

Abstract
Objective:
Copy number variants (CNVs) were analyzed from next-generation sequencing data, with the aim of improving diagnostic yield in skeletal muscle disorder cases.
Methods:
Four publicly available bioinformatic analytic tools were used to analyze CNVs from sequencing data from patients with muscle diseases. The patients were previously analyzed with a targeted gene panel for single nucleotide variants and small insertions and deletions, without achieving final diagnosis. Variants detected by multiple CNV analysis tools were verified with either array comparative genomic hybridization or PCR. The clinical significance of the verified CNVs was interpreted, considering previously identified variants, segregation studies, and clinical information of the patient cases.
Results:
Combining analysis of all different mutation types enabled integration of results and identified the final cause of the disease in 9 myopathy cases. Complex effects like compound heterozygosity of different mutation types and compound disease arising from variants of different genes were unraveled. We identified the first large intragenic deletion of the titin (TTN) gene implicated in the pathogenesis of a severe form of myopathy. Our work also revealed a “double-trouble” effect in a patient carrying a single heterozygous insertion/deletion mutation in the TTN gene and a Becker muscular dystrophy causing deletion in the dystrophin gene.
Conclusions:
Causative CNVs were identified proving that analysis of CNVs is essential for increasing the diagnostic yield in muscle diseases. Complex severe muscular dystrophy phenotypes can be the result of different mutation types but also of the compound effect of 2 different genetic diseases.
Next-generation sequencing (NGS) methods have become the most common method for the genetic diagnosis of genetically heterogeneous disorders.1,2 We have previously developed a targeted NGS gene panel, MyoCap.1 An updated version used here includes probes for the exons of nearly 300 myopathy genes and candidate genes. Similar platforms are currently in use in many laboratories.2 The reported diagnostic success rates are significantly higher than those obtained by traditional gene-by-gene sequencing.2 However, over 50% patients remain undiagnosed when only concentrating on single nucleotide variants (SNVs) and small insertions and deletions (indels).2
Copy number variants (CNVs) are defined as genomic deletions or duplications greater than 1 kb in size.3 CNVs cause microdeletion and microduplication syndromes, and they have also been associated with several complex diseases.3,4 Generally, studies aiming for the identification of causative disease variants in skeletal muscle disorders have not systematically used CNV screening. Multiplex ligation-dependent probe amplification has a lower throughput when the amount of investigated genes increases.5 Array comparative genomic hybridization (aCGH) has long been considered the only reliable and robust platform for CNV discovery.4 However, in NGS studies, the diagnostic evaluation may end with a discovery of a single pathogenic or likely pathogenic mutation before the utilization of complementary methods, which may lead to an underestimation of CNV contribution to diseases. Recently, several CNV analysis tools for NGS data have been developed and are in use for routine diagnosis.3,4 Here, we describe the detection of CNVs from NGS data with a combination of already available bioinformatic tools.
METHODS
Standard protocol approvals, registrations, and patient consents.
DNA samples of muscle disease patients and healthy family members were obtained from clinicians in different countries. The study was approved by the Coordinating Ethics Committee of the Hospital District of Helsinki and Uusimaa. The samples were obtained according to the Helsinki declaration. Written informed consent was obtained from all patients.
CNV assessment from NGS data.
CNVs were analyzed in smaller batches from NGS data alignment files (.bam) obtained by analyzing DNA from 791 myopathy patients with MyoCap.1 We used 4 CNV analysis programs: Copy Number Inference From Exome Reads (CoNIFER) v0.2.2,6 eXome-Hidden Markov Model (XHMM) v1.1,7 ExomeDepth v1.1.10,8 and COpy number Detection by EXome sequencing (CODEX) v1.4.0,9 with the recommended default settings of each program. A minimum of 1-bp overlap was used to determine whether calls intersecting between different programs originated from the same CNV. In this article, we prioritize the specific cases with a very high clinical interest, focusing on 7 variants detected by multiple programs and verified by using independent tools.
CNV validation.
PCR was performed to confirm CNVs in patients I, IIIa, IIIb, IV, V, VI, and VII. Primers were designed using Primer3 v4.0.0 (primer3.ut.ee) (table e-1, http://links.lww.com/NXG/A12), and PCR was performed with DreamTaq DNA Polymerase (Thermo Fisher Scientific, Waltham, MA) (figure e-1). A custom aCGH (manuscript in preparation), investigating 187 of the genes included in MyoCap, was used to confirm CNV detected in patient IIa. Segregation study in the family of patient IIa was performed by PCR (table e-1).
RESULTS
Table 1 shows the clinical features of patients in this study, genetic findings, amount of programs that detected the CNV, and the CNV verification method.
Table 1.
Patient and variant features

Patient I was identified to have a heterozygous FINmaj mutation, an 11-bp insertion/deletion in the titin (TTN) gene.10 This variant causes dominant tibial muscular dystrophy, characterized by a late age at onset, normal or slightly elevated creatine kinase (CK) levels, and a mild distal phenotype. However, this patient has proximal weakness and a very high CK level. We excluded the presence of other deleterious variants in TTN. Surprisingly, the patient was found to have a previously reported dystrophin (DMD) deletion (exons 45–55) known to cause Becker muscular dystrophy (BMD).11
Patient IIa has a previously reported Iberian frameshift mutation, p.(Lys35963Asnfs*), in the last exon of TTN usually determining 1 component of the recessive distal titinopathy phenotype.10 Compared with the other carriers of this variant, this patient has a more severe disease progression with proximal weakness, loss of ambulation before the age of 40 years, and marked hyperCKemia. The patient had no further causative SNVs or indels in the TTN gene. We identified a large deletion in TTN (exons 34–41; figure 1A) in trans with the Iberian frameshift variant in the proband IIa as well as in patient IIb, a similarly affected brother. Their healthy relatives are heterozygous for only one of the aforementioned TTN mutations demonstrating the recessive effect of the detected deletion. The severe distal and proximal titinopathies were thus caused by the compound heterozygosity of the Iberian frameshift and the deletion (figure 2).
Figure 1. Visualizations of copy number variants of patients IIa and IIIa.

CoNIFER visualizations of a heterozygous deletion in the TTN in patient IIa (A) and a homozygous deletion in the SGCD in patient IIIa (B) accompanied with corresponding regions visualized with Integrative Genomics Viewer (IGV). In CoNIFER visualizations, the red line corresponds to the read depths for sample with deletion and black lines correspond to control samples. In the IGV visualization, the first row shows the sample with deletion and the second row shows a control sample without deletion.
Figure 2. Pedigree of patients IIa and IIb.
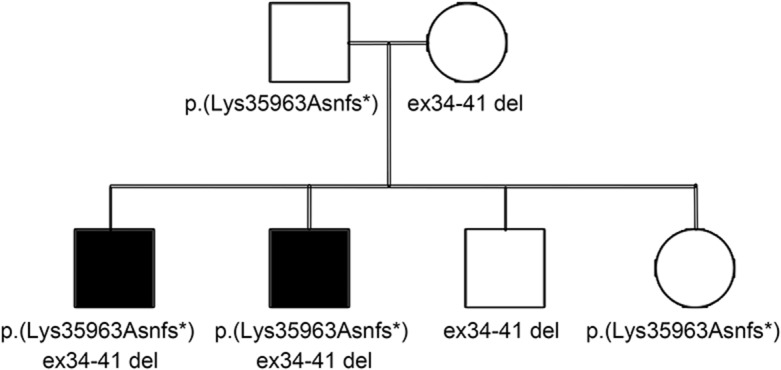
Segregation of the TTN Iberian tibial muscular dystrophy variant, p.(Lys35963Asnfs*), and deletion in the TTN (exons 34–41) in the family of patients IIa and IIb. The affected brothers IIa and IIb (black squares) are compound heterozygous for the variants. Their healthy parents and siblings are heterozygous for only one of the variants. Genetic findings are described using the reference transcript NM_001267550.1 for TTN.
Patients IIIa and IIIb, with a severe limb-girdle muscular dystrophy (LGMD) phenotype, are the daughters of first-cousin parents. Sanger sequencing of candidate genes (CAPN3 and ANO5), MyoCap, and whole-exome sequencing had been performed without identifying the causative variant. All the CNV detection programs identified a homozygous deletion in the SGCD gene (exons 1–5) (figure 1B).
Patient IV is the child of consanguineous parents, and he has been suffering from lower limb muscular weakness in the lower limbs since the age of 7. Deletions and duplications in the DMD gene had been excluded. Immunochemistry showed normal staining for dystrophin as well as for the sarcoglycans. A homozygous deletion in CAPN3 (exons 2–8) was detected.
In 2 males with an LGMD-like phenotype (patients V and VI), we found previously reported DMD deletions explaining the observed proximal muscular weakness. Both patients have in-frame deletions (exons 45–55 and exons 45–48) causing BMD.11
Patient VII with a Duchenne phenotype was also included in our screening. As expected, a previously reported out-of-frame DMD deletion (exons 42–43) was identified.11
DISCUSSION
Genomes are usually analyzed for SNVs and indels, but studies of CNVs are often underrepresented. CNVs in muscle diseases have previously been studied with complementary methods like targeted aCGH, which still remains the gold standard technique for CNV detection.4,5 Here, we show that combining analysis of all different mutation types enables integration of results and identifies the final cause of the disease in several cases. Complex effects like compound heterozygosity in patients IIa and IIb and compound genetic disease arising from variants of different genes in patient I can be unraveled. It has been suggested that the number of patients with a combination of 2 or more genetic diseases is probably underestimated.12 The phenotypic complexity in these patients may erroneously be interpreted as a new genetic disease with unidentified genetic defect or as a phenotypic expansion for a single disease.12 The proximal phenotype seen in patient I is mainly due to BMD, as the typical anterior lower-leg muscle lesions of the FINmaj TTN mutation may develop only after age 60. However, the identification of multilocus genomic variants is crucial for a proper genetic counseling in the family.
The combination of 4 analysis tools aided us to identify already known and previously unknown CNVs. CNVs can explain some of the missing heritability and undiagnosed cases in skeletal muscle disorders. An NGS-based strategy for CNV detection will be of great value for increasing the diagnostic yield in patients affected by mendelian muscle diseases. Current technology does not adequately capture repeat expansion diseases such as myotonic dystrophy or diseases related to other repetitive elements. Long-read sequencing technologies may further help the identification and mapping of CNVs as well as of repeat expansions. However, the clinical interpretation of CNVs remains challenging, in particular for CNVs identified in genes without previously reported disease causing deletions or duplications. The inclusion of CNV data in public databases, e.g., ExAC, could help in pathogenicity assessment of CNVs.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Merja Soininen and Helena Luque for their technical help and Sini Penttilä, Tiina Suominen, and Sara Lehtinen for acquisition of samples. They also thank all the patients and family members as well as the clinicians who provided samples.
GLOSSARY
- aCGH
array comparative genomic hybridization
- BMD
Becker muscular dystrophy
- CK
creatine kinase
- CNV
copy number variant
- indels
insertions and deletions
- LGMD
limb-girdle muscular dystrophy
- NGS
next-generation sequencing
- SNV
single nucleotide variant
AUTHOR CONTRIBUTIONS
Salla Välipakka: study concept and design, acquisition of data, analysis and interpretation of data, and drafting the manuscript for intellectual content. Marco Savarese and Mridul Johari: study concept and design, acquisition of data, analysis and interpretation of data, and revising the manuscript for intellectual content. Lydia Sagath, Meharji Arumilli, and Kirsi Kiiski: acquisition of data, analysis and interpretation of data, and revising the manuscript for intellectual content. Amets Sáenz, Adolfo Lopez De Munain, and Ana-Maria Cobo: analysis and interpretation of data. Katarina Pelin, Bjarne Udd, and Peter Hackman: study concept and design and revising the manuscript for intellectual content.
STUDY FUNDING
This study was supported by the Folkhälsan Research Foundation, the Jane and Aatos Erkko Foundation, the Academy of Finland (no. 138491, B.U.), the Sigrid Jusélius Foundation, the Association Française contre les Myopathies, and the Orion Research Foundation sr.
DISCLOSURE
S. Välipakka has received research support from the Folkhälsan Research Foundation. M. Savarese has received research support from the Association Française contre les Myopathies and Orion Research Foundation. M. Johari, L. Sagath, M. Arumilli, and K. Kiiski report no disclosures. A. Sáenz has received research support from Health Research Fund (PI13-00722) of the Spanish Ministry of Economy and Competitiveness, the European Unión (European Regional Development Fund). A. Lopez De Munain has received travel funding from Sanofi. A. Cobo and K. Pelin report no disclosures. B. Udd has served on the editorial board of Neuromuscular Disorders and has received grants from Finska Läkaresällskapet (20,000 euros 2017), the Sigrid Juselius Foundation (general grant for the study of myopathies), the Jane and Aatos Erkko Foundation, and the Vasa Central Hospital Research Foundation (grant partly for myopathies). P. Hackman reports no disclosures. Go to Neurology.org/ng for full disclosure forms.
REFERENCES
- 1.Evila A, Arumilli M, Udd B, Hackman P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord 2016;26:7–15. [DOI] [PubMed] [Google Scholar]
- 2.Nigro V, Savarese M. Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr Opin Neurol 2016;29:621–627. [DOI] [PubMed] [Google Scholar]
- 3.Pirooznia M, Goes FS, Zandi PP. Whole-genome CNV analysis: advances in computational approaches. Front Genet 2015;6:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tan R, Wang Y, Kleinstein SE, et al. An evaluation of copy number variation detection tools from whole-exome sequencing data. Hum Mutat 2014;35:899–907. [DOI] [PubMed] [Google Scholar]
- 5.Piluso G, Dionisi M, Del Vecchio Blanco F, et al. Motor chip: a comparative genomic hybridization microarray for copy-number mutations in 245 neuromuscular disorders. Clin Chem 2011;57:1584–1596. [DOI] [PubMed] [Google Scholar]
- 6.Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res 2012;22:1525–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fromer M, Moran JL, Chambert K, et al. Discovery and statistical genotyping of copy-number variation from whole-exome sequencing depth. Am J Hum Genet 2012;91:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Plagnol V, Curtis J, Epstein M, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012;28:2747–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang Y, Oldridge DA, Diskin SJ, Zhang NR. CODEX: a normalization and copy number variation detection method for whole exome sequencing. Nucleic Acids Res 2015;43:e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hackman P, Marchand S, Sarparanta J, et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul Disord 2008;18:922–928. [DOI] [PubMed] [Google Scholar]
- 11.Bladen CL, Salgado D, Monges S, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 duchenne muscular dystrophy mutations. Hum Mutat 2015;36:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 2017;376:21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.