

Abstract
Defective interfering (DI) genomes, or defective viral genomes (DVGs), are truncated viral genomes generated during replication of most viruses, including live viral vaccines. Among these, “panhandle” or copy-back (cb) and “hairpin” or snap-back (sb) DI genomes are generated during RNA virus replication. 5′ cb/sb DI genomes are highly relevant for viral pathogenesis since they harbor immunostimulatory properties that increase virus recognition by the innate immune system of the host. We have developed DI-tector, a user-friendly and freely available program that identifies and characterizes cb/sb genomes from next-generation sequencing (NGS) data. DI-tector confirmed the presence of 5′ cb genomes in cells infected with measles virus (MV). DI-tector also identified a novel 5′ cb genome, as well as a variety of 3′ cb/sb genomes whose existence had not previously been detected by conventional approaches in MV-infected cells. The presence of these novel cb/sb genomes was confirmed by RT-qPCR and RT-PCR, validating the ability of DI-tector to reveal the landscape of DI genome population in infected cell samples. Performance assessment using different experimental and simulated data sets revealed the robust specificity and sensitivity of DI-tector. We propose DI-tector as a universal tool for the unbiased detection of DI viral genomes, including 5′ cb/sb DI genomes, in NGS data.
Keywords: copy-back, panhandle, NGS, defective interfering viral genome, viral replication, snap-back
INTRODUCTION
Defective interfering (DI) genomes, also called defective viral genomes (DVGs), are truncated forms of viral genomes generated by most viruses during viral replication. DI genomes possess the minimum nucleotide sequence required for their replication but are unable to replicate in the absence of the replicative viral machinery provided by full-length genomes (Lazzarini et al. 1981; Marriott and Dimmock 2010; Dimmock and Easton 2014; López 2014; Frensing 2015; Manzoni and López 2018). Moreover, DI genomes retain a packaging signal for efficient encapsidation that allows them to be later assembled in viral particles and transmitted between hosts (Ke et al. 2013). In vitro, DI genomes have been detected during replication at high multiplicity of infection (MOI) of most RNA and DNA viruses (Huang 1973; Perrault 1981; Frensing 2015). Importantly, they have been identified in patients infected with influenza virus (Saira et al. 2013; Vasilijevic et al. 2017), dengue virus (Li et al. 2011), hepatitis C virus (Iwai et al. 2006; Ohtsuru et al. 2013), and respiratory syncytial virus (Sun et al. 2015), as well as in live attenuated viral vaccine batches that are used safely and efficiently on a large scale for human vaccination (McLaren and Holland 1974; Wiktor et al. 1977; Bellocq et al. 1990; Holland 1990).
DI genome production during viral replication plays an important role in viral pathogenesis since they posses immunostimulatory properties that activate host innate immune response (López 2014; Sun et al. 2015; Vasilijevic et al. 2017). Therefore, there is a need for accessible tools to detect DI genomes during viral infection, in viral stocks or live attenuated vaccine batches.
Four main classes of DI genomes exist: simple and multiple internal deletions, mosaic or complex DI genomes (including insertions), 5′ snap-back (sb) DI genomes or “hairpin” structure, and 5′ copy-back (cb) or “panhandle” structure DI genomes (Fig. 1; Lazzarini et al. 1981; Dimmock and Easton 2014). In 5′ cb/sb DI genomes a portion of the original virus genome is repeated in a reverse complement form and can be represented as a stem-like structure (Fig. 1D,E). 5′ cb/sb DI genomes are described mainly for RNA viruses belonging to the order Mononegavirales (Paramyxoviridae, Rhabdoviridae, and Filoviridae families). cb/sb DI genomes share similar RNA structure, i.e., a double-stranded stem formed by the opposite sensed 5′ and 3′ ends of the DI genome and with complementary blunt ends bearing a 5′-triphosphate (Fig. 1D,E; Rao and Huang 1982; Re et al. 1983; Baum and García-Sastre 2011). For reading clarity, sb DI genomes will be mentioned later also as cb DI genomes, as they only differ in the length of their loop. Of note, representation in Figure 1 is schematic, and hairpin and panhandle structures in a cellular context are expected to be more complex. Only 5′ cb DI genomes, containing a copy of the antigenomic promoter at both RNA ends, have been identified so far (Lazzarini et al. 1981). This could be due to the 5′ end antigenomic promoter (assigned as trailer) of Mononegavirales genomes being stronger than the 3′ end genomic promoter (assigned as leader) (Finke and Conzelmann 1999). 5′ cb DI genomes are generated when the viral RNA-dependent RNA polymerase (RdRp), due to a yet unknown mechanism, detaches from the positive-sense antigenome template and reattaches itself to the negative-sense strand nascent product to copy it back, forming a 5′ complementary sequence (Fig. 1D,E; Lazzarini et al. 1981; Dimmock and Easton 2014; López 2014). 5′ cb DI genomes give an excellent example of interference with viral replication since on both 3′ and 5′ ends they possess promoters with high affinity for viral polymerase to favor their replication and to compete with the full-length viral genome replication.
FIGURE 1.
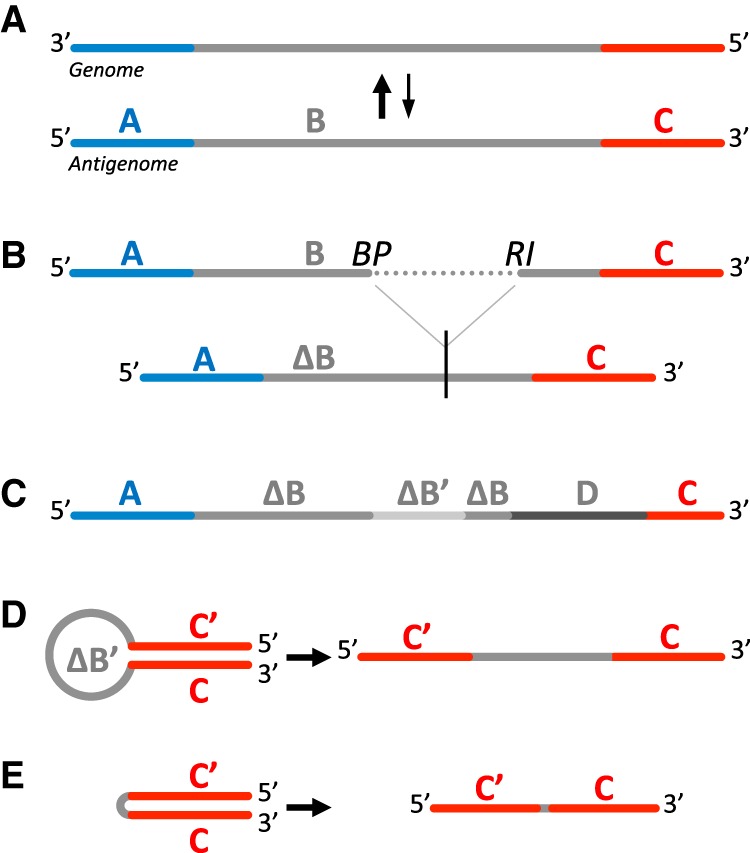
Schematic representation of the main classes of DI genomes that may arise from (A) a full-length negative-sense viral RNA genome and antigenome: (B) DI genomes with deletion, (C) mosaic genomes, (D) copy-back and (E) snap-back DI genomes. C′ represents C complementary region. ΔB and ΔB′ represent shorter B sequence and its complementary region. D represents sequence from other origin. (BP) Breakpoint site, (RI) reinitiation site.
The position at which the RdRp detaches from the antigenome defines the breakpoint site (BP), and the position at which the RdRp reattaches to the nascent strand defines the reinitiation site (RI). The stem and loop sizes of the schematic hairpin structure varies from one cb DI genome to another (Pfaller et al. 2015; Mura et al. 2017). Thus, a way to identify cb DI genomes is to detect a switch of strand polarity at different positions on the genome. We have previously detected, by classical PCR amplification and Sanger sequencing approaches, the presence of 5′ cb DI genomes of length varying from 402 to 2094 nt in cells infected with differently modified recombinant measles viruses (MV) but not with low passage MV Schwarz vaccine strain (Komarova et al. 2013; Sanchez David et al. 2016; Mura et al. 2017). We and others have shown that 5′ cb DI genomes efficiently activate type-I interferon (IFN) production via recognition by the cytosolic RIG-I-like receptors (RLR) (Strahle et al. 2006; Shingai et al. 2007; Baum et al. 2010; Komarova et al. 2013; Mercado-López et al. 2013; Runge et al. 2014; Sanchez David et al. 2016; Mura et al. 2017).
Next-generation sequencing (NGS) provides useful data sets to detect various DI genomes. Mappers that identify aberrant junctions might be exploited to identify deletion type of DI genomes, which resembles mRNA splice variants. However, such analyses require some bioinformatics knowledge to correctly search for signs of the presence of these types of DI genomes in the alignment. Several methods aiming at robustly and accurately detecting DI genomes harboring deletion from deep sequencing data sets have been proposed (Killip et al. 2013; Saira et al. 2013; Timm et al. 2014). In addition, diverse tools have been developed like Paparazzi (Vodovar et al. 2011) or ViReMa (Routh and Johnson 2014). However, none of these tools can easily detect or classify cb DI genomes.
We present here DI-tector, a user-friendly and freely available program, that enables detection of deletion/insertion DI genomes but also of cb/sb DI genomes in deep sequencing data sets.
We exploited published NGS data sets obtained from human cells infected with two members of the Paramyxoviridae family: MV and Sendai virus (SeV), which are both known to produce 5′ cb DI genomes. We validated DI-tector algorithm in silico and in cellulo and showed its superiority to conventional approaches for detection of cb DI genomes. In addition, DI-tector identified for the first time 3′ cb DI genomes in both MV and SeV samples. We validated the existence of one of these novel MV 3′ cb DI genomes using conventional approaches.
RESULTS
DI-tector detects various types of DI genomes
The DI-tector protocol consists of four steps: (i) alignment of reads against the host and viral genomes, (ii) unmatched reads segmentation, (iii) second alignment against the viral genome, and (iv) analyses of the matched reads (Figs. 2 and 3). Data analysis with DI-tector does not require any pretreatment of the original deep sequencing data set beyond standard adapter sequences trimming and quality-filtering. Alignment against the host genome in the first step is optional but is recommended since it may improve specificity results and accelerate the analytic process by decreasing the number of reads and discarding potential host contamination. Mapping against the viral genome is another way to reduce the data set size. Unmapped, or not perfectly mapped reads with mismatches, insertions, or clips, are considered as reads potentially containing recombinant junctions corresponding to DI genomes. Removal of duplicated reads speeds up the analysis. For the second step, a high number of segments positively correlates with a higher number of detected DI genomes but slows down the analysis (Supplemental Fig. S1). For faster overview analyses, we believe that a few segmentations of the reads are more than sufficient to detect highly represented DI genomes. DI-tector generates as output several files containing all the information needed to define DI genome sequences.
FIGURE 2.
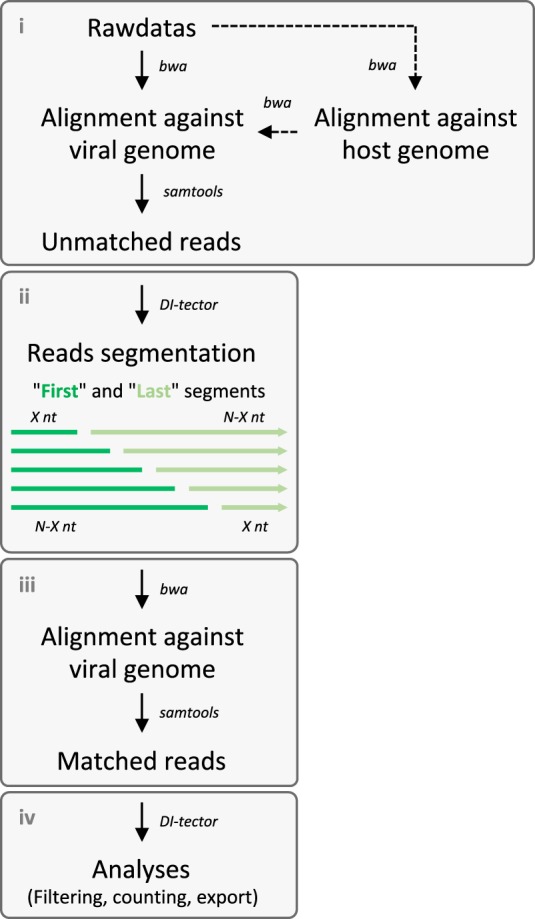
Schematic representation of the DI-tector workflow, which includes four main steps: (i) alignment against host and viral genomes to collect unmatched reads, (ii) segmentation, (iii) second alignment against viral genomes, and (iv) analyses of the matched segmented reads. Dashed lines represent optional steps.
FIGURE 3.

Schematic representation of DI-tector DI genome group clustering on a positive sense viral genome reference.
Characterization of DI genomes identified by DI-tector in MV and rMV-ΔV samples
During our previous studies, we generated several NGS data sets from human cells infected with either MV Schwarz strain or with a recombinant version of this strain that is unable to express the V protein (rMV-ΔV). The latter virus is known to produce 5′ cb DI genomes, contrary to the Schwarz strain (Sanchez David et al. 2016; Mura et al. 2017). In order to study viral RNA signatures on RLR receptors, we have previously performed affinity chromatography purification of RIG-I using cells overexpressing recombinant RIG-I protein fused to One-STrEP-tag (ST) (Sanchez David et al. 2016). As a control for RNA-nonspecific binding, cells overexpressing a ST-Cherry protein were used. This previous work generated data sets of RNA samples before (Total) or after purification (Beads) on ST-Cherry or ST-RIG-I proteins performed in triplicate. We analyzed these data sets using DI-tector and mainly focused on cb DI genomes. The two 5′ cb DI genomes exhibiting the most reads were a 336-nt-long (DI_336) and a 1236-nt-long DI genome (DI_1236) (Fig. 4A). The presence of other cb DI genomes was supported by very small numbers of reads. These species might be poorly represented in the sample or be false positive and were thus not analyzed. DI-tector found both DI_336 and DI_1236 in RNAs specifically associated with RIG-I upon infection with rMV-ΔV (Fig. 4). DI_1236 is a previously identified 5′ cb DI genome that results from a BP at position 14,861 and a RI at 15,694, with one nucleotide insertion (Sanchez David et al. 2016). Both positive and negative strands of DI_1236 were detected with DI-tector. DI_336, which was not previously identified using conventional techniques, resulted from a BP at position 15,651 and a RI at 15,803 and again both strands were detected in the data set. To our knowledge, this is the first identification of such a short MV DI genome. Of note, this newly identified DI_336 follows the “rule of six” postulating that for the paramyxovirus subfamily Paramyxovirinae, only panhexameric-length (6n + 0) genomes are replicated efficiently (Kolakofsky et al. 2005), reinforcing the potential existence of DI_336.
FIGURE 4.
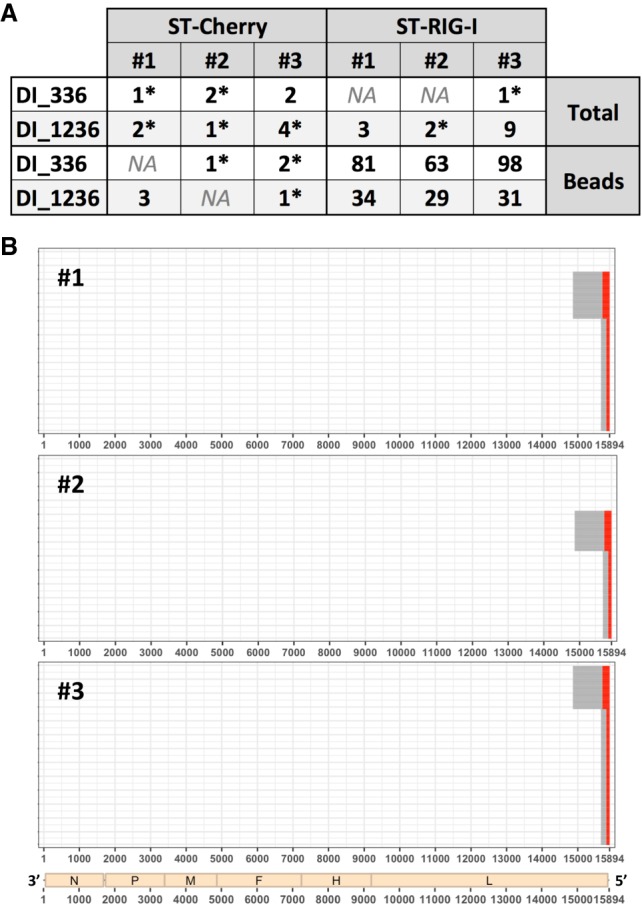
Characterization of DI genomes identified by DI-tector in RNA samples generated from rMV-ΔV infected cells. (A) Counts of reads overlapping junction for DI_336 and DI_1236 in several rMV-ΔV samples. Values are followed by a star when reads only align on one strand. Data sets were generated from RNA samples obtained from ST-Cherry or ST-RIG-I rMV-ΔV infected cells before (Total) or after purification (Beads) (# 1 to 3 represent three biological triplicates). (B) Example of results’ representation of 5′ cb DI genomes of rMV-ΔV samples after RIG-I affinity purification (# 1 to 3 represent three biological triplicates). Red and gray lines represent, respectively, cb DI genome stem and loop.
Surprisingly, several 3′ cb DI genomes were detected in our data sets. In contrast to 5′ cb DI genomes, which were characterized by a first read segment mapping to the reverse strand and a last read segment on the forward strand, 3′ cb DI genomes were identified by a first and a last segment mapping the forward and reverse strands, respectively (Fig. 3). However, 3′ cb DI genomes’ detection was generated from a low number of reads and these could thus be false positives.
Confirmation of newly discovered junctions in viral 5′ cb DI genomes
The existence of the putative DI_336 genome revealed by DI-tector was further examined by depicting number of reads overlapping its recombination junction (Supplemental Fig. S2). Reads obtained by sequencing RNA purified on RIG-I from cells infected with rMV-ΔV (sample #1) were mapped to the newly discovered DI_336 (Supplemental Fig. S2a) and, as a positive control, to the well-characterized DI_1236. Reads mapping to the recombination junction on both strands of the viral genome were observed, confirming the nature of the 5′ cb DI genomes associated with RIG-I (Supplemental Fig. S2b). Of note, by analyzing CIGAR information from sam files generated by mapping the human-depleted reads from rMV-ΔV to the viral genome (or using, for example, Tablet [Milne et al. 2013]), we observed some soft clip (i.e., bases in 5′ or 3′ of the read that are not part of the alignment) at positions 14,861, 15,694, 15,651, and 15,803. These positions correspond to BP and RI sites for DI_1236 and DI_336 (data not shown). We also observed several other clips but, after manual analysis, the unmapped part of the read was composed of only a few nucleotides. These clips were most likely due to the lower quality of the read extremities. Analyzing the reads covering the recombination junction also allowed the identification of a single-nucleotide insertion in the DI_1236 genome sequence, as previously reported by conventional approaches (Sanchez David et al. 2016).
Validation of the presence of DI_336 by RT-qPCR in rMV-ΔV infected cells
In order to test the robustness and accuracy of our DI-detector approach and to validate the existence of the 5′ cb DI_336 in rMV-ΔV infected cells, we performed RT-qPCR analysis on total RNA extracted from HEK293T cells infected with rMV-ΔV or with MV as negative control (Table 1). As expected, DI_336 genome was detected only in RNA extracted from HEK293T cells infected with rMV-ΔV (Table 1). These experiments validate the presence of DI_336 in rMV-ΔV infected cells by classical approaches and thus highlight the power of DI-tector to reveal the landscape of the DI genome population.
TABLE 1.
Validation of the presence of DI_336 by RT-qPCR in rMV-ΔV infected cells

Validation of the presence of a 3′ cb DI genome in MV infected cells
As 3′ cb DI genomes had not been previously characterized, we performed RT-PCR analysis of MV infected cells to attempt to validate their existence (Fig. 5). An amplicon of around 1200 bp was observed on agarose gel. Sanger sequencing of the PCR product allowed us to identify a 2202-nt-long 3′ cb DI genome (DI_2202) generated from a BP at position 1728 and a RI at 474, which is compatible with the expected amplicon size of 1218 bp observed on the gel. These results confirmed the existence of the 3′ cb DI genome species identified by DI-tector. Importantly, the identified MV DI_2202 respected the “rule of six.”
FIGURE 5.

Validation of the presence of a 3′ cb DI genome (DI_2202) by RT-PCR in MV infected cells. A RT-PCR was performed on total RNA extracted from HEK293T cells infected with MV. A 3′ cb DI genome of 2202 bp was detected. The amplicon of the expected size of 1219 bp was sequenced.
Impact of ribosomal RNA depletion on SeV DI genomes’ detection
MV DI_1236 and DI_336 were easily detected in RNA samples after enrichment on RIG-I (Fig. 4A), however their junctions remained poorly covered in total RNA samples purified from infected cells. A classical way to increase proportion of viral reads in NGS analysis is to treat the samples with RiboZero, which removes all ribosomal RNAs (Mertes et al. 2011). Such treatment is obviously restrictive to analysis of cells infected with RNA viruses. We took advantage of published data sets generated from total RNA extracted from SeV infected-cells to evaluate the impact of viral RNA enrichment over total RNA by ribosomal RNA depletion. After filtering against host and viral genomes, around five times more unmatched and mismatched reads were detected in RiboZero treated samples compared to nontreated ones (Fig. 6; Supplemental Table 1). Therefore, increasing the proportion of viral reads improves the sensitivity of DI genomes’ detection, which is useful since DI genomes entities are often poorly represented in total RNA populations.
FIGURE 6.
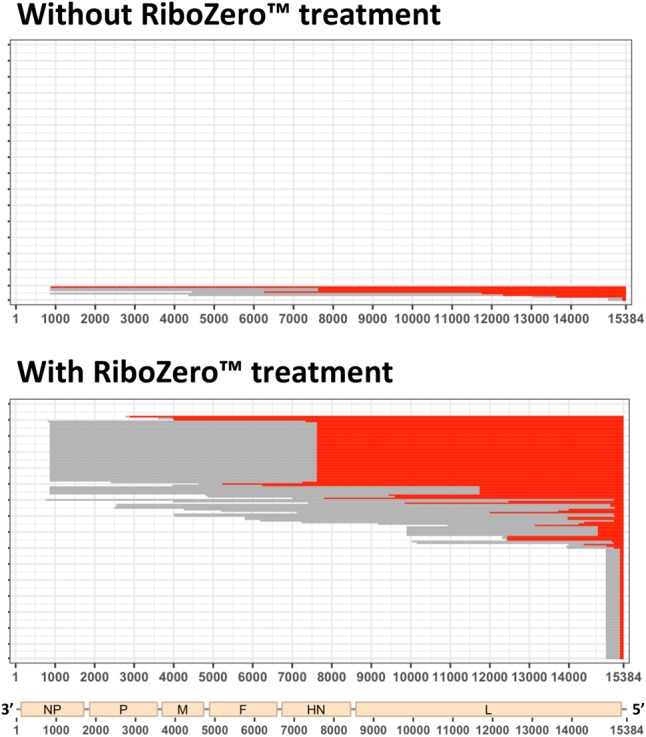
Impact of RiboZero treatment on SeV DI genomes’ detection. Example of results’ representation of 5′ cb DI genomes of SeV with and without RiboZero treatment (“--Min_Segment” = 15). Red and gray lines represent, respectively, cb DI genome stem and loop.
Sensitivity and specificity of DI-tector
To assess the sensitivity and specificity of DI-tector, we first analyzed data sets from noninfected cells. We failed to detect any DI genomes in these data sets, suggesting that reads covering DI genome junctions predicted from DI-tector analysis were not generated from aberrant host or viral sequences or any other contaminant sequences (data not shown).
We also generated three simulated data sets, each containing 500,000 reads of 51 nt harboring a junction at a position ranging from 1 to 50. The first data set, which covers junctions of 5′ cb DI sequences, was used to assess the sensitivity of DI-tector, and the two other data sets, which cover, respectively, junctions of 3′ cb DI genomes and DI genomes with insertion/deletion, were used to determine DI-tector specificity (Fig. 7). Analysis of the 3′ cb DI genomes’ data set failed to predict any 5′ cb DI sequences, resulting in a specificity of 100% for this data set (Supplemental Fig. S3). To investigate further the specificity of DI-tector, we used a less favorable data set, which is composed of 500,000 reads covering junctions of genomes with insertion/deletion (250,000 each) (Fig. 7). We observed a sensitivity decreasing from around 65% and 60% to 15% and 10%, respectively, for “--Min_MAPQ” parameters of 25 and 26, when “--Min-Segment” parameter ranges from 5 to 25 (half of the read length). A “--Min-Segment” of 15 was providing a satisfactory combination of sensitivity and specificity. We thus decided to set it up as a default value in DI-tector.
FIGURE 7.
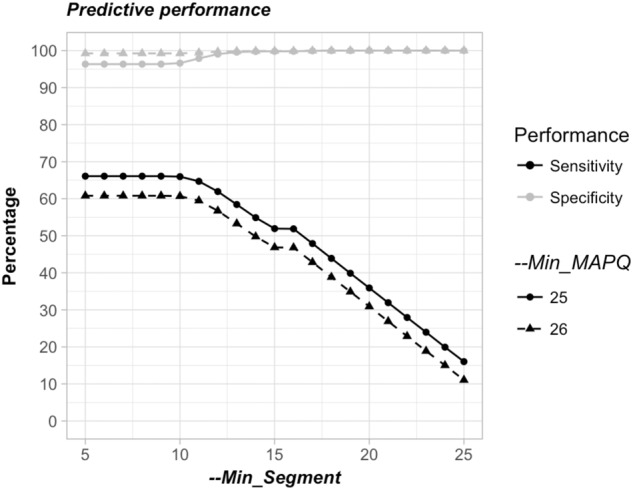
Predictive performance assessment of DI-tector. Sensitivity and selectivity values using different “--Min_Segment” and “--Min_MAPQ”.
It is important to note that sensitivity and specificity will be highly dependent on the data set used. For instance, sensitivity drastically increased with read length and representation of each DI specific read in the simulated data set (Supplemental Fig. S4).
For a more accurate characterization of the DI genome present in a data set, a “--Min_MAPQ” value of 26 seemed appropriate. However, as it may result in the omission of true DI genomes’ detection, we decided to use a “--Min_MAPQ” value of 25 as default.
Online DI-tector as a graphic tool for DI genomes’ representation
Once DI genomes are characterized, using either DI-tector or other methods, their RNA sequences may be graphically represented using online DI-tector (www.di-tector.cyame.eu). This graphic tool allows users to represent predicted DI genomes and their BP and RI positions on the viral genome. The list of DI genomes from the“*_ counts.txt” file or a custom list can be used to generate a scatter-plot, chord diagram, sequence logos or representation as in Figures 4 and 8. In the scatter-plot, BP and RI positions are respectively used for the x and y axis (Fig. 8A). Color and dot size change according to DI genome type and frequency. The closer the dot is to the diagonal of the scatter plot, the smaller the distance between BP and RI. This representation is useful to define replicative DI as the two entities (genome and antigenome) will be symmetrically plotted around the diagonal. Another option is to represent the data as a chords diagram (Fig. 8B). For each DI genome identified by DI-tector, sequence starts at the viral genome extremity, jumps from BP to RI (represented as an arc), and terminates from RI to genome extremity again. If the viral sequence is provided, sequence logos may be represented in frequency or bits units (Crooks et al. 2004). Graphics for the x nucleotides (defined by the user) before and after the BP or the RI may be generated (Fig. 8C). This representation allows the analysis of specific sequence patterns that may exist before and/or after BP and RI sites. All graphs are created as pictures and are easy to export without any programming language knowledge, either by copy/paste or saving as image.
FIGURE 8.

Example of graphical outputs available using DI-tector online tool with rMV-ΔV sample (replicate #1) data (www.di-tector.cyame.eu). (A) Scatter-plot, (B) chord diagram, or (C) sequence logo for 5′ cb DI and DI genomes with deletion.
DISCUSSION
For decades detection and characterization of DI viral genomes have been performed by northern blotting, RT-PCR, and Sanger sequencing (Lazzarini et al. 1981; López 2014). These approaches can be now combined with more recent techniques such as NGS. Indeed, methods to detect DI genomes from deep sequencing data sets have been already proposed (Killip et al. 2013; Saira et al. 2013; Timm et al. 2014) and diverse tools have been developed like, for example, Paparazzi (Vodovar et al. 2011) or ViReMa (Routh and Johnson 2014). They allow detection of DI genomes harboring deletion or mosaic recombination. Nevertheless, to our knowledge, none of these tools are able to easily detect or classify cb DI genomes. We therefore propose DI-tector as a tool that detects BP and RI of DI genomes from deep sequencing data sets in an unbiased manner. This program characterizes all potential DI genomes in the population, contrary to classical PCR amplification, which requires specific primers, assuming that the type of DI genomes searched is known. Moreover, DI-tector allows the use of a unique data set to detect various types of DI genomes, for instance from coinfected samples. DI-tector is available as a free python script at www.di-tector.cyame.eu. Graphical outputs, such as scatter-plot, chord diagram, sequence logo and linear representation of DI genomes, can be generated using the complementary online DI-tector (www.di-tector.cyame.eu).
One of the key steps of DI-tector is unmatched reads segmentation (Fig. 2ii). During this segmentation step, reads are split to detect the recombination junction, which occurred at least after the first X, and before the last X, nucleotides of the read (where X is defined by the user). This obviously excludes some reads overlapping the junction closer to extremities. This is mostly the case for the last read segment as this region is known to have lower quality compared to the beginning of the reads with Illumina technology. The exact site of recombination is sometimes tricky to identify, for instance when the nucleotides immediately after the BP are identical in the reference sequence and in the DI sequence at the junction site. Therefore, several possible combinations of BP and RI can be observed for the same junction site. Nevertheless, the distance between BP and RI, DI size and sequence will remain unchanged, which allows a correct characterization of the identified DI.
Putative DI genomes’ sequences determined by DI-tector must be confirmed in silico. One approach is to map all reads to the DI genome sequence. This allows the user to determine how many reads overlap the recombination junction and to identify the potential presence of reads mapping to both positive and negative strands, which is expected since DI genomes possess minimal signals for their replication. Stacked ladder-like patterns of short reads across the fusion point should be observed (Supplemental Fig. S2). Verifying that mapping does not occur in repetitive region must be done, as this would explain why the distance between the first and last read segment is not as expected and allow exclusion of mapping artifacts. Potential duplicated read and read quality can also be checked to reject ambiguous mapping.
During this in silico validation step, we observed that coverage at the recombinant junction was poor in certain conditions, mainly in total RNA samples purified from infected cells. This is likely due to the fact that around 99% of the reads of our data sets mapped to the human genome. Moreover, some cb DI genomes with junctions located near the extremities of the genomes are less covered. A recommendation is to have enough depth to avoid nondetection of certain DI genome sequences (mostly cb DI genomes) and increase confidence for detection of poorly covered recombination junctions. Starting from purified viral stocks may be envisaged to drastically reduce host read contaminants, but it may change the pattern of DI genomes.
DI-tector can be used to detect DI genomes in clinical samples, at the condition that the data set is rich enough in DI specific reads. Importantly, multiple reference sequences can be provided to DI-tector to perform a more global analysis on several genomes at the same time. Several enrichment techniques may be used to increase sequencing depth (Mertes et al. 2011), and therefore improve DI genomes’ detection sensitivity. Avoiding multiplexing can also improve the sequencing depth and exclude index hopping. Finaly, treating samples with RiboZero is another way to improve DI RNA genomes’ detection (Fig. 6).
In this study, we focused on cb DI genomes’ detection. DI_1236 was detected only in rMV-ΔV infected cells and was highly enriched after RIG-I purification. Another DI genome of 336 nt was also detected in RIG-I RNA samples. This 5′ cb DI genome had never been detected before due to RT-PCR technical limitations (position of 5′ cb DI genome specific primers). We used RT-qPCR to confirm the presence of DI_336 in our sample. This 5′ cb DI genome followed the rule of six, a hallmark shared by a large number of MV DI genomes (Mura et al. 2017).
In addition, DI-tector revealed the existence of 3′ cb DI genomes in MV and SeV samples. We confirmed the presence of 3′ cb DI genomes in MV infected cells by RT-PCR followed by Sanger sequencing analysis of the amplified PCR fragment. Therefore, we uncovered a novel aspect of Paramyxovirus biology by demonstrating that, similarly to 5′ cb DI genomes, which are generated when the viral RdRp detaches from the positive-sense antigenome template and reattaches itself to the nascent negative-sense strand, 3′ cb DI genomes are generated during the replication of the antigenome from the genome (Finke and Conzelmann 1999). These data highlight the power of the unbiased analysis performed by DI-tector.
Due to the nature of the samples, quantification of DI genomes detected by DI-tector is not reliable. For instance, in MV- and SeV-infected cell samples, reads may come from viral genomes or viral transcripts. Analysis of nontranslated regions may be used for DI genome quantification. This did not apply to our data since the coverage at genome extremity was lower than the average coverage. Using nontranscribed intergenic regions may also be a solution, but should be addressed for each virus and no automatic estimation of the quantity can be done. Moreover, only relative abundance may be assessed. A solution to quantify the DI genomes detected with DI-tector is to perform qPCR on the samples using specific probes overlapping the junction (Komarova et al. 2013; Mura et al. 2017), as the exact sequence of the recombination junction is given by DI-tector.
Predictive performance assessment of DI-tector using different experimental and simulated data sets revealed a robust specificity and sensitivity (Fig. 7; Supplemental Fig. S3). DI-tector prediction remains nevertheless poor when the junction is localized in the 10th first or last nucleotides of the reads. This is the trade-off to avoid a higher number of false-positive detections. To increase the power of detection we therefore recommend to use reads no shorter than 30 nt. This will not be an issue when DI genomes are plentiful (i.e., represented by several reads) as the probability that the junction span in the middle of the reads in at least some reads will exist. In case of rare events, we recommend to check the predicted junction position (see “*_output_sorted.txt” file). If the junction is closed to the read extremities (i.e., ∼10 nt), users should keep in mind that they have a higher chance to be false positive than when being further away from the read extremities. Classical techniques with high sensitivity, such as RT-qPCR, should be used to confirm the existence of these rare entities.
In conclusion, DI-tector provides a sensitive tool for DI viral genomes’ discovery, including cb DI genomes. It characterizes the BP and RI sites, as well as potential inserted nucleotides between recombination junctions. It applies to a variety of samples, from purified viral particles to infected whole cell lysates, if sufficient sequencing read depth is provided, allowing researchers to quickly identify and characterize different species of DI genomes. This method is useful to characterize DI genomes of viral populations in various viral and vaccine stocks.
MATERIALS AND METHODS
Our DI genome detection protocol comprises four main steps: (i) alignment of reads against host and viral genomes, (ii) unmatched and mismatched reads segmentation, (iii) second alignment of reads against viral genomes, and (iv) analyses of the matched reads, as detailed below (Figs. 2 and 3). These steps involved commonly used preexisting tools such as BWA (Li and Durbin 2009) and SAMtools (Li et al. 2009), which demand the minimum hardware requirement. Analyses and graphical representation of the results are proposed, respectively, as a python script freely available at www.di-tector.cyame.eu and a webtool at the same address. Figures 4, 6, 7, 8 and Supplemental Figures S1, S3, and S4 have been generated using a custom R script and R Studio (RStudio Team 2016). ggplot2 (Wickham 2009), readr (Wickham et al. 2017), and gridExtra (Auguie 2017) packages are required. Confirmation of newly discovered viral 5′ cb DI genomes implied usage of BWA, SAMtools, and BEDTools (Quinlan and Hall 2010).
Raw data
DI-tector pipeline proceeds with adaptor sequence trimmed fastq files generated from the sequencer. Reads quality may be evaluated by FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/) and reads can be quality trimmed when necessary. Fastq files (Single-end Read) for MV and recombinant MV that is unable to express the V protein (rMV-ΔV) have been previously generated in our laboratory (Sanchez David et al. 2016). Briefly, infected HEK-293 cells stably expressing One-STrEP-tagged RIG-I or Cherry protein were lysed with MOPS lysis buffer (20 mM MOPS-KOH pH7.4, 120 mM of KCl, 0.5% Igepal, 2 mM ß-Mercaptoethanol), supplemented with 200 unit/mL RNasin (Promega) and Complete Protease Inhibitor Cocktail (Roche). An aliquot of each cell lysate was used to perform total RNA purification using TRI Reagent LS (Sigma). The remaining cell lysate was kept to purify the RIG-I/RNA, or Cherry/RNA complexes by affinity chromatography using StrepTactin Sepharose High Performance beads (GE Healthcare). RNAs that had co-precipitated with either RIG-I or Cherry protein were isolated using TRI Reagent LS. RNA was dissolved in DNase-free and RNase-free ultrapure water. Extracted RNAs were analyzed using the Nanovue (GE Healthcare) and Bioanalyser RNA Nano Kit (Agilent). 300 ng of each RNA sample was treated for library preparation using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina). Sequencing was performed on the Illumina Hiseq2000 platform to generate single-end 51 bp reads bearing strand specificity (Sanchez David et al. 2016). Three biological experiments were performed.
Data sets of Sendai virus (SeV) infected Huh-7 cells were retrieved on NCBI (www.ncbi.nlm.nih.gov/sra, accession numbers SRX2600182 and SRX2600183). Briefly, RNA extraction of SeV infected cells was treated with DNase. Directional RNA-seq libraries were generated using the PrepX RNA-seq for Illumina Library Kit (WaferGen Biosystems). Libraries were pooled and sequenced on the NextSeq 500 (Illumina) targeting 10M PE reads at 150 read length. Reads were demultiplexed, which removed barcodes and sequencing adapters. The latter sample named SeV_RZ was treated with the Ribo-Zero Gold rRNA Removal Kit (Human/Mouse/Rat) (Illumina) prior to library preparation.
Reference sequences used in this study were GRCh38.p11 for Human genome (GCF_000001405.37_GRCh38.p11_genomic), NCBI reference sequence FJ211590.1 for MV (strain Schwarz), and NC_001552.1 for SeV. Fasta files containing human and viral genome sequences were indexed using the bwa index tool.
Alignment against host and viral genomes
The first step of the workflow consists of an alignment of the reads against the host genome (Fig. 2i). This step aims at discarding reads that map to the host genome and may partially map to the viral genome after segmentation, and at reducing the working file size. For example, MV and rMV-ΔV data sets were generated from total RNA samples of infected cells and contained mostly reads mapping the human genome (≈99%). This step uses a combination of bwa mem and samtools view with the parameters –bS –f4. An additional step consists of an alignment of the reads against the viral genome of interest, in order to exclude reads perfectly mapping the viral genome. Therefore, only unmapped reads are further analyzed. Of note, clipped reads (i.e., CIGAR motif contain S or H) are also conserved. Some of these reads may map to viral genome recombination junctions that are present in DI genomes.
Reads segmentation
The second step consists of a segmentation of each unmapped read into two pieces, assigned “first” and “last” segments (Fig. 2ii). This step allows the mapping of reads that contain junctions of the DI genomes, with first and last segments mapping each side of the junction. Moreover, in the case of cb DI genomes, each part of the initial read maps to forward and reverse strands of the viral RNA.
As the position of the junction is randomly covered by the reads, for each single read the first segment size will vary from X to N-X nucleotides and the last segment from N-X to X (where N is the initial length of the read and X the minimum segment length defined by the user using the parameter “--Min_Segment”) (Fig. 2ii). Increasing the range of segmentation (i.e., decreasing the X value) may increase the number of DI genomes detected, at the expense of working file size and time of analysis (Supplemental Fig. S1).
Alignment of segmented reads against the viral genome, analyses of matched reads, and characterization of DI genome sequences
The third step consists of an alignment of the segments against the viral genome of interest using bwa aln, bwa samse (Fig. 2iii). For the last step, only QNAME (read names), FLAG, RNAME (reference names), POS (position), MAPQ (mapping quality), CIGAR, SEQ (sequence of the segment), and MD tag values (string for mismatching positions) are collected (Fig. 2iv). Reads for which the first and/or the last segments are unmapped or multimapped are not further processed. Then FLAG values for both read segments are compared to allow DI genomes’ clustering by the nature of their junctions (Fig. 3). If the two fragments map to different reference strands, these reads will be processed to uncover cb DI genomes. If the two fragments map to the same reference strand, positions of the two fragments will be compared to characterize deletion/insertion forms of the DI genome. To define BP and RI sites, positions between first and last segments of the reads are compared. These positions are adjusted to take into consideration the MD tag value (i.e., mismatch, deletion, or clip). The user can define filtering values allowing to discard reads covering junction with low mapping quality or small insertion/deletion. The size filtering step is not applied to cb DI genomes’ detection to avoid exclusion of sb DI genomes, as they are characterized by small loops, i.e., a small delta between fragment positions.
Individual discovered DI genome entities are counted and sorted by type (5′ cb DI, 3′ cb DI genomes, insertions, or deletions). Results are exported in four main text format files. “*_summary.txt”, which recapitulates initial command line parameters and the total number of reads mapping a junction of each type of DI genomes. “*_counts.txt” lists all DI genomes with their respective counts and percentage, type, length, BP, and RI sites. Information concerning the length of the insert/deletion or loops is also mentioned. “*_output_sorted.txt” lists all reads overlapping a junction. For each read, DI genome length, BP, RI, delta position between BP and RI, segmentation of the read, MAPQ, RNAME, CIGAR, MD, POS (for both segments), QNAME values are mentioned. The sequence of the read is printed in upper-case letter before the junction and lower-case letter after. The sequence of each DI genome can be generated and exported as a multifasta file “*_fasta.fa”. Intermediate fastq and sam format files are also generated for potential further manual analyses. “*_Error.txt” and “*_Ali.txt” contain, respectively, error messages and a list of putatively correctly aligned segments that passed previous filters, mostly due to too many mismatches during alignment against the viral genome.
DI-tector parameters
DI-tector uses several parameters to process data. “Virus_Ref” and “Input_Data” are mandatory strings to define paths to files for, respectively, the indexed viral genome sequence and raw data file in fastq format (or fasta format if parameter “-f” or “—Fasta” is used). In addition, users can provide host genome reference indexed sequence, using “-g” or “--Host_Ref”, to perform optional alignment against host genome (Fig. 2i).
Two important parameters are “-s” or “--Min_Segment” and “-n” or “--Nb_Reads”, which respectively allow to set up minimum segment length (Default is 15) and show only DI genomes with count reads superior to “--Nb_Reads” value (Default is 1).
Several other parameters can be set up by the users, such as “-m” or “--Min_MAPQ” to skip alignments with MAPQ smaller than the mentioned value (Default is 25), “-l” or “--InDel_Length” to skip alignments with size of InDels smaller or equal to the mentioned value (Default size is 1), “-x” or “--Nb_threads” for the number of threads to use (Default is 1), “-o” or “--Output_Directory” for directory name where all compiled output files will be saved, “-t” or “--Tag” for tagging file names, or “-k” or “--Keep_files” to keep or not intermediate files (i.e., alignment, etc.). Basic help on parameters can be accessed using “-h” or “--help”.
Assessment of DI-tector detection performance
The detection performance of DI-tector was evaluated using three simulated data sets generated independently. They respectively contain 500,000 simulated reads covering junctions of 5′ copy-back (“5′ cb”), 3′ copy-back (“3′ cb”) DI genomes, or DI genomes with insertion/deletion (“INDEL”). These data sets were generated as follows: Two times 500,000 random values from 1 to 15,894 (MV genome length) were generated, respectively, for BP and RI sites. This step was repeated individually for each data set. For each combination of BP and RI values, a random value (J) between 1 and 50 (51 nt reads length −1) was generated. We generated 51-nt-long reads since the experimental data set for MV and rΔV-MV samples contained 51-nt-long reads. For the data set “INDEL,” sequences of MV genome from BP to BP + J and RI to RI + (51 − J) were combined. For the data set “5′ cb”, the first part of the read (i.e., from BP to BP + J) was reverse complemented. In a similar manner, the second part of the simulated reads [i.e., RI to RI + (51 − J)] was reverse complemented to generate “3′ cb” data set. Sensitivity (Sn) and specificity (Sp) were calculated as follows: Sn = TP/(TP + FN); Sp = TN/(TN + FP). TP (true positive) and FN (false negative) are respectively correctly and incorrectly detected 5′ copy-back junctions in “5′ cb” data set. FP (false positive) and TN (true negative) are respectively clustered as 5′ copy-back junction or not in “INDEL” data set. Data sets from noninfected cells (total or beads) (unpublished data) were also analyzed to estimate putative FP from host material.
Data sets used in Supplemental Figure S4 were similarly generated. The three simulated data sets contain a total of 5 × 105 reads covering 2.5 × 105, 1.6 × 105, and 1.3 × 105 junctions, respectively. This represents two, three, and four reads per unique junction for the corresponding data set. We also generated two simulated data sets containing reads of 100 or 150 nt.
Confirmation of newly discovered viral DI genome
To confirm the existence of reads mapping to the newly discovered recombination junction (BP and RI sites for DI_336), data sets were mapped to DI_336 and, in parallel, to DI_1236 indexed sequence using bwa mem, sorted and indexed using samtools sort and samtools index. Only reads mapping the recombination junction were represented (Supplemental Fig. S2).
RT-qPCR and RT-PCR detection of cb DI genomes
HEK293T cells were seeded into T25 flasks one day before infection. Recombinant MV infections were carried out at MOI of 1. Virus stocks were diluted with Opti-MEM to obtain a final inoculum volume of 2 mL. Cells were incubated with virus for 2 h at 37°C. Then, 4 mL of DMEM containing 10% FBS was added in each T25 flask, and cells were incubated at 37°C until infections were stopped by cell lysis 24 h later. Total RNA was extracted with the RNeasy Mini Kit (Qiagen). Quality and quantity of extracted RNAs were analyzed using the Nanovue and Bioanalyser RNA Nano Kit. RT-qPCR analysis was performed using Applied Biosystems StepOnePlus technology. DI and viral genome primers and probes were designed using Primer Express software (Applied Biosystems). Reactions were performed on 400 ng of total RNA by use of TaqMan RNA-to-Ct 1-Step Kits (Thermo Fisher Scientific) for one-step RT-qPCR analyses according to the manufacturer's protocol. Reactions were performed in a final volume of 20 µL in the presence of 100 nM TaqMan DI_336 (TGA CTT GGA TAG ATT CTT-FAM) or Genome (CAT CAG AAT TAA GAA AAA CGT AG-VIC) -specific probe and 100 nM DI_336 or Genome-specific forward (DI_336: CCC CCG TCA TAA TAA TCT GTT TCT; Genome: TCA GGC ATA CCC ACT AGT GTG AA) and reverse (DI_336 : ACC ACC TAG GGC AGG ATT AGG; Genome: TGA CAG ATA GCG AGT CCA TAA CG) primers.
3′ cb DI genome detection by RT-PCR was performed on RNA samples from MV infected cells prepared as described above. First-strand complementary DNA (cDNA) synthesis was performed on 500 ng of total RNA in a final volume of 20 µL with the RevertAid H Minus M-MuLV Reverse Transcriptase (Thermo Scientific) using primer F1 (AAA GTT GGG TAA GGA TAG) according to the manufacturer's protocol. Two microliters of the above RT reaction and Phusion High-Fidelity DNA Polymerase (New England Biolabs) was used for PCR reaction and in accordance with the manufacturer's protocol. Amplicon of DI_2202 was generated using primers F307 (CCA AAC TAA CAG GGG CAC TAA TAG G) and F677 (TTC GGA GCT AAG AAG GTG GA) and sent for Sanger sequencing for sequence confirmation.
DATA DEPOSITION
Python script used in this article is available at www.di-tector.cyame.eu. Graphical output can be generated using the freely available online DI-tector.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Elias Hage, Dr. Mickael Orgeur, Dr. Guillaume Cornelis, the Transcriptome and Epigenome Platform of Institut Pasteur, specifically Rachel Legendre, and all members of “Unité de Génomique Virale et Vaccination” for advice and discussions. We thank Dr. Bernd Jagla (“Hub Bioinformatique et Biostatistique,” Institut Pasteur) and Dr. Etienne Simon-Loriere (“Génomique évolutive des virus à ARN,” Institut Pasteur) for critical reading of the manuscript. We also thank Ségolène Gracias for help in choosing the online DI-tector logo. This work was supported by the Institut Pasteur, the “Centre National de la Rercherche Scientifique,” and funding from Agence Nationale de la Recherche (ANR-16-CE15-0025-01 to N.J.). M.M. was supported by the Institut de Recherche Biomédicale des Armées (IRBA).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.066910.118.
REFERENCES
- Auguie B. 2017. gridExtra: miscellaneous functions for “grid” graphics. R package version 2.3. https://CRAN.R-project.org/package=gridExtra.
- Baum A, García-Sastre A. 2011. Differential recognition of viral RNA by RIG-I. Virulence 2: 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum A, Sachidanandam R, Garcia-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc Natl Acad Sci 107: 16303–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellocq C, Mottet G, Roux L. 1990. Wide occurrence of measles virus subgenomic RNAs in attenuated live-virus vaccines. Biologicals 18: 337–343. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res 14: 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimmock NJ, Easton AJ. 2014. Defective interfering influenza virus RNAs: time to reevaluate their clinical potential as broad-spectrum antivirals? J Virol 88: 5217–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke S, Conzelmann KK. 1999. Virus promoters determine interference by defective RNAs: selective amplification of mini-RNA vectors and rescue from cDNA by a 3′ copy-back ambisense rabies virus. J Virol 73: 3818–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frensing T. 2015. Defective interfering viruses and their impact on vaccines and viral vectors. Biotechnol J 10: 681–689. [DOI] [PubMed] [Google Scholar]
- Holland JJ. 1990. Defective viral genomes. In Virology, 2nd ed. (ed. Fields BN, et al. ), pp. 151–165. Raven Press, New York. [Google Scholar]
- Huang AS. 1973. Defective interfering viruses. Annu Rev Microbiol 27: 101–118. [DOI] [PubMed] [Google Scholar]
- Iwai A, Marusawa H, Takada Y, Egawa H, Ikeda K, Nabeshima M, Uemoto S, Chiba T. 2006. Identification of novel defective HCV clones in liver transplant recipients with recurrent HCV infection. J Viral Hepat 13: 523–531. [DOI] [PubMed] [Google Scholar]
- Ke R, Aaskov J, Holmes EC, Lloyd-Smith JO. 2013. Phylodynamic analysis of the emergence and epidemiological impact of transmissible defective dengue viruses. PLoS Pathog 9: e1003193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killip MJ, Young DF, Gatherer D, Ross CS, Short JAL, Davison AJ, Goodbourn S, Randall RE. 2013. Deep sequencing analysis of defective genomes of parainfluenza virus 5 and their role in interferon induction. J Virol 87: 4798–4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakofsky D, Roux L, Garcin D, Ruigrok RWH. 2005. Paramyxovirus mRNA editing, the “rule of six” and error catastrophe: a hypothesis. J Gen Virol 86: 1869–1877. [DOI] [PubMed] [Google Scholar]
- Komarova AV, Combredet C, Sismeiro O, Dillies MA, Jagla B, Sanchez David RY, Vabret N, Coppée JY, Vidalain PO, Tangy F. 2013. Identification of RNA partners of viral proteins in infected cells. RNA Biol 10: 943–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzarini RA, Keene JD, Schubert M. 1981. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 26: 145–154. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Lott WB, Lowry K, Jones A, Thu HM, Aaskov J. 2011. Defective interfering viral particles in acute dengue infections. PLoS One 6: e19447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López CB. 2014. Defective viral genomes: critical danger signals of viral infections. J Virol 88: 8720–8723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni TB, López CB. 2018. Defective (interfering) viral genomes re-explored: impact on antiviral immunity and virus persistence. Future Virol 10.2217/fvl-2018-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott AC, Dimmock NJ. 2010. Defective interfering viruses and their potential as antiviral agents. Rev Med Virol 20: 51–62. [DOI] [PubMed] [Google Scholar]
- McLaren LC, Holland JJ. 1974. Defective interfering particles from poliovirus vaccine and vaccine reference strains. Virology 60: 579–583. [DOI] [PubMed] [Google Scholar]
- Mercado-López X, Cotter CR, Kim WK, Sun Y, Muñoz L, Tapia K, López CB. 2013. Highly immunostimulatory RNA derived from a Sendai virus defective viral genome. Vaccine 31: 5713–5721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertes F, ElSharawy A, Sauer S, van Helvoort JMLM, van der Zaag PJ, Franke A, Nilsson M, Lehrach H, Brookes AJ. 2011. Targeted enrichment of genomic DNA regions for next-generation sequencing. Brief Funct Genomics 10: 374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne I, Stephen G, Bayer M, Cock PJA, Pritchard L, Cardle L, Shaw PD, Marshall D. 2013. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform 14: 193–202. [DOI] [PubMed] [Google Scholar]
- Mura M, Combredet C, Najburg V, Sanchez David RY, Tangy F, Komarova AV. 2017. Non-encapsidated 5′ copy-back defective-interfering genomes produced by recombinant measles viruses are recognized by RIG-I and LGP2 but not MDA5. J Virol 91: e00643-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuru S, Ueda Y, Marusawa H, Inuzuka T, Nishijima N, Nasu A, Shimizu K, Koike K, Uemoto S, Chiba T. 2013. Dynamics of defective hepatitis C virus clones in reinfected liver grafts in liver transplant recipients: ultradeep sequencing analysis. J Clin Microbiol 51: 3645–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrault J. 1981. Origin and replication of defective interfering particles, pp. 151–207. Springer, Berlin. [DOI] [PubMed] [Google Scholar]
- Pfaller CK, Mastorakos GM, Matchett WE, Ma X, Samuel CE, Cattaneo R. 2015. Measles virus defective interfering RNAs are generated frequently and early in the absence of C protein and can be destabilized by adenosine deaminase acting on RNA-1-like hypermutations. J Virol 89: 7735–7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao DD, Huang AS. 1982. Interference among defective interfering particles of vesicular stomatitis virus. J Virol 41: 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Re GG, Gupta KC, Kingsbury DW. 1983. Sequence of the 5′ end of the Sendai virus genome and its variable representation in complementary form at the 3′ ends of copy-back defective interfering RNA species: identification of the L gene terminus. Virology 130: 390–396. [DOI] [PubMed] [Google Scholar]
- Routh A, Johnson JE. 2014. Discovery of functional genomic motifs in viruses with ViReMa—a Virus Recombination Mapper—for analysis of next-generation sequencing data. Nucleic Acids Res 42: e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team. 2016. RStudio: integrated development environment for R. Boston, MA: http://www.rstudio.com/. [Google Scholar]
- Runge S, Sparrer KMJ, Lässig C, Hembach K, Baum A, García-Sastre A, Söding J, Conzelmann KK, Hopfner K-P. 2014. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog 10: e1004081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V, et al. 2013. Sequence analysis of in vivo defective interfering-like RNA of influenza A H1N1 pandemic virus. J Virol 87: 8064–8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez David RY, Combredet C, Sismeiro O, Dillies MA, Jagla B, Coppée JY, Mura M, Galla MG, Despres P, Tangy F, et al. 2016. Comparative analysis of viral RNA signatures on different RIG-I-like receptors. Elife 5: e11275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shingai M, Ebihara T, Begum NA, Kato A, Honma T, Matsumoto K, Saito H, Ogura H, Matsumoto M, Seya T. 2007. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J Immunol 179: 6123–6133. [DOI] [PubMed] [Google Scholar]
- Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351: 101–111. [DOI] [PubMed] [Google Scholar]
- Sun Y, Jain D, Koziol-White CJ, Genoyer E, Gilbert M, Tapia K, Panettieri RA, Hodinka RL, López CB. 2015. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog 11: e1005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timm C, Akpinar F, Yin J. 2014. Quantitative characterization of defective virus emergence by deep sequencing. J Virol 88: 2623–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilijevic J, Zamarreño N, Oliveros JC, Rodriguez-Frandsen A, Gómez G, Rodriguez G, Pérez-Ruiz M, Rey S, Barba I, Pozo F. 2017. Reduced accumulation of defective viral genomes contributes to severe outcome in influenza virus infected patients. PLoS Pathog 13: e1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodovar N, Goic B, Blanc H, Saleh MC. 2011. In silico reconstruction of viral genomes from small RNAs improves virus-derived small interfering RNA profiling. J Virol 85: 11016–11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer, New York. [Google Scholar]
- Wickham H, Hester J, Francois R. 2017. readr: read rectangular text data. R package version 1.1.1. https://CRAN.R-project.org/package=readr. [Google Scholar]
- Wiktor TJ, Dietzschold B, Leamnson RN, Koprowski H. 1977. Induction and biological properties of defective interfering particles of rabies virus. J Virol 21: 626–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.