

Abstract
Mitochondrial DNA (mtDNA) is a core non-nuclear genetic material found in all eukaryotic organisms, the size of which varies extensively in the eumycota, even within species. In this study, mitochondrial genomes of six isolates of Annulohypoxylon stygium (Lév.) were assembled from raw reads from PacBio and Illumina sequencing. The diversity of genomic structures, conserved genes, intergenic regions and introns were analyzed and compared. Genome sizes ranged from 132 to 147 kb and contained the same sets of conserved protein-coding, tRNA and rRNA genes and shared the same gene arrangements and orientation. In addition, most intergenic regions were homogeneous and had similar sizes except for the region between cytochrome b (cob) and cytochrome c oxidase I (cox1) genes which ranged from 2,998 to 8,039 bp among the six isolates. Sixty-five intron insertion sites and 99 different introns were detected in these genomes. Each genome contained 45 or more introns, which varied in distribution and content. Introns from homologous insertion sites also showed high diversity in size, type and content. Comparison of introns at the same loci showed some complex introns, such as twintrons and ORF-less introns. There were 44 short fragment insertions detected within introns, intergenic regions, or as introns, some of them located at conserved domain regions of homing endonuclease genes. Insertions of short fragments such as small inverted repeats might affect or hinder the movement of introns, and these allowed for intron accumulation in the mitochondrial genomes analyzed, and enlarged their size. This study showed that the evolution of fungal mitochondrial introns is complex, and the results suggest short fragment insertions as a potential factor leading to larger mitochondrial genomes in A. stygium.
Keywords: intron insertion site, Twintron, ORF-less intron, small inverted repeat, mitochondrial genome
Introduction
The mitochondrial organelle is found in most eukaryotic organisms (Karnkowska et al., 2016). Mitochondria are thought to have had a monophyletic origin from a vestigial endosymbiotic proteobacterial ancestor, but the size and the gene content of mitochondria are much less than those of free-living proteobacteria (Adams and Palmer, 2003). Some mitochondrial genes have been transferred to the host nucleus, while other genes have been lost (compared to extant proteobacteria), likely because there are genes with similar function in the nuclei of eukaryotes (Adams and Palmer, 2003). Common characteristics of mitochondrial DNA (mtDNA) are the high A+T content, lack of methylation, conservation of the gene function, and high copy number (Campbell et al., 1999).
With the rapid development of high-throughput sequencing technologies, increasingly more fungal mitochondrial genomes have been sequenced and characterized. The reported genomes usually contain a similar set of conserved genes, including 14 protein-coding genes (nad1, nad2, nad3, nad4, nad4L, nad5, nad6, atp6, atp8, atp9, cox1, cox2, cox3, and cob), a set of tRNAs, and small and large ribosomal RNA (rns and rnl). However, the size of fungal mtDNAs varies greatly. The smallest mitochondrial genomes, such as those of Harpochytrium sp. and Candida orthopsilosis, are around 20 kb in size with few or no introns and few or no non-conserved genes. Several large mitochondrial genomes in fungi (more than 150 kb) have been released in the organelle genome database in NCBI recently, including those of Rhizoctonia solani AG-1 and AG3 (Losada et al., 2014), Sclerotinia borealis F-4128 (Mardanov et al., 2014), Pyronema omphalodes CBS 100304 (Nowrousian, 2016), Chrysoporthe austroafricana (Kanzi et al., 2016), Ustilago bromivora UB2112, Phellinus noxius FFPRI411160, Ophiocordyceps sinensis (Kang et al., 2017), and Phlebia radiata 79 (Salavirta et al., 2014). One common features among large fungal mtDNAs is that they contain many introns. Comparison of other mtDNAs also reveals that intron number and length is positively correlated to mtDNA size, and is the primary source of size variation (Deng et al., 2016b). At present, the mechanisms of why and how larger mtDNAs evolved and more introns were retained remains unknown.
Mitochondrial introns usually consist of a catalytically active intron RNA and a small intron-encoded protein (IEP; Michel and Ferat, 1995). The RNA functions as a ribozyme to catalyze its own splicing, yielding spliced exons and an excised intron. Based on RNA secondary structure, introns in fungal mtDNAs are classified into two major groups: I and II (Saldanha et al., 1993). Group I introns are subdivided into seven specific types: IA, IA3, IB, IC1, IC2, ID, and group I derived (I*) (Lang et al., 2007). Group I introns are widely distributed, probably through horizontal transfer, and the driving force is presumed to be supplied by homing endonuclease (HE) genes, which they carry or are from the host (Gonzalez et al., 1998). The HE genes encode enzymes to promote intron mobility via introducing doubled stranded breaks at specific target sites. Based on conserved amino acid motifs, two families of HE genes are usually encoded by the open reading frame (ORF) of Group I introns, namely the LAGLIDADG and GIY-YIG proteins. The IEP of the group II intron is a multifunctional reverse transcriptase, which stabilizes the catalytically active RNA structure when splicing, and reverse transcribes its hosts intron into new DNA (Lambowitz and Zimmerly, 2011).
Twintron refers to a intron complex with an existing intron containing an insertion of a mobile intron (Bullerwell et al., 2003). This term was first proposed by Copertino and Hallick (Copertino and Hallick, 1991) based on chloroplast psbF gene research in Euglena gracilis. Since then, several categories of twintrons have been identified. According to the group types of internal and external introns, a twintron can be group I internal to group I (Einvik et al., 1998), group II internal to group II (Copertino and Hallick, 1991), group I internal to group II (Hafez et al., 2013), and group II internal to group I. A twintron can also be a complex of multiple introns, with excisions yielding three or more individual introns (Drager and Hallick, 1993). Although major twintrons have been reported in the chloroplast genome of Euglena, the genetic elements have also been uncovered recently in fungal mitochondrial genomes. Two twintrons have been detected in the positions of mS917 and mS1247 in the rns gene of the Cryphonectria parasitica mitochondrial genome. The mS917 twintron is an ORF-less group ID intron invaded by another ID intron encoding LAGLIDADG in the ORF. The mS1247 twintron was the first recorded mixed twintron among fungal mitochondrial genomes, and could be excised into a group II internal intron and a group I external intron (Hafez et al., 2013). Cox3-i2 in mtDNA of Hypomyces aurantius H.a 0001, is a new twintron complex (Deng et al., 2016b). Instead of one invaded by the other, the two group IA introns within this twintron complex are arranged side by side (Deng et al., 2016b).
The genus Annulohypoxylon, previously referred to as Hypoxylon sect. Annulata (Suwannasai et al., 2013), belongs to Xylariaceae, Ascomycota. Among its 45 currently accepted species, many have been isolated from plants as endophytes or as pathogens (Stone et al., 2004). Annulohypoxylon, as well as other genera in Xylariaceae has attracted natural products researchers in recent decades for the high diversity of secondary metabolites, which are highly specific for certain taxa or taxonomic groups as chemotaxonomic markers (Whalley and Edwards, 1995). Aside from identification of natural products for taxonomic and medicinal objectives (Maciel et al., 2017), A. stygium has been reported to be a potential candidate for glycol hydrolase production due to its high pectinase, xylanase and β-glucosidase activities under specific conditions (Robl et al., 2015). A. stygium is also identified as associated with Tremella fuciformis, providing nutrition for growth and development of the commercially valued fruit bodies (Deng et al., 2016a).
In this study, the complete mitochondrial genome sequencing of six isolates of A. stygium was conducted. Mitochondrial reads were isolated, and assembled into complete circular molecules. Mitochondrial genome comparisons were performed to analyze the diversity of genomic structures, conserved genes, intergenic regions and introns. The purpose of the study was to elucidate the evolution of complex introns, and elucidate possible forces driving mitochondrial genome enlargements in A. stygium.
Materials and methods
Isolates and genome sequencing
Six isolates of A. stygium were used in this study. Isolate As31 was obtained from the Edible Fungal Germplasm Resources Management Center of Fujian province, Fuzhou, China. It is a companion fungus of a major T. fuciformis commercial cultivar Tr21. The other five isolates, As03, As15, As23, As24, and As28 were isolated from rotting wood substrates associated with naturally growing T. fuciformis fruit bodies in different areas of Fujian, China. Following Hsieh et al. (2005), concatenation of beta-tubulin and actin gene sequences was used for species designations of the isolates. Corresponding sequences of Annulohypoxylon urceolatum (AY951670 and AY951782), A. stygium 90081906 (AY951667 and AY951775), A. stygium 90041409 (AY951666 and AY951776), A. stygium var. annulatum 3 (AY951669 and AY951777) and A. stygium var. annulatum 91042205 (AY951668 and AY951778) were employed as controls. Sequences were aligned using Clustal W with gap opening penalty of 5 and gap extension penalty of 2. The distances were computed using the Neighbor-joining method. All positions containing gaps and missing data were eliminated from phylogenetic analysis. Evolutionary analyses were conducted in Mega 7 (Kumar et al., 2016). After growth in potato dextrose broth at 28°C and 180 rpm for 48 h, the hyphae were collected by filtration through Whatman #1 filter paper, and washed three times with sterilized distilled water. Total genomic DNA was isolated using the CTAB method (Manicom et al., 1987).
PacBio Sequencing was used to obtain a complete mitochondrial genome of As31 as well as the nuclear genome. Three SMRT cells were used in the PacBio RS II system, to obtain 3.5 GB raw data with a mean read length of 4.8 kb. In addition, an Illumina sequencing strategy was used to sequence all six isolates. Paired-end sequencing libraries with an insert size of ~500 bp were constructed, and then sequenced to 3–4 Gb raw data per isolate with 250 bp paired-end reads using the Illumina Hiseq 2500 system. All sequencing was done at the Genome and Biotechnology Research Center, Fujian Agriculture and Forestry University.
Genome assembly
The PacBio reads of As31 were assembled into contigs using the software Canu 1.2 (Koren et al., 2017). Mitochondrial related contigs were identified using local BLASTX against a database of known mitochondrial proteins from closely related species. The contigs were assembled into a finished circle molecule by overlap manually. Some small indels or polymorphisms were corrected by mapping short reads from Illumina sequencing onto the PacBio genome. Illumina reads of the five other isolates were assembled into draft genome sequences using the software SPAdes-3.8.1 (Bankevich et al., 2012). Mitochondrial related contigs were isolated via doing BLASTN of whole genomic contigs against mtDNA of As31, and then assembled into circular molecules using paired-end read relationships. Remaining gaps were resolved by conventional PCR with Sanger sequencing.
Genome annotation
Annotation of mitochondrial genomes was performed using the automated annotation program MFannot (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl). Borders between exons and introns were confirmed manually using BLASTP against NCBI protein databases and codon usage table 4 (Mold, Protozoan, and Coelenterate Mitochondrial Code) following Haridas and Gantt (2010), or by comparing with intron-less mtDNA in corresponding insertion sites. The intron types were identified using the RNA-Weasel software (http://megasun.bch.umontreal. ca/RNAweasel) (Lang et al., 2007). The types of intron-encoded ORFs were identified by searching the NCBI conserved domain database. Comparison of introns within same insertion site was performed using the online programs Kalign or Clustal Omega (both can be found at https://www.ebi.ac.uk/Tools/msa/; Li et al., 2015). Insertions of short fragments among these six tested mtDNAs were identified by multiple sequence alignments using Kalign or Clustal Omega. Hairpin structures of short inserted fragments were detected by using the online program Mfold (Zuker et al., 1999). A DNA sequence of hairpin structure with continuous matched stem length >5 and a loop size < 11 was regarded as a small inverted repeat (SIR; Lavrov et al., 2012), and the SIR counts for each genome was tabulated.
Results
Species designation
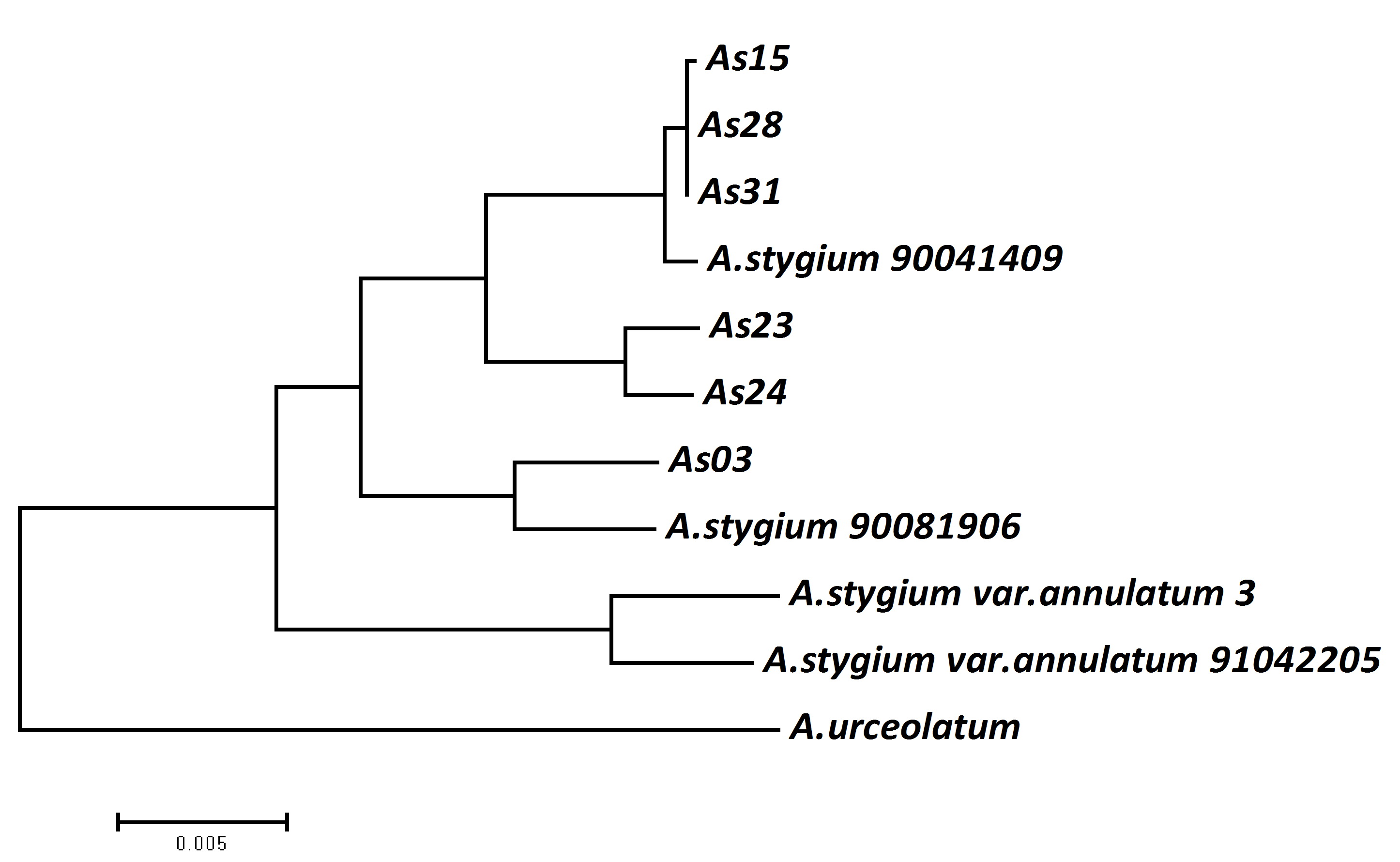
Based on the most parsimonious phylogenetic tree of the concatenation of beta-tubulin and actin gene sequences, 11 strains were clustered into three clades; the A. stygium clade, the A. stygium var. annulatum clade, and the A. urceolatum clade. All six isolates were located in the A. stygium clade (Figure S1).
Assembly of mtDNA
A 144,034 bp mitochondrial contig was assembled from the 3.5 Gb PacBio reads of As31. Sequence comparison of the ends of the contig showed a 10,285-bp repeat, allowing for joining to a 133,749-bp circular molecule. Twelve singleton indel differences were detected by mapping Illumina reads onto the PacBio assembly. All these differences existed in single nucleotide repeat areas and were corrected to match the Illumina data, finally yielding a mitochondrial genome of As31 with a circular DNA of 133,761 bp (Figure 1).
Figure 1.
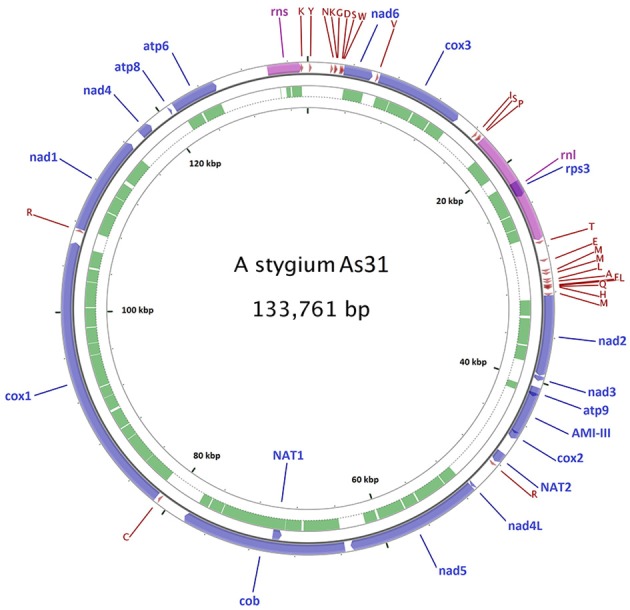
Diagram of the As31 mitochondrial genome. Fifteen conserved protein-coding genes, 26 tRNAs, small and large subunit rRNAs, and three transferase genes are shown. Blue arrows represent protein coding genes; pink arrows represent rRNAs, dark red arrows represent tRNA; green arcs represent introns of conserved protein-coding genes and rRNAs. The other five mtDNAs share similar diagrams as As31, including gene positions, orientation and order.
Using the mtDNA genome of As31 as a reference, mitochondria-related contigs of the five other isolates were extracted from the whole draft genome Illumina assemblies generated using SPAdes-3.8.1. The contigs of As03, As15, As23, As24, and As28 were assembled into circular mtDNAs of 145,449, 131,996, 147,325, 140,328, and 143,806 bp, respectively, leaving one gap in As03 and two gaps in As24 (Figure S2). Primers were designed at the gap ends to bridge the gaps by PCR sequencing. However, the Sanger sequencing did not provide complete sequences, perhaps due to special secondary structures, and the gaps have been filled with N's in the presumed circular sequences.
Gene content
From the sequence comparisons, the same sets of 15 conserved protein-coding genes, 26 tRNAs, and small and large subunit rRNAs were found in mitochondrial DNAs of all six isolates (Table 1). The conserved protein-coding genes included ATP synthase subunits 6, 8, and 9 (atp6, atp8, and atp9), the cytochrome oxidase subunits 1, 2, and 3 (cox1, cox2, and cox3), NADH dehydrogenase subunits 1, 2, 3, 4, 5, 6, and 4L (nad1, nad2, nad3, nad4, nad5, nad6, and nad4L), apocytochrome b (cob), and one ribosomal protein (rps3 in rnl). Twenty-six tRNA genes were comprised of three copies of tRNA-met encoding genes, two for each of tRNA-ser, tRNA-arg, tRNA-leu, and tRNA-lys and the other 15 standard tRNAs, most of which were located between rnl-nad2 (11), rns-nad6 (8), and cox3-rnl (3). All of these genes were predicted from the same forward strand (Figure 1 for As31). The syntenic order of the conserved genes was identical for all isolates. Most intergenic regions were homogeneous and had similar sizes except for the region between cytochrome b (cob) and cytochrome c oxidase I (cox1) genes which ranged from 2,998 to 8,039 bp among the six isolates.
Table 1.
Comparison of mitochondrial genomes of six isolates of Annulohypoxylon stygium.
| Isolates | Mitochondrial genome size (bp) | GC % | Conserved protein-coding genes | tRNA | rRNA | intron | Intergenic region (bp) | Accession | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number | Length | Number | Size (bp) | Number | Size (bp) | Number | Size (bp) | |||||
| As03 | 145,449 | 30.17 | 15 | 15,003 | 26 | 1,938 | 2 | 4,341 | 49 | 87,530 | 38,059 | MH620790 |
| As15 | 131,996 | 29.81 | 15 | 15,012 | 26 | 1,938 | 2 | 4,426 | 47 | 79,669 | 32,373 | MH620791 |
| As23 | 147,325 | 29.77 | 15 | 15,015 | 26 | 1,938 | 2 | 4,431 | 47 | 92,443 | 34,920 | MH620792 |
| As24 | 140,328 | 30.05 | 15 | 15,015 | 26 | 1,938 | 2 | 4,430 | 47 | 85,627 | 34,740 | MH620793 |
| As28 | 143,806 | 30.18 | 15 | 15,012 | 26 | 1,938 | 2 | 4,424 | 48 | 88,863 | 34,991 | MH620794 |
| As31 | 133,761 | 29.86 | 15 | 15,012 | 26 | 1,938 | 2 | 4,427 | 45 | 81,437 | 32,369 | NC_023117 |
rps3 (1,422 bp) located in intron of rnl, which was counted both as conserved protein-coding gene and intron in the table.
Three transferase genes as well as a transferase-related fragment were predicted from mtDNA of A. stygium As31 (Figure 1). There were two copies of putative N-acetyl-transferase genes located downstream of cox2 and the third intron of cob (cob-i3). An aminotransferase class-III like gene (containing two short introns) was in the region between atp9 and cox2, and a fragment encoding AAT_I super family domain was located within the area between atp6 and rns. These genes and fragments were identified in mtDNAs of all six sequenced isolates.
Intron content and distribution
Different from the conserved genes and transferase genes, the content and distribution of introns within conserved genes varied greatly among isolates (Table 2). Introns within the same insertion site sometimes differed from each other in size and content. Sixty-five intron insertion sites were found in 13 conserved genes: cox1 was found to have 17 sites; rnl had eight; nad5 had seven each; cob and cox3 had six; rns and nad2 had five each; nad1 had four; atp6 had three; and single sites were also found in nad4, nad6, atp9, and cox2. According to the length and similarity (92% identity), nad1-i4 had five different intron types; cox1-i2 and cox1-i11 contained four types; five introns had three types; 14 introns had two types; and the other 43 introns were singletons. As a result a total of 99 different introns were identified from the six isolates. They varied greatly in size, ranging from 12 bp for rns-i2 (As03) to 6,259 bp for cob-i3 (As31).
Table 2.
Information on introns.
| Name | Position (aa/bp) | Type | Presence | Length (bp) | Lowest similarity | Intron type | Conserved domain | ORF | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| As03 | As15 | As23 | As24 | As28 | As31 | ||||||||
| nad6-i1 | 80 | I | + | + | + | + | + | + | 1,911–1,923 | 97.3 | ID | L1 | |
| cox3-i1 | 73 | I | + | + | – | – | + | + | 1,330–1,337 | 94 | IB | L1 | orf262 |
| cox3-i2 | 73–76 | I | – | – | + | + | – | – | 2,496 | 100 | IB | L1; L1 | orf262; orf241 |
| cox3-i3 | 92 | I | – | + | – | – | + | + | 2,378 | 100 | II | RT | orf547 |
| cox3-i4 | 143 | I | + | + | + | + | + | + | 1,681–1,689 | 99 | ID | double L1 | orf405 |
| cox3-i5 | 183 | I | + | + | + | + | + | + | 1,640–1,649 | 98.5 | IA | double L1 | orf362 |
| cox3-i6 | 214 | I | – | – | + | + | – | – | 1,779 | 99.9 | IA | L1 | orf266 |
| II | + | – | – | – | – | – | 1,743 | – | IA | L1 | orf254 | ||
| rnl-i1 | 305 | I | – | – | + | + | – | – | 2,562 | 99.96 | II | RVT | orf489 |
| II | + | – | – | – | – | – | 3,048 | – | II | RVT | orf625 | ||
| rnl-i2 | 510 | I | + | – | – | + | – | – | 1,530 | 99.74 | I(derived) | GIY-YIG | orf225 |
| rnl-i3 | 743 | I | + | + | – | – | + | – | 1,862–1,863 | 99.7 | IC1 | GIY-YIG | orf253 |
| II | – | – | – | + | – | – | 521 | – | IC1 | – | – | ||
| rnl-i4 | 1,086 | I | – | – | – | – | + | + | 2,289 | 100 | IC1 | double L1 | – |
| II | – | + | + | + | – | – | 464–466 | 99.1 | – | – | – | ||
| III | + | – | – | – | – | – | 1,022 | – | IC1 | – | |||
| rnl-i5 | 1,277 | I | + | – | – | – | – | – | 1,317 | – | IB | L1 | orf182 |
| rnl-i6 | 2,291 | I | + | + | + | + | + | + | 2,709–2,717 | 97.7 | rps3 | – | rps3 |
| rnl-i7 | 2,372 | I | + | – | – | – | – | – | 1,640 | – | IC2 | double L1 | orf450 |
| II | – | + | + | – | + | + | 1,314 | 99.8 | IC2 | double L1 | orf382 | ||
| III | – | – | – | + | – | – | 1,362 | – | IC2 | double L1 | orf382 | ||
| rnl-i8 | 2,428 | I | – | + | + | + | + | + | 1,144–1,148 | 99.6 | IA | L2 | orf261 |
| nad2-i1 | 125 | I | – | + | + | + | + | + | 1,487–1,490 | 98.7 | IC2 | double L1 | orf416 |
| II | + | – | – | – | – | – | 1,454 | – | IC2 | double L1 | – | ||
| nad2-i2 | 210 | I | – | + | + | + | + | + | 2,548 | 98.9 | II | RT | orf697 |
| nad2-i3 | 253 | I | + | + | + | + | + | + | 1,397 | 99.6 | – | double L1 | – |
| nad2-i4 | 329 | I | – | – | – | + | – | – | 2,403 | – | II | RT | orf711 |
| nad2-i5 | 566 | I | + | – | – | – | – | – | 855 | – | – | L1 | orf207 |
| atp9-i1 | 60 | I | + | – | – | – | – | – | 519 | – | IA | – | – |
| II | – | + | – | – | + | + | 667 | 100 | IA | – | – | ||
| III | – | – | + | + | – | – | 673–675 | 99.7 | IA | – | – | ||
| cox2-i1 | 77 | I | – | + | – | – | + | – | 1,134 | 100 | IB | GIY-YIG | orf250 |
| nad5-i1 | 83 | I | + | – | – | – | – | – | 2,012 | – | – | double L1 | orf485 |
| nad5-i2 | 108 | I | + | + | + | + | + | + | 1,372 | 99.4 | IC2 | double L1 | orf376 |
| nad5-i3 | 138 | I | – | + | – | – | – | + | 2,895–2,896 | 99.9 | ID | double L1; double L2 | – |
| II | – | – | + | + | + | – | 4,523–4,524 | 99.9 | – | double L1; double L2; GIY–YIG | orf405 | ||
| nad5-i4 | 187 | I | + | + | + | + | + | + | 1,307–1,312 | – | IB | L1 | orf340 |
| nad5-i5 | 233 | I | + | + | + | + | + | + | 2,566–2,574 | 97.7 | ID; ID | – | orf165; orf132 |
| nad5-i6 | 304 | I | + | + | + | + | + | + | 1,210 | 99.6 | IB | L1 | org380 |
| nad5-i7 | 468 | I | + | – | – | – | – | – | 42 | – | – | – | – |
| cob-i1 | 67 | I | – | + | + | – | + | + | 3,482–3,489 | 99.5 | IB(5') | L1; L1 | orf529; orf461 |
| cob–i2 | 134 | I | + | + | + | + | + | + | 1,562–1,564 | 99.3 | ID | GIY-YIG | orf216 |
| cob-i3 | 143 | I | – | – | + | + | – | – | 4,153–4,155 | 99.9 | ID; IB | double L1 | – |
| II | + | + | – | – | + | + | 6,251–6,259 | 99.5 | ID; IB | double L1 | – | ||
| cob-i4 | 167 | I | + | + | + | + | + | + | 1,244–1,260 | 94.7 | IA | – | orf222 |
| cob-i5 | 207 | I | + | + | + | + | + | + | 933–934 | 96 | ID | L2 | orf219 |
| cob-i6 | 277 | I | + | – | – | – | – | – | 2,156 | – | IB | L2 (b); GIY-YIG | orf296 |
| cox1-i1 | 59 | I | + | – | – | – | + | – | 2,858 bp | 99.8 | II | RVT | orf852 |
| cox1-i2 | 73 | I | + | + | – | – | + | – | 1,263 | 99.4 | IB | L1 | – |
| II | – | – | – | + | – | – | 2,561 | – | ID; IB | double L1; L1 | orf309; – | ||
| III | – | – | – | – | – | + | 2,693 | – | IC2; IB | double L1; L1 | orf401; orf194 | ||
| cox1-i3 | 82 | IV | – | – | + | – | – | – | 4,039 | – | ID; IC2; IB | double L1; double L1; L1 | orf309; orf417; orf194 |
| I | + | – | – | – | – | – | 2,606–2,822 | – | II | RT | orf675 | ||
| II | – | + | + | – | + | + | 2,676–2,679 | 100 | II | RT | orf675 | ||
| III | + | 2,822 | – | II | RT | orf675 | |||||||
| cox1-i4 | 93 | I | + | – | – | – | – | – | 2,596 | – | IB | double L1 | – |
| cox1-i5 | 98 | I | + | + | + | + | + | + | 1,138 | 100 | IB | L1 | orf316 |
| cox1-i6 | 131 | I | + | + | + | + | + | + | 1,717–1,779 | 92.9 | IB | GIY-YIG | orf275 |
| cox1-i7 | 149 | I | – | – | + | – | – | – | 2,682 | – | IB | L1; L1 | orf326 |
| cox1-i8 | 174 | I | + | – | + | + | – | – | 2,465–2,468 | 96.3 | IB | – | orf718 |
| cox1-i9 | 205 | I | – | + | – | – | + | + | 1,300 | 100 | ID | double L1 | orf173 |
| II | + | – | + | + | – | – | 1,368 | 100 | ID | double L1 | orf173 | ||
| cox1-i10 | 214 | I | + | + | + | + | + | + | 1,062 | 99.3 | IB | double L1 | orf316 |
| cox1-i11 | 243 | I | + | – | – | – | – | – | 4,817 | – | IB; ID | L1; double L1 | orf404 |
| II | – | + | – | – | – | – | 1,576 | – | IB | L1 | orf404 | ||
| III | – | – | + | – | – | – | 3,212 | – | IB | L1 | orf404 | ||
| IV | – | – | – | + | + | + | 3,084–3,086 | 99.2 | IB; ID | L1; double L1 | orf404 | ||
| cox1-i12 | 257 | I | + | + | + | + | + | + | 1,128 | 99.3 | IB | L1 | orf346 |
| cox1-i13 | 269 | I | + | – | – | – | – | – | 1,427 | – | IB | L1 | – |
| II | – | + | – | – | + | + | 1,495 | 100 | IB | L1 | – | ||
| cox1-i14 | 294 | I | + | – | – | – | – | – | 1,984 | 100 | – | L2 | orf501 |
| II | – | + | + | + | + | + | 1,841–1,847 | 99.5 | IB | L1; L2 | orf445 | ||
| cox1-i15 | 323 | I | – | + | + | + | + | + | 2,427 | 98.3 | I(derived) | GIY-YIG; GIY-YIG | orf268; orf331 |
| II | + | – | – | – | – | – | 2,474 | – | I(derived) | GIY-YIG; GIY-YIG | orf430; orf331 | ||
| cox1-i16 | 339 | I | + | + | + | + | + | + | 1,206–1,229 | 96.9 | I(derived) | double L1 | orf277 |
| cox1-i17 | 397 | I | – | + | – | – | + | + | 1,607 | 100 | IB | GIY-YIG | orf445 |
| nad1-i1 | 49 | I | – | + | – | – | + | + | 2,231 | 100 | IB(3') | GIY-YIG | orf320 |
| nad1-i2 | 84 | I | + | – | – | – | – | – | 1,467 | – | – | double L1 | – |
| II | – | + | + | + | + | + | 1,594–1,596 | 97.6 | – | double L1 | – | ||
| nad1-i3 | 99 | I | + | + | + | + | + | + | 1,445–1,480 | 97.6 | IC2 | double L1 | – |
| nad1-i4 | 214 | I | – | + | – | – | – | + | 2,471 | 100 | IB | GIY-YIG (b) | – |
| II | – | – | + | – | – | – | 3,903 | – | IB | GIY-YIG (b); double L1(b) | |||
| III | – | – | – | + | – | – | 3,878 | – | IB | GIY-YIG (b); double L1 | orf392 | ||
| IV | – | – | – | – | + | – | 3,843 | – | IB | GIY-YIG (b); double L1 | orf392 | ||
| V | + | – | – | – | – | – | 3,896 | – | IB | GIY-YIG (b); double L1 | orf 327 | ||
| nad4-i1 | 152 | I | – | – | + | + | – | – | 2,138 | 100 | – | double L1 | orf302 |
| atp6-i1 | 120 | I | + | + | + | + | + | + | 1,490–1,511 | 96.9 | IB | double L1 | orf346 |
| atp6-i2 | 181 | I | – | + | – | – | + | + | 1,918 | 100 | – | GIY-YIG | orf382 |
| II | – | – | + | – | – | – | 3,269 | – | – | GIY-YIG; GIY-YIG (b) | – | ||
| atp6-i3 | 198 | I | – | – | + | – | – | – | 1,946 | – | IC2 | GIY-YIG | – |
| II | + | – | – | – | – | – | 1,905 | – | – | GIY-YIG | – | ||
| rns-i1 | 860 | I | – | – | + | + | – | – | 2,389–2,390 | 99.9 | II | double L1 | orf366 |
| rns-i2 | 910 | I | + | – | – | – | – | – | 12 | – | – | – | – |
| II | – | + | + | + | + | + | 50 | 100 | – | – | – | ||
| rns-i3 | 1,108 | I | – | – | – | + | – | – | 1,406 | – | IC2 | double L1 | orf418 |
| rns-i4 | 1,440 | I | – | + | – | – | + | + | 432 | 100 | ID | – | – |
| rns-i5 | 1,557 | I | + | + | – | – | + | + | 960–972 | 97 | – | GIY-YIG | – |
| II | – | – | + | – | – | – | 1,007 | – | – | GIY-YIG | – | ||
| III | – | – | – | + | – | – | 1,061 | – | – | GIY-YIG | – | ||
Column 2: “position” represents the insertion site of introns into conserved genes; the unit for protein coding genes is aa; the unit for rRNA sequences is bp; interval number means that the insertion site is uncertain, but should be within the interval. Column 3: “type” represents intron type located in same insertion site. Introns with more than 90 % nucleotide identity and similar length are classified in same type. Column 4–9: +/– indicates the presence or not of a corresponding intron. Column 11: “lowest similarity” means the lowest one of similarity values from pairwise intron alignments within same type. Column 13: L1, LAGLIDADG_1 domain; L2, LAGLIDADG_2 domain; RT, reverse transcriptase domain; RVT, RNA-dependent DNA polymerase domain; (b) means degraded domain. Column 14: “ORF” represents intact conserved domain sequences.
Most introns (86%) belonged to group I or contained group I-characterized conserved domains, which included LAGLIDADG_1, double LAGLIDADG_1, LAGLIDADG_2, GIY-YIG motifs, or mixtures (Table 2). These group I introns were included in all subgroups except IA3. Some introns contained two or more subgroup I specific sequences. For example, subgroup I specific sequences of IB, IC2 and ID were detected in cox1-i2 of As23. Among nine group II introns, three carried genes encoding RNA-dependent-DNA polymerase. Interestingly, the group II intron rns-i1 only had an orf366 encoding double LAGLIDADG_1 domain. In addition to group I and group II introns, the other five introns were not typical mitochondrial introns. These included rnl-i6 (containing the rps3 gene), rnl-i4 (type II) and three short introns (nad5-i7 with 42 bp, rns-i2 (type I) with 12 bp, and rns-i2 (type II) with 50 bp).
The mtDNAs of six isolates shared 19 “common introns,” which appeared in all isolates with similar size and more than 90% identity. Except for rnl-i6 (containing conserved gene rps3) and nad5-i5 (containing two IDs), other “common introns” were group I with the length ranging from 900 to 2 kb. Each contained one conserved domain-encoded ORF or fragment. Different introns from the same insertion sites at 15 positions had different sizes, but shared high sequence similarity (>90%) among partial fragments. Different introns from the same insertion sites of cox1-i9, atp6-i3, and atp9-i1 shared less similarity (80–90% identity) among partial fragments. Two completely different introns were also detected from each insertion site of cox1-i14, rns-i2, and rnl-i1, and rnl-i7 (Table 2).
Differences of short fragments
Forty-four types of short fragments were different among the six tested mtDNAs (Supplemental Table 1). Those fragments ranged from 12 to 106 bp in length, but most (75%) had the size from 30 to 60 bp. Three of them were short introns of nad5 or rns, while 23 fragments were located within introns of the conserved genes. The other 18 fragments were in intergenic regions. Among these short fragments within introns, As23:87773-87824, As24:28235-28282, and As24:37117-37158 were found in LAGLIDADG_1 domain regions; As28:124339-124385, As24:138958-139006 and As24:139007-139060 were in GIY-YIG domain regions. There were three sets of tandem short fragments: As24:138958-139006 and As24:139007-139060; As23:16465-16482 and As23:16483-16545; and As23:118651-118685 and As23:118686-118723.
Out of 44 short fragments, 43 were found in either As03 or one or more of the other five isolates. Seven were found only in mtDNA of As03, whereas 13 co-existed in the other five mtDNAs. Furthermore, three short fragments were found in mtDNAs of As15, As28 and As31, 15 were in As23 and As24, and another five just in As23 or As24. Based on the distribution of short fragments, six mtDNAs were clustered into three groups: group I (AS15, As28 and As31), group II (As23 and As24), and group III (As03). An exception was As28:124339-124385, which was found in mtDNAs of As03, As23, As24, and As28. Twenty seven short fragments had a single SIR (small inverted repeat), three had two SIRs, and one had three SIRs. No typical SIR structures were found in 14 other short fragments.
Some short fragments were multi-copy in individual mtDNAs. They included As03:124881-124916 (8 copies in As03), As23:118651-118685 (7 copies in As23 and As24), As23:118686-118723 (5 copies in As23 and As24), As23:7552-7599 (3 copies in As23 and 2 copies in As24), As24:16469-16531(3 copies in As23 and As24), As24:37117-37158 (3 copies in As23 and As24), As28:35488-35535 (3 copies in As15, As28 and As31). In addition, there were two copies of As23:137527-137604, As23:140496-140543, As23:47023-47067, and As23:80096-80143 in the corresponding mitochondrial genomes. The copy fragments showed more than 90% identity with their corresponding insertions.
Analysis of twintrons
Different introns from the same insertion sites shared some parts of similar sequences. Typical cases were located in the sites of nad1-i4, nad5-i3, cox1-i2, and cox1-i11. Five different introns found in nad1-i4 (Table 1). The nad1-i4 of As31 (and As15) was the smallest one (2,471 bp), and contained a GIY-YIG domain encoded sequence, which was interrupted, forming three segments. There were two more fragments by comparing nad1-i4 of As28 with that of As31: a 46-bp SIR and a 1329-bp parasitic intron encoding a double LAGLIDADG_1 domain. Parasitic introns are self-splicing introns which insert into another host introns (Gogarten and Hilario, 2006). Both insertions were found in the third GIY-YIG domain segment (Figure 2A). In addition, four small insertions were found in these introns among the isolates: two in As03, one in As23 and As24, and another one in five isolates except As03.
Figure 2.

Possible evolution of complex introns. Del operators point to the insertion sites of introns into the conserved genes. Black bars indicate conserved sequences of introns. Other color bars represent new insertion elements. Co-linearity between introns is indicated by dotted lines. Conserved domain regions are shown by solid lines under bars.
Two introns were detected in nad5-i3 (Table 1 and Figure 2B). A smaller one was in each of As15 and As31; a larger one was in each of As23, As24, and As28; and no intron was at that site in As03. The smaller one contained two fragments encoding LAGLIDADG_1 and LAGLIDADG_2 homologs, respectively, indicating that it was a twintron. The larger intron contained an additional 1,500-bp intron, which was GIY-YIG located in a LAGLIDADG_2 coding region. Furthermore, a 13-bp fragment flanked insertion site was found to be lost in larger introns. The larger intron contained the information of at least three introns (Figure 2B).
Four introns varying widely in size were detected in cox1-i2, specifically 1,263 bp (As03, As15 and As28), 2,561 bp (As24), 2,693 bp (As31), and 4,039 bp (As23) (Table 1 and Figure 2C). A member of the IC2 intron group with double LAGLIDADG_1 had been inserted into a cox1-i2 intron sequence, such as that of As28, to form an intron sequence as found in As31. There were three additional fragments of cox1-i2 in As23 compared with that of As31: a 393-bp fragment at its 5′-end, a 909 bp intron with double LAGLIDADG_1 domain, and a SIR in LAGLIDADG_1 domain region. Loss of the fragment from 1,642 to 3,119 bp of a cox1-i2 sequence such as in As23, formed a corresponding intron in As24. As a result, three out of four introns in cox1-i2 were twintrons found in AS23, As24, and As31, all of which contained the full original intron content of As28.
Four different introns were detected in cox1-i11, which were 1,576 bp (As15), 3,086 bp (As24, As28, and As31), 3,212 bp (As23), and 4,817 bp (As03) in length (Figure 2D). They carried a common fragment of approximately 1,300 bp, which contained a LAGLIDADG_1 domain region. Compared with the introns of As15, a parasitic intron with a 1510-bp sequence was inserted into cox1-i11 of As28. Similarly, an additional fragment of 1,548-bp was found in cox1-i11 of As03 compared to that of As23. The two parasitic introns shared 96.5% similarity.
Formation of ORF-less intron
Comparison of introns at the same loci also showed the formation of three ORF-less introns (Figure 3). The rnl-i4 of As28 was a 2,289-bp intron with a subgroup IC2-characterized sequence and a fragment encoding double LAGLIDADG_1 domain. Loss of the fragment from 344 to 2,167 bp to form a 456-bp intron of As15 (without IC2 and double LAGLIDADG_1); loss of the fragment from 717 to 1,938 bp and short fragment from 20 to 64 bp to form a 1,022-bp intron of As03 (without double LAGLIDADG_1). A similar case was found in rnl-i3 of As23. A fragment with orf253 encoding a GIY-YIG domain was lost from rnl-i3 of As28 to form a 521-bp one of As23.
Figure 3.
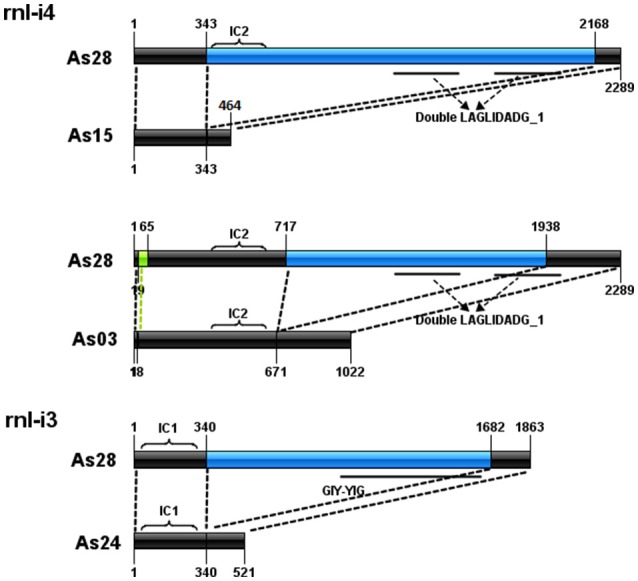
Formation of three orf-less introns. Black bars indicate conserved sequences of introns. Blue bars represent lost parts of sequences. Bright green bars represent short insertion fragments. Conserved domain regions are represented by solid lines under bars.
Discussion
In this study, we analyzed mitochondrial genomes of six isolates of A. stygium. Sixty-five insertion sites and 99 different introns were identified. Introns from each of 16 insertion sites varied greatly in size, but shared high similarity among partial fragments. The similar sequences were inferred to be shared in the common ancestor, and each intron represented one of its evolutionary descendants. Intron comparisons may demonstrate their possible evolution. For cox1-i2, as an example, the intron of As28 was a typical mitochondrial intron, with a group IB RNA structure and a LAGLIDAGE_1-like gene. It was inferred to be the ancestor of all introns in cox1-i2, since it carried the least content. An additional parasitic IC2 intron was detected in the intron of As31. A 52-bp SIR-like fragment was detected within double LAGLIDADG_1 region of the parasitic IC2 intron of As23, indicating that the intron of As23 may have originated from the ancestral intron currently conserved in As31. Similar to the intron of As23, that of As24 contained two additional fragments at the 5′ end, but lacked the parasitic IC2 intron. The intron of As24 was a presumed descendant of the one from As23, but with the loss of the IC2 intron. As a result, four introns in cox1-i2 may have derived from the ancestral homolog from the As28 form, to the As31 form, to the As23 form, and then to the As24 form (Figure 2C).
In a similar fashion, nine twintrons were identified, namely cox3-i1 of As24, nad5-i3 of As28, cob-i3 of As31, cox1-i2 of As24, As23, As31, and cox1-i11 of As28 and As03 and nad1-i4 of As28. Although twintrons are referred to as an intron inserted by one or more parasitic introns (Bullerwell et al., 2003), both insertions of parasitic introns, and activities of short fragments (such as SIRs) or long non-intron fragments can contribute to the formation of twintrons. Activities of non-intronic elements have made the structure of twintrons more complex, which was hard to identify accurately based on the secondary structure of RNA in the host intron. For example, formation of nad1-i4 in As28 resulted from at least two insertions: a 46-bp SIR and a 1,329-bp parasitic intron which are found close to each other (9 bp distance; Figure 2A). It was difficult to distinguish the 46-bp SIR and 9-bp host intron fragment from 1,329-bp parasitic intron without comparison with nad1-i4 of As31. Mitochondrial introns generally have conserved insertion sites (Swithers et al., 2009). Formation of larger introns in nad5-i3 involved an insertion of a 1,500-bp parasitic intron into a smaller intron, as well as the loss of a 13-bp fragment flanking the insertion site. It is unclear if loss of the short fragment was related to the parasitic intron insertion event.
Introns without ORFs have been widely found in fungal mitochondrial genomes (Mullineux et al., 2010; Hafez et al., 2013). HEG is an independent mobile element, which breaks away from intact introns, and results in ORF-less introns (Sethuraman et al., 2013). Intron comparisons in this study revealed two sets of introns with each set containing an intact intron and a corresponding ORF-less one: rnl-i3 of As28/As24 and rnl-i4 of As28/As03. Compared with rnl-i3 of As28, a 1,341-bp fragment containing a 762-bp GIY-YIG-encoded ORF was lost in As24. The ORF was supposed to be an intact HEG. However, the lost fragment from rnl-i4 in As28 just contained part of the double LAGLIDADG_1 motif region, and the other part of which can be found in rnl-i4 in As03. Deletion of the fragment was perhaps driven not by mobility of the interrupting HEG, but by other unknown mechanisms.
Several group II introns with ORFs encoding putative LAGLIDADG homing endonucleases have been identified in mitochondrial small or large subunit rRNA genes of both Ascomycetes and Basidiomycetes (Monteiro-Vitorello et al., 2009; Pfeifer et al., 2012). These composite elements were reported to function as group I/HEG composite elements (Mullineux et al., 2010). Here, we also identified a group II (domain V) intron with ORF366 encoding putative double LAGLIDADG HEG in mitochondrial rns gene of As23 and As24.
Aside from introns, the six mtDNA genomes contained the same sets of conserved protein-coding, tRNA and rRNA genes. Arrangement and orientation of these genes were identical. Additionally, intergenic regions except for the region between cob and cox1, were homogenous, and of similar size. Conserved genome profiles, large intron number, and high diversity of intron type, size and number are unique characteristics of mtDNAs of A. stygium reported so far, which make them the ideal material for the study of the evolution of fungal mitochondrial introns.
The presence of transferase encoding genes was another feature of mtDNA in A. stygium. Transferase encoding genes are rare in fungal mitochondrial genomes, since only three copies were detected from ~500 fungal mtDNAs released in the organelle genome database of NCBI, including an N-acetyl-transferase in Phialocephala subalpina (YP_004733051.1), a hypothetical protein of Pestalotiopsis fici (YP_009317063.1), and an N-acetyl-transferase of P. fici (YP_009317072). However, there were three transferase genes as well as a transferase-related fragment in all six mtDNAs of A. stygium. They included two copies of putative N-acetyl-transferase genes and an aminotransferase class-III-like gene. Corresponding transferase genes from different mtDNAs shared high similarity. P. fici and A. stygium belong to same order of Xylariales, and these transferase genes likely evolved via vertical gene transfer from their ancestor, rather than horizontal gene transfer.
The six mtDNAs were 131,996–147,325 bp in length. To our knowledge, only eight completely sequenced and well annotated fungal mtDNAs are larger than 150 kb (Losada et al., 2014; Mardanov et al., 2014; Salavirta et al., 2014; Kanzi et al., 2016; Nowrousian, 2016; Kang et al., 2017). One of the common features for those large mtDNAs was that all of them contain an abundance of introns, many of which carry homing endonuclease genes (group I type) or reverse transcriptase genes (group II type). Those introns occupy a large proportion of their genome, for example up to 67.6% of O. sinensis (Kang et al., 2017). Similarly, six mtDNAs in this study contained 45–49 introns, most of which carried self-mobility related genes. Some introns even contained two or more homing endonuclease genes, which made their host intron huge. For example, cox1-i2 in A. stygium was a large intron of 4,039 bp, which harbored three ORFs with LAGLIDAGE HE genes. Altogether, introns accounted for the range from 59.6 to 62.2% of the full mitochondrial genome sizes among the size isolates. Our findings as well as the previous studies demonstrated that the number and length of introns are two key factors that contribute to the size of large fungal mtDNAs.
Comparative mitochondrial genomic analyses among isolates within intraspecies or closely related species might enhance our understanding of recent fungal mitochondrial evolution. Related researche on several genera has been performed in the past few years. Nine mitochondrial genomes from Aspergillus and Penicillium species (Joardar et al., 2012), and three from Cordyceps militaris (Zhang et al., 2015) represented the small genome sizes (<40 kb); six mtDNAs from Rhizophagus irregularis (Formey et al., 2012), five from Rhynchosporium species (Torriani et al., 2014), and three from Fusarium species (Al-Reedy et al., 2012) represented moderate genome sizes (40 kb to 100 kb); and three out of four mitochondrial genomes from Chrysoporthe species (Kanzi et al., 2016) represented large genome sizes (more than 100 kb). Comparisons of isolates from the same genera showed that the mtDNA size of closely related isolates were similar, retaining almost identical synteny of coding gene regions. Most genome size variation can be attributed to the gain/loss of mobile elements such as introns, mobile open reading frames, and mitochondrial plasmid-like DNA polymerase genes. In this study, all six tested mtDNAs were large, and also ranged in size. Similar with previous studies, the main source for size differences of the six mtDNAs was due to mobility of introns plus group II like fragments located at between cob and cox1.
Common introns were found in all tested mtDNAs, and they could be derived from their ancestors. These introns should be stable in their host genomes and have little or no activity of mobility. Mitochondrial genomes of nine Aspergillus and Penicillium species have zero common introns (Joardar et al., 2012), of three Cordyceps militaris strains have four common introns (Zhang et al., 2015), and of six Rhizophagus irregularis isolates have five common introns (Formey et al., 2012). Common intron number (19) of mtDNAs in A. stygium was much greater than those species with small mtDNA size, indicating that there might be one or more mechanisms in A. stygium which accelerate degradation, and hinder the mobility of their mitochondrial introns.
Small inverted repeats are short elements <100 bp that can fold into hairpin-like structures. These hairpin-like elements have been widely found in the organellar genomes of plants, aquatic animals (Lukić-Bilela et al., 2008; Lavrov, 2010) and fungi (de Zamaroczy and Bernardi, 1986; Dieckmann and Gandy, 1987; Paquin et al., 2000; Formey et al., 2012). Previous studies suggested that small inverted repeats may function in DNA recombination (de Zamaroczy and Bernardi, 1986; Dieckmann and Gandy, 1987), mRNA processing, translation, and stabilization (Yin et al., 1981; André et al., 1992). In this study, 31 out of 44 short insertion fragments contained one or more hairpin structures. There were 18 short insertions among the introns. Six of these were in conserved domain regions in HE genes. The remaining 12 were not found in the conserved domain, but they may still affect the structure of catalytically active introns of RNA, of intron-encoding proteins or of intron insertion sites.
A possible factor for so many introns in mtDNA of A. stygium is the presence of short fragment insertions in introns. Insertion of short fragments may result in the change of secondary structure of RNA, altered structure of the conserved domain of HE genes, or interruption of ORF integrity in introns, thus reducing or hindering the activity of RNA or/and HE. Broken introns may increase the frequency of different types of mutations, including insertions/deletions of parasitic introns, loss of HEGs, single base mutation and insertions of other short fragments. All these mutations can give rise to a great number of high-diversity introns, which may contribute to the large genomic size of A. stygium mitochondria.
Conclusions
MtDNAs of six isolates from A. stygium ranged from 132 to 147 kb in size and contained the same sets of conserved protein-coding, tRNA and rRNA genes, and shared the same gene arrangement and orientation. Those six genomes varied greatly in intron distribution and content. Comparison of introns within the same insertion site demonstrated formation of some complex introns, such as twintrons and ORF-less introns. Complex introns underwent not only insertion/deletion of introns, but also insertion/deletion of short fragments such as small inverted repeats. Movements of short fragment within an intron might affect or hinder the activity of introns, which could cause intron accumulation in mitochondrial genomes, and enlarge their size.
Author contributions
BX and RM conceived this study. YD analyzed the data, prepared figures and drafted the manuscript. TH participated in early analysis of preliminary data and manuscript writing and revision. SL provided suggestion for the research, contributed to the data interpretation, writing, and revising the manuscript critically. QC participated in the data analysis. LL and QW sequenced and analyzed the mitochondrial genome. All authors have read and approved the final version of the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. David Lightfoot for editorial review of the manuscript. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture. USDA is an equal opportunity provider and employer.
Footnotes
Funding. This work was supported by Natural Science Foundation of China (grant number: 31670021).
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02079/full#supplementary-material
Phylogenetic tree of six isolates of Annulohypoxylon stygium based on partial beta-tubulin and actin gene sequences. Concatenation of beta-tubulin and actin gene sequences was used for species designations of the isolates. Corresponding sequences of Annulohypoxylon urceolatum (AY951670 and AY951782), A. stygium 90081906 (AY951667 and AY951775), A. stygium 90041409 (AY951666 and AY951776), A. stygium var. annulatum 3 (AY951669 and AY951777) and A. stygium var. annulatum 91042205 (AY951668 and AY951778) were used. Sequences were aligned using Clustal W with gap opening penalty of 5 and gap extension penalty of 2. The distances were computed using the Neighbor-joining algorithm as implemented in Mega 7, and all positions containing gaps and missing data were eliminated from phylogenetic analysis.
{kind=link}
Diagram of six mitochondrial genomes. Fifteen conserved protein-coding genes, 26 tRNAs, small and large subunit rRNAs, and three transferase genes are shown. Blue arrows represent protein coding genes; pink arrows represent rRNAs, dark red arrows represent tRNA; green arcs represent introns of conserved protein-coding genes and rRNAs.
{kind=link}
References
- Adams K. L., Palmer J. D. (2003). Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol. Phylogenet. Evol. 29, 380–395. 10.1016/S1055-7903(03)00194-5 [DOI] [PubMed] [Google Scholar]
- Al-Reedy R. M., Malireddy R., Dillman C. B., Kennell J. C. (2012). Comparative analysis of Fusarium mitochondrial genomes reveals a highly variable region that encodes an exceptionally large open reading frame. Fungal Genet. Biol. 49, 2–14. 10.1016/j.fgb.2011.11.008 [DOI] [PubMed] [Google Scholar]
- André C., Levy A., Walbot V. (1992). Small repeated sequences and the structure of plant mitochondrial genomes. Trends Genet. 8, 128–132. 10.1016/0168-9525(92)90370-J [DOI] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullerwell C. E., Forget L., Lang B. F. (2003). Evolution of monoblepharidalean fungi based on complete mitochondrial genome sequences. Nucleic Acids Res. 31, 1614–1623. 10.1093/nar/gkg264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell A., Mrázek J., Karlin S. (1999). Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 96, 9184–9189. 10.1073/pnas.96.16.9184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copertino D. W., Hallick R. B. (1991). Group-II twintron-an intron within an intron in a Chloroplast cytochrome-B-559 Gene. EMBO J. 10, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Zamaroczy M., Bernardi G. (1986). The GC clusters of the mitochondrial genome of yeast and their evolutionary origin. Gene 41, 1–22. 10.1016/0378-1119(86)90262-3 [DOI] [PubMed] [Google Scholar]
- Deng Y., van Peer A. F., Lan F. S., Wang Q. F., Jiang Y., Lian L. D., et al. (2016a). Morphological and molecular analysis identifies the associated fungus (“ Xianghui”) of the medicinal white jelly mushroom, Tremella fuciformis, as Annulohypoxylon stygium. Int. J. Med. Mushrooms 18, 253–260. 10.1615/IntJMedMushrooms.v18.i3.80 [DOI] [PubMed] [Google Scholar]
- Deng Y., Zhang Q., Ming R., Lin L., Lin X., Lin Y., et al. (2016b). Analysis of the mitochondrial genome in Hypomyces aurantius reveals a novel twintron complex in fungi. Int. J. Mol. Sci. 17:E1049. 10.3390/ijms17071049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieckmann C. L., Gandy B. (1987). Preferential recombination between GC clusters in yeast mitochondrial DNA. EMBO J. 6, 4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drager R. G., Hallick R. B. (1993). A complex twintron is excised as four individual introns. Nucleic Acids Res. 21, 2389–2394. 10.1093/nar/21.10.2389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einvik C., Elde M., Johansen S. (1998). Group I twintrons: genetic elements in myxomycete and schizopyrenid amoeboflagellate ribosomal DNAs. J. Biotechnol. 64, 63–74. 10.1016/S0168-1656(98)00104-7 [DOI] [PubMed] [Google Scholar]
- Formey D., Molès M., Haouy A., Savelli B., Bouchez O., Bécard G., et al. (2012). Comparative analysis of mitochondrial genomes of Rhizophagus irregularis-syn. Glomus irregulare-reveals a polymorphism induced by variability generating elements. New Phytol. 196, 1217–1227. 10.1111/j.1469-8137.2012.04283.x [DOI] [PubMed] [Google Scholar]
- Gogarten J. P., Hilario E. (2006). Inteins, introns, and homing endonucleases: recent revelations about the life cycle of parasitic genetic elements. BMC Evol. Biol. 6:94. 10.1186/1471-2148-6-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez P., Barroso G., Labarère J. (1998). Molecular analysis of the split cox1 gene from the Basidiomycota Agrocybe aegerita: relationship of its introns with homologous Ascomycota introns and divergence levels from common ancestral copies. Gene 220, 45–53. 10.1016/S0378-1119(98)00421-1 [DOI] [PubMed] [Google Scholar]
- Hafez M., Majer A., Sethuraman J., Rudski S. M., Michel F., Hausner G. (2013). The mtDNA rns gene landscape in the Ophiostomatales and other fungal taxa: Twintrons, introns, and intron-encoded proteins. Fungal Genet. Biol. 53, 71–83. 10.1016/j.fgb.2013.01.005 [DOI] [PubMed] [Google Scholar]
- Haridas S., Gantt J. S. (2010). The mitochondrial genome of the wood-degrading basidiomycete Trametes cingulata. FEMS Microbiol. Lett. 308, 29–34. 10.1111/j.1574-6968.2010.01979.x [DOI] [PubMed] [Google Scholar]
- Hsieh H. M., Ju Y. M., Rogers J. D. (2005). Molecular phylogeny of Hypoxylon and closely related genera. Mycologia 97, 844–865. 10.1080/15572536.2006.11832776 [DOI] [PubMed] [Google Scholar]
- Joardar V., Abrams N. F., Hostetler J., Paukstelis P. J., Pakala S., Pakala S. B., et al. (2012). Sequencing of mitochondrial genomes of nine Aspergillus and Penicillium species identifies mobile introns and accessory genes as main sources of genome size variability. BMC Genomics. 13:698. 10.1186/1471-2164-13-698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X., Hu L., Shen P., Li R., Liu D. (2017). SMRT Sequencing revealed mitogenome characteristics and mitogenome-wide DNA modification pattern in Ophiocordyceps sinensis. Front. Microbiol. 8:1422. 10.3389/fmicb.2017.01422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanzi A. M., Wingfield B. D., Steenkamp E. T., Naidoo S., van der Merwe N. A. (2016). Intron derived size polymorphism in the mitochondrial genomes of closely related chrysoporthe species. PLoS ONE 11:e0156104. 10.1371/journal.pone.0156104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnkowska A., Vacek V., Zubáčová Z., Treitli S. C., PetrŽelková R., Eme L., et al. (2016). A eukaryote without a mitochondrial organelle. Curr. Biol. 26, 1274–1284. 10.1016/j.cub.2016.03.053 [DOI] [PubMed] [Google Scholar]
- Koren S., Walenz B. P., Berlin K., Miller J. R., Bergman N. H., Phillippy A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. 10.1101/gr.215087.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambowitz A. M., Zimmerly S. (2011). Group II Introns: Mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol. 3:a003616. 10.1101/cshperspect.a003616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang B. F., Laforest M. J., Burger G. (2007). Mitochondrial introns: a critical view. Trends Genet. 23, 119–125. 10.1016/j.tig.2007.01.006 [DOI] [PubMed] [Google Scholar]
- Lavrov D. V. (2010). Rapid proliferation of repetitive palindromic elements in mtDNA of the endemic Baikalian sponge Lubomirskia baicalensis. Mol. Biol. Evol. 27, 757–760. 10.1093/molbev/msp317 [DOI] [PubMed] [Google Scholar]
- Lavrov D. V., Maikova O. O., Pett W., Belikov S. I. (2012). Small inverted repeats drive mitochondrial genome evolution in Lake Baikal sponges. Gene 505, 91–99. 10.1016/j.gene.2012.05.039 [DOI] [PubMed] [Google Scholar]
- Li W., Cowley A., Uludag M., Gur T., McWilliam H., Squizzato S., Lopez R. (2015). The EMBL-EBI bioinformatics web and programmatic tools framework. Nucleic Acids Res. 43, W580–W584. 10.1093/nar/gkv279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada L., Pakala S. B., Fedorova N. D., Joardar V., Shabalina S. A., Hostetler J., et al. (2014). Mobile elements and mitochondrial genome expansion in the soil fungus and potato pathogen Rhizoctonia solani AG-3. FEMS Microbiol. Lett. 352, 165–173. 10.1111/1574-6968.12387 [DOI] [PubMed] [Google Scholar]
- Lukić-Bilela L., Brandt D., Pojskić N., Wiens M., Gamulin V., Müller W. E. (2008). Mitochondrial genome of Suberites domuncula: palindromes and inverted repeats are abundant in non-coding regions. Gene 412, 1–11. 10.1016/j.gene.2008.01.001 [DOI] [PubMed] [Google Scholar]
- Maciel O. M. C., Tavares R. S. N., Caluz D. R. E., Gaspar L. R., Debonsi H. M. (2017). Photoprotective potential of metabolites isolated from algae-associated fungi Annulohypoxylon stygium. J Photoch Photobio B. 178, 316–322. 10.1016/j.jphotobiol.2017.11.018 [DOI] [PubMed] [Google Scholar]
- Manicom B. Q., Bar-Joseph M., Rosner A., Vigodsky-Haas H., Kotze J. M. (1987). Potential applications of random dna probes and restriction-fragment-length-polymorphisms in the taxonomy of the fusaria. Phytopathology 77, 669–672. 10.1094/Phyto-77-669 [DOI] [Google Scholar]
- Mardanov A. V., Beletsky A. V., Kadnikov V. V., Ignatov A. N., Ravin N. V. (2014). The 203 kbp mitochondrial genome of the phytopathogenic fungus Sclerotinia borealis reveals multiple invasions of introns and genomic duplications. PLoS ONE 9:e107536. 10.1371/journal.pone.0107536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel F., Ferat J. L. (1995). Structure and activities of group II introns. Annu. Rev. Biochem. 64, 435–461. 10.1146/annurev.bi.64.070195.002251 [DOI] [PubMed] [Google Scholar]
- Monteiro-Vitorello C. B., Hausner G., Searles D. B., Gibb E. A., Fulbright D. W., Bertrand H. (2009). The Cryphonectria parasitica mitochondrial rns gene: plasmid-like elements, introns and homing endonucleases. Fungal Genet. Biol. 46, 837–848. 10.1016/j.fgb.2009.07.005 [DOI] [PubMed] [Google Scholar]
- Mullineux S. T., Costa M., Bassi G. S., Michel F., Hausner G. (2010). A group II intron encodes a functional LAGLIDADG homing endonuclease and self-splices under moderate temperature and ionic conditions. RNA 16, 1818–1831. 10.1261/rna.2184010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowrousian M. (2016). Complete mitochondrial genome sequence of the Pezizomycete pyronema confluens. Genome A 2016:e00355–16. 10.1128/genomeA.00355-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquin B., Laforest M. J., Lang B. F. (2000). Double-hairpin elements in the mitochondrial DNA of Allomyces: evidence for mobility. Mol. Biol. Evol. 17, 1760–1768. 10.1093/oxfordjournals.molbev.a026274 [DOI] [PubMed] [Google Scholar]
- Pfeifer A., Martin B., Kämper J., Basse C. W. (2012). The mitochondrial LSU rRNA group II intron of Ustilago maydis encodes an active homing endonuclease likely involved in intron mobility. PLoS ONE 7:e49551. 10.1371/journal.pone.0049551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robl D., Pdos Santos Costa P., Büchli F., da Silva Lima D. J., da Silva Delabona P., Squina F. M., et al. (2015). Enhancing of sugar cane bagasse hydrolysis by Annulohypoxylon stygium glycohydrolases. Bioresource Technol. 177, 247–254. 10.1016/j.biortech.2014.11.082 [DOI] [PubMed] [Google Scholar]
- Salavirta H., Oksanen I., Kuuskeri J., Mäkelä M., Laine P., Paulin L., et al. (2014). Mitochondrial genome of Phlebia radiata is the second largest (156 kbp) among fungi and features signs of genome flexibility and recent recombination events. PLoS ONE 9:e97141. 10.1371/journal.pone.0097141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha R., Mohr G., Belfort M., Lambowitz A. M. (1993). Group-I and group-Ii introns. Faseb J. 7, 15–24. 10.1096/fasebj.7.1.8422962 [DOI] [PubMed] [Google Scholar]
- Sethuraman J., Rudski S. M., Wosnitza K., Hafez M., Guppy B., Hausner G. (2013). Evolutionary dynamics of introns and their open reading frames in the U7 region of the mitochondrial rnl gene in species of Ceratocystis. Fungal Biol. 117, 791–806. 10.1016/j.funbio.2013.10.002 [DOI] [PubMed] [Google Scholar]
- Stone J. K., Polishook J. D., White J. F. (2004). Endophytic Fungi. Biodiversity of Fungi. Burlington, NJ: Elsevier Academic Press, 241–270. [Google Scholar]
- Suwannasai N., Martín M. P., Phosri C., Sihanonth P., Whalley A. J., Spouge J. L. (2013). Fungi in thailand: a case study of the efficacy of an ITS barcode for automatically identifying species within the Annulohypoxylon and Hypoxylon genera. PLoS ONE 8:e54529. 10.1371/journal.pone.0054529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swithers K. S., Senejani A. G., Fournier G. P., Gogarten J. P. (2009). Conservation of intron and intein insertion sites: implications for life histories of parasitic genetic elements. BMC Evol. Biol. 9:303. 10.1186/1471-2148-9-303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torriani S. F., Penselin D., Knogge W., Felder M., Taudien S., Platzer M., Brunner P. C. (2014). Comparative analysis of mitochondrial genomes from closely related Rhynchosporium species reveals extensive intron invasion. Fungal Genet. Biol. 62, 34–42. 10.1016/j.fgb.2013.11.001 [DOI] [PubMed] [Google Scholar]
- Whalley A. J. S., Edwards R. L. (1995). Secondary metabolites and systematic arrangement within the Xylariaceae. Can. J. Bot. 73, S802–S810. 10.1139/b95-325 [DOI] [Google Scholar]
- Yin S., Heckman J., RajBhandary U. L. (1981). Highly conserved GC-rich palindromic DNA sequences flank tRNA genes in Neurospora crassa mitochondria. Cell 26, 325–332. 10.1016/0092-8674(81)90201-4 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Zhang S., Zhang G., Liu X., Wang C., Xu J. (2015). Comparison of mitochondrial genomes provides insights into intron dynamics and evolution in the caterpillar fungus Cordyceps militaris. Fungal Genet. Biol. 77, 95–107. 10.1016/j.fgb.2015.04.009 [DOI] [PubMed] [Google Scholar]
- Zuker M., Mathews D. H., Turner D. H. (1999). Algorithms and thermodynamics for RNA secondary structure prediction: a practical guide. Rna Biochemistry and Biotechnology. 70, 11–43. 10.1007/978-94-011-4485-8_2 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phylogenetic tree of six isolates of Annulohypoxylon stygium based on partial beta-tubulin and actin gene sequences. Concatenation of beta-tubulin and actin gene sequences was used for species designations of the isolates. Corresponding sequences of Annulohypoxylon urceolatum (AY951670 and AY951782), A. stygium 90081906 (AY951667 and AY951775), A. stygium 90041409 (AY951666 and AY951776), A. stygium var. annulatum 3 (AY951669 and AY951777) and A. stygium var. annulatum 91042205 (AY951668 and AY951778) were used. Sequences were aligned using Clustal W with gap opening penalty of 5 and gap extension penalty of 2. The distances were computed using the Neighbor-joining algorithm as implemented in Mega 7, and all positions containing gaps and missing data were eliminated from phylogenetic analysis.
Diagram of six mitochondrial genomes. Fifteen conserved protein-coding genes, 26 tRNAs, small and large subunit rRNAs, and three transferase genes are shown. Blue arrows represent protein coding genes; pink arrows represent rRNAs, dark red arrows represent tRNA; green arcs represent introns of conserved protein-coding genes and rRNAs.