

Abstract
Snyder-Robinson syndrome, also known as spermine synthase deficiency, is an X-linked intellectual disability syndrome (OMIM #390583). First described by Drs. Snyder and Robinson in 1969, this syndrome is characterized by an asthenic body habitus, facial dysmorphism, broad-based gait, and osteoporosis with frequent fractures. We report here a pediatric autopsy of a 4 year old male with a history of intellectual disability, gait abnormalities, multiple fractures, and seizures previously diagnosed with Snyder-Robinson syndrome with an SMS gene mutation (c.831G>T:p.L277F). The cause of death was hypoxic-ischemic encephalopathy secondary to prolonged seizure activity. Although Snyder-Robinson syndrome is rare, the need to recognize clinical findings in order to trigger genetic testing has likely resulted in under diagnosis.
Keywords: Spermine Synthase; Snyder-Robinson syndrome; Mental Retardation, X-linked; Intellectual Disability
INTRODUCTION
In 1969, Drs. Snyder and Robinson reported a family with a sex-linked mental retardation disorder characterized by hypotonia and dysequilibrium.1 Further study of this family led to the identification of a genetic defect in the SMS gene located on the short arm of the X chromosome. The SMS gene (NM_004595.4) encodes for spermine synthase, which is involved in polyamine biosynthesis.2
Currently the clinical features of SRS are based on twenty patients and include intellectual disability, hypotonia, asthenic body build/diminished body bulk, and bone abnormalities (osteoporosis, fractures, kyphoscoliosis).3 Other features of SRS include ambulation abnormalities, facial dysmorphism, mild short stature, seizures, genital abnormalities, and renal complications. A diagnosis of SRS is confirmed or established based on laboratory testing of spermine synthase enzyme activity or identification of a hemizygous loss-of-function pathogenic variant in SMS.3 The prevalence of SRS is unknown, although cases have been reported in the Americas and Europe.4-7
METHODS
Inpatient and outpatient medical records were reviewed. Patient demographics, clinical history, clinical presentation, radiographic findings, autopsy findings, and neuropathology were recorded and analyzed. Written informed consent was obtained from the next of kin.
Whole exome sequencing (GeneDx, Gaithersburg, MD) was performed using genomic DNA and the Agilent Clinical Research Exome kit. Targeted regions were sequenced by massively parallel sequencing on an Illumina HiSeq system with 100 bp paired-end reads. The sequence was aligned to a reference human genome (GRCh37/UCSC hg 19) and analyzed (Xome Analyzer). Potentially pathogenic sequences were confirmed using another method.
CASE REPORT
A 4-year-old male had a past medical history of Snyder-Robinson syndrome, with mild cognitive and adaptive intellectual disability, gait abnormalities requiring a walker or holding hands to walk, multiple fractures, and treatment resistant myoclonic epilepsy. He lived at home and attended preschool. He did not display any behavioral concerns and was able to use lots of words and some signs for communication.
Whole exome sequence analysis identified a de novo mutation in the SMS gene, c.831G>T: p.L277F. This variant corresponds to SNP rs1064794901, which has not been found in more than 60,000 controls in the Exome Aggregation Consortium (ExAC) database. Although this variant had not been previously reported, it is a strong candidate for pathogenicity based on the following considerations: this variant was not identified in the NHLBI Exome Sequencing Project as a benign SNP; it is a de novo mutation not seen in either parent; this substitution occurs at a conserved site across species; and finally, in silico analysis predicts this substitution to damage protein structure or function.
He received regular clinical care for developmental delay and epilepsy. He was in his usual state of health, seizure free for four months, when he developed symptoms of congestion and restlessness. The following day he had a myoclonic seizure lasting 30 minutes, accompanied by a fever of 102.5°F (39,1°C). The seizure episode lasted approximately 30 minutes requiring a visit to the emergency center for two doses of lorazepam (0.05mg/kg) and a paracetamol suppository. A chest x-ray showed bilateral perihilar infiltrates concerning for a viral infection. He did not return to baseline behavior and was sleepy and not speaking or walking. He had signs of possible new seizure activity, which included tongue thrusting, bilateral leg pulling toward his core, and bilateral arm movements. He was admitted to a University hospital two days later.
His persistent altered mental status prompted a sepsis workup with blood cultures and cerebral spinal fluid analysis. Aerobic and anaerobic cultures were negative for any growth and a meningitis/encephalitis panel was negative for any organisms. He received empiric antibiotics and antiviral medications. Due to concern for raised intracranial pressure, an invasive intracranial pressure monitor was placed. Brain imaging showed cerebral edema consistent with hypoxic-ischemic encephalopathy; the edema progressed with uncal herniation observed by imaging. After discussion with healthcare providers and family, the decision was made to withdraw support and proceed with organ donation after cardiac death.
AUTOPSY PRESENTATION
Autopsy examination showed a tall male child (18 kg 75th percentile for weight and 115 cm >95 percentile for height) with dysmorphic features including prominent cupped ears, protruding lower lip, and a high arched palate (Figure 1).
Figure 1. Dysmorphic features included macrocephaly, prominent cupped ears, and protruding lower lip.

He had an asthenic build and kyphosis. The heart, lungs, aortic arch, kidneys and corneas were recovered for organ donation. The testes were small (measured 1.3 cm, reference range; 2.0 cm) and histology shows absence of Leydig cells and an expanded interstitium (Figure 2).
Figure 2. Photomicrography of the testes showing regularly arranged tubules. No Leydig cells are present (H&E, 20X).

Prominent macrocephaly (54.4 cm; >97th percentile WHO head circumference-for-age Boys) with corresponding megalencephaly (2,840 grams; expected weight for age = 1,191 grams) and cerebral edema was present (Figure 3). Consistent with the imaging findings of uncal herniation, there was softening and discoloration of the left uncus. Microscopic examination of the brain revealed diffuse, predominantly acute, hypoxic-ischemic changes and hippocampal pathology consistent with the patient's extensive seizure history as well as evidence of incipient central herniation (Figure 4).
Figure 3. Coronal sections of the frontal lobe (A) and at the level of the caudate and putamen (B) from the current case (top) and a normal control (bottom) demonstrate diffuse enlargement of all structures in an otherwise structurally normal brain including normal cortical lamination and neuronal organization.
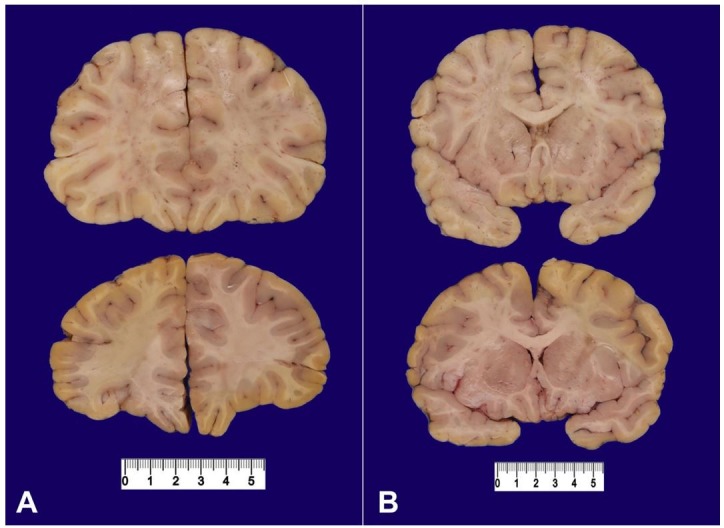
Figure 4. A – section of the hippocampus shows acutely hypoxic-ischemic neurons and reactive vessels with underlying neuronal loss; B – the area of infarct corresponding to incipient uncal herniation is shown (H&E, 10X for both).

CLINICAL DISCUSSION
The history of chronic seizures and recent episode of congestion and fever likely contributed to the seizure event leading up to his hospitalization. The protracted seizure resulted in diffuse acute hypoxic-ischemic neuronal injury with cerebral edema and herniation resulting in his death.
CONCLUSION
Previously published cases of SRS were mainly familial (Table 1) while the current case is a de novo mutation.1,2,4-11 This patient had both clinical signs and symptoms of SRS with a likely pathogenic variant of SMS. Although Kesler et al.,9 have described slightly enlarged brain volumes by MRI brain imaging of two SRS patients, our patient had severe megalencephaly, which has not been previously reported. Based on the limited data regarding brain volumes in SRS patients, it is not clear yet the degree of penetrance of megalencephaly or enlarged brain volumes is present in this population. Additionally, while genital abnormalities such as low testicular volume, hypospadias, and undescended testes have been reported in 15% of cases, here we identified a Leydig cell deficiency in the setting of microrchidia.3
Table 1. Literature review of reported cases of SRS.
| Author and Year | # new patients | Mutation | Specific findings | Notes |
|---|---|---|---|---|
| Snyder and Robinson 1969 | Family K8145 | X-linked | Likely x-linked MR, hypotonia, disequilibrium. No characteristic facial appearance or seizures | Several affected members had >average head circumference 1 with abnormal EEG |
| Arena 1996 | Family K8145 | Xp21.3-p22.12. | Diminished muscle mass, osteoporosis, kyphoscoliosis, slight facial asymmetry with a prominent lower lip, nasal speech, high narrow/cleft palate, long great toes | autopsy results for 2-year-old presumed SRS patient |
| Cason 2003 | 11 - Family K8145 (from 4 generations) | c.329+5 G>A | Unsteady gait, movement d/o, seizures, | 2/4 patients with abnormal EEG |
| De Alencastro 2008 | 3 | p.G56S | Severe epilepsy, cognitive impairment | Normal head CT & EEG |
| Bercerra-Solano 2009 | 2 | c.496T>G p.V132G | Fractures, develop delay, cognitive impairment, kyphoscoliosis | |
| Kesler 2009 | 0 | Total brain volumes were somewhat enlarged | ||
| Schwartz 2011 | 0 | Spermine synthase assay | ||
| Zhang 2013 |
4 (from 3 generations) | c.1084A>G p.Y328C | Milder phenotype | No neurological symptoms |
| Peron 2013 | 1 | c.200G>A p.G67X | Ectopic kidney and epilepsy (focal motor seizures, negative myoclonus). | |
| Abela 2016 | 2 (Twin boys) | c.388C>T p.R130C | Early onset epileptic encephalopathy |
Although SRS is rare, it is likely underdiagnosed. The numbers of SRS patients, particularly with de novo mutations, will likely increase with the increasing role of molecular testing in patients who present with an undefined developmental delay syndrome.
ACKNOWLEDGEMENTS
We would like to acknowledge Terry Anderson, DCS for expert assistance in conducting the autopsy and Stephanie Stauffer, MD for assistance performing a literature search.
Footnotes
How to cite: Starks R, Kirby P, Ciliberto M, Hefti M. Snyder-Robinson syndrome. Autops Case Report [Internet]. 2018;8(3):e2018031. http://dx.doi.org/10.4322/acr.2018.031
Financial support: None
REFERENCES
- 1.Snyder RD, Robinson A. Recessive sex-linked mental retardation in the absence of other recognizable abnormalities: report of a family. Clin Pediatr (Phila). 1969;8(11):669-74. 10.1177/000992286900801114. [DOI] [PubMed] [Google Scholar]
- 2.Cason AL, Ikeguchi Y, Skinner C, et al. . X-linked spermine synthase gene (SMS) defect: the first polyamine deficiency syndrome. Eur J Hum Genet. 2003;11(12):937-44. 10.1038/sj.ejhg.5201072. [DOI] [PubMed] [Google Scholar]
- 3.Albert J, Schwartz CE, Boerkoel CF, Stevenson RE. Snyder-Robinson syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington; 2013. [cited 2013 Jun 27]. p. 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK144284/ [PubMed] [Google Scholar]
- 4.Alencastro G, McCloskey DE, Kliemann SE, et al. . New SMS mutation leads to a striking reduction in spermine synthase protein function and a severe form of Snyder-Robinson X-linked recessive mental retardation syndrome. J Med Genet. 2008;45(8):539-43. 10.1136/jmg.2007.056713. [DOI] [PubMed] [Google Scholar]
- 5.Becerra-Solano LE, Butler J, Castaneda-Cisneros G, et al. . A missense mutation, p.V132G, in the X-linked spermine synthase gene (SMS) causes Snyder-Robinson syndrome. Am J Med Genet. 2009;149A(3):328-35. 10.1002/ajmg.a.32641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z, Norris J, Kalscheuer V, et al. . A Y328C missense mutation in spermine synthase causes a mild form of Snyder–Robinson syndrome. Hum Mol Genet. 2013;22(18):3789-97. 10.1093/hmg/ddt229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peron A, Spaccini L, Norris J, et al. . Snyder-Robinson syndrome: a novel nonsense mutation in spermine synthase and expansion of the phenotype. Am J Med Genet. 2013;161A(9):2316-20. 10.1002/ajmg.a.36116. [DOI] [PubMed] [Google Scholar]
- 8.Arena JF, Schwartz C, Ouzts L, et al. . X-linked mental retardation with thin habitus, osteoporosis, and kyphoscoliosis: linkage to Xp21.3-p22.12. Am J Med Genet. 1996;64(1):50-8. . [DOI] [PubMed] [Google Scholar]
- 9.Kesler SR, Schwartz C, Stevenson RE, Reiss AL. The impact of spermine synthase (SMS) mutations on brain morphology. Neurogenetics. 2009;10(4):299-305. 10.1007/s10048-009-0184-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz CE, Wang X, Stevenson RE, Pegg AE. Spermine synthase deficiency resulting in X-linked intellectual disability (Snyder-Robinson syndrome). Methods Mol Biol. 2011;720:437-45. 10.1007/978-1-61779-034-8_28. [DOI] [PubMed] [Google Scholar]
- 11.Abela L, Simmons L, Steindl K, et al. . N(8)-acetylspermidine as a potential plasma biomarker for Snyder-Robinson syndrome identified by clinical metabolomics. J Inherit Metab Dis. 2016;39(1):131-7. 10.1007/s10545-015-9876-y. [DOI] [PubMed] [Google Scholar]