

ABSTRACT
The advent of immune checkpoint blockade as a new strategy for immunotherapy has changed the outlook for many aggressive cancers. Although complete tumor eradication is attainable in some cases, durable clinical responses are observed only in a small fraction of patients, underlining urgent need for improvement. We previously showed that RON, a receptor tyrosine kinase expressed in macrophages, suppresses antitumor immune responses, and facilitates progression and metastasis of breast cancer. Here, we investigated the molecular changes that occur downstream of RON activation in macrophages, and whether inhibition of RON can cooperate with checkpoint immunotherapy to eradicate tumors. Activation of RON by its ligand, MSP, altered the gene expression profile of macrophages drastically and upregulated surface levels of CD80 and PD-L1, ligands for T-cell checkpoint receptors CTLA-4 and PD-1. Genetic deletion or pharmacological inhibition of RON in combination with anti-CTLA-4, but not with anti-PD-1, resulted in improved clinical responses against orthotopically transplanted tumors compared to single-agent treatment groups, resulting in complete tumor eradication in 46% of the animals. Positive responses to therapy were associated with higher levels of T-cell activation markers and tumor-infiltrating lymphocytes. Importantly, co-inhibition of RON and anti-CTLA-4 was also effective in clearing metastatic breast cancer cells in lungs, resulting in clinical responses in nearly 60% of the mice. These findings suggest that RON inhibition can be a novel approach to potentiate responses to checkpoint immunotherapy in breast cancer.
Keywords: immunotherapy, breast cancer, metastasis, RON receptor tyrosine kinase, anti-CTLA-4, anti-PD-1, MMTV-PyMT
Introduction
The ability of tumors to evade the immune system is achieved through a variety of mechanisms. Engagement of co-inhibitory T-cell receptors, also known as checkpoint molecules, is a common event in tumor immunoevasion. Two well-studied checkpoint receptors on T-cell surfaces are CTLA-4 and PD-1, for which clinically approved inhibitors are now available.1,2 CTLA-4 and PD-1 can bind to CD80/CD86 and PD-L1/L2, respectively, to counteract activation signals initiated by the T-cell receptor (TCR). Blocking immune checkpoints with “checkpoint inhibitor” therapy was shown to be a powerful approach to release T cell inhibition in preclinical models and is now approved by the FDA for the treatment of certain cancers.3,4 In some cases, long-term durable responses and complete remissions are achieved with checkpoint inhibition, but only a fraction of patients mount a productive anti-tumor immune response and benefit from the treatment. To this end, numerous approaches have been proposed to potentiate responses to immunotherapy including combination treatment with various immune-modulating drugs, or co-treatment with chemotherapy or radiotherapy.5–7 A better understanding of the context in which checkpoint inhibitors are successful, and new strategies to make them more effective, are needed to fully realize the potential of these promising new drugs.
Breast cancer is the most common form of invasive cancer in women, and it is the second leading cause of cancer-related deaths.8 Although the emergence of targeted therapies has resulted in improved clinical outcomes overall, more than 40,000 people succumb to this disease every year in the U.S. alone, highlighting an urgent need for developing better treatments. It is now appreciated that the immune system plays important roles in determining breast cancer outcomes.9 Bolstered by the success of checkpoint inhibitors in other types of cancer, numerous immunotherapy trials have been launched for hormone receptor-positive and triple-negative subtypes of breast cancer. Preliminary results in trials involving inhibitors of PD-1 or its ligand PD-L1 suggest that some breast cancer patients can benefit from checkpoint immunotherapy, although response rates were only 10–20%.10–13
In preclinical studies, immune-competent transgenic mouse models recapitulate many key features of human cancers and are most appropriate for immunotherapy experiments. The MMTV-PyMT transgenic mouse is a well-studied experimental model in which the Polyomavirus Middle T (PyMT) oncogene is expressed under the control of the tissue-restricted MMTV promoter in an immunocompetent background. MMTV-PyMT mice develop mammary tumors with high penetrance and mimic the molecular features of hormone receptor-negative breast adenocarcinomas.14 This model has been instrumental for studying tumor-host interactions and anti-tumor immune responses.15–18 The MMTV-PyMT model has been used to test CTLA-4 or PD-1 checkpoint inhibition in combination with other therapeutic approaches such as irradiation or inhibition of the tyrosine kinase Axl.19–21 In these studies, combination approaches provided clinical benefit, whereas single-treatment with checkpoint inhibitors was not efficacious. These data suggest the utility of the PyMT model in discovering synergistic immunotherapeutic drug combinations.
RON (also known as Macrophage Stimulating-1 Receptor, MST1R) is an understudied receptor tyrosine kinase that shares similar structure with its well-studied relative c-MET.22 RON is expressed in resident tissue macrophages, and in tumor cells of various origins.23,24 RON can be activated by aberrant overexpression or by binding the active form of its ligand, macrophage stimulating protein (MSP), a constitutively secreted pro-protein found in serum. In tumor cells, overexpression of RON results in proliferation, migration, and a more aggressive phenotype.25–28 Particularly, overexpression of RON in breast tumors is associated with increased metastasis and poor clinical outcomes.16,26 In macrophages, activation of RON by MSP results in attenuation of immune responses.29–33 Previously, we and others have shown that host RON signaling negatively regulates antitumor immunity in mouse models of cancer.31,34,35 Importantly, host RON was critical for conversion of micrometastatic lesions into overt clinical metastases, a step that is thought to be rate-limiting in the deadly metastatic cascade.34 Inhibition of RON activity with a MET/RON dual kinase inhibitor BMS777607 (also known as ASLAN002)36 also blocked metastatic outgrowth in a manner that was dependent on CD8 + T cells.34 BMS777607/ASLAN002 has been in clinical trials and exhibited a good tolerability profile and demonstrated biological effects on RON-mediated activities.37–39
Based on the known immunosuppressive role of RON in tumor models, we posited that RON inhibition might provide a novel therapeutic modality in combination with checkpoint blockade in cancer. In this study, we discovered control of checkpoint ligand molecules by RON, and determined that inhibition of RON functions in combination with checkpoint inhibitor immunotherapy to attain better clinical responses.
Results
MSP-RON signaling activates an immunomodulatory gene expression signature
Within the immune system, RON expression is restricted to macrophages, where it regulates inflammatory phenotypes,23,31,32 but a comprehensive understanding of the consequences of RON activation is lacking. To elucidate specific changes in gene expression downstream of MSP-RON signaling in macrophages, we performed RNA sequencing (RNAseq) analysis on magnetically-sorted naïve F4/80+ resident peritoneal macrophages from wild-type (WT) mice or syngeneic mice that lack the RON tyrosine kinase domain (RON TK-/-),40 after 7 hours of treatment with recombinant MSP. As expected, RON TK-/- macrophages did not respond to MSP treatment and displayed gene expression that was very similar to untreated WT macrophages. Biological replicates of MSP-treated WT macrophages clustered together and exhibited a distinct gene expression signature, as shown by the RNAseq sample distance matrix (Figure 1a). With a threshold of a 2-fold change and FDR q < 0.01, 2934 genes were found to be differentially expressed in MSP-treated WT macrophages in comparison with MSP-treated RON TK-/- macrophages (1193 upregulated, 1741 downregulated). With a 4-fold change threshold (and FDR q < 0.01), this number was reduced to 823 differentially expressed genes (396 upregulated, 427 downregulated) (Figure 1b). Pathway enrichment analysis revealed that many of these differentially expressed genes belonged to immune cell trafficking and inflammatory response pathways (Fig S1a,b). When MSP-treated RON TK-/- macrophages were compared with untreated WT macrophages, the gene expression pattern was largely similar, confirming that our results were dependent on the intact kinase domain of RON (Figure 1a and S1c).
Figure 1.

MSP-RON signaling activates an immunomodulatory gene expression signature in macrophages. A. RNA sequencing was performed on F4/80 positive-sorted peritoneal macrophages treated with MSP for 7 hours (n = 3). Euclidian distance matrix shows distinct clustering of MSP-treated wild-type (WT) macrophages. MSP-treated (+) and untreated (-) RON TK-/- macrophages exhibited similar gene expression profiles. In this plot, darker color indicates higher similarity between samples. Color key and overlaid histogram indicates the frequency of observations at a given Euclidian distance value. B. Numbers of differentially-expressed genes as determined by different fold-change thresholds. Gene sets were obtained by comparing MSP-treated WT macrophages with MSP-treated RON TK-/- macrophages (FDR q-value <0.01). C. Changes in mRNA expression levels of selected immune costimulatory and coinhibitory ligands. Linear fold change was calculated in MSP-treated WT macrophages compared to MSP-treated RON TK-/- macrophages. p-values were obtained with Benjamini-Hochberg multiple testing correction. ND: not detected. D. Representative flow cytometry histograms of WT and RON TK-/- peritoneal macrophages after 24 hours of treatment with (+) MSP or with vehicle control, (-)MSP. Histograms were pre-gated on CD11b(hi) F4/80(hi) RON(+) resident macrophages (see Fig S5 for representative flow cytometry data). Dashed lines indicate the fluorescence values at the population peak in MSP-stimulated WT samples. E. Quantification of mean fluorescence intensity (MFI) of indicated markers in WT or RON TK-/- macrophages (n = 5 for both) with and without MSP treatment by flow cytometry. F. Flow cytometric analysis of expression levels of PD-L1 and CD80 in MSP-treated macrophages with or without RON-selective kinase inhibitors BMS777607 and merestinib (LY2801653). Two-tailed t-test was used to compare staining intensity. (ns) p > 0.05, (*) p ≤ 0.05, (**) p ≤ 0.01, (***) p ≤ 0.001, (****) p ≤ 0.0001.
Macrophage polarization is conceptually divided into proinflammatory (M1) and immunomodulatory (M2) phenotypes.41 RON activation has previously been reported to skew macrophages to the M2 phenotype and attenuate inflammation.42 When we compared expression levels of differentially regulated genes in our study with previously published datasets corresponding to defined M1 versus M2 states,43 we found that RON activation did not strictly correlate with either the M1 or M2 state (Fig S2). However, we found that RON activity was associated with an overall immunosuppressive state. For example, macrophage genes known to be upregulated by the inflammatory stimulus lipopolysaccharide (LPS)44 were downregulated in the presence of MSP stimulation (Fig S3a,b). Differentially expressed genes in our study were mostly in agreement with findings reported by Chaudhuri et al., who used microarray technology to investigate gene expression changes in macrophages 20 hours after MSP treatment31 (Fig S4). In sum, data gathered to date suggest that MSP-RON signaling is not a simple molecular switch for an M2 macrophage state. Rather, it activates a complex immunomodulatory gene expression signature that ultimately suppresses CD8 + T cell activity to facilitate tumor progression and metastasis.31,34,35
MSP-RON upregulates CD80 and PD-L1 expression through MAPK signaling
In the course of our analysis, we noted that MSP-RON signaling resulted in upregulation of several immune checkpoint ligand mRNAs, with CD80 and PD-L1 being the most differentially expressed (9.7- and 3.6-fold, respectively; Figure 1c). We analyzed protein levels of CD80 and PD-L1, as well as the related ligands PD-L2 and CD86, with flow cytometry (Fig S5). MSP-treatment resulted in significant upregulation of surface levels of PD-L1 and CD80 on macrophages, whereas PD-L2 remained unchanged and CD86 was downregulated (Figure 1d,e). MSP-RON-mediated upregulation of CD80 and PD-L1 was blocked when either of two RON-selective kinase inhibitors, BMS777607 or merestinib (LY2801653),36,45 were added (Figure 1f).
To investigate the kinetics and biological requirements for CD80 and PD-L1 protein upregulation downstream of MSP-RON, we performed a time-course analysis of MSP-treated macrophages. Upregulation of CD80 was evident by 6 hours, while increases in PD-L1 were not detected until 12 hours after treatment (Figure 2a). Short-term treatment of the macrophages with inhibitors of transcription or translation (actinomycin D or cycloheximide, respectively) prior to MSP stimulation completely abrogated MSP-mediated upregulation of CD80 and PD-L1 (Figure 2a). Since RON can signal through both MAPK and PI3K pathways to control gene expression,26,31 we tested the activity of, and the requirement for, these two signaling nodes in the regulation of CD80 and PD-L1. Western blot analysis of MSP-treated macrophages revealed phosphorylated forms of ERK and AKT as surrogates of MAPK and PI3K pathway activation, respectively (Figure 2b). MSP-RON driven phosphorylation of ERK and AKT was blocked when MEK1/2 or PI3K inhibitors were added to the culture (Figure 2b). MAPK pathway inhibitors abrogated the upregulation of both CD80 and PD-L1, whereas PI3K inhibition was only effective to fully block the upregulation of CD80 (Figure 2c).
Figure 2.

MSP-RON signaling upregulates PD-L1 and CD80 molecules on macrophage surfaces in a MAPK-dependent manner. A. Time-course flow cytometric analysis of mean fluorescence intensity (MFI) of PD-L1 and CD80 on macrophages (+) or (-) MSP, and in the presence or absence of actinomycin D (ActD) or cycloheximide (CHX) (n = 3). Two-way ANOVA with Tukey’s correction was used to compare differences. B. Western blot analysis of macrophages 15 minutes after treatment with or without MSP in the presence or absence of MAPK and PI3K pathway inhibitors, PD0325901 (MEK1/2i) and BKM120 (PI3Ki). C. Flow cytometric analysis of surface levels of PD-L1 and CD80 on macrophages treated with MSP for 24 hours in the presence or absence of BKM120 (PI3Ki), PD0325901 (MEK1/2i), or SCH772984 (ERK1/2i) (n = 3). One-way ANOVA with Tukey’s correction was used to compare staining intensity. (ns) p > 0.05, (*) p ≤ 0.05, (**) p ≤ 0.01, (***) p ≤ 0.001, (****) p ≤ 0.0001.
Activation of JAK-STAT signaling has been shown to upregulate PD-L1 and CD80 expression in various cell types including cancer cells and macrophages.46–49 We assessed induction of STAT1/3/5 phosphorylation in MSP-treated macrophages in the presence of MEK1/2 or JAK inhibitors. Upon MSP stimulation, we detected higher levels of STAT1 serine-727 phosphorylation, a residue known to be a direct target of MAPK signaling,50 (Figure 2d). Phosphorylation of Ser727 was unaffected by JAK inhibition, but was reduced to background levels in the presence of MEK1/2 inhibitor. MSP-stimulation did not activate phosphorylation of STAT1-Tyr701, STAT3-Tyr705, and STAT5-Tyr694 residues, which are phosphorylated by JAK kinases51 and were relatively faint (Figure 2d). We then assessed whether inhibition of JAK and/or MEK1/2 could block MSP-mediated upregulation of CD80 and PD-L1 in macrophages in culture. Interestingly, either the MEK inhibitor or the JAK inhibitor reduced the PD-L1 upregulation to near background levels, whereas the JAK inhibitor had a statistically significant but smaller effect on CD80 and CD86 regulation (Figure 2e). These data suggest that both JAK-mediated tyrosine phosphorylation and MAPK-mediated serine phosphorylation of STAT proteins may contribute to regulation of checkpoint ligands downstream of MSP/RON – but only the MAPK-mediated effects are a direct effect of RON activation.
Inhibition of RON cooperates with anti-CTLA4 to boost antitumor immunity
We previously demonstrated that host RON signaling suppresses CD8 + T-cell responses against breast cancer cells, resulting in metastatic progression in mice.34 The ability of RON signaling to upregulate CD80 and PD-L1, amongst a large collection of other immunosuppressive molecules, led us to hypothesize that inhibition of RON might cooperate with approved checkpoint inhibitors such as anti-CTLA-4 (aCTLA-4) and anti-PD-1(aPD-1) to enhance anti-tumor responses. We utilized our previously described MMTV-PyMT model16,34 to investigate this question. However, to be able to track antigen-specific CD8 + T-cell responses we also engineered the tumor cells to express a model antigen: a fragment of Lymphocytic Choriomeningitis Virus (LCMV) nucleoprotein that produces an immunodominant MHC-I associated peptide, NP118 (RPQASGVYM) in FVB hosts;52 hereafter referred to as PyMT-NP tumor cells. We confirmed immunodominance of NP118 in FVB hosts by flow cytometric analysis of IFNγ production in peripheral blood CD8 + T-cells from LCMV-infected mice (Fig S6).
We first studied the effects of Ron inhibition and immunotherapy on established tumors growing in the mammary fat pad. We transplanted PyMT-NP cells orthotopically into the mammary fat pad and waited until tumors reached 100 mm3 before randomizing mice into four experimental groups: vehicle (DMSO), RON inhibitor (BMS777607/ASLAN002; hereafter referred to as RONi), aCTLA-4, and RONi+aCTLA-4 combination treatment. Treatments were applied for three weekly cycles in which RONi was administered orally five days of the week, and aCTLA-4 immunotherapy was delivered intraperitoneally on a bi-weekly schedule. The presence of NP118 did not cause problems with tumor growth in immunocompetent hosts, as evidenced by aggressive tumor growth in vehicle-treated mice (Figure 3a, black lines). Response to therapy was assessed using two metrics: tumor growth rate and the number and proportion of mice experiencing clinical benefit (complete or partial response to treatment; see Methods).
Figure 3.

Pharmacological inhibition or genetic ablation of RON cooperates with aCTLA4, but not with aPD1, to control breast cancer progression in mice. A. Growth curves of orthotopically transplanted PyMT-NP tumors in wild type (WT) mice following treatment with vehicle control (black; n = 30); RONi alone (green; n = 17); aCTLA-4 alone (blue; n = 13); or RONi+aCTLA-4 (red; n = 13). Each line represents tumor growth in an individual mouse. The vehicle-treated group is overlaid on each graph for comparison purposes. All treatment was initiated when the tumors reached 100 mm3 in size (day 0). The dashed line indicates the partial response cutoff of 300% increase in tumor size (two tumor doublings). Color-coded arrows indicate average percent change in tumor size at the endpoint (day 24) B. Percent of mice in each treatment group categorized as refractory to treatment or showing partial or complete response. Statistical analysis was performed via Fisher’s exact test, comparing the numbers of refractory tumors and tumors with clinical benefit (complete response and partial response combined). Tumor growth curves for aPD-1 experiments are shown in Fig S7b (aPD-1; n = 14) (RONi+aPD-1; n = 16) C. Change in tumor growth rates with treatment, compared between groups using one-way ANOVA and Tukey’s correction. Log-transformed growth curves for each mouse are shown in Fig S7a,c. D. Growth curves of orthotopically transplanted PyMT-NP tumors in RON TK-/- hosts: WT vehicle (black; n = 4); RON TK-/- vehicle (green; n = 12); RON TK-/- aCTLA-4 (blue; n = 13); RON TK-/- RONi+aCTLA-4 (red; n = 12). Mice were treated as in panel A. E. Percent of mice in each RON TK-/- treatment group categorized as in panel B. F. Change in the growth rate of tumors following treatment in RON TK-/- hosts. Log-transformed growth curves for each mouse are shown in Fig S7d. (ns) p > 0.05, (*) p ≤ 0.05, (**) p ≤ 0.01, (***) p ≤ 0.001, (****) p ≤ 0.0001.
Mirroring findings in the clinic,4,10 some subjects (mice) did not respond to immunotherapy at all, while others experienced slower tumor growth or eradication of the tumor altogether. RON inhibition alone (RONi single agent) did not result in appreciable clinical benefit, and it did not significantly reduce tumor growth rate compared to the vehicle group (Figure 3a-c). Treatment with aCTLA-4 as a single agent was more effective in controlling tumor growth, and it resulted in 46% of the mice having clinical benefit. 23% of the mice in aCTLA-4 single agent-treatment group experienced eradication of the tumor (i.e., complete response). Strikingly, combining the RON inhibitor and aCTLA-4 therapy doubled the frequency of complete responders to 46% and provided clinical benefit in 92% of the animals (Figure 3a-c). Moreover, the combination-treated group demonstrated tumor shrinkage in most mice, while the aCTLA-4 single agent-treated group exhibited positive tumor growth rate as a whole, albeit at a significantly lower magnitude than vehicle or RONi single agent treated groups (Figure 3c and S7a). We also tested whether RONi could cooperate with aPD-1 treatment in the same model. Treatment with aPD-1 as a single agent was mostly ineffective at reducing PyMT-NP tumor growth, and combining RONi with aPD-1 did not result in any enhancement of tumor control (Figure 3b,c and S7a,b).
To investigate the generality of RONi-mediated potentiation of aCTLA-4 immunotherapy, we utilized the MC38 colon adenocarcinoma model in the C57BL/6 genetic background.53,54 In this model, single agent RONi or aCTLA-4 treatment did not affect subcutaneous tumor growth (Fig S8a-c). However, while tumor shrinkage was not observed in any of the mice, combination of RONi and aCTLA-4 significantly reduced the tumor growth rate (Fig S8c), suggesting RON inhibition can potentiate immunotherapy responses in other types of cancers.
It should be noted that BMS777607/ASLAN002 can also inhibit MET and a few other kinases, and/or could potentially affect tumor growth by acting on tumor cells directly.36 To definitively determine whether the antitumor responses in combination with aCTLA-4 were specifically due to blockade of host RON signaling, we transplanted PyMT-NP cells (containing wild-type RON) into syngeneic RON TK-/- recipients34,40 and followed the same treatment schedule. Treatment of RON TK-/- mice with vehicle did not have a significant effect on tumor growth, which was consistent with our data in WT recipients treated with RONi (Figure 3d-f). However, the single agent aCTLA-4 immunotherapy in RON TK-/- hosts exhibited a remarkable clinical response, mirroring our findings with RONi+aCTLA-4 combination treatment in WT hosts (Figure 3d-f and S7a-c). Further supporting the role of host RON as the target of RONi in these experiments, we noted that treatment of RON TK-/- mice with the combination of RONi+aCTLA-4 did not have any more effect than RON TK-/- mice treated with aCTLA-4 alone. These findings indicate that the RONi BMS777607/ASLAN002 functions through blocking host RON kinase to cooperate with aCTLA-4 immunotherapy.
Therapeutic efficacy in roni+actla-4 treated mice is associated with improved CD8+ t-cell responses
To investigate the immune landscape of mice in each treatment group, we analyzed secondary lymphoid organs and tumor-infiltrating lymphocytes. The frequency of CD8+ T cells in the spleen was significantly higher in mice treated with RONi+aCTLA-4 compared with vehicle or single treatment groups (Figure 4a and S9). Within the CD8+ population, the proportion of PD-1 expressing cells was also higher, suggesting that the cells were antigen-experienced. Percentages of activated PD-1(+)CD62L(low) CD4+ and CD8 + T-cells were also greater in mice treated with RONi+aCTLA-4 (Figure 4b,c and S9). At the experimental endpoint (day 24 post-treatment), intratumoral CD8+ T-cell infiltration was analyzed by immunofluorescence in cases where there was tumor remaining to analyze. Despite lack of data from the best responders due to tumor eradication, we found that intratumoral CD8+ T cell infiltration was significantly elevated when the animals were treated with aCTLA-4 single therapy or with RONi+aCTLA-4 combination therapy (Figure 4d and S10). The frequency of intratumoral CD8+ cells trended higher in the combination treated-mice compared to aCTLA-4 single treatment, although this difference was not statistically significant, perhaps due to the inability to acquire data from complete responders. It is important to note that highest CD8+ cell infiltration was observed in mice that responded to therapy in both treatment groups; there were two mice in the aCTLA-4 group that had very high CD8+ T cell infiltration into the tumors. These mice had partial responses (Figure 4d).
Figure 4.
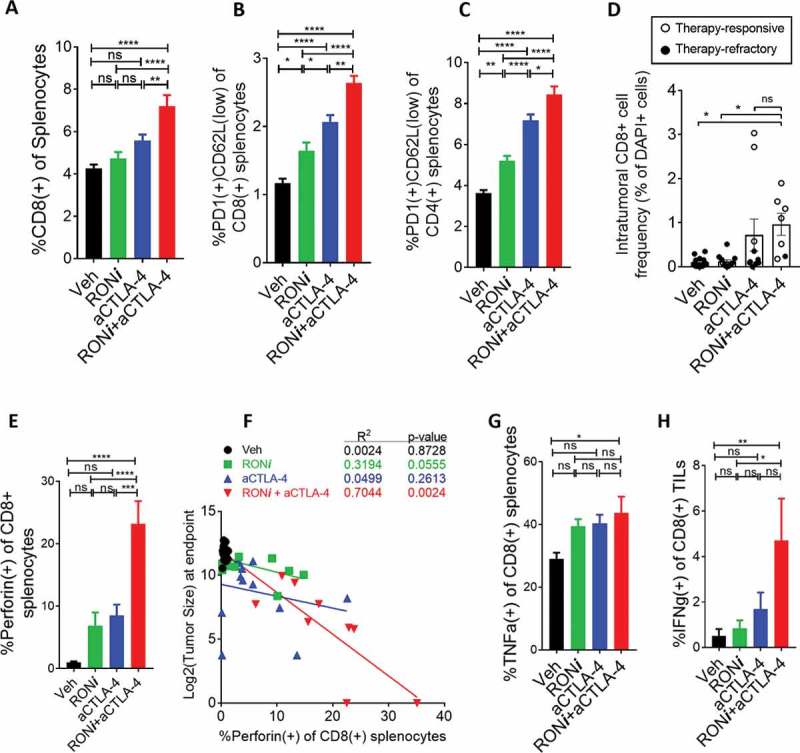
RONi+aCTLA4 therapeutic efficacy is associated with improved CD8 + T-cell responses. A. Flow cytometric analysis of CD8 + T cell frequency in spleens at the experimental endpoint, shown for each treatment group. CD8+ cells were pre-gated on single live cells; complete flow cytometry plots are shown in Fig S8. B-C. Frequency of PD1-expressing CD62L(low) cells within the splenic CD8+ (B) and CD4+ (C) T-cell compartments. D. Immunofluorescence analysis of CD8+ cells within tumors that remained after treatment. The percentage of CD8+ cells within the tumor was calculated as a ratio to DAPI+ cells. Data points represent individual mice. Unfilled points represent tumors that responded to therapy and black filled points depict refractory cases as shown in Figure 3. Representative images of immunofluorescence are shown in Fig S9. E. Frequency of perforin-expressing CD8+ T-cells in spleens at the experimental endpoint, shown for each treatment group. F. Correlation plots showing log2-transformed tumor size versus perforin+CD8+ splenocyte frequency at the endpoint. G. Frequency of TNFα+ expressing splenic CD8+ cells following restimulation with PMA and ionomycin. H. Frequency of IFNγ+ tumor-infiltrating CD8 + T-cells following restimulation with the tumor-specific NP118 peptide. Data in G and H were normalized to unstimulated controls for each mouse, and flow cytometry plots and gating for intracellular stains are shown in Fig S10. Statistical comparisons were performed with one-way ANOVA with Tukey’s correction. (ns) p > 0.05, (*) p ≤ 0.05, (**) p ≤ 0.01, (***) p ≤ 0.001, (****) p ≤ 0.0001.
Cytotoxic T-cells eliminate tumor cells through perforin/granzyme B-mediated lysis.55 Intracellular staining for perforin also revealed significantly higher levels of perforin+ CD8+ T-cells in spleens of mice treated with RONi+aCTLA-4 (Figure 4e and S11). The proportion of perforin-expressing cells within the splenic CD8+ compartment inversely correlated with tumor size at the endpoint in the treatment groups (Figure 4f), particularly in the RONi+aCTLA-4 combination treatment group. PMA-ionomycin restimulation of splenocytes also revealed more TNFα-producing CD8+ T cells in the combination treatment group (Figure 4g). Importantly, tumor-infiltrating CD8+ T-cells from mice treated with RONi+aCTLA4 displayed higher levels of antigen-specific IFNγ production following restimulation with the NP118 peptide, demonstrating enhanced anti-tumor immune responses (Figure 4h). These findings demonstrate that efficacious RONi+aCTLA-4 combination therapy correlates with improved anti-tumor T cell responses in mice.
Inhibition of RON cooperates with aCTLA-4 immunotherapy to prevent metastatic outgrowth
Metastasis is the deadliest feature of aggressive cancers, due to drug resistance of metastatic tumors. Many tumors have spawned micrometastases to other organs before the primary tumor is diagnosed. These micrometastases can then grow into overt metastatic disease at distant sites,56 and adjuvant chemotherapy is intended to eliminate microscopically seeded cells. Distant recurrence rates for breast cancer are still between 20–30%,57 indicating the need for better therapies to kill previously seeded micrometastases. Our previous work revealed that host RON signaling suppressed anti-tumor CD8+ T-cell responses, allowing metastatic outgrowth of seeded tumor cells, but RON inhibitor therapy did not completely prevent outgrowth.34 Our present results prompted us to examine the combination of RONi and aCTLA-4 in the micrometastatic setting as a potential therapeutic regimen for adjuvant therapy. MMTV-PyMT tumor cells were injected via the lateral tail vein into wild-type or RON TK-/- hosts to seed breast cancer cells in the lungs. Again, RON TK-/- hosts were used to control for off-target effects of RONi, and as a test for complete loss of host RON tyrosine kinase activity. To model established micrometastatic disease, tumor cells were allowed to grow for seven days before starting drug treatment. Mice were euthanized after three weekly cycles of treatment with RONi and aCTLA-4 as single agents or in combination. At the endpoint, analysis of the clinical response was performed by assessing tumor burden in the lungs and by quantifying the number of mice that had visible metastatic lesions versus those that had no apparent macrometastases. Histology was then performed to determine the extent of tumor clearance.
Wild-type animals in the vehicle treatment group exhibited aggressive metastatic tumor growth, with 100% of mice having overt metastasis in which approximately 40% of the lung area was covered with tumors (Figure 5a-c and S12a). Single-agent RONi treatment did not result in complete tumor clearance in any of the mice (Figure 5a,b and S12b). The metastatic tumor area was lower with RONi treatment, but this difference did not reach statistical significance (Figure 5b; p = 0.0555). Unlike our findings in the orthotopic tumor transplantation model, aCTLA-4 immunotherapy did not provide a significant clinical benefit in wild-type animals over the vehicle control, with only 1/12 (8%) of mice protected from metastasis (Figure 5a-c and S12c). In contrast, although combining RONi and aCTLA-4 was not sufficient to completely eliminate metastases in any of the wild-type animals, it significantly reduced the area occupied by metastases (Figure 5a-c and S12d). To assess the effect of complete RON kinase loss of function in this setting, and to control for any off-target effects of RONi, we carried out similar experiments with RON TK-/- host animals. Similar to our findings in wild-type mice treated with RONi, lack of host RON signaling alone (RON TK-/- hosts treated with vehicle control) was not enough to provide tumor clearance, although it reduced metastatic area significantly (Figure 5a-c and S12e). This observation is consistent with our previously published results.34 Importantly, treatment of RON TK-/- animals with RONi did not make a significant difference, indicating that off-target effects are not a concern in this model (Figure 5a-c and S12f). Strikingly, treatment of RON TK-/- mice with aCTLA-4 (dual inhibition of RON and CTLA-4) resulted in 42% of mice experiencing complete clearance of macrometastases, and significantly lowered tumor burden in the remaining mice, to under 2% of lung area (Figure 5a-c and S11g). Upon histological analysis, we could find occasional micrometastases in some of these animals (see Fig S13 for representative images), suggesting that, although dual inhibition of RON and CTLA-4 led to remarkable tumor control in most mice, some of the tumor cells were not killed. As expected, combination treatment with RONi and aCTLA-4 in RON TK-/- hosts did not significantly enhance responses compared to aCTLA-4 single-agent treatment in RON TK-/- mice (Figure 5a-c and S12h).
Figure 5.
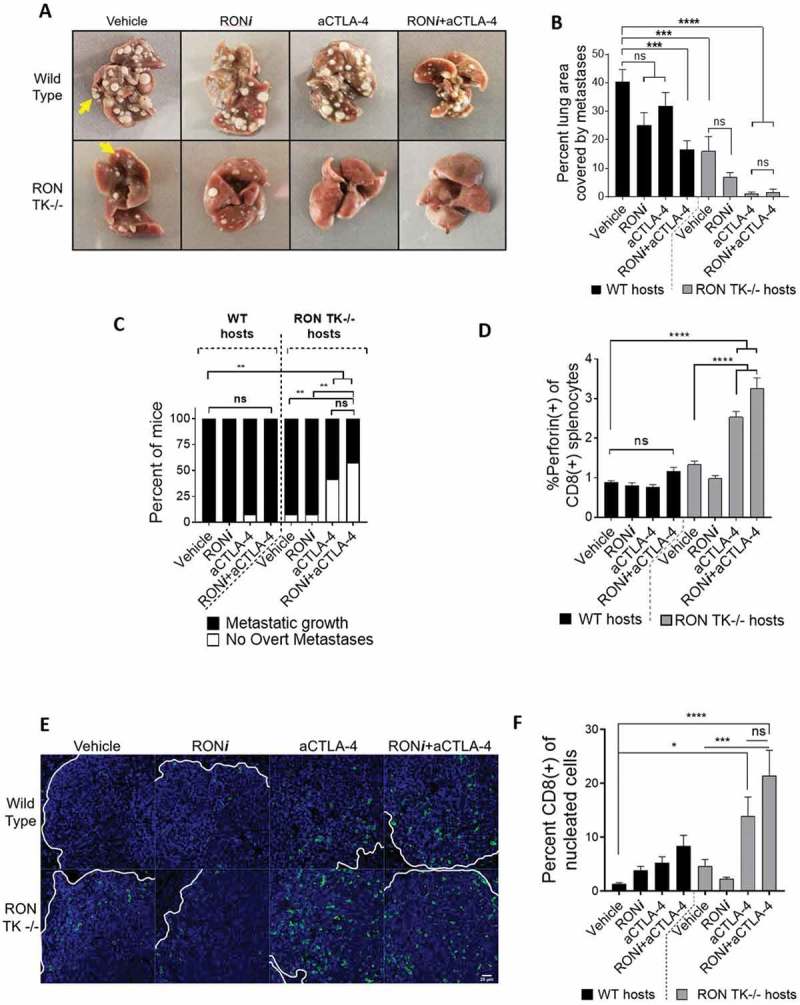
Pharmacologic inhibition or genetic ablation of host RON cooperates with aCTLA-4 immunotherapy to block the metastatic growth of breast cancer cells. A. Representative images of fixed lungs from mice treated with three weekly cycles of vehicle, RONi, and/or aCTLA4 (n = 12 per group). Micrometastatic cells were allowed to grow for 7 days prior to treatment initiation to model adjuvant therapy. Yellow arrows show metastatic tumors on the red background of the lung. Images from each mouse are shown in Fig S11a-h. B. Analysis of the percent lung area covered by macrometastatic colonies in wild-type (WT; black bars) and RON TK-/- hosts (gray bars). C. Frequency of mice exhibiting overt metastasis in lungs (black bars) versus no visible tumors (white bars). Statistical analyses were done using Fisher’s exact test. D. Frequency of perforin+ CD8+ splenic T-cells in each treatment group. E. Representative images of lung metastatic tumor-infiltrating CD8 + T cells (green) overlaid with the nuclear DAPI staining (blue). White lines outline the margin of the metastatic nodule in the lung sections. Scale bar indicates 25 μm in length. F. Quantification of metastatic tumor-infiltrating CD8+ selective kinase inhibitors T cells. 4 mice per each treatment group and 5 fields of vision per each mouse were analyzed. Amount of CD8+ cell infiltration is reported as the frequency of nucleated cells within the metastatic nodule. Statistical analyses were performed using one-way ANOVA with Tukey’s correction. (ns) p > 0.05, (*) p ≤ 0.05, (**) p ≤ 0.01, (***) p ≤ 0.001, (****) p ≤ 0.0001.
We proceeded to analyze systemic and local immune responses in these mice via flow cytometry and immunofluorescence. Similar to our findings in the orthotopic tumor experiments, intracellular staining of CD8+ splenocytes revealed the highest frequencies of perforin expression in RON TK-/- mice treated with aCTLA-4 (or with RONi+aCTLA-4) (Figure 5d), which correlated with tumor eradication (Figure 5a-c). Wild-type and RON TK-/- mice treated with vehicle had similar frequencies of activated, PD-1 expressing CD62L(low) splenic CD4+ and CD8 + T cells (Fig S14a,b). However, treatment with aCTLA-4 immunotherapy alone, or RONi+aCTLA-4, resulted in significantly elevated levels of PD-1(+)CD62L(low) CD4 and CD8+ splenocytes (Fig S14a,b). Interestingly, combination therapy in RON TK-/- mice resulted in slightly higher levels of these populations, suggesting potential off-target or on-target non-RON effects of BMS777607 inhibitor in the immune system (Fig S14a,b) that did not improve tumor control (Figure 5a-c). When we analyzed the infiltration of CD8+ cells into the metastatic nodules (despite lack of data from the best responders due to complete tumor eradication), we observed higher levels of infiltration when the mice were treated with RONi+aCTLA-4 combination therapy (Figure 5e,f). The highest levels of CD8+ cell infiltration was observed in RON TK-/- animals treated with aCTLA-4 alone, or in combination with RON inhibitor. As expected, RON inhibitor did not provide an advantage over aCTLA-4 single therapy in these mice, suggesting off-target effects of RON inhibitor is not a concern in this model. To determine whether aCTLA-4 immunotherapy was working through effects on T cells as expected, we performed a similar experimental metastasis study using NOD-SCID mice, which lack functional T and B cells. As expected, aCTLA-4 was ineffective in these mice, and combination of aCTLA-4 with RON inhibitor did not provide a significant therapeutic advantage over RON inhibitor single-therapy (Fig S15a,b). In summary, combining the RON inhibitor BMS777607 with aCTLA-4 immunotherapy in the PyMT model of aggressive breast cancer resulted in remarkable control of metastatic breast tumor growth in the adjuvant setting.
Discussion
The considerable excitement around success with cancer immunotherapy centers on activating a dynamic and adaptable immune response against heterogeneous and rapidly evolving tumors, in order to provide a sustained positive clinical outcome. Indeed, patients who benefit from immune checkpoint inhibitor therapy often have long-term benefit and even cures, although they represent only a small fraction of treated patients. Significant effort is currently focused on improving responses to immunotherapy through combining various treatment approaches.5 Combining two immune checkpoint inhibitors, ipilimumab (aCTLA-4) and nivolumab (aPD-1), was approved by the FDA for the frontline treatment of melanoma in 2015. Unfortunately, grade 3 and 4 treatment-related adverse events were observed in 55% of patients, twice as frequently compared to single-treatment arms.4 These data emphasize that discovering cooperating immune pathways that can be safely modulated to provide synergistic clinical benefit will be a key step in improving immunotherapy outcomes.
In this study, we show that RON, a non-essential receptor tyrosine kinase with potent immunosuppressive functions, induces an immunomodulatory gene expression signature in macrophages upon ligand-mediated activation. Among many differentially upregulated genes were several immune checkpoint ligands, including PD-L1 and CD80, both of which required MAPK signaling for expression downstream of RON. Pharmacological inhibition of RON with BMS777607/ASLAN002, or complete loss of host RON signaling through genetic means, improved clinical responses to aCTLA-4 therapy in an immunocompetent mouse model of breast cancer. Combining RON inhibition with aCTLA-4 immunotherapy provided clinical benefit for primary tumors in 92% of the animals (46% complete response), and was significantly better at preventing progression from micrometastatic to macrometastatic disease in the adjuvant setting. This was especially true when complete loss of RON kinase activity is achieved (RON TK-/- model + aCTLA-4), where there were remarkable effects on clearance of metastases. Positive clinical outcomes in RONi+aCTLA-4 treated animals were associated with higher frequencies of splenic and intratumoral CD8 + T-cells and higher effector cytokine production by these cells, suggesting improvements in tumor-specific immune activation. Importantly, RON inhibition also improved response to aCTLA-4 therapy in a separate colon cancer model, suggesting that our findings are not limited to a single model or to breast cancer.
RON’s role in upregulating checkpoint ligands in a MAPK-dependent manner raises an interesting possibility of combining checkpoint blockade with MEK inhibitors that are approved or currently in clinical trials. The MAPK signaling pathway is a key regulator of cell growth and survival, and it is overactivated in nearly 30% of human cancers.58 Two MEK inhibitors, trametinib and cobimetinib,59,60 are currently approved for the treatment of melanoma and more than 10 other MEK1/2 inhibitors are in various stages of clinical testing across a spectrum of cancer types including breast cancer.61 In addition to its cell-intrinsic protumorigenic effects, MAPK signaling has also been implicated in the upregulation of checkpoint ligands such as PD-L1.62,63 Therefore, combination of immunotherapy with MEK1/2 inhibitors has great potential to block both tumor-intrinsic and immune-mediated protumorigenic effects of the MAPK signaling pathway. Although MAPK signaling is important for early steps of T cell activation,64,65 several studies have shown that MEK inhibitors reprogram the tumor microenvironment and potentiate responses to checkpoint immunotherapy.66–68 In these studies, tumor growth was reduced by the combination of MEK inhibitors and various forms of immunotherapy, but tumors were not completely eradicated. It would be interesting to test whether RON and MEK inhibitors can cooperate to provide a better therapeutic outcome in the context of checkpoint immunotherapy.
Our findings reveal that MSP-RON signaling not only upregulates PD-L1, but also upregulates CD80 while downregulating CD86. CD80 and CD86 can each bind to CTLA-4 to suppress T cell activation69 or bind to CD28 to stimulate T cell activation.70 These contrasting effects warrant further investigation into the mechanism of how RON inhibitors provide a therapeutic benefit when combined with aCTLA-4 immunotherapy. One possible explanation may lie in the fact that CD80 is the preferred molecule over CD86 in suppressing T cell responses. Although both CD80 and CD86 can bind to CTLA-4, they have distinct binding characteristics and biological effects on T cells.71 CD80 was shown to have a higher affinity to CTLA-4, when compared to CD86.70,72 Using in vivo models, others have shown that the molecular signals delivered by CD80 and CD86 are not necessarily redundant. For instance, CD80 expressed in leukemic cells was found to suppress T cell immunity while CD86 was unable to do so.73 Similarly, CD80, but not CD86, was found to induce allograft tolerance, demonstrating that CD86 cannot replace all biological functions of CD80.74,75 The profound upregulation of CD80 downstream of MSP-RON signaling, therefore, may potently drive CTLA-4 mediated immunosuppression even when its other known ligand, CD86, is concomitantly downregulated. Further, blocking RON would prevent upregulation of CD80 and PD-L1, and downregulation of CD86, potentially allowing CD86 to perform its co-stimulatory function to activate T cells in the presence of anti-CTLA-4 immunotherapy. Our ongoing studies focus on further delineating the mechanisms of MSP-RON mediated immunosuppression and this possibility will be formally investigated in our future studies.
Our results are largely consistent with previous investigations utilizing immune checkpoint therapy in the PyMT breast cancer model. Two other studies showed no effects of aPD-1 therapy on PyMT tumor growth.20,21 However, in these reports, aCTLA-4 single agent treatment was also shown to be ineffective in controlling tumor growth, unlike our results, which showed 50% clinical benefit. This difference may be due to variations in the treatment regimens and/or the utilization of models of different genetic backgrounds (C57BL/6 mice as opposed to FVB mice used in this study). The latter possibility is especially intriguing since it was previously reported that host genetic makeup can affect tissue associated macrophage responses and antitumor immunity.31 In this context, evaluating new immune-targeting therapies in various mouse models is essential for informing clinical development of new compounds and biologics.
Currently, several clinical trials are underway that aim to elucidate clinical safety and efficacy of immune-checkpoint inhibition in breast cancer. At least 15 of these trials involve aCTLA-4, and more than 50 others involve inhibition of aPD-1 as single agents or in combination with other treatment approaches (www.clinicaltrials.gov). Importantly, the RON-selective tyrosine kinase inhibitor used in this study, BMS777607 (also known as ASLAN002), has completed a Phase I trial with a good tolerability profile in patients with advanced solid tumors. Analysis of serum samples from patients treated with this RON inhibitor in the Phase I study revealed reduced levels of bone turnover markers, which is a surrogate for RON-dependent activity in osteoclasts.39 These results suggest that therapeutic doses of BMS777607/ASLAN002 can be achieved without significant side effects, and support further clinical investigations in various cancer settings, now to include combination studies with aCTLA-4.
The mechanism by which host RON signaling regulates antitumor immunity still has not been fully elucidated. In the immune system, RON expression is restricted to terminally-differentiated macrophages. RON is reported to be expressed by resident macrophages in the bone, peritoneal cavity, and the lungs, and also in tumor-associated macrophages (TAMs).23,35 TAMs were reported to suppress anti-tumor CD8 + T-cell responses in a mouse model of prostate cancer via RON signaling.35 On the other hand, RON can also attenuate inflammatory responses in alveolar macrophages and protect mice from mortality following lung injury,30 suggesting potent control of the immune responses in a localized manner. Supporting this view, local expression of MSP by breast cancer cells has been shown to recruit TAMs and polarize them into an immunosuppressive phenotype, resulting in enhanced tumor growth in mice.76 Therefore, while mechanisms of action have not been fully elucidated due to our inability to selectively inhibit or knockout RON in specific macrophage populations (e.g., TAMs vs. resident lung macrophages), it is clear that inhibition of RON has strong potential in the cancer setting through its dual roles in tumor cells and in the tumor microenvironment.31,34,35 In particular, the present study shows that targeting RON signaling in combination with anti-CTLA-4 immunotherapy may slow or prevent breast tumor growth, including the emergence of metastatic disease.
Materials and methods
Mice and tumors
All animal procedures were carried out in accordance with the University of Utah Institutional Animal Care and Use Committee approval. RON TK-/- mice in FVB background were described previously.40 4–6 week old wild-type (FVB) and FVB RON TK-/- female mice were used in immunotherapy experiments. For macrophage studies, 6–8 week old wild-type (FVB) and FVB RON TK-/- female mice were used. Tumor cells were derived from spontaneous tumors arising in transgenic FVB MMTV-PyMT mice, and were engineered to express mouse MSP and the NP118 immunodominant peptide through retroviral transduction and subsequent positive selection with 5 μg/ml puromycin (PyMT-NP cells). Tumor cells were cultured in DME:F12 medium (Gibco, Invitrogen) supplemented with fetal bovine serum (10%) (Gibco, Invitrogen), insulin-transferrin-selenium-ethanolamine (1x) (Gibco, Invitrogen), recombinant murine EGF (10 ng/ml) (Invitrogen), hydrocortisone (1 μg/ml) (Sigma), penicillin-streptomycin-gentamycin (Gibco, Invitrogen) (1x) for a maximum of 4 days using previously described methods16,34 prior to injection into mice. MC38 colon carcinoma cells were purchased from Kerafast (Boston, USA), and maintained in Dulbecco’s Modified Eagle Medium (DMEM) containing high glucose and GlutaMAX (Gibco), supplemented with heat-inactivated fetal bovine serum (10%), nonessential amino acids (0.1 mM), sodium pyruvate (1 mM), penicillin/streptomycin (100 U/ml), gentamycin (50 mg/ml) and HEPES (10 mM).
Macrophage isolation and analysis
F4/80+ peritoneal macrophages were magnetically sorted (Miltenyi Biotec, GmbH) from peritoneal lavage fluid from female wildtype and RON TK-/- mice, into chilled tubes pre-coated with 5% FBS in PBS overnight. For RNAseq experiments, 750,000 F4/80+ macrophages per well were cultured in 24 well-plates in 500 μL of DMEM medium supplemented with 10% FBS and penicillin/streptomycin (Gibco). RON signaling was activated by the addition of 100 ng/mL recombinant human MSP (R&D Systems). After 7 hours of culture in MSP- or vehicle-containing medium, total RNA was isolated using the RNeasy Mini Kit (Qiagen, MD, USA) according to manufacturer’s protocol. RNA quality was measured by using the TapeStation system (Agilent, CA, USA). RNAseq library preparation was performed with polyA selection (TruSeq stranded mRNA library preparation kit, Illumina) prior to 50-cycle single-end sequencing on the Illumina HiSeq 2500 sequencing platform. Sequence reads were mapped to the mouse genome, and DESeq2 algorithm77 was used to assess differentially-expressed genes. Differentially-expressed genes in our study were compared with published gene sets by using Gene Set Enrichment Analysis (http://www.broad.mit.edu/gsea/).78 Pathway enrichment was assessed by using Ingenuity Pathway Analysis software (IPA, Qiagen, MD, USA).
For assessment of checkpoint ligand protein regulation, macrophages were stimulated with 100 ng/mL MSP for 24 hours. To test the requirement for RON kinase activity in this setting, cells were pre-incubated with 1μM BMS777607 or merestinib (LY2801653) (Selleck Chem) for 1 hour prior to addition of MSP. To test the requirement for transcription or translation in ligand upregulation, cells were preincubated with actinomycin D (1 μg/mL) or cycloheximide (10 μg/mL) 30 minutes prior to addition of MSP and cells were collected for flow cytometry analysis at time points between 0 and 24 hours later. To assess effects of MAPK and PI3K activity downstream of MSP/RON, cells were lysed with Pierce IP buffer (Thermo Fisher) 15 minutes after stimulation with 100 ng/mL MSP. Western blot analysis was performed with antibodies specific for pAKT, panAKT, pERK1/2, panERK1/2 and GAPDH (1:1000 primary antibody dilution, 1:5000 HRP-conjugated secondary antibody dilution) (Cell Signaling Technology). Antibodies recognizing phosphorylated forms or pan STAT1/3/5 were used at 1:250 and 1:500, respectively. To test the involvement of various signaling pathways downstream of RON, inhibitors BKM120 (PI3Ki), PD0325901 (MEK1/2i), and SCH772984 (ERK1/2i) were each added at 0.5 μM. Ruxolitinib (JAKi) was used at a final concentration of 1 μM. Cells were pre-conditioned with inhibitors for 1 hour prior to stimulation with 100 ng/mL MSP for Western blot and flow cytometric analysis.
Flow cytometry and histology
For macrophage experiments, cells were isolated from tissue culture plates by treating with 5 mM EDTA in PBS (pH 7.4) for 20 minutes at 37°C and by vigorous pipetting. For immunotherapy experiments, splenocytes were prepared directly from mice by physical dissociation via pressing between two microscope slides followed by red blood cell lysis with ammonium-chloride-potassium (ACK) buffer and filtering through a 100 μm nylon mesh filter. Tumors at the endpoint were collected, minced with razor blades, and then digested with 1 mg/mL collagenase IV (Sigma) for 1 hour at 37°C. After enzymatic digestion, tumor-infiltrating lymphocytes were isolated by centrifugation in a 44%-56% discontinuous Percoll gradient with no brakes. Contaminating red blood cells were lysed with ACK buffer and samples were filtered through a 100 μm nylon mesh filter. Surface antigens were stained with fluorophore-conjugated antibodies on ice, in PBS supplemented with 2% FBS and 0.01% sodium azide. Intracellular antigens were stained after 4 hours of re-stimulation with PMA (50 ng/mL) and Ionomycin (250 ng/mL), or with NP118 (0.1 μM), in the presence of brefeldin A (Cytofix/Cytoperm Kit, BD Biosciences). Stained cells were analyzed using a LSRFortessa cytometer (BD Biosciences, USA) and FlowJo software (TreeStar, USA). Prior to staining with fluorophore-conjugated antibodies, samples were treated with aCD16/32 Fc-blocking antibodies to reduce non-specific binding. Antibodies were purchased from BD Pharmingen and eBioscience (Thermo Fisher) and were used at 1:400 dilution: CD8a (53.67)-PE-Cy7, CD4 (GK1.5)-eFluor450, PD-1 (RMP1-30)-APC, CD62L (MEL-14)-Brilliant Violet 510; CD11b (M1/70)-eFluor506, F4/80 (BM8)-eFluor450, PD-L1 (MIH5)-PE-Cy7, PD-L2 (TY25)-PE, CD80 (16-10A1)-PerCp-Cy5.5, CD86 (GL1)-eFluor450, TNFα(MP6-XT22)-PerCp-eFluor710, IFNγ (XMG1.2)-APC-Cy7, Perforin-PE (eBioOMAK-D), Fixable Viability Dye eFluor506/eFluor780. During analysis, cellular debris was excluded by gating forward scatter vs side scatter parameters, followed by gating on live, single cells and lineage-specific subpopulations. Frequencies of cell populations were compared using the student’s T-test or with one-way ANOVA, where applicable.
Tumor-infiltrating CD8+ cells were assessed by immunofluorescence microscopy of tumor sections after 48-hour fixation in formalin-free zinc fixative (BD Biosciences). Tumor tissue mircoarrays (TMA) were generated by punching paraffin-embedded tumor blocks with a 1.5 mm hollow needle at intact tumor core (excluding necrotic areas and non-tumorous tissues as determined by a prior hematoxylin/eosin staining). Two representative regions from each tumor sample were selected to be transferred to recipient paraffin blocks as a TMA, unless the tumor was too small to be sampled twice. TMA blocks were then sectioned into 3 μm-thick sections. TMA sections were deparaffinized with CitriSolve solution, rehydrated with serial dilutions of ethanol, followed by heat-inactivated epitope retrieval (HIER) with 10mM Sodium Citrate, pH 6.0. Non-specific blocking was performed by incubating the section with 5% bovine serum albumin and 10% goat serum, together with FcR blocking reagent (Miltenyi Biotec, GmbH). After the blocking, the sections were incubated with 1:400 CD8a primary antibody (clone 4SM16) (eBioscience, Thermo Fisher) overnight at 4°C followed by corresponding secondary antibody conjugated with Alexa Fluor 488 fluorophore. Autofluorescence blocking was obtained by immersing the slide in 0.1% Sudan black. Samples were counterstained with DAPI and mounted with 80% glycerol. Image acquisition was performed with an inverted wide-field microscope (ECLIPSE Ti-E, Nikon) integrated with an Andor Clara CCD camera, and the entire TMA punches were analyzed using ImageJ-based FIJI software. CD8+ cells were counted manually and quantified per number of DAPI+ nuclei in non-necrotic areas. Quantification of tumor infiltrating CD8 + T cells in experimental metastasis experiments was performed on 4 samples randomly selected from each treatment group, except in cases where treatment was so effective that any remaining tumor had to be specifically searched for and selected for quantification. Lungs were processed and stained similar to primary tumors and 5 separate tumor fields-of-vision containing the infiltrated CD8 + T cells were imaged via Leica SP8 white light laser confocal microscope at 20x magnification. Computer-assisted quantification of CD8+ signal was performed by setting a common threshold for all the images by using Fiji software.79
Actla-4 and aPD-1 immunotherapy
20,000 PyMT-NP cells were transplanted unilaterally into the fourth inguinal mammary fat pads of 4–6 week old mice. When the tumor reached 100 mm3, mice were randomized to the following four groups in a rolling enrollment: 1) Vehicle (DMSO); 2) RONi (BMS777607; 50 mg/kg orally; 5 days on and 2 days off per weekly cycle); 3) aCTLA-4 (9D9; 10 mg/kg intraperitoneally; twice per weekly cycle), and 4) RONi+aCTLA4 combination (same dose regimens). In applicable experiments, 4 mg/kg aPD-1 (4H2) was delivered intraperitoneally three times a week until the experimental end-point. Tumors were palpated and measured every other day with digital calipers. Treatments continued for 4 cycles, or until the tumor reached the IACUC-approved ethical endpoint of 3000 mm3. Mice were generally sacrificed on the 24th day of treatment. Tumor burden at the endpoint was assessed in individual mice using two metrics: 1) clinical response classification (complete response/partial response/refractory), and 2) tumor growth rate. For the first metric, “complete response” was defined as eradication of tumor at the endpoint. “Partial Response” was defined to be up to 300% increase in tumor size compared to the start of treatment (this group includes partial responders with some tumor shrinkage, stable disease, and mice with slow-growing tumors). “Refractory disease” was defined as any tumor exhibiting more than 300% increase in size. In the vehicle-treated group, the average increase in tumor size was 2316% (±291%). To calculate the change in tumor growth rate, exponential tumor growth curves were log2-transformed to obtain linear tumor growth versus time. The slope of the linear regression was calculated for each mouse individually (ΔLog2(tumor size)/Δdays of treatment) and averaged. Statistical differences were assessed by using one-way ANOVA and Tukey’s multiple comparison correction.
For experiments with the MC38 colon carcinoma model, 100,000 cells were injected subcutaneously into 5–8 week old C57BL/6 mice. RON inhibitor and aCTLA-4 treatment was initiated when the tumor became palpable (~7 days post-injection) using the same treatment regimen as in PyMT experiments. Treatment continued for 2 weekly cycles and mice were euthanized at day 14. Tumor growth rate was calculated using the same methods as in PyMT experiments, and statistical analysis was performed by using one-way ANOVA and Tukey’s multiple comparison correction.
For metastasis studies, we modeled the adjuvant therapy setting by seeding tumor cells in the lung, waiting 7 days, and then initiating treatment. Mice were injected with 100,000 MMTV-PyMT cells suspended in HBSS via the lateral tail vein. After 7 days, treatment was initiated using the same doses and schedules that were used for the treatment of orthotopically transplanted tumors. Mice were euthanized on day 32 and lungs were harvested and fixed in formalin-free zinc fixative. Photographs of the metastatic lungs were taken and images were imported to Image J for quantification. Metastatic tumor area was quantified as a ratio of normal lung using Image J software.
Funding Statement
This work was funded by an Innovator and Scholar Concept award from the DOD Breast Cancer Research Program (BC113194 to A.L.W.) and a Leadership Award from the Susan G. Komen Foundation (SAC160078 to A.L.W.);Susan G Komen Foundation [SAC160078];DOD Breast Cancer Research Program [BC113194];
Acknowledgments
BMS777607, aCTLA-4 and aPD-1 were kindly provided by Bristol Meyers Squibb. Shared resources such as High Throughput Genomics and Bioinformatics and Biostatistics Cores were supported by the HCI Cancer Center Support Grant (5P30CA042014-24; the content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health). We are grateful for use of the Flow Cytometry Facility (James Marvin) and the Fluorescence Microscopy Core Facility (Mike Bridge) at the University of Utah Health Sciences, both funded by the National Center for Research Resources (NCRR) of the National Institutes of Health under Award Number 1S10RR026802-01. Microscopy equipment was obtained with a NCRR Shared Equipment Grant # 1S10RR024761-01. We thank Andrew Stephen Baessler for his assistance in Western blots. We also thank members of Welm Lab for their assistance in rapid and systematic tissue processing for large in vivo experiments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplementary materials
Supplementary materials for this article can be accessed here
Author contributions
H.A.E. and S-C.A.L. designed and performed experiments; acquired, analyzed and interpreted data; and prepared the manuscript. H.G. performed preliminary optimization experiments. F.H. analyzed the intratumoral T-cell infiltration. M.A.W. provided means to generate model antigen-expressing tumor cells and advised on immunophenotyping. A.L.W. oversaw the experiments, data analysis and prepared the manuscript. All authors reviewed the manuscript in its final form.
Data and material availability
All data are provided in the manuscript. Utah materials used in the experiments are available through material transfer agreements with the University of Utah.
References
- 1.Schildberg FA, Klein SR, Freeman GJ, Sharpe AH.. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity. 2016;44(5):955–972. doi: 10.1016/j.immuni.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J ImmunTher Cancer. 2014;2(1):3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. New Engl J Med. 2015;373(1):23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ott PA, Hodi FS, Kaufman HL, Wigginton JM, Wolchok JD. Combination immunotherapy: a road map. J ImmunTher Cancer. 2017;5(1):16. doi: 10.1186/s40425-017-0218-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, Formenti SC. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11(2):728–734. [PubMed] [Google Scholar]
- 7.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31(1):51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 8.National Cancer Institute Bethesda MD. SEER Cancer Stat Facts: Female Breast Cancer 2017. [accessed 2017 July 2]. https://seer.cancer.gov/statfacts/html/breast.html.
- 9.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rugo H, Delord J-P, Im S-A, Ott P, Piha-Paul S, Bedard P, Sachdev J, Le Tourneau C, van Brummelen E, Verga A, et al. Preliminary efficacy and safety of pembrolizumab (MK-3475) in patients with PD-L1–positive, estrogen receptor-positive (ER+)/HER2-negative advanced breast cancer enrolled in KEYNOTE-028. San Antonio Breast Cancer Symp. 2015;76:S5–7. doi: 10.1158/1538-7445.SABCS15-S5-07. [DOI] [Google Scholar]
- 11.Nanda R, Chow LQM, Dees EC, Berger R, Gupta S, Geva R, Pusztai L, Pathiraja K, Aktan G, Cheng JD, et al. Pembrolizumab in patients with advanced triple-negative breast cancer: phase Ib keynote-012 study. J Clin Oncol. 2016;34(21):2460–2467. doi: 10.1200/JCO.2015.64.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McArthur HL, Page DB. Immunotherapy for the treatment of breast cancer: checkpoint blockade, cancer vaccines, and future directions in combination immunotherapy. Clin Adv Hematol Oncol: H&O. 2016;14(11):922–933. [PubMed] [Google Scholar]
- 13.Emens L, Braiteh F, Cassier P, Delord J, Eder J, Shen X, Xiao Y, Wang Y, Hegde P, Chen D, et al. Inhibition of PD-L1 by MPDL3280A leads to clinical activity in patients with metastatic triple-negative breast cancer. San Antonio Breast Cancer Symposium vol. 75 2014. p. PD1–6. doi: 10.1517/14656566.2014.904288. [DOI] [Google Scholar]
- 14.Guy CT, Cardiff RD, Muller WJ. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol Cell Biol. 1992;12(3):954–961. doi: 10.1128/MCB.12.3.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Welm AL, Sneddon JB, Taylor C, Nuyten DSA, van de Vijver MJ, Hasegawa BH, Bishop JM. The macrophage-stimulating protein pathway promotes metastasis in a mouse model for breast cancer and predicts poor prognosis in humans. Proc Natl Acad Sci U S A. 2007;104(18):7570–7575. doi: 10.1073/pnas.0702095104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strachan DC, Ruffell B, Oei Y, Bissell MJ, Coussens LM, Pryer N, Daniel D. CSF1R inhibition delays cervical and mammary tumor growth in murine models by attenuating the turnover of tumor-associated macrophages and enhancing infiltration by CD8(+) T cells. Oncoimmunology. 2013;2(12):e26968. doi: 10.4161/onci.26968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, Coussens LM. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102. doi: 10.1016/j.ccr.2009.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young KH, Baird JR, Savage T, Cottam B, Friedman D, Bambina S, Messenheimer DJ, Fox B, Newel P, Bahjat KS, et al. Optimizing timing of immunotherapy improves control of tumors by hypofractionated radiation therapy. PLoS ONE. 2016;11(6):1–15. doi: 10.1371/journal.pone.0157164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aguilera TA, Rafat M, Castellini L, Shehade H, Kariolis MS, Hui AB-Y, Stehr H, von Eyben R, Jiang D, Ellies LG, et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun. 2016;7:13898. doi: 10.1038/ncomms13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210(11):2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ronsin C, Muscatelli F, Mattei MG, Breathnach R. A novel putative receptor protein tyrosine kinase of the met family. Oncogene. 1993;8(5):1195–1202. [PubMed] [Google Scholar]
- 23.Iwama A, Wang MH, Yamaguchi N, Ohno N, Okano K, Sudo T, Takeya M, Gervais F, Morissette C, Leonard EJ, et al. Terminal differentiation of murine resident peritoneal macrophages is characterized by expression of the STK protein tyrosine kinase, a receptor for macrophage-stimulating protein. Blood. 1995;86(9):3394–3403. [PubMed] [Google Scholar]
- 24.Wagh PK, Peace BE, Waltz SE. Met-related receptor tyrosine kinase Ron in tumor growth and metastasis. Adv Cancer Res. 2008;100:1–33. doi: 10.1016/S0065-230X(08)00001-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu X, Zhao L, DeRose YS, Lin Y-C, Bieniasz M, Eyob H, Buys SS, Neumayer L, Welm AL. Short-form ron promotes spontaneous breast cancer metastasis through interaction with phosphoinositide 3-kinase. Genes Cancer. 2011;2(7):753–762. doi: 10.1177/1947601911421924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faham N, Welm AL. RON signaling is a key mediator of tumor progression in many human cancers. Cold Spring Harb Symp Quant Biol. 2016;81:177–188. doi: 10.1101/sqb.2016.81.031377 [DOI] [PubMed] [Google Scholar]
- 27.Yao H-P, Zhou Y-Q, Zhang R, Wang M-H. MSP-RON signalling in cancer: pathogenesis and therapeutic potential. Nat Rev Cancer. 2013;13(7):466–481. doi: 10.1038/nrc3545. [DOI] [PubMed] [Google Scholar]
- 28.Wang MH, Yao HP, Zhou YQ. Oncogenesis of RON receptor tyrosine kinase: A molecular target for malignant epithelial cancers. Acta Pharmacol Sin. 2006;27(6):641–650. doi: 10.1111/j.1745-7254.2006.00361.x. [DOI] [PubMed] [Google Scholar]
- 29.Morrison AC, Correll PH. Activation of the Stem Cell-Derived Tyrosine Kinase/RON Receptor Tyrosine Kinase by Macrophage-Stimulating Protein Results in the Induction of Arginase Activity in Murine Peritoneal Macrophages. J Immunol. 2002;168(2):853–860. doi: 10.4049/jimmunol.168.2.853. [DOI] [PubMed] [Google Scholar]
- 30.Nikolaidis NM, Gray JK, Gurusamy D, Fox W, Stuart WD, Huber N, Waltz SE. Ron receptor tyrosine kinase negatively regulates tnfα production in alveolar macrophages by inhibiting Nf-Κb activity and adam17 production. Shock. 2010;33(2):197–204. doi: 10.1097/SHK.0b013e3181ae8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chaudhuri A, Wilson NS, Yang B, Martinez AP, Liu J, Zhu C, Bricker N, Couto S, Modrusan Z, French D, et al. Host genetic background impacts modulation of the TLR4 pathway by RON in tissue-associated macrophages. Immunol Cell Biol. 2013;91(7):451–460. doi: 10.1038/icb.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang M-H, Zhou Y-Q, Chen Y-Q. Macrophage-stimulating protein and RON receptor tyrosine kinase: potential regulators of macrophage inflammatory activities. Scand J Immunol. 2002;56(6):545–553. doi: 10.1046/j.1365-3083.2002.01177.x. [DOI] [PubMed] [Google Scholar]
- 33.Ray M, Yu S, Sharda DR, Caleph B, Liu Q, Kaushal N, Prabhu KS, Hankey PA, Wilson CB, Liu Q, et al. Inhibition of TLR4-induced IκB kinase activity by the RON receptor tyrosine kinase and its ligand, macrophage-stimulating protein. J Immunol. 2010;185(12):7309–7316. doi: 10.4049/jimmunol.1000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eyob H, Ekiz HA, DeRose YS, Waltz SE, Williams MA, Welm AL. Inhibition of Ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013;3(7):751–760. doi: 10.1158/2159-8290.CD-12-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gurusamy D, Gray JK, Pathrose P, Kulkarni RM, Finkleman FD, Waltz SE. Myeloid-specific expression of Ron receptor kinase promotes prostate tumor growth. Cancer Res. 2013;73(6):1752–1763. doi: 10.1158/0008-5472.CAN-12-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schroeder GM, An Y, Cai ZW, Chen XT, Clark C, Cornelius LAM, Dai J, Gullo-Brown J, Gupta A, Henley B, et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4- ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the met kinase superfamily. J Med Chem. 2009;52(5):1251–1254. doi: 10.1021/jm801586s. [DOI] [PubMed] [Google Scholar]
- 37.Multiple Ascending Dose Study of BMS-777607 in Subjects With Advanced or Metastatic Solid Tumors Clinicaltrials.Gov. 2008. [accessed 2017 July 2]. 3/1/2008-3/1/2009 http://clinicaltrials.gov/show/NCT00605618. doi:NCT00605618.
- 38.A Phase I Multiple Ascending Dose Study of ASLAN002 in Subjects With Advanced or Metastatic Solid Tumours Clinicaltrials.Gov. 2012. [accessed 2017 July 2]. 20121001–20131201 http://clinicaltrials.gov/show/NCT01721148. doi:NCT01721148.
- 39.Andrade K, Fornetti J, Zhao L, Miller SC, Randall RL, Anderson N, Waltz SE, McHale M, Welm AL. RON kinase: A target for treatment of cancer-induced bone destruction and osteoporosis. Sci Transl Med. 2017;9:374. doi: 10.1126/scitranslmed.aai9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waltz SE, Eaton L, Hess KA, Peace BE, Ihlendorf JR, Wang MH, Kaestner KH, Degen SJF, Toney-Earley K, Hess KA, et al. Ron-mediated cytoplasmic signaling is dispensable for viability but is required to limit inflammatory responses. J Clin Investig. 2001;108(4):567–576. doi: 10.1172/JCI200111881.Introduction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front Immunol. 2014;5(OCT):1–22. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, Morris SM, Hankey PA. Regulation of macrophage arginase expression and tumor growth by the ron receptor tyrosine kinase. J Immunol. 2011;187(5):2181–2192. doi: 10.4049/jimmunol.1003460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado JDD, Popovich PG, Partida-Sanchez S, Guerau-De-arellano M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. 2015;10(12):5–11. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buxadé M, Lunazzi G, Minguillón J, Iborra S, Berga-Bolaños R, Del Val M, Aramburu J, López-Rodríguez C. Gene expression induced by Toll-like receptors in macrophages requires the transcription factor NFAT5. J Exp Med. 2012;209(2):379–393. doi: 10.1084/jem.20111569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan SB, Peek VL, Ajamie R, Buchanan SG, Graff JR, Heidler SA, Hui YH, Huss KL, Konicek BW, Manro JR, et al. LY2801653 is an orally bioavailable multi-kinase inhibitor with potent activity against MET, MST1R, and other oncoproteins, and displays anti-tumor activities in mouse xenograft models. Invest New Drugs. 2013;31(4):833–844. doi: 10.1007/s10637-012-9912-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doi T, Ishikawa T, Okayama T, Oka K, Mizushima K, Yasuda T, Sakamoto N, Katada K, Kamada K, Uchiyama K, et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol Rep. 2017;37(3):1545–1554. doi: 10.3892/or.2017.5399. [DOI] [PubMed] [Google Scholar]
- 47.Mimura K, Teh JL, Okayama H, Shiraishi K, Kua L-F, Koh V, Smoot DT, Ashktorab H, Oike T, Suzuki Y, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109(1):43–53. doi: 10.1111/cas.13424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q, Wang HY, Wei F, Liu X, Paterson JC, Roy D, Mihova D, Woetmann A, Ptasznik A, Odum N, et al. Cutaneous T cell lymphoma expresses immunosuppressive CD80 (B7-1) cell surface protein in a STAT5-dependent manner. J Immunol. 2014;192(6):2913–2919. doi: 10.4049/jimmunol.1302951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang X, Zeng Y, Qu Q, Zhu J, Liu Z, Ning W, Zeng H, Zhang N, Du W, Chen C, et al. PD-L1 induced by IFN-γ from tumor-associated macrophages via the JAK/STAT3 and PI3K/AKT signaling pathways promoted progression of lung cancer. Int J Clin Oncol. 2017;22(6):1026–1033. doi: 10.1007/s10147-017-1161-7. [DOI] [PubMed] [Google Scholar]
- 50.Wen Z, Zhong Z, Darnell JE. Maximal activation of transcription by statl and stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82(2):241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- 51.Ihle JN. The Janus protein tyrosine kinase family and its role in cytokine signaling. Adv Immunol. 1995;60:1–35. [DOI] [PubMed] [Google Scholar]
- 52.Schulz M, Aichele P, Schneider R, Hansen TH, Zinkernagel RM, Hengartner H. Major histocompatibility complex binding and T cell recognition of a viral nonapeptide containing a minimal tetrapeptide. Eur J Immunol. 1991;21(5):1181–1185. doi: 10.1002/eji.1830210513. [DOI] [PubMed] [Google Scholar]
- 53.Corbett TH, Griswold DP, Roberts BJ, Peckham JC, Schabel FM. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35(9):2434–2439. doi: 10.1158/0008-5472.can-05-1299. [DOI] [PubMed] [Google Scholar]
- 54.Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, Arancibia-Cárcamo CV, Sobel RA, Rudensky AY, Kuchroo VK, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med. 2015;212(10):1603–1621. doi: 10.1084/jem.20141030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kilinc MO, Gu T, Harden JL, Virtuoso LP, Egilmez NK. Central role of tumor-associated CD8+ T effector/memory cells in restoring systemic antitumor immunity. J Immunol (Baltimore, Md 1950). 2009;182(7):4217–4225. doi: 10.4049/jimmunol.0802793. [DOI] [PubMed] [Google Scholar]
- 56.Uhr JW, Pantel K. Controversies in clinical cancer dormancy. Proc Natl Acad Sci. 2011;108(30):12396–12400. doi: 10.1073/pnas.1106613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abe O, Abe R, Enomoto K, Kikuchi K, Koyama H, Masuda H, Nomura Y, Sakai K, Sugimachi K, Tominaga T, et al. Effects of radiotherapy and of differences in the extent of surgery for early breast cancer on local recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;366(9503):2087–2106. doi: 10.1016/S0140-6736(05)67887-7. [DOI] [PubMed] [Google Scholar]
- 58.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 59.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P, et al. Improved Survival with MEK Inhibition in BRAF-Mutated Melanoma. New Engl J Med. 2012;367(2):107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 60.Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, et al. Combined Vemurafenib and Cobimetinib in BRAF -Mutated Melanoma. New Engl J Med. 2014;371(20):1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- 61.Cheng Y, Tian H. Current development status of MEK inhibitors. Molecules. 2017;22:10. doi: 10.3390/molecules22101551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Atefi M, Avramis E, Lassen A, DJL Wong, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, TG Graeber, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res. 2014;20(13):3446–3457. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sumimoto H, Takano A, Teramoto K, Daigo Y. RAS-mitogen-activated protein kinase signal is required for enhanced PD-L1 expression in human lung cancers. PLoS ONE. 2016;11:11. doi: 10.1371/journal.pone.0166626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gupta S, Weiss A, Kumar G, Wang S, The NA. T-cell antigen receptor utilizes Lck, Raf-1, and MEK-1 for activating mitogen-activated protein kinase. Evidence for the existence of a second protein kinase C-dependent pathway in an Lck-negative Jurkat cell mutant. J Biol Chem. 1994;269(25):17349–17357. [PubMed] [Google Scholar]
- 65.Weiss A, Littman DR. Signal transduction by lymphocyte antigen receptors. Cell. 1994;76(2):263–274. doi: 10.1016/0092-8674(94)90334-4. [DOI] [PubMed] [Google Scholar]
- 66.Dushyanthen S, Teo ZL, Caramia F, Savas P, Mintoff CP, Virassamy B, Henderson MA, Luen SJ, Mansour M, Kershaw MH, et al. Agonist immunotherapy restores T cell function following MEK inhibition improving efficacy in breast cancer. Nat Commun. 2017;8:1. doi: 10.1038/s41467-017-00728-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poon E, Mullins S, Watkins A, Williams GS, Koopmann JO, Di Genova G, Cumberbatch M, Veldman-Jones M, Grosskurth SE, Sah V, et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J ImmunTher Cancer. 2017;5:1. doi: 10.1186/s40425-017-0268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, Gould SE, Maecker H, Irving BA, Kim JM, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity. 2016;44(3):609–621. doi: 10.1016/j.immuni.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 69.McCoy KD, Le Gros G. The role of CTLA-4 in the regulation of T cell immune responses. Immunol Cell Biol. 1999;77(1):1–10. doi: 10.1046/j.1440-1711.1999.00795.x. [DOI] [PubMed] [Google Scholar]
- 70.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1(9):793–801. doi: 10.1016/S1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 71.Sansom DM. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology. 2000;101(2):169–177. doi: 10.1046/j.1365-2567.2000.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ikemizu S, Gilbert RJC, Fennelly JA, Collins AV, Harlos K, Jones EY, Stuart DI, Davis SJ. Structure and dimerization of a soluble form of B7-1. Immunity. 2000;12(1):51–60. doi: 10.1016/S1074-7613(00)80158-2. [DOI] [PubMed] [Google Scholar]
- 73.LaBelle JL, Hanke CA, Blazar BR, Truitt RL. Negative effect of CTLA-4 on induction of T-cell immunity in vivo to B7-1 +, but not B7-2 +, murine myelogenous leukemia. Blood. 2002;99(6):2146–2153. doi: 10.1182/blood.V99.6.2146. [DOI] [PubMed] [Google Scholar]
- 74.Judge TA, Wu Z, Zheng XG, Sharpe AH, Sayegh MH, Turka LA. The role of CD80, CD86, and CTLA4 in alloimmune responses and the induction of long-term allograft survival. J Immunol (Baltimore, Md 1950). 1999;162(4):1947–1951. [PubMed] [Google Scholar]
- 75.Yamada A, Kishimoto K, Dong VM, Sho M, Salama AD, Anosova NG, Benichou G, Mandelbrot DA, Sharpe AH, Turka LA, et al. CD28-independent costimulation of T cells in alloimmune responses. J Immunol. 2001;167(1):140–146. doi: 10.4049/jimmunol.167.1.140. [DOI] [PubMed] [Google Scholar]
- 76.Benight NM, Wagh PK, Zinser GM, Peace BE, Stuart WD, Vasiliauskas J, Pathrose P, Starnes SL, Waltz SE. HGFL supports mammary tumorigenesis by enhancing tumor cell intrinsic survival and influencing macrophage and T-cell responses. Oncotarget. 2015;6(19):17445–17461. doi: 10.18632/oncotarget.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mesirov J, Gillette M, Paulovich A, Pomeroy S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide. October. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hulsen T, de Vlieg J, Alkema W. BioVenn - a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics. 2008;9(1):488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.