

ABSTRACT
Immunogenic cell death is characterized by the emission of danger signals that facilitate activation of an adaptive immune response against dead-cell antigens. In the case of cancer therapy, tumor cells undergoing immunogenic death promote cancer-specific immunity. Identification, characterization, and optimization of stimuli that induce immunogenic cancer cell death has tremendous potential to improve the outcomes of cancer therapy. In this study, we show that non-thermal, atmospheric pressure plasma can be operated to induce immunogenic cell death in an animal model of colorectal cancer. In vitro, plasma treatment of CT26 colorectal cancer cells induced the release of classic danger signals. Treated cells were used to create a whole-cell vaccine which elicited protective immunity in the CT26 tumor mouse model. Moreover, plasma treatment of subcutaneous tumors elicited emission of danger signals and recruitment of antigen presenting cells into tumors. An increase in T cell responses targeting the colorectal cancer-specific antigen guanylyl cyclase C (GUCY2C) were also observed. This study provides the first evidence that non-thermal plasma is a bone fide inducer of immunogenic cell death and highlights its potential for clinical translation for cancer immunotherapy.
Keywords: Non-thermal plasma, immunogenic cell death, colorectal cancer, plasma cancer immunotherapy
Introduction
Cancer treatment strategies in the past have focused on reducing tumor burden through delivery of cytotoxic agents. These methods often do not rely on the patient’s adaptive immune responses for the resolution of cancer. Thus, once cells escape treatment, they continue to grow, resulting in tumor recurrence and resistance to therapy.1–3 Immunogenic cell death (ICD), initially described by Zitvogel, Kroemer, and co-workers, is a modality of death where dying cells stimulate immune responses against dead-cell antigens.4–6 In cancer therapy, this is advantageous as tumor cells undergoing ICD activate an anti-tumor immune response that is specific for that cancer. Therefore, developing treatments that elicit immunogenic cell death and facilitate the active participation of the patient’s adaptive immune system, offer the potential to improve clinical outcomes of cancer therapy.
Plasma, the fourth state of matter, is ionized gas composed of charged particles, active neutral gas species, electric fields, and low amounts of ultraviolet (UV) light.7–11 Development of plasma systems that can be sustained in atmospheric pressure and at room temperature has opened doors for biomedical applications, including, but not limited to, cancer therapy.12–14 Mounting evidence demonstrates that these ‘non-thermal plasmas’ (NTP) can be optimized to destroy tumors with minimal damage to neighboring healthy tissue.14–17 Decreased tumor burden and prolonged animal survival following direct plasma treatment have been reported, suggesting that plasma should be further explored as a viable candidate for cancer treatment.18
The potential of NTP to induce immunogenic cancer cell death is only recently being explored. Bekeschus, et al. has demonstrated that NTP treatment of two murine cell lines in vitro, the B16F10 melanoma cells and the CT26 colorectal cancer cells, increased immunogenic cell surface molecules such as major histocompatibility complex I (MHC-I) and surface-exposed calreticulin (ecto-CRT).19,20 We have reported successful in vitro ICD induction in two human cell lines, a radiation-resistant primary nasopharyngeal carcinoma cell line (CNE-1) and the A549 lung carcinoma cell line in response to NTP exposure.21,22 The mechanism is postulated to be reactive oxygen and nitrogen species (RONS) dependent. NTP-generated RONS rapidly change the oxidative status of cells and induce endoplasmic reticulum (ER) stress pathways in these cells.19–22 Upregulation of two proteins associated with ER stress and upstream of CRT emission, activating transcription factor 4 (ATF4) and stanniocalcin (STC2), was also demonstrated.21 Moreover, abrogation of NTP-generated and cell-stimulated RONS tempered the effect of NTP on CRT emission. These reports indicate that NTP-induced ICD is not specific to a single cancer cell type, and merits further investigation into its clinical relevance as an anti-cancer modality. Plasma treatment in animal models of cancers is needed to assess if plasma-induced ICD could benefit patient outcome.
In this study, we used the CT26 murine colorectal tumor model to explore the potential of NTP to induce ICD in vivo. NTP generated by a nanosecond-pulsed dielectric barrier discharge (nspDBD) plasma system induced the expression of two key surrogate markers of ICD in these cancer cells: ecto-CRT and secreted adenosine triphosphate (ATP). A vaccination assay, used to determine if a stimulus is a bone fide ICD-inducer, showed partial protective immunity against tumor challenge in syngeneic Balb/c mice immunized with NTP-treated CT26 cells. Furthermore, treatment of subcutaneous colorectal tumors expressing the cancer antigen guanylyl cyclase C (GUCY2C) resulted in higher expression of ICD markers in tumors, recruitment of antigen presenting cells (APCs), and generation of more GUCY2C-specific T cells. Together, our findings are the first report that establish the potential of plasma for cancer immunotherapy via ICD.
Results
Plasma induces emission of surrogate markers of ICD
To measure cell death in response to nspDBD plasma, the CT26 colorectal carcinoma cell line was exposed to several plasma energies. Cell viability, quantified with a Muse Cell Analyzer 24 hours after plasma treatment, decreased in an energy dependent manner (Figure 1(a)). As previously described, not all modalities of cell death are immunogenic and capable of initiating anti-tumor effects. The identification of ICD in vitro mainly relies on detection of associated damage associated molecular patterns (DAMPs). Therefore, we examined the effect of plasma on cell viability and two DAMP signals in CT26 cells: externalization of CRT and secretion of ATP.23
Figure 1.
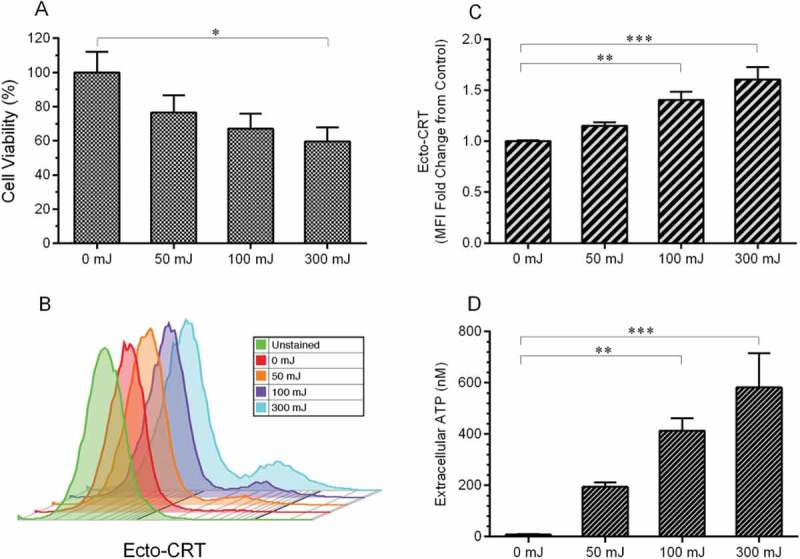
Plasma-induced cell death, surface emission of CRT, and secretion of ATP in CT26 cells. A) Cell viability is indicated by the percentage of live CT26 cells normalized to untreated (0 mJ), 24 hours after plasma treatment. B, C) CRT was detected on the surface of intact CT26 cells 24 hours after plasma exposure. B) Representative histograms and C) mean fluorescence intensity showed increased surface CRT following plasma treatment. D) ATP content was detected in the media 10 minutes after plasma treatment using a chemiluminescent kit. CRT, ATP, and viability data are presented as means ± S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001 (one-way ANOVA, Dunnett’s multiple comparison test).
Immunogenicity of dying cancer cells is strongly dictated by surface exposure of CRT.24 Normally located on the ER membrane, exposed CRT on the cell surface acts as an ‘eat me’ DAMP signal that facilitates recognition, engulfment, and processing of tumor cells by APCs,25–27 a critical step for the initiation of an adaptive anti-cancer response.28–30 Surface-exposure of CRT in response to 10 second plasma exposure was measured 24 hours after treatment. Intact cells were labeled with anti-CRT antibodies, stained with fluorescent secondary antibodies and analyzed using flow cytometry. Our results show that the emission of ecto-CRT on CT26 cells increased in an energy dependent manner, suggesting plasma may increase the immunogenicity of tumor cells (Figure 1(b,c)).
ATP, the most abundant intracellular molecule required for metabolism, is secreted from cells undergoing ICD.31 It has even been suggested that secretion of ATP follows overlapping pathways with externalization of CRT.32 Once ATP reaches the extracellular space, it becomes another hallmark of ICD and functions as a ‘find me’ DAMP signal for recruitment and activation of APCs.32–34 To detect this secreted DAMP by cells exposed to plasma, the cell culture media was collected 10 minutes after treatment and extracellular ATP was quantified. ATP levels were low at baseline (8.2 nM) and increased 70-fold (582.1 nM) following 300 mJ plasma treatment (Figure 1(d)).
Vaccination with plasma-induced ICD cells provides protection against tumor challenge in mice
To ascertain whether the DAMP signals elicited by plasma could enhance immune responses against cancer, we performed a vaccination assay. Balb/c mice were immunized with CT26 cells treated in vitro with plasma at the ICD-inducing regime (29 kV, 30 Hz, 1 mm gap distance, 10 seconds). Cells were prepared for inoculation and injected into the left flank as a whole-cell vaccine to allow an immune response to develop. One week after immunization, mice were challenged with live CT26 cancer cells on the opposite flank and tumor growth was monitored twice a week until day 26 when the study was terminated as a subset of the animals reached IACUC-approved endpoints (Figure 2(a)). CT26 cells treated with media only or Cisplatin (50 μM for 24 hours), a non-ICD inducer,23 were used as controls.
Figure 2.
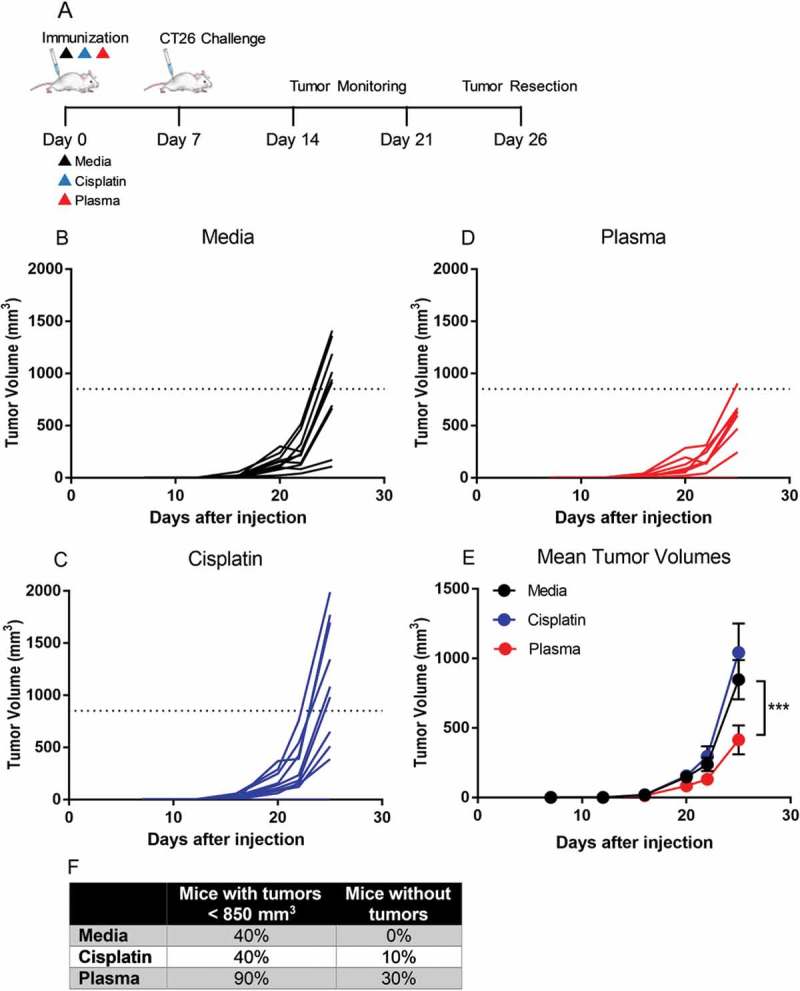
Immunization of mice with cancer cells plasma-treated at ICD-inducing regimes reduced the growth rate in challenge tumor. A) Balb/c mice were injected with media-, Cisplatin-, or plasma-treated CT26 cells subcutaneously into the left flank (n = 10 per group). One week later, mice were challenged with live CT26 cells on the opposite flank and monitored for 26 days. Challenge tumors grew more rapidly in mice immunized with cells treated with media (B) or Cisplatin (C) compared to mice immunized with plasma-treated cells (D). Data in B-D indicate tumor growth of each mouse. A dotted line is plotted to indicate the mean tumor volume of the control group (media treatment). E) Mean tumor volumes in the mice vaccinated with plasma-treated cells were significantly smaller compared to those immunized with untreated cells. Data are presented in E as mean value ± S.E.M. ***p < 0.001 (Two-way ANOVA, Dunnett’s multiple comparisons test). F) The percentage of mice with tumor volumes less than the mean volume of the media group and the percentage of mice that did not develop subcutaneous tumors at the challenge site are displayed.
Challenge tumors in the media and Cisplatin groups grew rapidly while tumors in the plasma group developed relatively slowly (Figure 2(b–d)). The mean tumor volume for the plasma immunized group was significantly smaller compared to that of the media group (414.7 ± 104.3 mm3 vs 847.4 ± 141.5 mm3; p < 0.001) or the Cisplatin group (1041.8 ± 208.3 mm3) at day 26 (Figure 2(e)). Indeed, 90% of the mice in the plasma immunized group had tumor volumes smaller than the mean tumor volume of the media group (850 mm3), suggesting that these mice were partially protected by vaccination. Moreover, 3 out of the 10 mice in the plasma group did not develop subcutaneous tumors on the challenge site (Figure 2(f)).
Plasma induces ICD in vivo and stimulates immune cell recruitment
To directly validate whether plasma could induce ICD in vivo, CT26 colorectal tumors were established subcutaneously in Balb/c mice and exposed to plasma when they became palpable (Figure 3). A safe operating plasma regimen was first identified by exposing subcutaneous CT26 tumors in Balb/c mice to plasma for various treatment times (10, 25, 50 sec) at set plasma parameters (29 kV and 750 Hz) once daily for five consecutive days. One day and 3 days after the final treatment, tumors were resected with overlaying skin, fixed, and stained with hematoxylin and eosin (H&E) to assess the structural changes in the dermal and epidermal layers of the skin. Ten seconds of plasma treatment resulted in minimal epidermal damage but no changes to the tumor were observed (Figure 4(b,f)). When plasma treatment duration was increased to 25 seconds, thermal and necrotic damage in the epidermis was observed one day after plasma (Figure 4(c)) but not in the underlying tumor. By the third day after treatment, the epidermis appears improved but not fully healed, suggesting that the damage was reversible (Figure 4(g)). The 50-second treatment resulted in considerable damage through all the layers of the skin, and, in fact, tumors just below the skin showed thermal and necrotic damage (Figure 4(d)). By the third day thermal damage was still apparent and changes in collagen had begun to appear. There were also more neutrophils in the skin signifying an inflammatory response (Figure 4(h)).
Figure 3.

In vivo plasma treatment system. A) A nanosecond-pulsed power supply generated 29 kV pulses and a function generator was used to control the frequency of pulses and the plasma exposure time. A z-positioner was used to hold the high voltage electrode in place during treatment, approximately 1 to 2 mm above the target. B) Plasma was generated directly above of the subcutaneous tumor.
Figure 4.
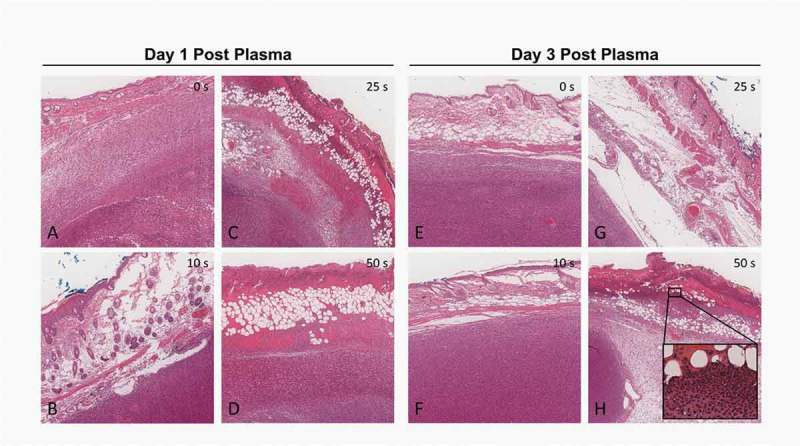
H & E staining of plasma-treated subcutaneous tumors. Tumors with overlaying skin were resected 1 day (A-D) or 3 days (E-H) following the final plasma treatment. Tumors received no treatment (A, E), 10-second treatment (B, F), 25-second treatment (C, G) or 50-second treatment (D, H) daily over the course of 5 days. Images were taken at 20x. Inset in H shows neutrophil infiltration.
Based on the results of our safety studies, we chose to investigate if the 10 second plasma treatment over five days induced ICD in the subcutaneous tumors. Tumors were exposed to plasma using three different treatment procedures: i) the same area each day (Plasma 1 Spot), ii) two areas of the tumor each day (Plasma 2 Spots), or iii) different areas of the tumor each day (Plasma Multiple Spots). Three days after the last plasma treatment, tumors were resected, fixed, and sectioned. Immunofluorescence microscopy was performed on tumor sections to identify ICD markers. CRT expression increased in all plasma treatment groups (Figure 5(a)) and was maximum following the “Plasma Multiple Spot” regimen (1.6 ± 0.2 fold, p < 0.05). Tumor sections were also stained for High mobility group box 1 (HMGB1), another DAMP signal much like ATP, that recruits inflammatory immune cells and mediates signals between APCs.35–38 HMGB1 has been observed to translocate from the nucleus to the cytoplasm and extracellular space.37–39 Here, we observed an increase in immunofluorescence intensity of HMBG1 in all plasma treatment groups (Figure 5(b)), suggesting HMGB1 protein concentration may be elevated following plasma exposure.
Figure 5.
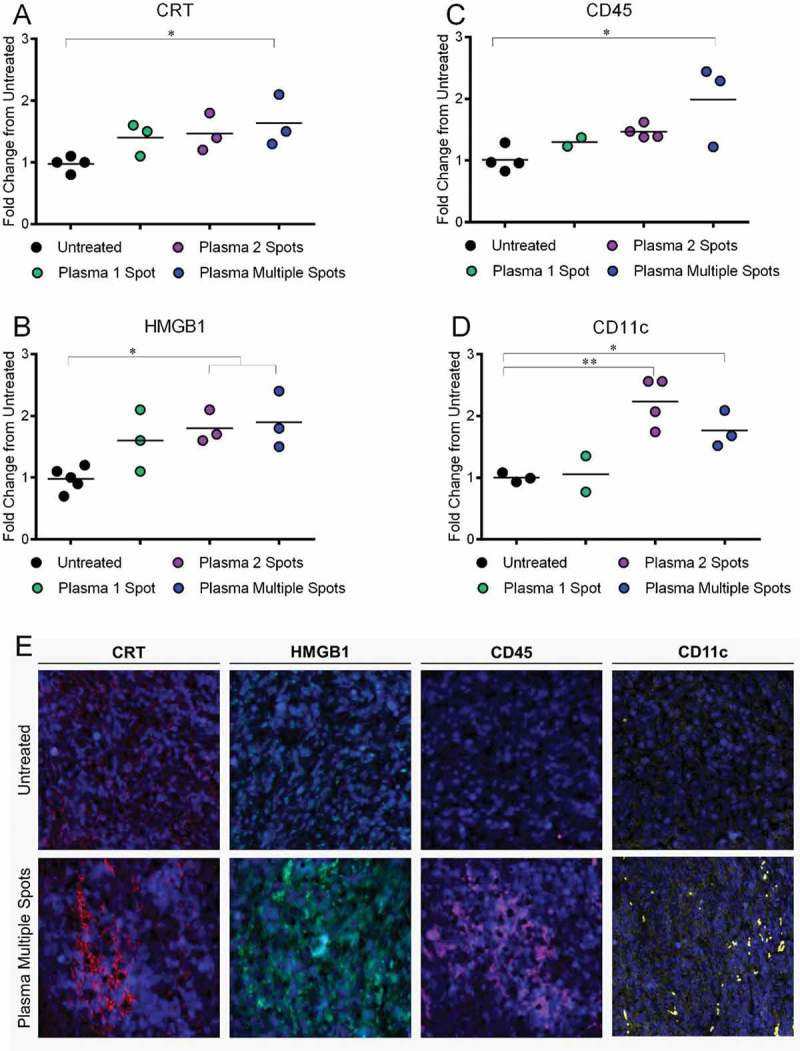
Plasma treatment of subcutaneous tumors in Balb/c mice induced DAMP emission and increased leukocytes and APCs in the tumor 3 days after final treatment. Tumor sections were fixed, stained, and imaged by fluorescence microscopy. Fluorescence intensity of representative sections of the tumor were quantified with ImageJ and normalized to untreated controls. The emission of A) CRT and B) HMGB1 increased in all plasma treatment groups by ~1.5-fold. The presence of leukocytes (C, CD45+ cells) and APCs (D, CD11c+ cells) increased compared to untreated tumors. Data are presented from individual resected tumors. *p < 0.05, **p < 0.05 (One-way ANOVA, Dunnett’s multiple comparisons test). E) Representative images of tumors subject to multi-spot plasma treatment compared to untreated controls are shown (10x, 400μm ᵡ 400μm).
To determine if emitted DAMPs stimulated the recruitment of immune cells into the tumor environment, we also stained for CD45+ (leukocytes) and CD11c+ (APCs) immune cells. Indeed, increased CRT and HMGB1 were associated with more CD45+ (2.0 ± 0.4 fold, p < 0.05) and CD11c+ cells (1.8 ± 0.2 fold, p < 0.05) following the Plasma Multiple Spots regimen (Figure 5(c,d)). Because CD45 is expressed by all leukocytes, including T cells, B cells, neutrophils, NK cells and others,40,41 it is possible that other immune cell subsets are also being recruited as a downstream consequence of ICD induction. Representative immunofluorescence images of the multi-spot treatment compared to the untreated are shown in (Figure 5(e)).
Altogether, Plasma Multiple Spots treatments enhanced both emission of DAMPs and recruitment of immune cells in the tumor compared to the other application methods. Therefore, treatment of different spots may be more beneficial compared to repeat treatment of the same area. This treatment condition was used to investigate plasma-induced ICD effects on downstream T-cell response.
Plasma amplifies specific T-cell responses against CT26-GUCY2C tumors
To analyze whether plasma-induced ICD could stimulate an adaptive anti-tumor response, we treated subcutaneous CT26 tumors expressing the colorectal cancer antigen GUCY2C42,43 (CT26-GUCY2C) in Balb/c mice. Mice were treated with either plasma alone or with plasma in combination with the Ad5-GUCY2C-S1 vaccine. The Ad5-GUCY2C-S1 vaccine was previously shown to safely induce GUCY2C-specific immune responses and antitumor immunity in mice44–51 and has been translated to human clinical trials.52 Mice were treated with plasma for five consecutive days and one group was vaccinated with the Ad5-GUCY2C-S1 vaccine, one week after the last plasma treatment (Figure 6(a)). An untreated group and a vaccine only group (Ad5-GUCY2C-S1 vaccination) served as our negative and vaccine controls, respectively.
Figure 6.
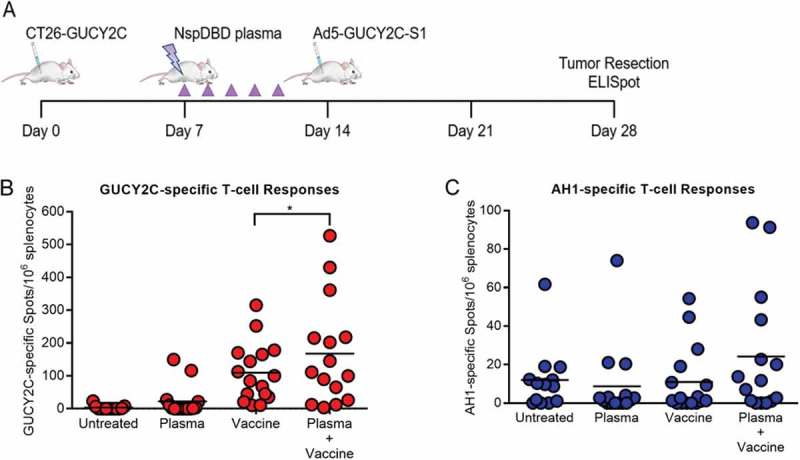
Plasma in combination with vaccination enhanced cancer-specific T-cell responses. A) Mice were challenged with CT26-GUCY2C cells (Day 0) and treated with plasma on day 7. A subgroup of the plasma-treated mice was vaccinated on day 14 with Ad5-GUCY2C-S1. B) GUCY2C-specific and C) AH1-specific T-cell responses were quantified by IFNγ ELISpot in the spleen o.
Splenic GUCY2C-specific T-cell responses were analyzed by IFNγ ELISpot assay 28 days after the initial tumor inoculation. Plasma treatment alone had a marginal effect on GUCY2C-specific responses (22.1 ± 11.9 spots vs 3.0 ± 1.8 spots in Untreated; p = ns). However, plasma treatment prior to vaccination amplified GUCY2C-specific T-cell responses (167.8 ± 41.5 spots vs 109.7 ± 22.3 spots with vaccine alone, p < 0.05) (Figure 6(b)). This observation supports the potential of plasma to increase the immunogenicity of cancer cells and stimulate canonical pathways required for tumor control.
Typically, immune responses are generated against one or two dominant epitopes of an antigen. When the specificity of immune responses spreads to include subdominant epitopes, new populations of T cells may be generated against the antigen.53,54 We tested whether plasma and vaccination treatment exposes neoantigens unrelated to the original vaccination target, GUCY2C.55 We measured T-cell responses against AH1, an endogenous immunodominant MHC class I-associated epitope from gp70 expressed by CT26 tumor cells (Figure 6(c)).55–57 Mice receiving both plasma and peptide vaccine exhibited a modest increase in the AH1-specific T-cell response (24.2 ± 8.3 vs 11.0 ± 4.3 spots; p = ns). While the study was not powered to detect a statistically-significant change, epitope spreading was observed. Further optimization of plasma treatment to alter tumor microenvironment may produce conditions favorable for epitope spreading. Altogether, we provide compelling evidence that plasma acts as an adjuvant for cancer therapy as shown by the amplification of GUCY2C-specific and AH1-specific T-cell responses in mice. Increased efficacy with the addition of vaccine suggests that plasma may prime the host’s immune system and may allow for its use in other combination therapies.
Discussion
Although significant advancement in conventional tumor-targeted cancer therapies (e.g. surgery, chemotherapy, radiation therapy, etc.) has reduced cancer related morbidity and mortality, this comes at the cost of significant toxicity. Another major challenge is relapse from cells that escape treatment and eventually develop resistance to therapy.58–60 In contrast, immunotherapy aims to activate the patient’s natural defenses to selectively target tumors for resolution of cancer60–63 with reduced non-specific damage to normal tissue. While current strategies (e.g. adoptive T-cell transfer, checkpoint inhibitors, etc.) are clinically efficacious, several instances of serious adverse effects, including pneumonitis and enterocolitis, have been reported.64,65 To address these, an ICD-mediated immunotherapy approach is being explored. These treatments stimulate the release of DAMP signals in cancer cells which engage APCs to expose neoantigens and facilitate the initiation of adaptive immune responses.66,67 The methods to induce ICD include certain chemotherapeutic agents, irradiation, photodynamic therapy with hypericin (PDT-hypericin), and high hydrostatic pressure.68
Here, we have demonstrated that non-thermal plasma may be operated for ICD induction and utilized in immunotherapeutic strategies. In vitro, nspDBD plasma elicited CRT emission and secretion of ATP from CT26 colon carcinoma cells. Our vaccination study showed that mice immunized with cancer cells treated with ICD-inducing plasma were partially protected against tumor challenge. In agreement with in vitro studies, in vivo plasma treatment of subcutaneous tumors in mice induced immunogenic cancer cell death. The treatment of multiple spots within the tumor elicited the greatest emission of DAMPs and recruitment of APCs into the tumor area. This treatment condition also led to a tumor-antigen-specific T-cell response. While this is the first report of plasma inducing ICD in an animal model, these results should be confirmed in other cancer models to validate, broaden, and enhance the relevance of plasma as an ICD inducer.
Overall, our results highlight the potential of plasma development for cancer immunotherapy. Further optimization of plasma parameters (applied voltage, pulse frequency, application time, etc.) and treatment schedules must be performed to improve its efficacy. This could be accomplished, in part, by determining the physical and chemical mechanism by which plasma elicits ICD. In vivo, it has been reported that anti-tumor effects of plasma are associated with plasma-generated RONS, but the link to immunogenic cell death has not been made.16 In vitro, RONS produced by plasma were demonstrated to be the major effectors for eliciting ecto-CRT and ATP secretion in cancer cells, although potential synergy with the associated pulsed electric fields or UV radiation should not be discounted.22 Therefore, an in-depth delineation of the specific RONS essential for ICD could help the development of improved plasma systems.
The study of the extracellular microenvironment generated by plasma should be paralleled with investigation of the intracellular mechanisms of induced ICD and the mode of cell death. In 2018, a comprehensive review on the molecular mechanisms of cell death was published by the Nomenclature Committee on Cell Death (NCCD).69 Twelve major cell death subroutines (intrinsic apoptosis, extrinsic apotosis, mitochondrial permeability transition (MPT)-driven necrosis, necroptosis, ferroptosis, pyroptosis, parthanatos, entotic cell death, NETotic cell death, lysosome-dependent cell death, autophagy-dependent cell death, and immunogenic cell death) were defined from morphological, biochemical, molecular, and functional perspectives. To date, six DAMPs have been linked with cell death that is immunogenic [CRT, ATP, HMGB1, type I interferon (IFN), cancer cell-derived nucleic acids, and annexin A1], but not all the underlying mechanisms are clear, and some are dependent on the specific ICD-inducer.70–76 As these mechanisms become elucidated, understanding the molecular processes following plasma-induced ICD will be possible and will help the development of this potential therapy.
The scheduling of plasma treatment will also influence clinical outcome, as plasma may also induce bystander effects on other resident or recruited cells in the tumor environment (e.g. macrophages, dendritic cells, effector T cells, etc.). Indeed, in a separate study where mini pigs were exposed to plasma, recruited myeloid cells were detected in the treated areas of the skin one week later,77 suggesting that local plasma exposure may influence immune cells even in the absence of cancer. Furthermore, several studies have demonstrated that within a defined range of treatment parameters, plasma may stimulate immune cell function (e.g. migration, secretion of cytokines, etc.) in vitro.21,78–81 In depth studies in animal models are required to determine direct plasma effect on cells of the immune system, as the host microenvironment may affect biological outcome. However, this could potentially provide an advantage over radiation or PDT, known ICD inducers, as they are reported to be detrimental to immune cells.82–84 An indicator that plasma may be immunomodulatory at physiologically safe doses was reported in a study where Drosophila exposure to plasma caused differentiation of hematocytes without affecting development or fecundity of the organism.85
While the present studies provide preclinical proof-of-concept for plasma immunotherapy in cancer, clinical administration of plasma will be a major challenge for its use in cancer treatment. For superficial cancers such as melanomas, administration of plasma is relatively straight-forward, as cancerous tumors/lesions are easily accessible for direct deposition of plasma-generated RONS. However, treatment of non-superficial cancers is a challenge for the plasma medicine field. One approach to deliver plasma species to deep tumors is through the use of plasma treated liquid (PTL),86,87 in which media is treated with NTP to enrich dissolved RONS and injected locally in the tumor or perfused through body cavities.88,89 Utsumi and co-workers demonstrated that injection of PTL locally into subcutaneous tumors can inhibit growth of malignant tumors in mice though the anti-tumor effects are not as prominent as direct plasma treatment.89 This is likely due to the instability of plasma-dissolved species in the media and the animals’ antioxidant capacity.90 Optimization of PTL generation and storage is required before it finds a role in clinical cancer treatment. A more direct, but invasive approach may involve intraoperative plasma treatment following surgical tumor excision to eliminate cancer cells remaining in the surgical margins. Physicists and engineers are designing different plasma sources and geometries for a less invasive and more focused approach to deliver plasma inside the body.91–93 Plasma has been shown to propagate along tubes up to several meters in length and with diameters as small as 15 μm.91,92 The effectiveness of some of these endoscopic plasma devices is being tested in an in vivo pancreatic cancer model.93 For successful clinical application with this approach, a detailed understanding of the RONS delivered to the target from the plasma aperture is critical. Finally, as we show here, immunization with a plasma-created whole-cell vaccine provided protective anti-tumor effects. With optimization of vaccine development and delivery this could be a feasible strategy for plasma-mediated cancer control where plasma acts as an adjuvant.
Ultimately, it is unlikely that a single treatment will be the solution to any type of cancer; a combination of different therapies may be required. Our data suggests that combining plasma with other immunotherapeutic agents may provide additional clinical value (Figure 6). Development of these strategies should be considered based on their effect on the different steps of adaptive immune response progression.94 For example, plasma-induced ICD could prime the host immune response against tumor antigens, which could be boosted by a targeted vaccine, while checkpoint inhibitor blockade may enhance the therapeutic effect of plasma immunotherapy.
Conclusion
We recently proposed a new paradigm of plasma treatment for cancer: ‘plasma onco-immunotherapy’.95 This approach not only debulks tumors, but also engages the innate immune system via ICD to initiate adaptive immune responses.95,96 In this study, we demonstrated that plasma is a bona fide ICD inducer and can be used alone or in combination with other immunotherapies to generate tumor-specific T-cell responses. With further development of plasma delivery systems and administration protocols, it has potential for clinical translation as a standalone treatment modality or an adjuvant for cancer immunotherapy.
Materials and methods
nspDBD plasma system and treatment parameters
NTP was generated in vitro by applying high voltage pulses to a dielectric barrier discharge (DBD) electrode. DBD electrodes used in this study were fabricated in our lab and have a quartz dielectric covering a copper electrode. This prevents current build-up and creates an electrically safe plasma without heating surrounding gas and tissue. A nanosecond pulser (FPG-20-05NM, FID GmbH, Germany) was used to generate high voltage pulses, characterized in our previous work.8 Briefly, our system produced: 29 kV pulses, 2 ns rise times, 20 ns total pulse duration and 0.9 mJ/pulse.
Cell culture and in vitro plasma treatment
Colorectal cancer cell line CT26.WT obtained from ATCC (CRL-2638). Generation of CT26-GUCY2C cells was described previously.97 Cells were cultured in complete media: DMEM with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (Corning Life Sciences, USA). All cells were grown in a humidified environment at 37°C with 5% CO2 (Panasonic, MCO-19AICUVH-PA, USA). Cells were plated one day prior to plasma treatment in 24-well plates at 3.0x105 cells/mL (0.5 mL/well). Before treatment with plasma, media was removed from each well and cells were washed twice with phosphate buffered saline (PBS). PBS from the second wash was removed from the well right before cells were exposed to plasma in the absence of any liquid. Fresh, complete media (0.5 mL) was immediately added back into the well following exposure to plasma.
For treatment with plasma, a DBD electrode (1.3 cm diameter) was placed 1 mm above cells in the 24-well plate on top of a grounded metal plate with a z-positioner. Since all liquid was removed, plasma was generated in the gap between the electrode and the plate, directly on the cells by applying high voltage pulses from the nanosecond pulser. Treatment time was fixed to 10 seconds, and pulse frequency was controlled by an external function generator (TTi, TG5011LXT, USA). A range of pulse frequencies were used (50, 15, 30 and 75 Hz), and the combination of plasma treatment parameters produced in the following plasma treatment energies: 50, 100, 300 and 700 mJ, respectively.8
Mice and in vivo plasma treatment
Balb/c mice were obtained from Jackson Laboratory (USA), and animal protocols were approved by The Thomas Jefferson University Institutional Animal Care and Use Committee. Subcutaneous tumors were established by injecting 1 × 105 CT26-GUCY2C cells in the flanks of mice, and monitored for growth. Prior to plasma treatment, hair over the tumor area was removed using a chemical depilatory agent to avoid obstruction with plasma generation and treatment. Tumors were treated once daily with plasma beginning on day 7 (for effector T cell development studies) or day 18 (for ICD and recruitment studies) and continued for 5 consecutive days. A smaller DBD electrode (3 mm diameter) was fabricated and used for treatment of mouse tumors. The nanosecond pulser and function generator used for in vitro treatment were also used here. Pulse frequency was adjusted to 750 Hz and treatment time was 10, 25 or 50 seconds. Mice were anesthetized with 5% isoflurane and treated on the grounding plate with the electrode positioned approximately 1 mm above the tumor with the z-positioner. Tumor volumes were monitored by measuring 3 orthogonal diameters and calculated using .
Cell viability assay
Cell viability was determined 24 hours after plasma treatment using a Muse Cell Analyzer (Millipore, USA). Cell suspensions were collected, diluted 1:20 with Muse Count & Viability Reagent (Millipore, USA) and analyzed according to the manufacturer’s instructions. The percent viability was determined for each sample.
ATP release assay
Cell supernatant was collected 10 minutes after plasma treatment and extracellular ATP was measured using a luciferin and luciferase-based chemiluminescent kit (Sigma-Aldrich, USA). All procedures were performed and reagents were prepared following manufacturer instructions. Luminescence value was measured by a Photon-Master luminometer (LuminUltra, USA) which was calibrated with the provided UltraClear calibration solution. The measured relative light units were converted into ATP concentration (pgATP/mL). Data were represented at ATP concentration (nM).
Fluorescence detection of surface-exposed calreticulin
CT26 cells were collected 24 hours after plasma treatment and washed twice with blocking buffer (PBS+ 1% heat-inactivated FBS). Cells were then incubated with rabbit anti-mouse calreticulin antibody (ThermoFisher Scientific, USA) in blocking buffer (1:200) for 30 minutes in the dark at room temperature. Following incubation, cells were washed twice with blocking buffer and stained with Alexa Fluor 488 conjugated goat anti-rabbit IgG secondary antibody (ThermoFisher Scientific, USA) at 1:500 in blocking buffer. Cells were incubated at room temperature, in the dark, for 40 minutes. Following staining, cells were washed and fixed with 4% PFA and analyzed by FACS.
Anti-tumor vaccination assay
To prepare the whole-cell vaccine, CT26 cells were treated with either: 1) 300 mJ of plasma, 2) Cisplatin (50 μM) or 3) complete media. Plasma-created vaccine was prepared from cells treated in 24-plates with plasma and cultured in regular media for 24 hours. Cisplatin-vaccine was prepared from cells incubated for 24 hours in Cisplatin media. Media-Vaccine was prepared from cells cultured in regular, complete media for 24 hours. Cisplatin- and media- vaccines were used as controls.
After a 24-hour incubation 3 × 106 cells in 100 μL of PBS were inoculated subcutaneously into the left flank of Balb/c mice. Seven days later, mice were challenged with 3 × 105 live CT26 cells subcutaneously injected into the right flank. Tumors were measured twice weekly with calipers and all mice were euthanized on day 28.
H&E staining of tissue sections and damage assessment
Tumors, with overlaying skin, were resected 1 or 3 days after the final plasma treatment and fixed in 10% formalin for at least 48 hours. Tissue was then paraffin-embedded, sectioned with a microtome, deparaffinized and stained with hematoxylin and eosin. Images of stained sections were captured using the EVOS FL Auto Cell imaging system. Sections were evaluated for damaged by a blinded pathologist.
Immunofluorescence staining of tumor tissue
Tumor sections from day 3 after final plasma treatment were used for fluorescence detection of DAMP signals and immune cell recruitment. For antigen retrieval, slides were transferred to a Dako Target Retrieval buffer pH9 (1:10 dilution in H2O) and boiled for 15 min in a pressure cooker. Slides were then cooled and blocked with blocking solution (10% milk in PBS + 0.3% v/v TritonX + 15 μL Fab donkey anti-mouse IgG (H + L) fragments) for 1 hour at room temperature in a humidified chamber. Following blocking, tumor sections were stained the following antibodies (1:100 in blocking solution) overnight at 4°C in a humidified environment: anti-mouse CRT (PA3-900, ThermoFisher Scientific, USA) and anti-mouse HMGB1 (MA5-16,264, ThermoFisher Scientific, USA) or anti-mouse CD45 (103,101, Biolegend, USA) and anti-mouse CD11c (33,483, Abcam, USA). Tissue samples were then washed four times with PBS + 0.1% v/v Tween (PBST). Secondary antibodies (1:1000 in blocking bluffer) were added: donkey anti-rabbit IgG Alexa Fluor 594 for CRT (A21207, Life Technologies, USA), goat anti-mouse IgG Alexa Fluor 488 for HMGB1 (115–545-205, Jackson Immuno, USA), donkey anti-rat IgG (H + L) Alexa Fluor 594 for CD45 (A21209, Life Technologies, USA) and goat anti-armenian hamster IgG (H + L) Alexa Fluor 488 for CD11c (127–545-160, Jackson Immuno, USA). Tissue sections were stained for 1.5 hours at room temperature in a humidified environment, protected from light. Following secondary staining, tissue sections were washed four times with PBST and mounted with DAPI (P36935, Molecular Probes, USA). A glass cover slip was placed on top of each tissue section and cured overnight. Sections were viewed under an EVOS FL Auto Imaging System (Life Technologies, USA). Using ImageJ software, mean fluorescence intensity of DAMPs and CD45+/CD11c+ signals were determined by measuring the intensity of three representative areas on the tissue. Data are presented as normalized mean fluorescence intensity of individual resected tumors.
ELISpot analysis
IFNγ ELISpot assays were previously described.97 Briefly, on Day 1, multiscreen filtration plates (Millipore, MSIPS4W10) plates were coated with 100 uL per well of anti-IFNγ antibody (BD Biosciences, USA; clone R4-6A2; 10 ug/mL) overnight at 4°C. On day 2, the coating antibody was discarded, the plate washed and blocked. Splenocyes were isolated via mechanical disruption with RBC lysis and added at 1 × 106 cells per well in cRPMI. The following stimulators were added: DMSO (negative control), 10 µg/mL GUCY2C254-262 peptide (JPT, Germany) or 10 µg/mL AH-1 peptide (AnaSpec, USA) for 24 hours in cRPMI. Plates were incubated at 37°C, 5% CO2 for 24 hours and spots were developed with 2 μg/mL biotinylated anti-IFNγ detection antibody (BD Biosciences, USA; clone XMG1.2) and 2 μg/mL alkaline phosphatase-conjugated streptavidin (ThermoFisher Scientific, USA), followed by NBT/BCIP substrate (ThermoFisher Scientific, USA). Spot forming cells were enumerated using the S6 Universal-V Analyzer automated reader system, software ImmunoSpot v5 (Cellular Technology Limited, USA). Spot parameters were established using automated gating and quantification. Data are presented as antigen-specific spots (normalized by subtracting the DMSO baseline negative control values).
Funding Statement
Financial support
Financial support was provided by: NIH to D. Merlino (F30 DK103492) and the Kimmel Cancer Center of Thomas Jefferson University (P30 CA56036); PhRMA Foundation to D. Merlino and A. Snook; the W.W. Smith Charitable Trust to A. Snook; and Margaret Q. Landenberger Research Foundation to A. Snook.
Disclosure statement
The authors declare no conflict of interest.
References
- 1.Group EBCTC Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: meta-analysis of individual patient data for 10 801 women in 17 randomised trials. The Lancet. 2011; 378(9804):1707–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg RM, Sargent DJ, Morton RF, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, Alberts SR.. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. Journal of Clinical Oncology. 2004; 22(1):23–30. [DOI] [PubMed] [Google Scholar]
- 3.Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, Gray MJ, Cheng H, Hoff PM, Ellis LM.. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res. 2006; 12(14):4147–4153. [DOI] [PubMed] [Google Scholar]
- 4.Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M.. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005; 202(12):1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Obeid M, Panaretakis T, Tesniere A, Joza N, Tufi R, Apetoh L, Ghiringhelli F, Zitvogel L, Kroemer G.. Leveraging the immune system during chemotherapy: moving calreticulin to the cell surface converts apoptotic death from “silent” to immunogenic. Cancer Research. 2007; 67(17):7941–7944. [DOI] [PubMed] [Google Scholar]
- 6.Green DR, Ferguson T, Zitvogel L, Kroemer G.. Immunogenic and tolerogenic cell death. Nature Reviews Immunology. 2009; 9(5):353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobrynin D, Fridman G, Friedman G, Fridman A.. Physical and biological mechanisms of direct plasma interaction with living tissue. New Journal of Physics. 2009; 11(11):115020. [Google Scholar]
- 8.Lin A, Chernets N, Han J, Alicea Y, Dobrynin D, Fridman G, Freeman TA, Fridman A, Non‐Equilibrium Dielectric Miller V.. Barrier Discharge Treatment of Mesenchymal Stem Cells: Charges and Reactive Oxygen Species Play the Major Role in Cell Death. Plasma Process Polym. 2015; 12(10):1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruggeman P, Leys C.. Non-thermal plasmas in and in contact with liquids. J Phys D: Appl Phys. 2009; 42(5):053001. [Google Scholar]
- 10.Babaeva NY, Kushner MJ.. Intracellular electric fields produced by dielectric barrier discharge treatment of skin. J Phys D: Appl Phys. 2010; 43(18):185206. [Google Scholar]
- 11.Babaeva NY, Kushner MJ.. Reactive fluxes delivered by dielectric barrier discharge filaments to slightly wounded skin. J Phys D: Appl Phys. 2013; 46(2):025401. [Google Scholar]
- 12.Ayan H, Staack D, Fridman G, Gutsol A, Mukhin Y, Starikovskii A, Fridman A, Friedman G.. Application of nanosecond-pulsed dielectric barrier discharge for biomedical treatment of topographically non-uniform surfaces. J Phys D: Appl Phys. 2009; 42(12):125202. [Google Scholar]
- 13.Isbary G, Morfill G, Schmidt H, Georgi M, Ramrath K, Heinlin J, Karrer S, Landthaler M, Shimizu T, Steffes B.. A first prospective randomized controlled trial to decrease bacterial load using cold atmospheric argon plasma on chronic wounds in patients. British Journal of Dermatology. 2010; 163(1):78–82. [DOI] [PubMed] [Google Scholar]
- 14.Brullé L, Vandamme M, Riès D, Martel E, Robert E, Lerondel S, Trichet V, Richard S, Pouvesle J-M, Le Pape A.. Effects of a non thermal plasma treatment alone or in combination with gemcitabine in a MIA PaCa2-luc orthotopic pancreatic carcinoma model. PloS one. 2012; 7(12):e52653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walk RM, Snyder JA, Srinivasan P, Kirsch J, Diaz SO, Blanco FC, Shashurin A, Keidar M, Sandler AD.. Cold atmospheric plasma for the ablative treatment of neuroblastoma. Journal of pediatric surgery. 2013; 48(1):67–73. [DOI] [PubMed] [Google Scholar]
- 16.Chernets N, Kurpad DS, Alexeev V, Rodrigues DB, Freeman TA.. Reaction Chemistry Generated by Nanosecond Pulsed Dielectric Barrier Discharge Treatment is Responsible for the Tumor Eradication in the B16 Melanoma Mouse Model. Plasma Process Polym. 2015; 12(12):1400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keidar M, Walk R, Shashurin A, Srinivasan P, Sandler A, Dasgupta S, Ravi R, Guerrero-Preston R, Trink B.. Cold plasma selectivity and the possibility of a paradigm shift in cancer therapy. Br J Cancer. 2011; 105(9):1295–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirst AM, Frame FM, Arya M, Maitland NJ, O’Connell D.. Low temperature plasmas as emerging cancer therapeutics: the state of play and thoughts for the future. Tumor Biology. 2016; 37(6):7021–7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bekeschus S, Mueller A, Gaipl U, Weltmann K-D.. Physical plasma elicits immunogenic cancer cell death and mitochondrial singlet oxygen. IEEE Transactions on Radiation and Plasma Medical Sciences. 2018; 2(2):138–146. [Google Scholar]
- 20.Bekeschus S, Rödder K, Fregin B, Otto O, Lippert M, Weltmann K-D, Wende K, Schmidt A, Gandhirajan RK.. Toxicity and Immunogenicity in Murine Melanoma following Exposure to Physical Plasma-Derived Oxidants Oxidative medicine and cellular longevity. 2017; 201712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin A, Truong B, Pappas A, Kirifides L, Oubarri A, Chen S, Lin S, Dobrynin D, Fridman G, Fridman A.. Uniform Nanosecond Pulsed Dielectric Barrier Discharge Plasma Enhances Anti‐Tumor Effects by Induction of Immunogenic Cell Death in Tumors and Stimulation of Macrophages. Plasma Process Polym. 2015; 12(12):1392–1399. [Google Scholar]
- 22.Lin A, Truong B, Patel S, Kaushik N, Choi EH, Fridman G, Fridman A, Nanosecond-Pulsed Miller V.. DBD Plasma-Generated Reactive Oxygen Species Trigger Immunogenic Cell Death in A549 Lung Carcinoma Cells through Intracellular Oxidative Stress. International Journal of Molecular Sciences. 2017; 18(5):966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N.. Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology. 2014; 3(9):e955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini J-L, Castedo M, Mignot G, Panaretakis T, Casares N.. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature medicine. 2007; 13(1):54–61. [DOI] [PubMed] [Google Scholar]
- 25.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg P-A, Michalak M, Henson PM.. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005; 123(2):321–334. [DOI] [PubMed] [Google Scholar]
- 26.Panaretakis T, Kepp O, Brockmeier U, Tesniere A, Bjorklund AC, Chapman DC, Durchschlag M, Joza N, Pierron G, van Endert P.. Mechanisms of pre‐apoptotic calreticulin exposure in immunogenic cell death. The EMBO journal. 2009; 28(5):578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chao MP, Jaiswal S, Weissman-Tsukamoto R, Alizadeh AA, Gentles AJ, Volkmer J, Weiskopf K, Willingham SB, Raveh T, Park CY.. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Science Translational Medicine 2010; 2(63):63ra94–63ra94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernandez NC, Lozier A, Flament C, Ricciardi-Castagnoli P, Bellet D, Suter M, Perricaudet M, Tursz T, Maraskovsky E, Zitvogel L.. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nature medicine. 1999; 5(4):405–411. [DOI] [PubMed] [Google Scholar]
- 29.Guermonprez P, Valladeau J, Zitvogel L, Théry C, Amigorena S.. Antigen presentation and T cell stimulation by dendritic cells. Annual review of immunology. 2002; 20(1):621–667. [DOI] [PubMed] [Google Scholar]
- 30.Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA.. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proceedings of the National Academy of Sciences of the United States of America 2005; 102(27):9571–9576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroemer G, Galluzzi L, Kepp O, Zitvogel L.. Immunogenic cell death in cancer therapy Annual review of immunology. 2013; 3151–72. [DOI] [PubMed] [Google Scholar]
- 32.Garg AD, Krysko DV, Verfaillie T, Kaczmarek A, Ferreira GB, Marysael T, Rubio N, Firczuk M, Mathieu C, Roebroek AJ.. A novel pathway combining calreticulin exposure and ATP secretion in immunogenic cancer cell death. The EMBO journal. 2012; 31(5):1062–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala A, Shen S, Kepp O, Métivier D, Galluzzi L, Perfettini J.. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death & Differentiation. 2014; 21(1):79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.la Sala A, Ferrari D, Di Virgilio F, Idzko M, Norgauer J, Girolomoni G.. Alerting and tuning the immune response by extracellular nucleotides. Journal of leukocyte biology. 2003; 73(3):339–343. [DOI] [PubMed] [Google Scholar]
- 35.Bianchi ME, Manfredi AA.. High‐mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunological reviews. 2007; 220(1):35–46. [DOI] [PubMed] [Google Scholar]
- 36.Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP.. A novel pathway of HMGB1‐mediated inflammatory cell recruitment that requires Mac‐1‐integrin. The EMBO journal. 2007; 26(4):1129–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L.. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010; 29(4):482–491. [DOI] [PubMed] [Google Scholar]
- 38.Bell CW, Jiang W, Reich CF, Pisetsky DS.. The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol, Cell Physiol. 2006; 291(6):C1318–C1325. [DOI] [PubMed] [Google Scholar]
- 39.Fucikova J, Moserova I, Truxova I, Hermanova I, Vancurova I, Partlova S, Fialova A, Sojka L, Cartron PF, Houska M.. High hydrostatic pressure induces immunogenic cell death in human tumor cells. Int J Cancer. 2014; 135(5):1165–1177. [DOI] [PubMed] [Google Scholar]
- 40.Altin JG, Sloan EK.. The role of CD45 and CD45-associated molecules in T cell activation. Immunol Cell Biol. 1997; 75(5): 430–445. [DOI] [PubMed] [Google Scholar]
- 41.Ledbetter JA, Tonks NK, Fischer EH, Clark EA.. CD45 regulates signal transduction and lymphocyte activation by specific association with receptor molecules on T or B cells. Proceedings of the National Academy of Sciences 1988; 85(22):8628–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Snook AE, Magee MS, Waldman SA.. GUCY2C-targeted cancer immunotherapy: past, present and future. Immunol Res. 2011; 51(2–3):161–169. doi: 10.1007/s12026-011-8253-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snook AE, Eisenlohr LC, Rothstein JL, Waldman SA.. Cancer mucosa antigens as a novel immunotherapeutic class of tumor-associated antigen. Clin Pharmacol Ther. 2007; 82(6):734–739. doi: 10.1038/sj.clpt.6100369 [DOI] [PubMed] [Google Scholar]
- 44.Xiang B, Baybutt TR, Berman-Booty L, Magee MS, Waldman SA, Alexeev VY, Snook AE.. Prime-Boost Immunization Eliminates Metastatic Colorectal Cancer by Producing High-Avidity Effector CD8⁺ T Cells. J Immunol. 2017; 198(9):3507–3514. doi: 10.4049/jimmunol.1502672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Snook AE, Baybutt TR, Hyslop T, Waldman SA.. Preclinical Evaluation of a Replication-Deficient Recombinant Adenovirus Serotype 5 Vaccine Expressing Guanylate Cyclase C and the PADRE T-helper Epitope. Hum Gene Ther Methods. 2016; 27(6):238–250. doi: 10.1089/hgtb.2016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Snook AE, Magee MS, Schulz S, Waldman SA.. Selective antigen-specific CD4(+) T-cell, but not CD8(+) T- or B-cell, tolerance corrupts cancer immunotherapy. Eur J Immunol. 2014; 44(7):1956–1966. doi: 10.1002/eji.201444539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Snook AE, Magee MS, Marszalowicz GP, Schulz S, Waldman SA.. Epitope-targeted cytotoxic T cells mediate lineage-specific antitumor efficacy induced by the cancer mucosa antigen GUCY2C. Cancer Immunol Immunother. 2012; 61(5):713–723. doi: 10.1007/s00262-011-1133-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Snook AE, Li P, Stafford BJ, Faul EJ, Huang L, Birbe RC, Bombonati A, Schulz S, Schnell MJ, Eisenlohr LC, et al. Lineage-specific T-cell responses to cancer mucosa antigen oppose systemic metastases without mucosal inflammatory disease. Cancer Res. 2009; 69(8):3537–3544. doi: 10.1158/0008-5472.CAN-08-3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Snook AE, Stafford BJ, Li P, Tan G, Huang L, Birbe R, Schulz S, Schnell MJ, Thakur M, Rothstein JL, et al. Guanylyl cyclase C-induced immunotherapeutic responses opposing tumor metastases without autoimmunity. J Natl Cancer Inst. 2008; 100(13):950–961. doi: 10.1093/jnci/djn178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snook AE, Huang L, Schulz S, Eisenlohr LC, Waldman SA.. Cytokine adjuvanation of therapeutic anti-tumor immunity targeted to cancer mucosa antigens. Clin Transl Sci. 2008; 1(3):263–264. doi: 10.1111/j.1752-8062.2008.00054.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Witek M, Blomain ES, Magee MS, Xiang B, Waldman SA, Snook AE.. Tumor radiation therapy creates therapeutic vaccine responses to the colorectal cancer antigen GUCY2C. Int J Radiat Oncol Biol Phys. 2014; 88(5):1188–1195. doi: 10.1016/j.ijrobp.2013.12.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snook A, Baybutt T, Mastrangelo M, Lewis N, Goldstein S, Kraft W, Oppong Y, Hyslop T, Myers R, Alexeev V, et al. A Phase I study of Ad5-GUCY2C-PADRE in stage I and II colon cancer patients. Journal for ImmunoTherapy of Cancer. 2015; 3(Suppl 2):P450. doi: 10.1186/2051-1426-3-s2-p450 [DOI] [Google Scholar]
- 53.Ribas A, Timmerman JM, Butterfield LH, Economou JS.. Determinant spreading and tumor responses after peptide-based cancer immunotherapy. Trends in immunology. 2003; 24(2):58–61. [DOI] [PubMed] [Google Scholar]
- 54.van der Most RG, Currie A, Robinson BW, Lake RA.. Cranking the immunologic engine with chemotherapy: using context to drive tumor antigen cross-presentation towards useful antitumor immunity. Cancer Research. 2006; 66(2):601–604. [DOI] [PubMed] [Google Scholar]
- 55.Facciponte JG, Ugel S, De Sanctis F, Li C, Wang L, Nair G, Sehgal S, Raj A, Matthaiou E, Coukos G.. Tumor endothelial marker 1–specific DNA vaccination targets tumor vasculature. The Journal of clinical investigation. 2014; 124(4):1497–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang AY, Gulden PH, Woods AS, Thomas MC, Tong CD, Wang W, Engelhard VH, Pasternack G, Cotter R, Hunt D.. The immunodominant major histocompatibility complex class I-restricted antigen of a murine colon tumor derives from an endogenous retroviral gene product. Proceedings of the National Academy of Sciences 1996; 93(18):9730–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rice J, Buchan S, Stevenson FK.. Critical components of a DNA fusion vaccine able to induce protective cytotoxic T cells against a single epitope of a tumor antigen. The Journal of Immunology. 2002; 169(7):3908–3913. [DOI] [PubMed] [Google Scholar]
- 58.Yu Z, Geng J, Zhang M, Zhou Y, Fan Q, Chen J.. Treatment of osteosarcoma with microwave thermal ablation to induce immunogenic cell death. Oncotarget. 2014; 5(15):6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zitvogel L, Apetoh L, Ghiringhelli F, Kroemer G.. Immunological aspects of cancer chemotherapy. Nature Reviews Immunology. 2008; 8(1):59–73. [DOI] [PubMed] [Google Scholar]
- 60.Palumbo MO, Kavan P, Jr WH Miller, Panasci L, Assouline S, Johnson N, Cohen V, Patenaude F, Pollak M, Jagoe RT.. Systemic cancer therapy: achievements and challenges that lie ahead. Frontiers in pharmacology. 2013; 4(57):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bentzen SM. Preventing or reducing late side effects of radiation therapy: radiobiology meets molecular pathology. Nature reviews Cancer. 2006; 6(9):702–713. doi: 10.1038/nrc1950 [DOI] [PubMed] [Google Scholar]
- 62.Burstein HJ. Side effects of chemotherapy. Journal of Clinical Oncology. 2000; 18(3):693–693. [DOI] [PubMed] [Google Scholar]
- 63.Mellman I, Coukos G, Dranoff G.. Cancer immunotherapy comes of age. Nature. 2011; 480(7378):480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Naidoo J, Wang X, Woo KM, Iyriboz T, Halpenny D, Cunningham J, Chaft JE, Segal NH, Callahan MK, Lesokhin AM.. Pneumonitis in Patients Treated With Anti–Programmed Death-1/Programmed Death Ligand 1 Therapy. Journal of Clinical Oncology. 2016JCO682005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE, Kammula US, Topalian SL, Sherry RM, Kleiner D.. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte–associated antigen 4. Journal of Clinical Oncology. 2006; 24(15):2283–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Krysko O, Aaes TL, Bachert C, Vandenabeele P, Krysko D.. Many faces of DAMPs in cancer therapy. Cell death & disease. 2013; 4(5):e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gameiro SR, Jammeh ML, Wattenberg MM, Tsang KY, Ferrone S, Hodge JW.. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014; 5(2):403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G.. Immunogenic cell death in cancer and infectious disease. Nature Reviews Immunology. 2016; 12(2):97. [DOI] [PubMed] [Google Scholar]
- 69.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW.. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death & Differentiation. 2018; 25486–541. [Google Scholar]
- 70.Obeid M, Panaretakis T, Joza N, Tufi R, Tesniere A, Van Endert P, Zitvogel L, Kroemer G.. Calreticulin exposure is required for the immunogenicity of γ-irradiation and UVC light-induced apoptosis. Cell Death & Differentiation. 2007; 14(10):1848–1850. [DOI] [PubMed] [Google Scholar]
- 71.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, Shen S, Kepp O, Scoazec M, Mignot G.. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science. 2011; 334(6062):1573–1577. [DOI] [PubMed] [Google Scholar]
- 72.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P.. Toll-like receptor 4–dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature medicine. 2007; 13(9):1050. [DOI] [PubMed] [Google Scholar]
- 73.Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C.. Cancer cell–autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nature medicine. 2014; 20(11):1301. [DOI] [PubMed] [Google Scholar]
- 74.Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD.. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nature immunology. 2012; 13(9):832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peschiaroli F, Businaro L, Gerardino A, Ladoire S, Apetoh L, Bravo-San JM. Chemotherapy-induced antitumor immunity requires formyl peptide receptor. 360 (6394):aad0779. doi: [DOI] [Google Scholar]
- 76.GARG A Krysko. D, Vandenabeele P, Agostinis P. Extracellular ATP and P2X7 receptor exert context-specific immunogenic effects after immunogenic cancer cell death. Cell death & disease. 2016; 7(e2097):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ranieri P, Shrivastav R, Wang M, Lin A, GregoryFridman, Fridmam A, Han L, Miller V. Nanosecond pulsed Dielectric Barrier Discharge induced Anti-Tumor Effects Propagate Through the depth of Tissue via Intracellular Signaling. Plasma Medicine 2017; 7(3):283–297. doi: 10.1615/PlasmaMed.2017019883 [DOI] [Google Scholar]
- 78.Miller V, Lin A, Fridman G, Dobrynin D, Fridman A.. Plasma Stimulation of Migration of Macrophages. Plasma Process Polym. 2014; 11(12):1193–1197. [Google Scholar]
- 79.Bekeschus S, Schmidt A, Bethge L, Masur K, von Woedtke T, Hasse S, Wende K.. Redox stimulation of human thp-1 monocytes in response to cold physical plasma Oxidative medicine and cellular longevity. 2015; 20165910695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaushik NK, Kaushik N, Min B, Choi KH, Hong YJ, Miller V, Fridman A, Choi EH.. Cytotoxic macrophage-released tumour necrosis factor-alpha (TNF-α) as a killing mechanism for cancer cell death after cold plasma activation. J Phys D: Appl Phys. 2016; 49(8):084001. [Google Scholar]
- 81.Bekeschus S, Moritz J, Schmidt A, Wende K.. Redox regulation of leukocyte-derived microparticle release and protein content in response to cold physical plasma-derived oxidants. Clinical Plasma Medicine. 2017; 7(8):24–35. [Google Scholar]
- 82.Kawase Y, Naito S, Ito M, Sekine I, Fujii H.. The Effect of Ionizing Radiation on Epidermal Langerhans Cells–A Quantitative Analysis of Autopsy Cases with Radiation Therapy–. J Radiat Res. 1990; 31(3):246–255. [DOI] [PubMed] [Google Scholar]
- 83.Liao Y-P, Wang C-C, Butterfield LH, Economou JS, Ribas A, Meng WS, Iwamoto KS, McBride WH.. Ionizing radiation affects human MART-1 melanoma antigen processing and presentation by dendritic cells. The Journal of Immunology. 2004; 173(4):2462–2469. [DOI] [PubMed] [Google Scholar]
- 84.Gollnick SO, Liu X, Owczarczak B, Musser DA, Henderson BW.. Altered expression of interleukin 6 and interleukin 10 as a result of photodynamic therapy in vivo. Cancer research. 1997; 57(18):3904–3909. [PubMed] [Google Scholar]
- 85.Lee A, Lin A, Shah K, Singh H, Miller V, Rao SG.. Optimization of Non-Thermal Plasma Treatment in an In Vivo Model Organism. PloS one. 2016; 11(8):e0160676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mohades S, Laroussi M, Sears J, Barekzi N, Razavi H.. Evaluation of the effects of a plasma activated medium on cancer cells. Physics of Plasmas. 2015; 22(12):122001. [Google Scholar]
- 87.Traylor MJ, Pavlovich MJ, Karim S, Hait P, Sakiyama Y, Clark DS, Graves DB.. Long-term antibacterial efficacy of air plasma-activated water. J Phys D: Appl Phys. 2011; 44(47):472001. [Google Scholar]
- 88.Yan D, Talbot A, Nourmohammadi N, Cheng X, Canady J, Sherman J, Keidar M.. Principles of using cold atmospheric plasma stimulated media for cancer treatment. Scientific reports. 2015; 518339. doi: 10.1038/srep18339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Utsumi F, Kajiyama H, Nakamura K, Tanaka H, Mizuno M, Ishikawa K, Kondo H, Kano H, Hori M, Kikkawa F.. Effect of indirect nonequilibrium atmospheric pressure plasma on anti-proliferative activity against chronic chemo-resistant ovarian cancer cells in vitro and in vivo. PloS one. 2013; 8(12):e81576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Judée F, Fongia C, Ducommun B, Yousfi M, Lobjois V, Merbahi N.. Short and long time effects of low temperature Plasma Activated Media on 3D multicellular tumor spheroids Scientific reports. 2016; 621421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Polak M, Winter J, Schnabel U, Ehlbeck J, Weltmann KD.. Innovative Plasma Generation in Flexible Biopsy Channels for Inner‐Tube Decontamination and Medical Applications. Plasma Process Polym. 2012; 9(1):67–76. [Google Scholar]
- 92.Kim JY, Ballato J, Foy P, Hawkins T, Wei Y, Li J, Kim S-O.. Apoptosis of lung carcinoma cells induced by a flexible optical fiber-based cold microplasma. Biosensors Bioelectron. 2011; 28(1):333–338. [DOI] [PubMed] [Google Scholar]
- 93.Robert E, Vandamme M, Brullé L, Lerondel S, Le Pape A, Sarron V, Riès D, Darny T, Dozias S, Collet G.. Perspectives of endoscopic plasma applications. Clinical Plasma Medicine. 2013; 1(2):8–16. [Google Scholar]
- 94.Vanneman M, Dranoff G.. Combining immunotherapy and targeted therapies in cancer treatment. Nature Reviews Cancer. 2012; 12(4):237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Miller V, Lin A, Fridman A.. Why Target Immune Cells for Plasma Treatment of Cancer. Plasma Chem Plasma Process. 2015; 36(1): 259–268. [Google Scholar]
- 96.Kepp O, Galluzzi L, Martins I, Schlemmer F, Adjemian S, Michaud M, Sukkurwala AQ, Menger L, Zitvogel L, Kroemer G.. Molecular determinants of immunogenic cell death elicited by anticancer chemotherapy. Cancer and Metastasis Reviews. 2011; 30(1):61–69. [DOI] [PubMed] [Google Scholar]
- 97.Snook AE, Stafford BJ, Li P, Tan G, Huang L, Birbe R, Schulz S, Schnell MJ, Thakur M, Rothstein JL.. Guanylyl Cyclase C–Induced Immunotherapeutic Responses Opposing Tumor Metastases Without Autoimmunity. Journal of the National Cancer Institute. 2008; 100(13):950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]