

Abstract
Spinal muscular atrophy (SMA) is caused by reduced levels of full-length SMN (FL-SMN). In SMA patients with one or two copies of the Survival Motor Neuron 2 (SMN2) gene there are a number of SMN missense mutations that result in milder-than-predicted SMA phenotypes. These mild SMN missense mutation alleles are often assumed to have partial function. However, it is important to consider the contribution of FL-SMN as these missense alleles never occur in the absence of SMN2. We propose that these patients contain a partially functional oligomeric SMN complex consisting of FL-SMN from SMN2 and mutant SMN protein produced from the missense allele. Here we show that mild SMN missense mutations SMND44V, SMNT74I or SMNQ282A alone do not rescue mice lacking wild-type FL-SMN. Thus, missense mutations are not functional in the absence of FL-SMN. In contrast, when the same mild SMN missense mutations are expressed in a mouse containing two SMN2 copies, functional SMN complexes are formed with the small amount of wild-type FL-SMN produced by SMN2 and the SMA phenotype is completely rescued. This contrasts with SMN missense alleles when studied in C. elegans, Drosophila and zebrafish. Here we demonstrate that the heteromeric SMN complex formed with FL-SMN is functional and sufficient to rescue small nuclear ribonucleoprotein assembly, motor neuron function and rescue the SMA mice. We conclude that mild SMN missense alleles are not partially functional but rather they are completely non-functional in the absence of wild-type SMN in mammals.
Introduction
Spinal muscular Atrophy (SMA) has an incidence of approximately 1/10,000 and is the leading genetic cause of infant death (1–4). SMA is caused by homozygous loss or mutation of the Survival Motor Neuron 1 (SMN1) gene with retention of the Survival Motor Neuron 2 (SMN2) gene (5,6). The SMN1 and SMN2 genes essentially differ by a single nucleotide transition in exon 7, which alters splicing regulation and results in SMN2 producing mostly messenger RNA (mRNA) that lacks exon 7 (7–11). In turn, the SMN protein lacking amino acids encoded by exon 7 does not oligomerize efficiently and is rapidly degraded (12–14). Thus, when SMN1 is lost SMN2 does not produce sufficient levels of SMN protein and SMA results (15,16). SMA has a range of severities where type 0 is the most severe and type 4 is the least severe. One correlate to severity is the copy number of SMN2 where more copies are present in milder SMA patients (17,18).
In the majority of SMA cases, the SMN1 gene is absent and the sole source of SMN mRNA is from SMN2 (19). Five percent of SMA patients have a small insertion or deletion in the SMN1 gene. In most cases, these insertions and deletions disrupt the ability of the SMN1 gene to make SMN protein (6,20–22). In ∼1% of SMA patients, the SMN1 gene contains a missense mutation (6). A missense mutation changes one of the amino acids in the SMN protein. Some of these missense mutations result in a mild SMA phenotype in the presence of one or two copies of SMN2. For example, patients with the SMNA2G missense mutation and one copy of SMN2 present with type 3 SMA (6,22). Typically an individual with one copy of SMN2 would present with severe type 0 or type 1 SMA. Therefore, we define this type of missense mutation as mild. Conversely, a severe missense allele is one that results in a type 1 SMA phenotype in the presence of two or more copies of SMN2 (6,23).
Interestingly, there has never been a report of a missense allele occurring in the absence of SMN2 although 10–15% of the healthy population lacks SMN2 (19). This raises the question as to the mode of action of missense alleles in SMA and whether they are functional by themselves. We have previously shown that the mild SMN missense mutations SMNA2G and SMNA111G act by complementing SMN. The mutations are not capable of even partial rescue of a mouse lacking SMN2 suggesting that missense alleles lack function when they are not paired with SMN2 (24,25). Moreover, SMN is known to oligomerize and some severe mutations in the C-terminal of SMN disrupt oligomerization which disrupts SMN’s function in small nuclear ribonucleoprotein (snRNP) assembly (12,14,26). Furthermore, some severe mutations occurring in the Tudor domain of SMN alter the ability of the arginine and glycine-rich tail of Sm proteins to interact with SMN (27). The SMN-Gemin2 complex forms oligomers that span the range of dimers to octamers (28,29). Mutations that lie in the inner surface of the YG domain are severe and disrupt oligomerization, whereas mild mutations such as SMNT274I that lie on the outside surface do not disrupt oligomerization (28,29). As such the YG box is sufficient to form oligomers composed of two tetramers. The exact functional unit in the cytoplasm for assembly of snRNPs can be a smaller unit, such as the dimer or tetramer, whereas the larger structure could occur in gems in the nucleus (29). SMN has been shown to function in the assembly of Sm proteins onto snRNAs as well as the assembly of Sm/Lsm proteins onto the U7 small nuclear ribonucleic acid (snRNA) (30–34). SMN has also been implicated in the assembly of RNP complexes that play a role in transport and translational control of mRNA in axons (30,35,36).
SMN mutations equivalent to those found in SMA patients have been examined in species other than mammals. The C. elegans equivalent of SMND44V is CeSmnD27N (37). This mutant resulted in a slightly decreased life span of 15 days versus 17 in the worm. Decreased movement was observed but animals with the mutation were viable and fertile, indicating that CeSmnD27N is a hypomorphic allele that functions at a reduced level (37). A series of SMN missense mutations have also been examined in Drosophila, in particular the severe mutation dSmnI93F and the mild mutation dSmnT205I that are equivalent to SMNI116F and SMNT274I, respectively. However, in Drosophila the alleles do not correlate with the severity of phenotype found in humans (38). Lastly, a synthetic mutation SMNQ282A did not have the ability to correct axonal defects caused by knockdown of SMN levels in the zebrafish (25,39).
In this study we sought to study the function of SMN missense mutations in the presence and absence of wild-type full-length SMN (FL-SMN) from SMN2 in vivo in mice. We examined the missense alleles SMND44V, SMNT274I, SMNI116F and SMNQ282A and determined that these alleles do not function the same in mammals and invertebrates. As with other mild SMN mutations capable of oligomerizing with SMN produced by SMN2, we find that all mild alleles (SMND44V, SMNT274I and SMNQ282A) behave in a similar fashion to SMNA2G and SMNA111G in mice (24,25). Here we demonstrate that mild missense alleles can rescue survival, function in snRNP assembly and result in normal motor neuron function (as determined by Compound Muscle Action Potential (CMAP) and Motor Unit Number Estimates (MUNE)) only in the presence of small amounts of wild-type FL-SMN SMN produced by SMN2. These alleles are not capable of rescuing the Smn null allele in SMA mice in the absence of SMN2. Thus, mild missense alleles of SMN can complement the small amount of SMN produced by SMN2 yielding a functional oligomeric complex. Moreover, missense alleles of SMN are not functional in the absence of wild-type FL-SMN protein in mice.
Results
Generation of transgenic mouse lines containing SMN missense alleles
Transgenes containing mild mutations SMND44V, SMNT274I, SMNQ282A and the severe mutation SMNI116F under the control of a 4.1 kb human SMN promoter were microinjected into FVB/N oocytes at the Genetically Engineered Mouse Modeling Core (GEMMC) of The Ohio State University. The location of each missense mutation in the SMN protein is shown in Fig. 1 and the DNA change is shown in Supplementary Table 1. Multiple founders were obtained for each missense mutation transgenic line. Every founder was tested for expression of the corresponding mRNA by reverse transcriptase-polymerase chain reaction (RT-PCR) and for expression of SMN protein by western blot analysis with the human-specific KH SMN antibody (generously provided by Dr Krainer) in brain and spinal cord tissue (40) (data not shown). We obtained eight founders for SMND44V, of which three lines (D44V #4, D44V #6 and D44V #10*) expressed SMND44V mRNA and protein. We obtained 6 founder lines for SMN T274I, of which three lines (T274I #1, T274I #4 and T274I #5*) expressed SMNT274I mRNA and protein. We obtained 12 founder mice for SMNQ282A, of which three lines (Q282A #2*, Q282A #5 and Q282A #6) expressed mutant SMNQ282A mRNA and protein. Finally, we obtained 13 founder mice for SMNI116F, with four lines (I116F #7, I116F #9, I116F #11* and I116F #13) expressing SMNI116F mRNA but low amounts of SMN mutant protein. SMND44V line #10, SMNT274I line #5, SMNQ282A line #2 and SMNI116F line #11 (indicated by an asterisk above) were used for further investigation as they produced the highest amount of SMN mRNA and protein.
Figure 1.
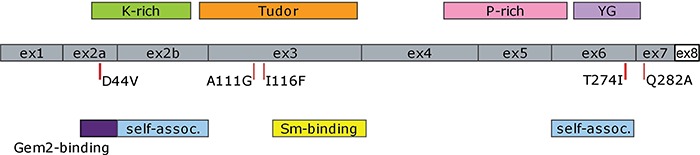
Diagrammatic representation of human SMN complementary DNA (cDNA) showing locations of exons and protein domains. The location of the missense mutations used in this study are indicated. Note that exon 8, the untranslated region represented as a white box, is not drawn to scale. (Self-assoc. = self-association domain).
The copy number of the missense SMN transgenes was determined by droplet digital polymerase chain reaction (ddPCR) (Bio-Rad) on genomic DNA for each of the lines selected for further experiments. The copy number of the transgenes was compared to an internal two-copy control gene (Smn). The following copy numbers were obtained by ddPCR: SMND44V line #10 has one copy (two when homozygous), SMNT274I line #5 has five copies (10 when homozygous), SMNQ282A line #2 has four copies (eight when homozygous) and SMNI116F line #11 has 55 copies (110 when homozygous). We have found that the copy number of the transgenic insertion does not always correlate with the amount of expression. The reference SMN∆7 mice have two copies of SMN2 and are homozygous for the SMN∆7 transgene (41). The total number of SMN∆7 copies in a homozygous animal is 14 as determined previously and in this study by ddPCR (41).
The mice were crossed onto the severe SMA mouse background (Smn+/−; SMN2+/+) to obtain mice that were heterozygous for the missense mutation transgene, homozygous for SMN2 and heterozygous for the mouse Smn knockout allele (Smn+/−; SMN2+/+; SMN transgene+/−). These mice were then backcrossed to obtain mice that were homozygous for the missense mutation transgene (Smn+/−; SMN2+/+; SMN transgene+/+). Lastly, these mice were intercrossed to determine if the SMN missense transgene could rescue the Smn−/− mouse in the presence of two copies of SMN2. Mice that were heterozygous for mouse Smn were used as controls in this study (Smn+/−; SMN2+/+; SMN transgene+/+). None of the mice containing the missense mutation contained the SMN∆7 allele. The SMN∆7 SMA mice, which lived 14 days on average, were used as a comparison for mRNA and protein expression levels (41).
Analysis of SMN mRNA expression in SMA mice with mild and severe SMN missense alleles
We performed RT-PCR on spinal cord tissue collected at 4 days of age and used ddPCR to determine the amount of FL-SMN mRNA produced by the homozygous transgenes in a SMA background (SMN2+/+, Smn−/−). The quantification of SMN mRNA expression for each of the transgenes used in this study is shown in Fig. 2. The primer and probe set used detects only FL-SMN and expression is measured relative to the housekeeping gene YWHAZ12. There is a marked increase in the expression of FL-SMN mRNA when compared to SMN∆7 SMA mice. The SMNQ282A and SMND44V lines show a ∼3-fold increase in SMN [69.9 ± 5.6 relative fluorescent units (RFU), P < 0.001 and 66.7 ± 6.5 RFU, P < 0.001 versus 20.5 ± 3 RFU] and SMNT274I showed a ∼6-fold increase (126.3 ± 12 RFU, P < 0.05). SMNI116F showed a ∼15-fold increase (317.7 ± 38 RFU, P < 0.001). RFU is defined as relative fluorescent units. Thus all missense mutation transgenes produce FL-SMN at levels greater than expression in SMN∆7 SMA mice ( Smn−/−; SMN2+/+; SMN∆7+/+).
Figure 2.
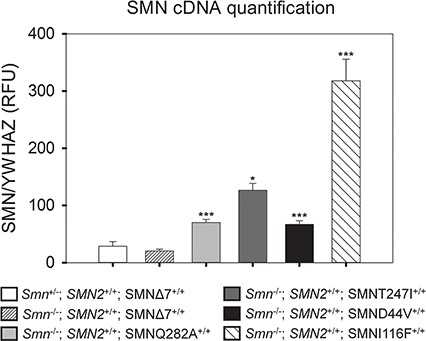
Quantification of FL-SMN cDNA using ddPCR indicates a significant increase in SMN expression in transgenic SMN missense lines as compared to the SMNΔ7 SMA mice at 4 days of age. SMN cDNA isolated from total spinal cord RNA was measured in the SMN missense lines. Expression of the housekeeping gene YWHAZ12 was used to normalize the amount of cDNA in the reaction. SMNQ282A and SMND44V mice exhibited an ∼3-fold increase in SMN expression as compared to SMNΔ7 SMA mice (69.9 ± 5.6 RFU and 66.7 ± 6.5 RFU versus 20.5 ± 3 RFU). SMN expression in the SMNT274I mice was 6-fold greater (126.3 ± 12 RFU) than the expression in SMNΔ7 SMA mice (Smn−/−; SMN2+/+; SMN∆7+/+). SMNI116F mice displayed the greatest amount of SMN expression with an ∼15-fold increase over SMN∆7 SMA mice (317.7 ± 38 RFU). There is equivalent expression of FL-SMN in SMN∆7 SMA and SMN∆7 heterozygous control mice (Smn+/−; SMN2+/+; SMNΔ7+/+). (n = 4 in each group, RFU = relative fluorescence units, error bars = standard error of mean (SEM), ***P < 0.001, *P < 0.05).
Analysis of SMN protein expression in SMA mice with mild and severe SMN missense alleles
The SMN protein expression for each of the missense mutation alleles was determined by an enzyme-linked immunosorbent assay (ELISA) (Fig. 3). This assay accurately measures the amount of SMN protein relative to total protein levels thus avoiding the use of a housekeeping gene for comparison. We isolated both brain and spinal cord tissue from 4-day-old animals. The mild missense alleles SMNQ282A and SMNT274I showed a ∼2–3-fold increase in protein expression in the brain (Fig. 3A) and an even greater increase (∼4–6-fold) in the spinal cord (Fig. 3B) as compared to the SMN∆7 SMA mouse. SMND44V protein expression was more modestly increased in the brain and spinal cord (∼1.5-fold) (Figs. 3A and 3B). The severe missense allele SMNI116F protein expression was similar to the SMN∆7 SMA expression as the protein is unstable and rapidly degraded similar to other severe missense alleles (13,16).
Figure 3.

Quantification of SMN protein measured by ELISA indicates a significant increase in protein expressed by the mild missense mutation alleles as compared to the SMNΔ7 SMA mice at 4 days of age. (A) SMN protein in brain tissue was increased ∼3-fold in SMNQ282A (14567.5 ± 905.3 pg/mg) and ∼2-fold in SMNT274I (11279.0 ± 337.3 pg/mg) versus SMNΔ7 SMA (4156.7 ± 301.1 pg/mg) mice. SMND44V expression was ∼1.5-fold increased (6509.3 ± 1135.6 pg/mg) and SMNI116F was less than the SMN∆7 SMA mice due to protein instability (1933.2 ± 140.2 pg/mg). SMNΔ7 heterozygous control mice had the greatest SMN protein expression (18266.4 ± 294.0 pg/mg) (n = 3 mice for each group). (B) SMN protein in spinal cord tissue was also increased in each of the SMN missense alleles. There was a greater than ∼6-fold increase in SMNQ282A (14181.3 ± 820.8 pg/mg, n = 3) and greater than ∼4-fold increase in SMNT274I (10779.6 ± 1588.9 pg/mg, n = 3) as compared to SMNΔ7 SMA mice (2312.8 ± 727.8 pg/mg, n = 2). SMND44V showed a more modest ∼1.5-fold increase (4269.6 ± 1681.0 pg/mg, n = 2). SMNΔ7 heterozygous control mice had the greatest amount of SMN protein expression (15475.5 ± 676.6 pg/mg, n = 3). (Error bars = SEM, ***P < 0.001, **P < 0.01, *P < 0.05).
Figure 4A shows the amount of SMN protein in each extract that was used for snRNP assembly. The total amount protein per assay is the same for each missense mutation allele. The amount of assembled U1 snRNA for each transgenic missense mutation line is also shown. All mild missense mutation alleles produced more SMN protein than the SMN∆7 SMA mice. However the severe mutation SMNI116F mice showed the same level of protein expression as the SMN∆7 mice. The SMNI116F protein appears unstable given the large increase in mRNA expression shown in Fig. 2 but low level of protein captured for the snRNP assembly assay. This result is similar to other severe SMN mutations when assayed in vivo (16,25).
Figure 4.
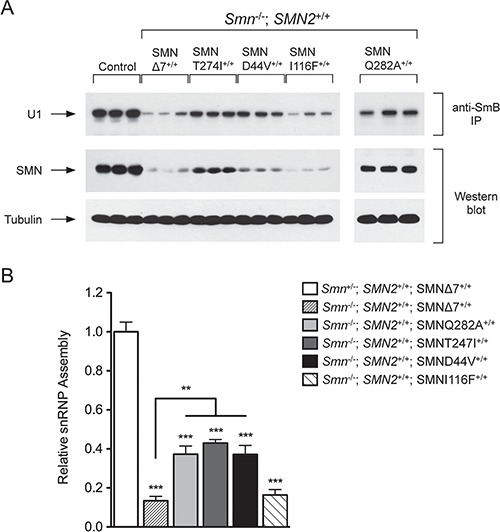
Increased snRNP assembly activity in transgenic mice containing mild SMN missense mutation alleles. (A) Western blot analysis shows the amount of SMN protein in the extract that was used for snRNP assembly. The amount of assembled U1 snRNA isolated with the anti-SmB antibody is also shown. The amount of captured SMN protein in the SMNI116F missense mutation line was similar to that isolated from the SMN∆7 SMA mice. (B) The snRNP assembly in total spinal cord tissue from each missense mutation line was assessed to determine the activity of the SMN complex. SMNQ282A and SMND44V showed an ∼2.6-fold increase while SMNT274I showed an ∼3-fold increase in snRNP assembly activity as compared to SMNΔ7 SMA mice (n = 3 for each group). The relative snRNP assembly in SMNI116F was similar to SMNΔ7 SMA mice (n = 3). SMNΔ7 SMA mice (n = 6) had ∼7-fold less snRNP assembly than heterozygous control mice (Smn+/−; SMN2+/+; SMNΔ7+/+) (n = 6). The activity of snRNP assembly in the heterozygous control mice was fixed at 1 and all other values were calculated accordingly. (Error bars = SEM, ***P < 0.001, **P < 0.01).
Analysis of snRNP assembly in SMA mice with mild and severe SMN missense alleles
We have shown previously that the mild alleles SMNA111G and SMNA2G can complex with wild-type FL-SMN from SMN2 and that the resulting SMN complexes can perform snRNP assembly (25,42), which is the most well characterized activity of SMN (43). We therefore assayed snRNP assembly activity in the spinal cord extracts of SMND44V, SMNT274I, SMNQ282A, SMNI1116F and SMN∆7 SMA mice containing two copies of SMN2 and no mouse Smn (Smn−/−; SMN2+/+; SMN transgene+/+). The control line contained two copies of SMN2, was heterozygous for Smn and homozygous for the SMN∆7 transgene (Smn+/−; SMN2+/+; SMN∆7+/+). The assay used in vitro transcribed radioactive U1 snRNA that was mixed with spinal cord extract from 4-day-old mice, followed by immunoprecipitation of assembled U1 snRNP with anti-SmB antibodies as previously described (Fig. 4A) (25,42) The snRNP assembly activity for each missense allele in the presence of SMN2 is shown in Fig. 4B. The level of snRNP assembly in spinal cord extracts from SMN∆7 heterozygous (Smn+/−) control mice was adjusted to 100% and used to normalize the activity in the extracts from the other transgenic missense alleles. All lines assayed included three biological replicates (three different mice). The assay as indicated showed good reproducibility. The transgenic lines SMND44V, SMNT274I and SMNQ282A on a background containing two copies of SMN2 and lacking mouse Smn showed a marked increase in snRNP assembly activity when compared to SMN∆7 SMA mice. However, the transgenic line SMNI116F showed levels of activity similar to SMN∆7 SMA mice, which is consistent with the level of SMNI116F protein in the extract.
Mild SMN missense mutation alleles do not rescue Smn null mice
Each mild missense allele transgene was first studied on an Smn null background (Smn−/−) in the absence of SMN2. None of the missense mutation transgenic lines were capable of rescuing the embryonic lethality of Smn−/− mice in the absence of SMN2 (Table 1). We performed a Chi-square analysis for each possible outcome using mild missense alleles SMNQ282A, SMNT274I and SMND44V. The mice were null for mouse Smn. No mice of genotype Smn−/−, SMNQ282A (expected = 22, observed = 0, n = 117), Smn−/−, SMNT274I (expected = 17, observed = 0, n = 88), Smn−/−, SMND44V (expected = 10, observed = 0, n = 170) were observed. Supplementary Table 2 shows Chi-square calculations assuming all Smn−/− mice are non-viable. These data demonstrate that SMNQ282A, SMNT274I or SMND44V are not functional on their own and cannot rescue survival of Smn knockout mice.
Table 1.
Survival of SMNQ282A, SMNT274I and SMND44V mice without SMN2
| (A) SMNQ282A | Expected | Observed | χ2 |
|---|---|---|---|
| Smn +/+; SMNQ282A | 22 | 29 | |
| Smn +/+ | 7 | 8 | |
| Smn +/−; SMNQ282A | 44 | 69 | |
| Smn +/− | 15 | 11 | |
| Smn −/− ; SMNQ282A | 22 | 0 | |
| Smn −/− | 7 | 0 | |
| n = 117 | n = 117 | χ2 = 36.2 | |
| (B) SMNT274I | Expected | Observed | χ2 |
| Smn +/+; SMNT274I | 17 | 19 | |
| Smn +/+ | 5 | 9 | |
| Smn +/−; SMNT247I | 33 | 47 | |
| Smn +/− | 11 | 13 | |
| Smn −/− ; SMNT274I | 17 | 0 | |
| Smn −/− | 5 | 0 | |
| n = 88 | n = 88 | χ2 = 25.9 | |
| (C) SMND44V | Expected | Observed | χ2 |
| Smn +/+; SMND44V | 32 | 44 | |
| Smn +/+ | 11 | 24 | |
| Smn +/−; SMND44V | 64 | 71 | |
| Smn +/− | 21 | 31 | |
| Smn −/− ; SMND44V | 32 | 0 | |
| Smn −/− | 10 | 0 | |
| n = 170 | n = 170 | χ2 = 51.0 |
aChi-squared test using a 6 x 2 contingency table with 5 degrees of freedom was used to determine if the lack of rescue animals was significant or due to chance. The genotypes and number of mice born with each missense allele (A) SMNQ282A, (B) SMNT274I and (C) SMND44V in a mouse Smn null background with no SMN2 were counted. No mice of genotype Smn−/−; SMNQ282A (e = 22, o = 0, n = 117), Smn−/−; SMNT274I (e = 17, o = 0, n = 88) or Smn−/−; SMND44V (e = 10, o = 0, n = 170) were observed. The expected and observed frequencies were significantly different (SMNQ282A, P = 0; SMNT274I, P = 0.0001; SMND44V, P = 0) showing that SMNQ282A, SMNT274I or SMND44V need some amount of FL-SMN to function and rescue survival of SMA mice. Chi-square calculations assuming all Smn−/− mice are non-viable are shown in Supplementary Table 2. (e = expected, o = observed, n = the number of animals in the group).
Mild but not severe SMN missense mutation alleles can rescue Smn null mice in the presence of SMN2
Weight and survival analysis was performed in mice containing mild SMN missense alleles on an Smn null background with two copies of SMN2 ( Smn−/−; SMN2+/+; SMN transgene+/+) (Fig. 5). In the presence of SMN2, all of the mild transgene alleles (SMND44V n = 14, SMNT274I n = 19 or SMNQ282A n = 8) increased survival beyond 200 days with no evidence of a SMA-like phenotype (Fig. 5B, D, F). In addition, the corrected animals displayed a normal weight distribution similar to heterozygous or wild-type Smn mice (Fig. 5A, C, E). The weight of all three SMN missense allele lines is not significantly different from that of heterozygous SMN∆7 control mice (Smn+/−; SMN2+/+; SMN∆7+/+) at 100 days of age (Supplementary Figure 1). Therefore, while none of the missense alleles are capable of rescuing Smn null mice on their own, the mild missense alleles can rescue Smn null mice in the presence of two copies of SMN2.
Figure 5.
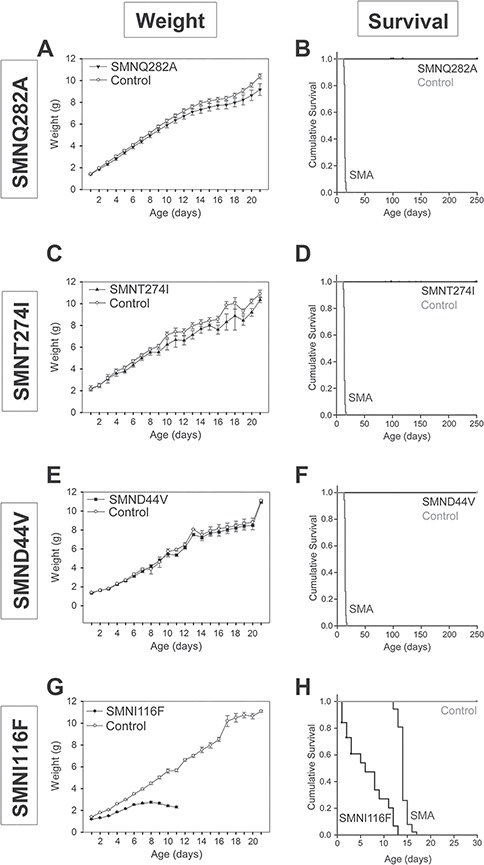
Weight and survival are normal in transgenic mice with mild SMN missense mutation alleles. Expression of SMNQ282A, SMNT274I or SMND44V in the Smn−/−; SMN2+/+ background completely rescued the weight of the mice (A–F, n = 14, n = 19 and n = 8, respectively). The severe allele SMNI116F does not rescue the (G) weight or (H) survival of severe SMA mice (n = 10). The Smn−/−; SMN2+/+; SMNI116F+/+ mice survive an average of 6.4 ± 1 days as compared to SMN∆7 SMA mice (Smn−/−; SMN2+/+; SMNΔ7+/+, n = 86) that survive 14.1 ± 0.1 days. (B, D, F, H) Control mice are defined as mice that contain the missense allele transgene and are heterozygous for mouse Smn (Smn+/−; SMN2+/+; SMN missense transgene+/+).
In contrast, the severe allele SMNI116F gave similar results to mice which lack mouse Smn and have two copies of SMN2 and live for 5.16 days (Smn−/−; SMN2+/+) (44). The SMNI116F showed survival of only 6.4 ± 1 days (Fig. 5H, n = 10) and severely impaired weight gain (Fig. 5G). Unlike the mild missense alleles, the severe missense allele SMNI116F cannot rescue Smn null mice even in the presence of SMN2. Overall, these results are consistent with phenotypic rescue requiring an increase in snRNP assembly to a critical threshold level. Any increase greater than this threshold does not result in further phenotypic effect. It is quite likely that different SMN missense mutations vary in their ability to restore activity to a heteromeric complex. Thus, explaining the lack of a direct correlation of snRNP activity with the SMN protein level of the mutant SMN transgene as determined by ELISA (Fig. 3). The SMNI116F allele displays low SMN expression levels, similar to SMN∆7 SMN levels, and this is likely due to the instability of this mutant and rapid protein turnover (13).
Normalization of SMA electrophysiology with expression of mild SMN mutations in the presence of SMN2
We have previously shown that SMA mice have reduced CMAP, SMUP and MUNE while displaying fibrillations that indicate denervation of muscle (45). Similar electrophysiological abnormalities are observed in SMA patients (45,46). Early restoration of SMN by either antisense oligonucleotides (ASOs) directed against ISS-N1, or gene therapy using scAAV9-SMN can correct these electrophysiological defects in SMA mice (45). Late administration of ASO at postnatal day 4 resulted in partial rescue of the phenotype, including reduced MUNE and development of fibrillations at later stages (46). Here, we assessed the electrophysiology of SMA mice containing the missense mutation transgene. We specifically assayed the SMA mice containing each missense mutation transgene (SMND44V+/+, SMNT274I+/+ or SMNQ282+/+) for correction of CMAP, SMUP, MUNE and lack of fibrillations at 100 days of age. The corrected mice displayed no electrophysiological abnormalities as shown in Fig. 6. The CMAP values for each transgenic line (SMNQ282A: 43.1 ± 1.6 mV, SMNT274I: 34.9 ± 2.0 mV, SMND44V: 36.7 ± 2.7 mV, n = 10 for each group) were not different from the heterozygous SMN∆7 control mice (Smn+/−; SMN2+/+; SMN∆7+/+), (39.8 ± 1.1 mV, n = 10,P = 0.1) (Fig. 6A). The single motor unit potentials (SMUP) were no different between the rescued lines and the control (Fig. 6B, P = 0.2). CMAP and SMUP values were used to determine the MUNE for each line. MUNE values for each rescued missense mutation line (SMNQ282A: 308.2 ± 14.67, SMNT274I: 291.43 ± 20.96, SMND44V: 255 ± 24.93, n = 10 for each group) were also not significantly different from heterozygous SMN∆7 control mice (Control: 314.67 ± 26.67, n = 10,P = 0.3) (Fig. 6C). Moreover, no fibrillations were detected in any of the lines (data not shown). The control used in this analysis at 100 days of age is the SMN∆7 heterozygous mouse (Smn+/−; SMN2+/+; SMN∆7+/+) as SMN∆7 SMA mice (Smn−/−; SMN2+/+; SMN∆7+/+) do not survive past 14 days of age. We conclude that the oligomers formed between each of the missense SMN mutant proteins and wild-type FL-SMN proteins are fully capable of rescuing motor neuron function in SMA mice.
Figure 6.
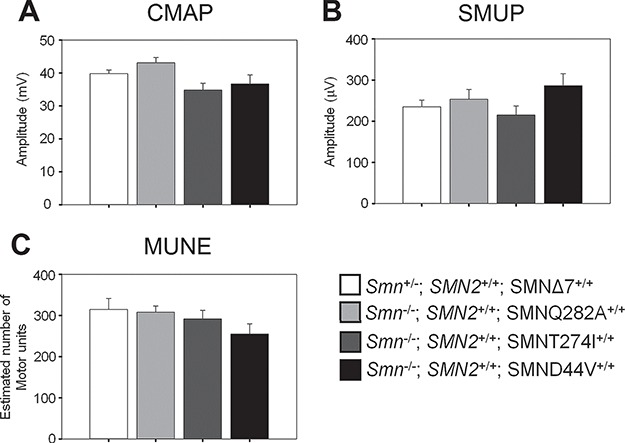
Motor unit function is completely rescued in mice containing mild SMN missense alleles and two copies of SMN2. Electrophysiological studies of motor function at 100 days of age show no significant difference motor function in the missense allele lines as compared to SMN∆7 heterozygous control mice. (A) CMAP in the missense allele lines (SMNQ282A: 43.1 ± 1.6 mV, SMNT274I: 34.9 ± 2.0 mV, SMND44V: 36.7 ± 2.7 mV, n = 10 for each group) were not different from the heterozygous SMN∆7 control mice (SMN2+/+, Smn+/−, SMN∆7+/+), (39.8 ± 1.1 mV, n = 10, P = 0.1) (B) The SMUP were no different between the missense allele lines and the control. (SMNQ282A: 253.7 ± 23.8 μV, SMNT274I: 215.3 ± 21.9 μV, SMND44V: 286.8 ± 29.0 μV, as compared to Control: 235.2 ± 16.1 μV, n = 10 for each group, P = 0.2) (C) MUNE (SMNQ282A: 308.2 ± 14.67, SMNT274I: 291.43 ± 20.96, SMND44V: 255 ± 24.93, n = 10 for each group) were also not significantly different from heterozygous SMN∆7 control mice (Control: 314.67 ± 26.67, n = 10,P = 0.3). SMNΔ7 SMA mice cannot be used as a control in this study because their average survival is 14 days. (Error bars = SEM).
Discussion
A number of SMA-linked missense mutations have been examined in biochemical assays in vitro, in tissue culture, in mice and in invertebrates including Drosophila and C. elegans. Missense mutations located in the C-terminus of SMN, such as SMNY272C, SMNG279V and the deletion of exon 7 in SMN∆7 disrupt the ability of SMN to oligomerize in vitro (12,14,28,29). These mutations are severe and can occur in cases of type 1 SMA with two copies of SMN2. SMN protein levels have been examined in lymphoblasts from a type 1 patient with the missense mutation SMNY272C that disrupts oligomerization and were found to be the same as in a typical type 1 patient with a SMN1 deletion (16), revealing the destabilizing effect of this specific mutation. Moreover, missense mutations that disrupt the ability to oligomerize or the loss of SMN exon7 results in rapid degradation of that SMN protein (13). The YG box encoded by exon 6 of SMN drives the oligomerization of SMN. A network of tyrosine-glycine packing between helices drives the formation of the SMN oligomers and severe missense mutations disrupt this driver of oligomerization resulting in monomeric SMN (28). The human SMN-Gemin2 dimer forms higher-order structures ranging from dimers to octamers (29). Interestingly, the SMN missense mutation SMNT274I that is located in the YG box and does not disrupt oligomerization of SMN is a mild SMA allele (28). Importantly, here we show that SMNT274I cannot rescue SMA mice unless SMN2 is present. This is completely consistent with SMNT274I oligomerizing and forming a heteromer with FL-SMN produced by SMN2. In all cases in the human population, the SMA causing missense mutations occur in SMN1 with intact SMN2 genes present (6,23). The loss of SMN2 occurs in about 10–15% of the normal population (47). Given this frequency one would expect that if SMN missense mutations had partial function on their own then SMA patients lacking SMN2 would be identified. To date, however, this has not yet been observed, suggesting that in humans the functional unit is the oligomer formed between the SMN produced by SMN2 and the mild missense mutation. Accordingly, all the mild alleles studied here and previously show the same feature when analyzed in mice: SMNA2G, SMNA111G, SMNT274I, SMND44V and SMNQ282A rescue Smn−/− mice containing SMN2 but never rescue Smn−/− mice in the absence of SMN2. We conclude that the mild SMN alleles have no function by themselves but retain their ability to oligomerize and function with FL-SMN produced by SMN2 (Fig. 7). This situation is similar to that of oligomeric enzymes such as argininosuccinate lyase (48) and propionyl-CoA carboxylase in which the interaction of two mutant proteins create an active site in the heteromer (49), resulting in a phenomenon known as allelic complementation.
Figure 7.
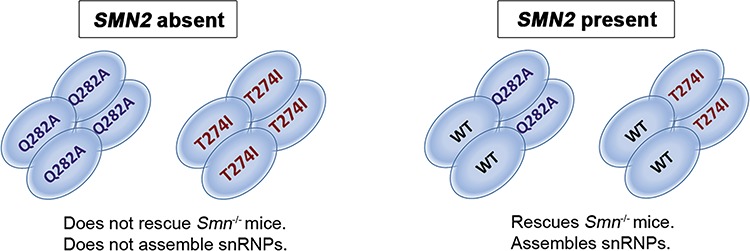
Model of SMN oligomerization and function in the presence of SMN missense mutation alleles. SMN missense mutations on their own cannot form a functional complex and fail to rescue Smn−/−; SMN2+/+ mice. In the presence of wild-type FL-SMN however, the missense mutation protein can complement the wild-type FL-SMN and oligomerize. Thus, the snRNP assembly as well as the survival of the mice is rescued. (A) An SMN homomeric complex is incapable of snRNP assembly and cannot rescue SMA mice. (B) Upon the addition of FL-SMN protein from SMN2, a heteromeric SMN complex is formed that can completely rescue the biochemical function of snRNP assembly and the survival of the mice.
In addition to furthering our understanding of the basic function of mild SMN missense alleles, these missense alleles can be used in a suppressor screen in mammalian cells to identify new potential therapeutic targets. Cells can be derived that possess a null Smn allele and a conditional Smn allele such as Smn exon 7 flanked by LoxP sites. Upon expression of Cre all functional SMN will be removed and only SMN protein from the SMN missense allele will be expressed. As these cells will not be viable, one can use mutagenesis to identify a second site suppressor that will allow the cells to survive. Identification of genes and mutations responsible for this suppression can both inform on the critical pathways used by SMN and identify new therapeutic targets.
In some instances, missense SMA mutations identified in humans occur in regions that are evolutionarily conserved in SMN from Drosophila or C. elegans (50). This is particularly true of the YG domain and the Tudor domain. In this case the equivalent missense mutations have been examined in these species (38,50,51). However, in C. elegans the missense mutant examined (CeSmnD27N) rescued viability and in Drosophila the missense mutations studied did not demonstrate the severity that was predicted from humans, perhaps due to the lack of critical as yet unidentified downstream targets of SMN (37,38). An additional important consideration when using model systems such as zebrafish, Drosophila and C. elegans is the contribution of maternal SMN from the egg. In Drosophila, an Smn null animal dies as a larva because the maternal SMN allows survival to this time point, but once maternal SMN is depleted the essential function of SMN is completely eliminated and death ensues (51,52). The early expression of a SMN missense mutation capable of oligomerization in the fly can result in protein oligomerization with maternal SMN (38). SMN missense mutations equivalent to SMND44V (dSmnD20V), SMNT274I (dSmn205I) and SMNI116F (dSmnI93F) as well as other mutations affecting oligomerization have been examined in Drosophila for their effect on development and survival, ranging from larval lethal to mature adult flies (38). However, these results do not parallel what is seen in SMA patients with one or two copies of SMN2 (6). The severe SMNI116F allele in fly (dSmnI93F) displayed similar rescue to wild-type Smn resulting in adult fly survival of 50% and 40%. Furthermore, the equivalent of SMNY130C (dSmnY107C), which occurs in type 3 patients with two copies of SMN2, results in less than 20% of adult fly survival despite being a mild allele in humans (38). Similarly, the mild mutation dSmnT205I, equivalent to SMNT274I, was less able to rescue than dSMNI93F, the equivalent of the severe SMNI116F allele in humans. The equivalent of the mild human mutation SMND44V in C. elegans (CeSmnD27N) has viability when bred without any alternative source of SMN (37). Thus, CeSmnD27N is viable and functional by itself however the equivalent allele in humans (SMND44V) does not rescue Smn−/− mice and only has function with small amounts of SMN from SMN2. Similar to SMND44V, other mild alleles such as SMNT274I, SMNA111G (25) and A2G (24), which are not conserved between species, all showed the same effect when expressed in mice with no rescue of Smn−/− mice unless SMN2 was present. To summarize, SMN missense alleles appear to behave differently in C. elegans and Drosophila when compared to mammals.
The exact reasons these mutants do not parallel the human situation in non-mammalian species is not known. It may be that some critical targets of SMN deficiency differ between mammals and invertebrates. Another non-mutually exclusive possibility is a higher requirement of SMN for splicing critical introns in mammals. These possibilities notwithstanding, our results indicate that mild SMN missense mutations are not partially functional on their own, but rather function in association with wild-type SMN produced by SMN2 in mammals. Furthermore, each functional oligomeric complex must contain at least one wild-type SMN monomer.
The SMNQ282A mutation is a synthetic mutation that cannot rescue axonal defects in Smn morpholino knockdown zebrafish (39). As such one would predict that SMA mice containing low levels of SMN protein produced by SMN2 and the SMNQ282A mutation would also have aberrant motor neuron function. We have previously reported that motor neuron dysfunction in SMA mice can be assayed by electrophysiological techniques, in a similar way to tests performed in SMA patients, by recording fibrillations on electromyography (EMG) as well as CMAP and MUNE reduction (45,46). These defects are rescued by expressing wild-type SMN in the motor neuron (53). We have found that all the SMN mild missense mutations (SMNQ282A, SMNT274I and SMND44V) assayed here in the presence of SMN2 completely rescue the CMAP and MUNE defects of SMA mice. We have also previously reported that there are no abnormal growth or pathfinding defects in SMA mice (54). It is possible that the SMN level in fish is so low that early developmental defects become apparent. Thus, we conclude that zebrafish and mice behave differently when SMN levels are reduced.
It has been notably difficult to create a mouse model of mild SMA as further addition of SMN copies to the severe SMA background results in significant correction of the disease phenotype (55). There are also differences in motor neuron deficits in different mouse models of SMA. For instance, the denervation of certain muscles such as the splenius capitis is marked in the SMN∆7 SMA mouse, but not in the Taiwanese SMA model (56,57). Caution is needed in interpretation of results using model mice where discernible muscle denervation is lacking. We suggest that the use of assays that monitor recovery of motor neuron survival, function and muscle denervation are optimal in analyzing SMA mice for correction.
SMA model mice are extremely sensitive to the amount of functional SMN complex that is present. For instance, on a pure C57Bl6 background four copies of SMN2 results in a completely normal mouse. Mice with three copies of SMN2 survive for a median of 15 to 22 days while a smaller number of animals have a normal phenotype (58). This contrasts with the human condition where three and four copies of SMN2 usually give rise to type 2 and type 3 SMA, respectively. Modeling in mice can be problematic in this regard. Osborne et al. have reported an allelic series with different FL-SMN levels arising from an SMN2 gene inserted into the mouse Smn locus. In this case the mice with two copies of the inserted SMN2 gene show a decrease in muscle size but neither loss of motor neurons nor a clear denervation phenotype. Again, this is unlike SMA in humans where even in type 3 or 4 SMA there is marked denervation (60–63). Type 3 SMA patients display motor neuron loss on autopsy (64) and imaging studies have revealed atrophy of the anterior horn (65). In the current study, the missense mutations SMNT274I, SMNQ282A and SMND44V all result in rescue in the presence of two copies of SMN2. There are three contributing reasons why these missense alleles result in a rescued mouse as opposed to a mild SMA phenotype. First, here there may be different developmental sensitivities to SMN levels between humans and mice. Specifically, the SMN complex is involved in formation of snRNPs that are important in splicing and as such developmental gene isoforms can be altered. Second, the mouse appears to be more sensitive to SMN levels where a relatively small SMN increase results in a normal life span and correction of any motor neuron phenotype. In this case the mild mutations interact with wild-type SMN produced by SMN2 to form more functional SMN complexes. This situation can overcome the requirement for SMN complexes in mouse motor neurons. Finally, in this study the missense mutation is overexpressed using transgenes thus there is a greater potential to produce a sufficient number of functional SMN complexes that are required to rescue the mice. The total number of functional SMN complexes present in the human or the mouse will determine the severity or rescue of the disease.
At least one major function of SMN is the assembly of RNPs, particularly spliceosomal snRNPs of the Sm class (32,66). It has also been suggested that SMN is important for the assembly of other RNA-protein complexes (30) and SMN has been shown to be required for the assembly of the U7 snRNP (31,67). However, it remains unclear which specific function(s) of SMN are critical to development of SMA. Given that SMN is important in snRNP assembly, reduced splicing of certain genes with important functions in neurons is a likely downstream effect of SMN deficiency that may contribute to SMA pathology (68–71). Further, species-specific differences in the efficiency of intron removal and their sensitivity to SMN reduction could be relevant elements distinguishing the functional consequences of low SMN in rodents versus humans.
In summary, all mild SMN missense SMA alleles examined to date (SMNA111G, SMNA2G, SMNT274I, SMND44V) and the synthetic allele SMNQ282A do not rescue an Smn null mouse on their own. However, in the presence of SMN2 all these mild mutations can rescue the SMA mouse by complementing SMN2 and rescuing snRNP assembly. Furthermore, the SMN missense mutations SMND44V, SMNT274I and SMNQ282A in the presence of SMN2 have normal motor neuron function and do not show any electrophysiological defects. In contrast, severe SMN missense mutations, such as SMNI116F, do not complement SMN2 and often result in a SMN protein that is quickly degraded (13,16). We suggest that the homomeric complexes containing only mild SMN missense mutations have no function in the absence of wild-type SMN. However heteromeric SMN complexes containing small amounts of wild-type FL-SMN and a missense mutation are functional and capable of rescuing the SMA motor neuron phenotype and survival in mice.
Materials and Methods
Generation of transgenic mice and breeding
The SMNQ282A, SMND44V, SMNT274I and SMNI116F transgenes were made in a similar manner. The previous vector containing SMNA111G (25) driven by a 4.1 kb SMN promoter (SMNp) fragment was cut with BamHI and EcoRI to release the SMN fragment and the pcDNA3 vector with the promoter band isolated. SMN mutant constructs were generated by site-directed mutagenesis using the QuikChange (Stratagene) kit. These new SMN mutant constructs were subcloned and sequenced to verify the mutation. The SMN complementary DNA (cDNA) construct can be released with BamHI and EcoRI as described previously described and subcloned into the pcDNA vector with the promoter. The clones were confirmed by PCR amplification restriction digestion and sequencing. 55 μg of each SMNp-SMN mutant (T274I, Q282A, I116F and D44V) pcDNA3 construct was linearized by cutting with PvuI and DraIII and sent to The Ohio State (GEMMC) for microinjection into FVB/N oocytes. Mice were screened by PCR for both SMNp and the SMN cDNA using three different sets of primers to ensure the transgene was intact. Allele specific primers SMNpF 5ʹTGGAGTTCGAGACGAGGCCTAAGC and SMN1.2R 5ʹCAGAATCATCGCTCTGGCCTGTGCC, SMN1F 5ʹGCGGCGGCAGTGGTGGCGGC and SMN4R 5ʹTGGAGCAGATTTGGGCTTGA, SMNex4F 5ʹGTGAGAACTCCAGGTCTCCTGG and SMN8R 5ʹCTACAACACCCTTCTCACAG. The presence of SMN2 and mouse Smn was screened as previously described (72). Founders were then backcrossed to SMA carrier mice (SMN2+/+, Smn+/−) or to Smn+/− lacking SMN2. Mice were weighed every day for 28 days until weaning and then once per week until 250 days. Survival was monitored daily and any deaths were noted. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the University Laboratory Animal Resources at The Ohio State University. Our protocol was approved by the Institutional Animal Care and Use Committee under Permit Number 2008A0089.
Genotyping of SMN missense allele transgenes
To genotype each transgenic line the following primers and restriction enzymes were used A111G: FP 5ʹGCTGTGGCTTCATTTAAGCAT and RP 5ʹAGTAGATCGGACAGATTTTGCT (59° annealing) followed by a KpnI digest. If the mutation is present the 290 bp will produce 117, 175 bp fragments. A2G: FP 5ʹTGGAGTTCGAGACGAGGCCTAAGC and RP 5ʹCAGAATCATCGCTCTGGCCTGTGCC (65° annealing) followed by a FokI digest. If the mutation is present the 438 bp product will produce 404, 34 bp fragments. D44V: FP 5ʹGCGGCGGCAGTGGTGGCGGC and RP 5ʹAGTAGATCGGACAGATTTTGCT (59° annealing). A digest was not used for this transgene. I116F: FP 5ʹGCGGCGGCAGTGGTGGCGGC and RP 5ʹAGTAGATCGGACAGATTTTGCT (59° annealing) followed by a MfeI digest. If the mutation is present the enzyme will not cut resulting in a 412 bp band. If the mutation is absent the product sizes are 331 and 81 bp. T274I: FP 5ʹATTTCATGGTACATGAGTGGCTATAATA and RP 5ʹCGCTTCACATTCCAGATCTGTCTG (57° annealing) followed by an SspI digest. If the mutation is present the 168 bp product will produce 141, 27 bp fragments. Q282A: FP 5ʹGAGTGGCTATCATACTGGCTATTATATGGGTTTCTGA and RP5ʹCGCTTCACATTCCAGATCTGTCTG (59° annealing) followed by a DdeI digest. If the mutation is present the 154 bp product will produce 120, 34 bp fragments. SMN2 and Smn were genotyped as previously described (72).
RT-PCR for identification of lines with transgene expression
Brain and spinal cord samples from 28 to 32-day-old mice were flash frozen in liquid nitrogen and stored at −80°C. 30 mg of tissue was homogenized in 500 μl of TRIzol, sonicated and incubated at room temperature for 5 min. 200 μl of chloroform was added to the sample and shaken vigorously. After incubation at room temperature for 2–3 min, samples were centrifuged for 15 min at 12,000 g at 4°C. The top layer was transferred to a new tube, and 1 volume of 70% ethanol was added. The QIAGEN RNeasy Mini Kit manufacturer’s protocol was followed from this step onward. The RNA was treated with the Turbo DNA-Free kit (Ambion) as per manufacturer’s protocol to remove any genomic DNA contamination. 2 μg of RNA was added to 250 ng of pdN6 random hexamers (GE Healthcare) in nuclease-free water. First strand cDNA synthesis was carried out by incubating this mix at 70°C for 10 min followed by cooling in ice for 10 min. A mix containing 2.5 μl of 10 mM dNTP (GE Healthcare), 0.625 μl of RNaseOUT (Invitrogen), 5 μl of 5x AMV Reverse Transcriptase Buffer (NEB) and 1 μl of AMV Reverse Transcriptase (NEB) was added to the tube. Similar control reactions were performed without the AMV Reverse Transcriptase. Samples were then incubated at 42°C for 1 hour followed by reaction termination at 85°C for 5 min. SMN was detected by PCR using SMNex4F 5ʹGTGAGAACTCCAGGTCTCCTGG and SMN8R 5ʹCTACAACACCCTTCTCACAG. Cyclophilin was used as control (CycloF 5ʹAGACGCCGCTGTCTCTTTTCG and CycloR 5’CCACAGTCGGAGATGGTGATC).
Transgene copy number quantification by ddPCR
Transgene copy number was determined using ddPCR (QX200 Bio-Rad) on tail DNA from a minimum of 10 heterozygous mice for each transgenic line. Tail biopsy DNA was diluted to 6 ng/μl in 500 μl of water. DNA was then digested using PstI to separate tandem copies of the transgene. DdPCR was performed using 1 μl of SMN-FL-FAM (FP 5ʹCAAAAAGAAGGAAGGTGCTCA, RP 5ʹTCCAGATCTGTCTGATCGTTTC, 5ʹFAM-TTAAGGAGAAATGCTGGCATAGAGCAGCAC-MGB), 1 μl mSmn-Intron1-VIC control (FP 5ʹTTTGGTCCTGTGTGACTGTGA, RP 5ʹAATCTGGAGCTTTCCAATGGCT, 5ʹVIC-AGGCTGGCTGAAGCAAGGCAACCAGATA-MGB), 10 μl ddPCR master mix (Bio-Rad) and 6 ng of digested tail DNA. The number of copies for each heterozygous transgene insertion was measured by dividing the SMN copy number by the mouse Smn intron one copy number. Thus all values are relative to two copies of Smn.
SMN cDNA quantification by ddPCR
Brain and spinal cord tissue were isolated from 4-day-old mice with the appropriate genotype. The tissue was flash frozen in liquid nitrogen and RNA was isolated as described above. cDNA was synthesized as described above. SMN cDNA quantification was obtained with ddPCR (QX200 BioRad) as previously described (73). FL-SMN was detected with SMN-FL-FAM (FP 5ʹCAAAAAGAAGGAAGGTGCTCA, RP 5ʹtccagatctgtctgatcgtttc, 5ʹFAM-TTAAGGAGAAATGCTGGCATAGAGCAGCAC-MGB), and normalized to the expression of YWHAZ12 (FP 5’AGGGTGACTACTACCGTTACTT, RP 5ʹTGCTTCTTGGTATGCTTGCT, 5ʹVIC-AGGTTGCTGCTGGTGATGACAAGA-MGB) in a multiplex assay.
SMN protein expression quantification by ELISA
SMN protein was measured at PharmOptima (Portage, MI) using the company’s proprietary electrochemiluminescence immunoassay based on Meso Scale Discovery technology. The assay is a quantitative sandwich immunoassay using mouse monoclonal antibody 2B1 (74) as the capture antibody and rabbit polyclonal anti-SMN antibody (Protein Tech, Cat. No. 11708-1-AP) labeled with a SULFO-TAG™ for detection. SMN levels are determined relative to a standard curve measured with recombinant SMN protein (Enzo Life Sciences, Cat. No. ADI-NBP-201-050) as calibrator. The dynamic range of the assay is 10 pg/ml to 20,000 pg/ml. Assay plates were read using a Meso Scale 6,000 sector imager.
Analysis of snRNP assembly activity
SMN protein was measured by western blot using SMN clone 8 (BD Transduction Laboratories, 610646, 1:10 000 dilution) and Tubulin DM1A (Sigma T9026, 1:10 000 dilution). In vitro snRNP assembly reactions with radioactive U1 snRNA and 4-days-of-age mouse spinal cord extracts were carried out essentially as described previously (42). Following addition of heparin and urea to a final concentration of 5 mg/ml and 2 M, respectively, reactions were incubated for 15 min. at room temperature and then analyzed by immunoprecipitation with anti-SmB antibodies SmB, clone 18F6 (75) was used for the IP assay. Immunoprecipitations were carried out in RSB-500 buffer (500 mM NaCl, 10 mM Tris–HCl pH 7.4, 2.5 mM MgCl2) containing 0.1% NP40, EDTA-free protease inhibitor cocktail (Roche) and phosphatase inhibitors (50 mM NaF, 0.2 mM Na3VO4) for 2 h at 4°C. Immunoprecipitated U1 snRNA was analyzed by electrophoresis on denaturing polyacrylamide gels and autoradiography. Quantification was performed using a STORM 860 Phosphorimager (Molecular Dynamics) and the ImageQuant version 4.2 software.
Electrophysiological analysis of motor unit function
Electrophysiological motor unit function was assessed by recording CMAP amplitude, and MUNE from the triceps surae muscle following stimulation of the sciatic nerve as previously described (45,46,76). Needle electrode electromyographic analysis for abnormal spontaneous activity (fibrillations) was performed in the triceps surae as previously described (45,46). Fibrillations are electrophysiological evidence of denervation of muscle fibers that occurs in SMA.
Statistical analysis
Chi-square analysis was performed using either a 4 x 2 contingency table with 3 degrees of freedom or a 6 x 2 contingency table with 5 degrees of freedom. Mice that were homozygous or heterozygous for the SMN missense transgene were grouped together. At a confidence interval of P < 0.05 we fail to reject the null hypothesis and the observed and expected outcomes are statistically different. A Fisher’s exact test of r x c contingency tables was also performed to confirm significance using the online calculator found at http://www.physics.csbsju.edu/stats/ (data not shown). Electrophysiological motor unit function test results were analyzed using one-way analysis of variance (ANOVA) and transgene cDNA and protein expression was analyzed using a T-test comparing groups to the SMN∆7 SMA control group with SigmaPlot. Growth curve analysis was analyzed with the compareGrowthCurves function from the Statistical Modeling package statmod available from the R Project for Statistical Computing: http://www.r-project.org (77,78).
Supplementary Material
Acknowledgements
We would like to thank Xiaohui Li for technical assistance and Anton J. Blatnik III for many interesting discussions. We also would like to thank Dr Xin-An Pu from the The Ohio State (GEMMC) for performing oocyte injections to create the transgenes reported.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health (R01NS0385650 to A.H.M.B., F31NS079002 to S.T., R01NS102451 to L.P.); The Marshall Heritage Foundation, Muscular Dystrophy Association (60027456 to A.H.M.B.); CureSMA to A.H.M.B. Transgenic mouse creation was supplemented by the National Institutes of Health (P30NS04578).
References
- 1. Roberts D.F., Chavez J. and Court S.D. (1970) The genetic component in child mortality. Arch. Dis. Child., 45, 33–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sugarman E.A., Nagan N., Zhu H., Akmaev V.R., Zhou Z., Rohlfs E.M., Flynn K., Hendrickson B.C., Scholl T., Sirko-Osadsa D.A. et al. (2012) Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of >72,400 specimens. Eur. J. Hum. Genet., 20, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pearn J. (1978) Incidence, prevalence, and gene frequency studies of chronic childhood spinal muscular atrophy. J. Med. Genet., 15, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pearn J.H. (1973) The gene frequency of acute Werdnig-Hoffmann disease (SMA type 1). A total population survey in North-East England. J. Oral Surg., 10, 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M. et al. (1995) Identification and characterization of a spinal muscular atrophy-determining gene. Cell, 80, 155–165. [DOI] [PubMed] [Google Scholar]
- 6. Burghes A.H. and Beattie C.E. (2009) Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci., 10, 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lorson C.L., Hahnen E., Androphy E.J. and Wirth B. (1999) A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A., 96, 6307–6311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H. and McPherson J.D. (1999) A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet., 8, 1177–1183. [DOI] [PubMed] [Google Scholar]
- 9. Cartegni L. and Krainer A.R. (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet., 30, 377–384. [DOI] [PubMed] [Google Scholar]
- 10. Kashima T. and Manley J.L. (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat. Genet., 34, 460–463. [DOI] [PubMed] [Google Scholar]
- 11. Gennarelli M., Lucarelli M., Capon F., Pizzuti A., Merlini L., Angelini C., Novelli G. and Dallapiccola B. (1995) Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem. Biophys. Res. Commun., 213, 342–348. [DOI] [PubMed] [Google Scholar]
- 12. Lorson C.L., Strasswimmer J., Yao J.M., Baleja J.D., Hahnen E., Wirth B., Le T., Burghes A.H. and Androphy E.J. (1998) SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet., 19, 63–66. [DOI] [PubMed] [Google Scholar]
- 13. Burnett B.G., Munoz E., Tandon A., Kwon D.Y., Sumner C.J. and Fischbeck K.H. (2009) Regulation of SMN protein stability. Mol. Cell. Biol., 29, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pellizzoni L., Charroux B. and Dreyfuss G. (1999) SMN mutants of spinal muscular atrophy patients are defective in binding to snRNP proteins. Proc. Natl. Acad. Sci. U.S.A., 96, 11167–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coovert D.D., Le T.T., McAndrew P.E., Strasswimmer J., Crawford T.O., Mendell J.R., Coulson S.E., Androphy E.J., Prior T.W. and Burghes A.H. (1997) The survival motor neuron protein in spinal muscular atrophy. Hum. Mol. Genet., 6, 1205–1214. [DOI] [PubMed] [Google Scholar]
- 16. Lefebvre S., Burlet P., Liu Q., Bertrandy S., Clermont O., Munnich A., Dreyfuss G. and Melki J. (1997) Correlation between severity and SMN protein level in spinal muscular atrophy. Nat. Genet., 16, 265–269. [DOI] [PubMed] [Google Scholar]
- 17. McAndrew P.E., Parsons D.W., Simard L.R., Rochette C., Ray P.N., Mendell J.R., Prior T.W. and Burghes A.H. (1997) Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am. J. Hum. Genet., 60, 1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arnold W.D. and Burghes A.H. (2013) Spinal muscular atrophy: development and implementation of potential treatments. Ann. Neurol., 74, 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prior T.W. and Nagan N. (2016) Spinal muscular atrophy: overview of molecular diagnostic approaches. Curr. Protoc. Hum. Genet., 88, Unit 9.27. [DOI] [PubMed] [Google Scholar]
- 20. Jedrzejowska M., Gos M., Zimowski J.G., Kostera-Pruszczyk A., Ryniewicz B. and Hausmanowa-Petrusewicz I. (2014) Novel point mutations in survival motor neuron 1 gene expand the spectrum of phenotypes observed in spinal muscular atrophy patients. Neuromuscul. Disord., 24, 617–623. [DOI] [PubMed] [Google Scholar]
- 21. Sun Y., Grimmler M., Schwarzer V., Schoenen F., Fischer U. and Wirth B. (2005) Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum. Mutat., 25, 64–71. [DOI] [PubMed] [Google Scholar]
- 22. Parsons D.W., McAndrew P.E., Iannaccone S.T., Mendell J.R., Burghes A.H. and Prior T.W. (1998) Intragenic telSMN mutations: frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am. J. Hum. Genet., 63, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burghes A.H.M. and McGovern V.L. (2017) Molecular and Cellular Therapies for Motor Neuron Diseases. Academic Press, London, UK. [Google Scholar]
- 24. Monani U.R., Pastore M.T., Gavrilina T.O., Jablonka S., Le T.T., Andreassi C., DiCocco J.M., Lorson C., Androphy E.J., Sendtner M. et al. (2003) A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J. Cell. Biol., 160, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Workman E., Saieva L., Carrel T.L., Crawford T.O., Liu D., Lutz C., Beattie C.E., Pellizzoni L. and Burghes A.H. (2009) A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum. Mol. Genet., 18, 2215–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shpargel K.B. and Matera A.G. (2005) Gemin proteins are required for efficient assembly of Sm-class ribonucleoproteins. Proc. Natl. Acad. Sci. U.S.A., 102, 17372–17377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Brahms H., Meheus L., Brabandere V., Fischer U. and Luhrmann R. (2001) Symmetrical dimethylation of arginine residues in spliceosomal Sm protein B/B' and the Sm-like protein LSm4, and their interaction with the SMN protein. RNA, 7, 1531–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin R., Gupta K., Ninan N.S., Perry K. and Van Duyne G.D. (2012) The survival motor neuron protein forms soluble glycine zipper oligomers. Structure, 20, 1929–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gupta K., Martin R., Sharp R., Sarachan K.L., Ninan N.S. and Van Duyne G.D. (2015) Oligomeric properties of survival motor neuron·Gemin2 complexes. J. Biol. Chem., 290, 20185–20199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li D.K., Tisdale S., Lotti F. and Pellizzoni L. (2014) SMN control of RNP assembly: from post-transcriptional gene regulation to motor neuron disease. Semin. Cell. Dev. Biol., 32, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tisdale S., Lotti F., Saieva L., Van Meerbeke J.P., Crawford T.O., Sumner C.J., Mentis G.Z. and Pellizzoni L. (2013) SMN is essential for the biogenesis of U7 small nuclear ribonucleoprotein and 3′-end formation of histone mRNAs. Cell. Rep., 5, 1187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pellizzoni L., Yong J. and Dreyfuss G. (2002) Essential role for the SMN complex in the specificity of snRNP assembly. Science, 298, 1775–1779. [DOI] [PubMed] [Google Scholar]
- 33. Meister G., Eggert C. and Fischer U. (2002) SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol., 12, 472–478. [DOI] [PubMed] [Google Scholar]
- 34. Pillai R.S., Grimmler M., Meister G., Will C.L., Lührmann R., Fischer U. and Schümperli D. (2003) Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev, 17, 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hao le T., Duy P.Q., An M., Talbot J., Iyer C.C., Wolman M. and Beattie C.E. (2017) HuD and the survival motor neuron protein interact in motoneurons and are essential for motoneuron development, function, and mRNA regulation. J. Neurosci., 37, 11559–11571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Donlin-Asp P.G., Fallini C., Campos J., Chou C.C., Merritt M.E., Phan H.C., Bassell G.J. and Rossoll W. (2017) The survival of motor neuron protein acts as a molecular chaperone for mRNP assembly. Cell Rep., 18, 1660–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sleigh J.N., Buckingham S.D., Esmaeili B., Viswanathan M., Cuppen E., Westlund B.M. and Sattelle D.B. (2011) A novel Caenorhabditis elegans allele, smn-1(cb131), mimicking a mild form of spinal muscular atrophy, provides a convenient drug screening platform highlighting new and pre-approved compounds. Hum. Mol. Genet., 20, 245–260. [DOI] [PubMed] [Google Scholar]
- 38. Praveen K., Wen Y., Gray K.M., Noto J.J., Patlolla A.R., Van Duyne G.D. and Matera A.G. (2014) SMA-causing missense mutations in survival motor neuron (Smn) display a wide range of phenotypes when modeled in Drosophila. PLoS Genet., 10, e1004489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carrel T.L., McWhorter M.L., Workman E., Zhang H., Wolstencroft E.C., Lorson C., Bassell G.J., Burghes A.H. and Beattie C.E. (2006) Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J. Neurosci., 26, 11014–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Le T.T., McGovern V.L., Alwine I.E., Wang X., Massoni-Laporte A., Rich M.M. and Burghes A.H. (2011) Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet., 20, 3578–3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J. and Burghes A.H. (2005) SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet., 14, 845–857. [DOI] [PubMed] [Google Scholar]
- 42. Gabanella F., Butchbach M.E., Saieva L., Carissimi C., Burghes A.H. and Pellizzoni L. (2007) Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE, 2, e921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tisdale S. and Pellizzoni L. (2015) Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci., 35, 8691–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Monani U.R., Sendtner M., Coovert D.D., Parsons D.W., Andreassi C., Le T.T., Jablonka S., Schrank B., Rossoll W., Prior T.W. et al. (2000) The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum. Mol. Genet., 9, 333–339. [DOI] [PubMed] [Google Scholar]
- 45. Arnold W.D., Porensky P.N., McGovern V.L., Iyer C.C., Duque S., Li X., Meyer K., Schmelzer L., Kaspar B.K., Kolb S.J. et al. (2014) Electrophysiological biomarkers in spinal muscular atrophy: preclinical proof of concept. Ann. Clin. Transl. Neurol., 1, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Arnold W., McGovern V.L., Sanchez B., Li J., Corlett K.M., Kolb S.J., Rutkove S.B. and Burghes A.H. (2016) The neuromuscular impact of symptomatic SMN restoration in a mouse model of spinal muscular atrophy. Neurobiol. Dis., 87, 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prior T.W., Nagan N., Sugarman E.A., Batish S.D. and Braastad C. (2011) Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med., 13, 686–694. [DOI] [PubMed] [Google Scholar]
- 48. Yu B. and Howell P.L. (2000) Intragenic complementation and the structure and function of argininosuccinate lyase. Cell. Mol. Life. Sci., 57, 1637–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rodriguez-Pombo P., Perez-Cerda C., Perez B., Desviat L.R., Sanchez-Pulido L. and Ugarte M. (2005) Towards a model to explain the intragenic complementation in the heteromultimeric protein propionyl-CoA carboxylase. Biochim. Biophys. Acta, 1740, 489–498. [DOI] [PubMed] [Google Scholar]
- 50. Talbot K., Ponting C.P., Theodosiou A.M., Rodrigues N.R., Surtees R., Mountford R. and Davies K.E. (1997) Missense mutation clustering in the survival motor neuron gene: a role for a conserved tyrosine and glycine rich region of the protein in RNA metabolism? Hum. Mol. Genet., 6, 497–500. [DOI] [PubMed] [Google Scholar]
- 51. Chan Y.B., Miguel-Aliaga I., Franks C., Thomas N., Trulzsch B., Sattelle D.B., Davies K.E. and Heuvel M. (2003) Neuromuscular defects in a Drosophila survival motor neuron gene mutant. Hum Mol Genet, 12, 1367–1376. [DOI] [PubMed] [Google Scholar]
- 52. Chang H.C., Dimlich D.N., Yokokura T., Mukherjee A., Kankel M.W., Sen A., Sridhar V., Fulga T.A., Hart A.C., Van Vactor D. et al. (2008) Modeling spinal muscular atrophy in Drosophila. PLoS One, 3, e3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McGovern V.L., Iyer C.C., Arnold W.D., Gombash S.E., Zaworski P.G., Blatnik A.J., Foust K.D. and Burghes A.H. (2015) SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNΔ7 mouse model of SMA. Hum. Mol. Genet., 24, 5524–5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McGovern V.L., Gavrilina T.O., Beattie C.E. and Burghes A.H. (2008) Embryonic motor axon development in the severe SMA mouse. Hum. Mol. Genet., 17, 2900–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Burghes A.H.M., DiDonato C.J., McGovern V.L. and Arnold W.D. (2017) Mammalian Models of Spinal Muscular Atrophy. Academic Press, London, UK. [Google Scholar]
- 56. Lin T.L., Chen T.H., Hsu Y.Y., Cheng Y.H., Juang B.T. and Jong Y.J. (2016) Selective neuromuscular denervation in Taiwanese severe SMA mouse can be reversed by morpholino antisense oligonucleotides. PLoS One, 11, e0154723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ling K.K., Gibbs R.M., Feng Z. and Ko C.P. (2012) Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet., 21, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Michaud M., Arnoux T., Bielli S., Durand E., Rotrou Y., Jablonka S., Robert F., Giraudon-Paoli M., Riessland M., Mattei M.G. et al. (2010) Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol. Dis., 38, 125–135. [DOI] [PubMed] [Google Scholar]
- 59. Osborne M., Gomez D., Feng Z., McEwen C., Beltran J., Cirillo K., El-Khodor B., Lin M.Y., Li Y., Knowlton W.M. et al. (2012) Characterization of behavioral and neuromuscular junction phenotypes in a novel allelic series of SMA mouse models. Hum. Mol. Genet., 21, 4431–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mazzei R., Gambardella A., Conforti F.L., Magariello A., Patitucci A., Gabriele A.L., Sprovieri T., Labate A., Valentino P., Bono F. et al. (2004) Gene conversion events in adult-onset spinal muscular atrophy. Acta Neurol. Scand., 109, 151–154. [DOI] [PubMed] [Google Scholar]
- 61. Clermont O., Burlet P., Lefebvre S., Bürglen L., Munnich A. and Melki J. (1995) SMN gene deletions in adult-onset spinal muscular atrophy. Lancet, 346, 1712–1713. [DOI] [PubMed] [Google Scholar]
- 62. Brahe C., Servidei S., Zappata S., Ricci E., Tonali P. and Neri G. (1995) Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet, 346, 741–742. [DOI] [PubMed] [Google Scholar]
- 63. Pearn J.H., Hudgson P. and Walton J.N. (1978) A clinical and genetic study of spinal muscular atrophy of adult onset: the autosomal recessive form as a discrete disease entity. Brain, 101, 591–606. [DOI] [PubMed] [Google Scholar]
- 64. Kuru S., Sakai M., Konagaya M., Yoshida M., Hashizume Y. and Saito K. (2009) An autopsy case of spinal muscular atrophy type III (Kugelberg-Welander disease). Neuropathology, 29, 63–67. [DOI] [PubMed] [Google Scholar]
- 65. El Mendili M.M., Lenglet T., Stojkovic T., Behin A., Guimarães-Costa R., Salachas F., Meininger V., Bruneteau G., Le Forestier N., Laforêt P. et al. (2016) Cervical spinal cord atrophy profile in adult SMN1-linked SMA. PLoS One, 11, e0152439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Meister G. and Fischer U. (2002) Assisted RNP assembly: SMN and PRMT5 complexes cooperate in the formation of spliceosomal UsnRNPs. Embo. J., 21, 5853–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pillai R.S., Grimmler M., Meister G., Will C.L., Luhrmann R., Fischer U. and Schumperli D. (2003) Unique Sm core structure of U7 snRNPs: assembly by a specialized SMN complex and the role of a new component, Lsm11, in histone RNA processing. Genes Dev., 17, 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao le T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D. et al. (2012) An SMN-dependent U12 splicing event essential for motor circuit function. Cell, 151, 440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M. and Dreyfuss G. (2008) SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell, 133, 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang Z., Pinto A.M., Wan L., Wang W., Berg M.G., Oliva I., Singh L.N., Dengler C., Wei Z. and Dreyfuss G. (2013) Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. U.S.A., 110, 19348–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Baumer D., Lee S., Nicholson G., Davies J.L., Parkinson N.J., Murray L.M., Gillingwater T.H., Ansorge O., Davies K.E. and Talbot K. (2009) Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet., 5, e1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gavrilina T.O., McGovern V.L., Workman E., Crawford T.O., Gogliotti R.G., DiDonato C.J., Monani U.R., Morris G.E. and Burghes A.H. (2008) Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet., 17, 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Porensky P.N., Mitrpant C., McGovern V.L., Bevan A.K., Foust K.D., Kaspar B.K., Wilton S.D. and Burghes A.H. (2012) A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet., 21, 1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Liu Q. and Dreyfuss G. (1996) A novel nuclear structure containing the survival of motor neurons protein. Embo. J., 15, 3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 75. Carissimi C., Saieva L., Gabanella F. and Pellizzoni L. (2006) Gemin8 is required for the architecture and function of the survival motor neuron complex. J. Biol. Chem., 281, 37009–37016. [DOI] [PubMed] [Google Scholar]
- 76. Arnold W.D., Sheth K.A., Wier C.G., Kissel J.T., Burghes A.H. and Kolb S.J. (2015) Electrophysiological motor enit number estimation (MUNE) measuring compound muscle action potential (CMAP) in mouse hindlimb muscles. J. Vis. Exp., 103, 52899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Elso C.M., Roberts L.J., Smyth G.K., Thomson R.J., Baldwin T.M., Foote S.J. and Handman E. (2004) Leishmaniasis host response loci (lmr1-3) modify disease severity through a Th1/Th2-independent pathway. Genes Immun., 5, 93–100. [DOI] [PubMed] [Google Scholar]
- 78. Baldwin T., Sakthianandeswaren A., Curtis J.M., Kumar B., Smyth G.K., Foote S.J. and Handman E. (2007) Wound healing response is a major contributor to the severity of cutaneous leishmaniasis in the ear model of infection. Parasite Immunol., 29, 501–513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.