032. [DOI] [PubMed] [Google Scholar]
- 175.Liu L, Wang W, Ju X-J, Xie R and Chu L-Y, Soft Matter, 2010, 6, 3759–3763. [Google Scholar]
- 176.Seiffert S, Thiele J, Abate AR and Weitz DA, J. Am. Chem. Soc, 2010, 132, 6606–6609. [DOI] [PubMed] [Google Scholar]
- 177.Jeong WC, Kim SH and Yang SM, ACS Appl. Mater. Interfaces, 2014, 6, 826–832. [DOI] [PubMed] [Google Scholar]
- 178.Yang XL, Ju XJ, Mu XT, Wang W, Xie R, Liu Z and Chu LY, ACS Appl. Mater. Interfaces, 2016, 8, 10524–10534. [DOI] [PubMed] [Google Scholar]
- 179.Zhang W, Abbaspourrad A, Chen D, Campbell E, Zhao H, Li Y, Li Q and Weitz DA, Adv. Funct. Mater, 2017, 27, 1700975. [Google Scholar]
- 180.Duarte AR, Unal B, Mano JF, Reis RL and Jensen KF, Langmuir, 2014, 30, 12391–12399. [DOI] [PubMed] [Google Scholar]
- 181.Radt B, Smith TA and Caruso F, Adv. Mater, 2004, 16, 2184–2189. [Google Scholar]
- 182.Weissleder R, Nat. Biotech, 2001, 19, 316–317. [DOI] [PubMed] [Google Scholar]
- 183.Kim B, Soo Lee H, Kim J and Kim SH, Chem. Commun, 2013, 49, 1865–1867. [DOI] [PubMed] [Google Scholar]
- 184.Lee MH, Hribar KC, Brugarolas T, Kamat NP, Burdick JA and Lee D, Adv. Funct. Mater, 2012, 22, 131–138. [Google Scholar]
- 185.Yu Z, Zheng Y, Parker RM, Lan Y, Wu Y, Coulston RJ, Zhang J, Scherman OA and Abell C, ACS Appl. Mater. Interfaces, 2016, 8, 8811–8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Liu Z, Liu L, Ju X-J, Xie R, Zhang B and Chu L-Y, Chem. Commun, 2011, 47, 12283–12285. [DOI] [PubMed] [Google Scholar]
- 187.Ge X-H, Huang J-P, Xu J-H and Luo G-S, Lab Chip, 2014, 14, 4451–4454. [DOI] [PubMed] [Google Scholar]
- 188.Huang G, Li F, Zhao X, Ma Y, Li Y, Lin M, Jin G, Lu TJ, Genin GM and Xu F, Chem. Rev, 2017, 117, 12764–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang H, Yu Y, Hu Y, He X, Berk Usta O and Yarmush ML, Lab Chip, 2017, 17, 1913–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shembekar N, Chaipan C, Utharala R and Merten CA, Lab Chip, 2016, 16, 1314–1331. [DOI] [PubMed] [Google Scholar]
- 191.Agarwal P, Wang H, Sun M, Xu J, Zhao S, Liu Z, Gooch KJ, Zhao Y, Lu X and He X, ACS Nano, 2017, 11, 6691–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Chung BG, Lee KH, Khademhosseini A and Lee SH, Lab Chip, 2012, 12, 45–59. [DOI] [PubMed] [Google Scholar]
- 193.Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA and Lewis JA, Adv. Mater, 2014, 26, 3124–3130. [DOI] [PubMed] [Google Scholar]
- 194.Selimović Š, Oh J, Bae H, Dokmeci M, and Khademhosseini A, Polymers, 2012, 4, 1554–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Zhang L, Chen K, Zhang H, Pang B, Choi CH, Mao AS, Liao H, Utech S, Mooney DJ, Wang H and Weitz DA, Small, 2018, 14, 1702955. [DOI] [PubMed] [Google Scholar]
- 196.Liu Z and Shum HC, Biomicrofluidics, 2013, 7, 44117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sun M, Durkin P, Li J, Toth TL and He X, ACS Sens, 2018, 3, 410–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Martinez CJ, Kim JW, Ye C, Ortiz I, Rowat AC, Marquez M and Weitz D, Macromol. Biosci, 2012, 12, 946–951. [DOI] [PubMed] [Google Scholar]
- 199.Li F, Truong VX, Thissen H, Frith JE and Forsythe JS, ACS Appl. Mater. Interfaces, 2017, 9, 8589–8601. [DOI] [PubMed] [Google Scholar]
- 200.Yamada M, Hori A, Sugaya S, Yajima Y, Utoh R, Yamato M and Seki M, Lab Chip, 2015, 15, 3941–3951. [DOI] [PubMed] [Google Scholar]
- 201.Wieduwild R, Krishnan S, Chwalek K, Boden A, Nowak M, Drechsel D, Werner C and Zhang Y, Angew. Chem. Int. Ed, 2015, 54, 3962–3966. [DOI] [PubMed] [Google Scholar]
- 202.Utech S, Prodanovic R, Mao AS, Ostafe R, Mooney DJ and Weitz DA, Adv. Healthc. Mater, 2015, 4, 1628–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wang Y, Zhao L, Tian C, Ma C and Wang J, Anal. Methods, 2015, 7, 10040–10051. [Google Scholar]
- 204.Yoon S, Kim JA, Lee SH, Kim M and Park TH, Lab Chip, 2013, 13, 1522–1528. [DOI] [PubMed] [Google Scholar]
- 205.Rossow T, Heyman JA, Ehrlicher AJ, Langhoff A, Weitz DA, Haag R and Seiffert S, J. Am. Chem. Soc, 2012, 134, 4983–4989. [DOI] [PubMed] [Google Scholar]
- 206.Headen DM, Aubry G, Lu H and Garcia AJ, Adv. Mater, 2014, 26, 3003–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Jiang Z, Xia B, McBride R and Oakey J, J. Mater. Chem. B, 2017, 5, 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Steinhilber D, Rossow T, Wedepohl S, Paulus F, Seiffert S and Haag R, Angew. Chem. Int. Ed, 2013, 52, 13538–13543. [DOI] [PubMed] [Google Scholar]
- 209.Rossow T, Bayer S, Albrecht R, Tzschucke CC and Seiffert S, Macromol. Rapid. Commun, 2013, 34, 1401–1407. [DOI] [PubMed] [Google Scholar]
- 210.Ma SH, Natoli M, Liu X, Neubauer MP, Watt FM, Fery A and Huck WTS, J. Mater. Chem. B, 2013, 1, 5128–5136. [DOI] [PubMed] [Google Scholar]
- 211.Henke S, Leijten J, Kemna E, Neubauer M, Fery A, van den Berg A, van Apeldoorn A and Karperien M, Macromol. Biosci, 2016, 16, 1524–1532. [DOI] [PubMed] [Google Scholar]
- 212.Choi C-H, Wang H, Lee H, Kim JH, Zhang L, Mao A, Mooney DJ and Weitz DA, Lab Chip, 2016, 16, 1549–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Allazetta S, Kolb L, Zerbib S, Bardy J and Lutolf MP, Small, 2015, 11, 5647–5656. [DOI] [PubMed] [Google Scholar]
- 214.Ferreira DS, Reis RL and Azevedo HS, Soft Matter, 2013, 9, 9237–9248. [Google Scholar]
- 215.Fu F, Shang L, Zheng F, Chen Z, Wang H, Wang J, Gu Z and Zhao Y, ACS Appl. Mater. Interfaces, 2016, 8, 13840–13848. [DOI] [PubMed] [Google Scholar]
- 216.Kim C, Chung S, Kim YE, Lee KS, Lee SH, Oh KW and Kang JY, Lab Chip, 2011, 11, 246–252. [DOI] [PubMed] [Google Scholar]
- 217.Chan HF, Zhang Y, Ho YP, Chiu YL, Jung Y and Leong KW, Sci. Rep, 2013, 3, 3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Su G, Zhao Y, Wei J, Han J, Chen L, Xiao Z, Chen B and Dai J, Biomaterials, 2013, 34, 3215–3222. [DOI] [PubMed] [Google Scholar]
- 219.Agarwal P, Zhao S, Bielecki P, Rao W, Choi JK, Zhao Y, Yu J, Zhang W and He X, Lab Chip, 2013, 13, 4525–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chan HF, Zhang Y and Leong KW, Small, 2016, 12, 2720–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Siltanen C, Diakatou M, Lowen J, Haque A, Rahimian A, Stybayeva G and Revzin A, Acta Biomater, 2017, 50, 428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Magin CM, Alge DL and Anseth KS, Biomed. Mater, 2016, 11, 022001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Chen Q, Utech S, Chen D, Prodanovic R, Lin JM and Weitz DA, Lab Chip, 2016, 16, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Griffin DR, Weaver WM, Scumpia PO, Di Carlo D and Segura T, Nat. Mater, 2015, 14, 737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Yu J, Huang TR, Lim ZH, Luo R, Pasula RR, Liao LD, Lim S and Chen CH, Adv. Healthc. Mater, 2016, 5, 2983–2992. [DOI] [PubMed] [Google Scholar]
- 226.Huang Q, Zou Y, Arno MC, Chen S, Wang T, Gao J, Dove AP and Du J, Chem. Soc. Rev, 2017, 46, 6255–6275. [DOI] [PubMed] [Google Scholar]
- 227.Geraili A, Jafari P, Hassani MS, Araghi BH, Mohammadi MH, Ghafari AM, Tamrin SH, Modarres HP, Kolahchi AR, Ahadian S and Sanati-Nezhad A, Adv. Healthc. Mater, 2017, 7, 1700426. [DOI] [PubMed] [Google Scholar]
- 228.Zhao X, Liu S, Yildirimer L, Zhao H, Ding R, Wang H, Cui W and Weitz D, Adv. Funct. Mater, 2016, 26, 2809–2819. [Google Scholar]
- 229.Yin H and Marshall D, Curr. Opin. Biotechnol, 2012, 23, 110–119. [DOI] [PubMed] [Google Scholar]
- 230.Hosic S, Murthy SK and Koppes AN, Anal. Chem, 2016, 88, 354–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Galler K, Brautigam K, Grosse C, Popp J and Neugebauer U, Analyst, 2014, 139, 1237–1273. [DOI] [PubMed] [Google Scholar]
- 232.Wang BL, Ghaderi A, Zhou H, Agresti J, Weitz DA, Fink GR and Stephanopoulos G, Nat. Biotech, 2014, 32, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Mazutis L, Gilbert J, Ung WL, Weitz DA, Griffiths AD and Heyman JA, Nat. Protoc, 2013, 8, 870–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Dolega ME, Abeille F, Picollet-D’hahan N and Gidrol X, Biomaterials, 2015, 52, 347–357. [DOI] [PubMed] [Google Scholar]
- 235.Ma Y, Neubauer MP, Thiele J, Fery A and Huck WTS, Biomater. Sci, 2014, 2, 1661–1671. [DOI] [PubMed] [Google Scholar]
- 236.Mao AS, Shin J-W, Utech S, Wang H, Uzun O, Li W, Cooper M, Hu Y, Zhang L, Weitz DA and Mooney DJ, Nat. Mater, 2017, 16, 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Lienemann PS, Rossow T, Mao AS, Vallmajo-Martin Q, Ehrbar M and Mooney DJ, Lab Chip, 2017, 17, 727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Kamperman T, Henke S, Visser CW, Karperien M and Leijten J, Small, 2017, 13, 1603711. [DOI] [PubMed] [Google Scholar]
- 239.Kamperman T, Henke S, van den Berg A, Shin SR, Tamayol A, Khademhosseini A, Karperien M and Leijten J, Adv. Healthc. Mater, 2017, 6, 1600913. [DOI] [PubMed] [Google Scholar]
- 240.Akbari S and Pirbodaghi T, Lab Chip, 2014, 14, 3275–3280. [DOI] [PubMed] [Google Scholar]
- 241.Fischlechner M, Schaerli Y, Mohamed MF, Patil S, Abell C and Hollfelder F, Nat. Chem, 2014, 6, 791–796. [DOI] [PubMed] [Google Scholar]
- 242.Novak R, Zeng Y, Shuga J, Venugopalan G, Fletcher DA, Smith MT and Mathies RA, Angew. Chem. Int. Ed, 2011, 50, 390–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Geng T, Novak R and Mathies RA, Anal. Chem, 2014, 86, 703–712. [DOI] [PubMed] [Google Scholar]
- 244.Allon Klein M, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, David Weitz A and Marc Kirschner W, Cell, 2015, 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Liu Q and Boyd BJ, Analyst, 2013, 138, 391–409. [DOI] [PubMed] [Google Scholar]
- 246.Kirsch J, Siltanen C, Zhou Q, Revzin A and Simonian A, Chem. Soc. Rev, 2013, 42, 8733–8768. [DOI] [PubMed] [Google Scholar]
- 247.Le Goff GC, Srinivas RL, Hill WA and Doyle PS, Eur. Polym. J, 2015, 72, 386–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kantak C, Zhu QD, Beyer S, Bansal T and Trau D, Biomicrofluidics, 2012, 6, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Liu EY, Jung S, Weitz DA, Yi H and Choi C-H, Lab Chip, 2018, 18, 323–334. [DOI] [PubMed] [Google Scholar]
- 250.Xie X, Zhang W, Abbaspourrad A, Ahn J, Bader A, Bose S, Vegas A, Lin J, Tao J and Hang T, Nano Lett, 2017, 17, 2015–2020. [DOI] [PubMed] [Google Scholar]
- 251.Zhao Y, Zhao X, Tang B, Xu W, Li J, Hu J and Gu Z, Adv. Funct. Mater, 2010, 20, 976–982. [Google Scholar]
- 252.Li H, Men D, Sun Y, Zhang T, Hang L, Liu D, Li C, Cai W and Li Y, Nanotechnology, 2017, 28, 405502. [DOI] [PubMed] [Google Scholar]
- 253.Shibata H, Heo YJ, Okitsu T, Matsunaga Y, Kawanishi T, Takeuchi S and Davis ME, Proc. Natl. Acad. Sci. USA, 2010, 107, 17894–17898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Choi TM, Park J-G, Kim Y-S, Manoharan VN and Kim S-H, Chem. Mater, 2015, 27, 1014–1020. [Google Scholar]
- 255.Kanai T, Lee D, Shum HC, Shah RK and Weitz DA, Adv. Mater, 2010, 22, 4998–5002. [DOI] [PubMed] [Google Scholar]
- 256.Lee SS, Kim B, Kim SK, Won JC, Kim YH and Kim SH, Adv. Mater, 2015, 27, 627–633. [DOI] [PubMed] [Google Scholar]
- 257.Lu AX, Oh H, Terrell JL, Bentley WE and Raghavan SR, Chem. Sci, 2017, 8, 6893–6903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Tan YC, Hettiarachchi K, Siu M and Pan YP, J. Am. Chem. Soc, 2006, 128, 5656–5658. [DOI] [PubMed] [Google Scholar]
- 259.Deshpande S, Caspi Y, Meijering AEC and Dekker C, Nat. Commun, 2016, 7, 10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Deng N-N, Yelleswarapu M and Huck WTS, J. Am. Chem. Soc, 2016, 138, 7584–7591. [DOI] [PubMed] [Google Scholar]
- 261.Elani Y, Law RV and Ces O, Nat. Commun, 2014, 5 5305. [DOI] [PubMed] [Google Scholar]
- 262.Matosevic S and Paegel BM, Nat. Chem, 2013, 5, 958–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Nuti N, Verboket PE and Dittrich PS, Lab Chip, 2017, 17, 3112–3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Deng NN, Yelleswarapu M, Zheng L and Huck WT, J. Am. Chem. Soc, 2017, 139, 587–590. [DOI] [PubMed] [Google Scholar]
- 265.Siontorou CG, Nikoleli GP, Nikolelis DP and Karapetis SK, Membranes, 2017, 7, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Pautot S, Frisken BJ and Weitz DA, P. Natl. Acad. Sci. USA, 2003, 100, 10718–10721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Martino C, Kim S-H, Horsfall L, Abbaspourrad A, Rosser SJ, Cooper J and Weitz DA, Angew. Chem. Int. Ed, 2012, 51, 6416–6420. [DOI] [PubMed] [Google Scholar]
- 268.Shum HC, Zhao Y-J, Kim S-H and Weitz DA, Angew. Chem. Int. Ed, 2011, 50, 1648–1651. [DOI] [PubMed] [Google Scholar]
- 269.Kim S-H, Nam J, Kim JW, Kim D-H, Han S-H and Weitz DA, Lab Chip, 2013, 13, 1351–1356. [DOI] [PubMed] [Google Scholar]
- 270.Weiss M, Frohnmayer JP, Benk LT, Haller B, Janiesch JW, Heitkamp T, Borsch M, Lira RB, Dimova R, Lipowsky R, Bodenschatz E, Baret JC, Vidakovic-Koch T, Sundmacher K, Platzman I and Spatz JP, Nat. Mater, 2018, 17, 89–96. [DOI] [PubMed] [Google Scholar]
- 271.Paterson DJ, Reboud J, Wilson R, Tassieri M and Cooper JM, Lab Chip, 2014, 14, 1806–1810. [DOI] [PubMed] [Google Scholar]
- 272.Elani Y, Solvas XC, Edel JB, Law RV and Ces O, Chem. Commun, 2016, 52, 5961–5964. [DOI] [PubMed] [Google Scholar]
- 273.Schlicht B and Zagnoni M, Sci. Rep, 2015, 5, 9951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Schwarz-Schilling M, Aufinger L, Muckl A and Simmel FC, Integr. Biol, 2016, 8, 564–570. [DOI] [PubMed] [Google Scholar]
- 275.Ho KK, Lee JW, Durand G, Majumder S and Liu AP, PLoS One, 2017, 12, e0174689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mark D, Haeberle S, Roth G, von Stetten F and Zengerle R, Chem. Soc. Rev, 2010, 39, 1153–1182. [DOI] [PubMed] [Google Scholar]
- 277.Amstad E, Datta SS and Weitz DA, Lab Chip, 2014, 14, 705–709. [DOI] [PubMed] [Google Scholar]
- 278.Chan HF, Ma S and Leong KW, Regen. Biomater, 2016, 3, 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Liu D, Zhang H, Fontana F, Hirvonen JT and Santos HA, Adv. Drug Deliv. Rev, 2017, 10.1016/j.addr.2017.08.003. [DOI] [PubMed]
- 280.Wang XY, Jin ZH, Gan BW, Lv SW, Xie M and Huang WH, Lab Chip, 2014, 14, 2709–2716. [DOI] [PubMed] [Google Scholar]
- 281.Armada-Moreira A, Taipaleenmaki E, Itel F, Zhang Y and Stadler B, Nanoscale, 2016, 8, 19510–19522. [DOI] [PubMed] [Google Scholar]