

Abstract
Duchenne muscular dystrophy (DMD), caused by mutations in the dystrophin gene, leads to severe muscle wasting and eventual death of the afflicted individuals, primarily due to respiratory failure. Deficit in myofiber regeneration, potentially due to an exhaustion of satellite cells, is one of the major pathological features of DMD. Myeloid differentiation primary response 88 (MyD88) is an adaptor protein that mediates activation of various inflammatory pathways in response to signaling from Toll-like receptors and interleukin-1 receptor. MyD88 also regulates cellular survival, proliferation and differentiation in a cell-autonomous manner. However, the role of MyD88 in satellite stem cell homeostasis and function in dystrophic muscle remains unknown. In this study, we demonstrate that tamoxifen-inducible deletion of MyD88 in satellite cells causes loss of skeletal muscle mass and strength in the mdx mouse model of DMD. Satellite cell-specific deletion of MyD88 inhibits myofiber regeneration and stimulates fibrogenesis in dystrophic muscle of mdx mice. Deletion of MyD88 also reduces the number of satellite cells and inhibits their fusion with injured myofibers in dystrophic muscle of mdx mice. Ablation of MyD88 in satellite cells increases the markers of M2 macrophages without having any significant effect on M1 macrophages and expression of inflammatory cytokines. Finally, we found that satellite cell-specific deletion of MyD88 leads to aberrant activation of Notch and Wnt signaling in skeletal muscle of mdx mice. Collectively, our results demonstrate that MyD88-mediated signaling in satellite cells is essential for the regeneration of injured myofibers in dystrophic muscle of mdx mice.
Introduction
Muscular dystrophy is a group of genetic disorders caused by mutations in genes encoding for various components of extracellular matrix, cytoskeletal, cytoplasmic enzymes and nuclear membrane proteins (1,2). Duchenne muscular dystrophy (DMD) is the most common X-linked lethal disorder, which afflicts ~1 in 5000 male births (3,4). DMD is caused by defects in dystrophin, a cytoskeletal protein, which provides mechanical reinforcement to the sarcolemma and also serves as a signaling module in skeletal muscle (5,6). Dystrophin-deficient myofibers undergo chronic muscle injury. During early stages of the disease progression, injury is counterbalanced by the ability of muscle fibers to regenerate. However, this capacity to regenerate is subsequently lost, potentially due to the exhaustion of muscle progenitor cells, leading to progressive muscle weakness and development of interstitial fibrosis that confines the patient to a wheelchair by the early teens and leads to death typically by the mid-twenties (1,6,7). In fact, augmentation of the regenerative potential of endogenous muscle stem cells or transplantation of muscle stem cells into dystrophic muscle has been suggested as one of the promising therapeutic approaches for DMD patients (8,9).
Skeletal muscle regeneration in adults is attributed to the presence of satellite cells, which reside between the sarcolemma and the basal lamina in a relatively dormant metabolic state (10). Upon injury to skeletal muscle, satellite cells become activated and undergo several rounds of cell division before differentiating into myoblasts, which ultimately fuse with injured myofibers to accomplish regeneration (10–12). Studies using genetic mouse models have shown that several signaling pathways regulate satellite cell fate and their ability to repair injured myofibers. For example, activation of Notch signaling promotes satellite cell self-renewal and inhibits their differentiation through repressing the levels of MyoD (13–17). In contrast, the activation of canonical Wnt signaling promotes differentiation and fusion of satellite cells to injured myofibers of mice (18,19). Interestingly, chronic activation of canonical Wnt signaling can lead to the development of interstitial fibrosis in skeletal muscle of mice (20). In addition, mitogen-activated protein kinases (MAPKs), especially ERK1/2 and JNK1, also play an important role in self-renewal and proliferation of satellite cells both in vitro and in vivo (21–23). It is notable that in response to acute injury, myofiber regeneration in animals proceeds at a finite rate through sequential activation and deactivation of specific signaling pathways (10). However, due to the chronic nature of myofiber injury, dystrophic muscle presents a highly complex microenvironment where a number of factors, including inflammatory immune cells, growth factors, and fibrosis can influence the regenerative potential of satellite stem cells (1). Indeed, due to the complexity of the process, the signaling mechanisms that regulate the regenerative potential of satellite cells in dystrophic muscle remain poorly understood.
Myeloid differentiation primary response 88 (MyD88) is the canonical adaptor protein that mediates numerous biologically important signal transduction pathways in innate immunity downstream of members of the Toll-like receptor (TLR) and interleukin-1 receptor (IL-1R) families (24–26). MyD88 links IL-1R–associated kinase (IRAK) family kinases through protein–protein interactions. Activation of IRAKs leads to a variety of functional outputs, including the activation of MAPKs and pro-inflammatory transcription factors such as nuclear factor-kappa B (NF-κB) and activator protein 1, making MyD88 a central regulator of inflammatory pathways (26–29). Because of the pivotal role of inflammation in the pathogenesis of DMD, a few previous studies have investigated the effect of ablation of MyD88 and/or specific TLRs in dystrophinopathy. It has been reported that whole body deletion of MyD88 in mdx (a mouse model of DMD) mice does not have an initial effect on the pathology, but ameliorates myopathy to some extent at later stages, potentially due to the attenuation of the inflammatory response (30). A more recent study has shown that the genetic ablation of TLR4 in mdx mice improves muscle histopathology and reduces macrophage infiltration and gene expression of pro-inflammatory molecules (31). While these studies suggest beneficial effects of inhibition of TLRs/MyD88 in DMD, especially for controlling inflammation, it remains unknown how MyD88 regulates the function of individual cell types present in the dystrophic microenvironment. We have recently reported that MyD88 promotes myoblast fusion in a cell-autonomous manner, independent of signaling from IL-1R/TLRs (32). However, the role of MyD88 in the regulation of satellite cell function in dystrophic muscle of mice remains completely unknown.
In this study, through the generation of satellite cell-specific inducible MyD88-knockout mdx mice, we have investigated the role and potential mechanisms through which MyD88 regulates satellite cell survival, proliferation and their regenerative potential in dystrophic muscle of mdx mice. Our results demonstrate that the ablation of MyD88 in satellite cells significantly reduces skeletal muscle mass and contractile function and exacerbates myopathy. MyD88 is essential for satellite cell proliferation and their fusion with injured myofibers of mdx mice. Satellite cell-specific ablation of MyD88 exacerbates interstitial fibrosis without having any major effect on the inflammatory milieu. Our results also demonstrate that deletion of MyD88 in satellite cells leads to an aberrant activation of Notch and Wnt signaling in dystrophic muscles.
Results
Generation of satellite cell-specific MyD88-knockout mdx mice and muscle functional analysis
We have previously reported that the levels MyD88 protein are increased in skeletal muscle of wild-type (WT) mice after acute injury (32). By performing immunoblotting, we first investigated how the levels of MyD88 are regulated in skeletal muscle of mdx mice. Results showed that the levels of MyD88 protein were drastically increased in the skeletal muscle of 8-week-old mdx mice compared to corresponding WT mice (Supplementary Material, Fig. S1A). Moreover, co-staining of muscle sections with Pax7 (a marker for satellite cells) and MyD88 proteins revealed that the levels of MyD88 are also considerably increased in satellite cells of mdx mice compared to WT mice (Supplementary Material, Fig. S1B).
We next sought to determine the role of MyD88 in satellite cell homeostasis and function in dystrophic muscle of mdx mice. In our initial studies, we found no difference in muscle pathology in 8-week-old mdx and mdx;Pax7-CreER mice (data not shown). To understand the satellite cell-specific role of MyD88 in dystrophic muscle, we first crossed floxed MyD88 (MyD88f/f) mice with Pax7-CreER mice to obtain MyD88;Pax7-CreER mice. MyD88;Pax7-CreER mice were then crossed with mdx mice to obtain Mdx;MyD88f/f and Mdx;MyD88;Pax7-CreER (henceforth Mdx;MyD88scko) mice (Supplementary Material, Fig. S2A). To induce Cre-mediated deletion of MyD88, 3-week-old Mdx;MyD88scko mice were given intraperitoneal (i.p.) injections of tamoxifen every day for 4 days. Littermate Mdx;MyD88f/f mice were also treated with tamoxifen and served as corresponding controls for Mdx;MyD88scko mice. The mice were maintained on a tamoxifen-containing chow for the entire duration of experimentation (Fig. 1A). Results showed that the average body weight of Mdx;MyD88scko mice was significantly lower compared to littermate Mdx;MyD88f/f mice after 5 weeks of starting tamoxifen injection (Fig. 1B). We next compared muscle force production during isometric contraction in vivo. Force recordings were taken from the posterior compartment of the lower extremity (Fig. 1C–J) after muscle length, positioning and current were optimized. Plantarflexion was stimulated at different frequencies and specific and maximum muscle forces produced in isometric contractions were measured. There was a significant reduction in the amount of force produced between 25–150 Hz in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 1C). Similarly, the corresponding calculated integral forces were also significantly reduced in Mdx;MyD88scko mice compared to littermate Mdx;MyD88f/f mice (Fig. 1D). Moreover, the Mdx;MyD88scko mice showed a significant reduction in specific twitch force (sPt) normalized by body weight compared to corresponding Mdx;MyD88f/f mice (Fig. 1E and F). There was also a trend toward reduction in specific work generated by twitch force in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 1G). Furthermore, maximum tetanic force and work normalized by body weight were significantly lower in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 1H–J). There was no significant difference in the fatigability of the muscles between the Mdx;MyD88f/f and Mdx;MyD88scko mice, which could be attributed to the fact that both mice are in the mdx background, which is highly susceptible to fatiguing (Supplementary Material, Fig. S3). Taken together, these results suggest that the deletion of MyD88 in satellite cells diminishes contractile properties of skeletal muscle in mdx mice.
Figure 1.
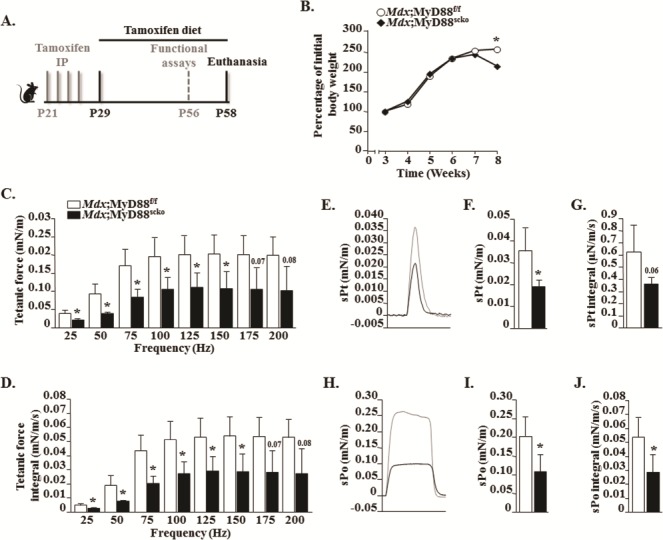
Ablation of MyD88 results in a decreased muscle contractile force production in male mdx mice. (A) Schematic representation of mice age and time of tamoxifen treatment, functional assays and analysis. (B) Percentage of initial body weight of male Mdx;MyD88f/f and Mdx;MyD88scko mice from week 3 to week 8. (C–J) Contractile properties of the posterior compartment of the lower limb were measured in vivo. (C) Normalized tetanic force to stimulation frequency relationship. (D) Calculated integral values for corresponding tetanic forces. (E) Representative traces of normalized sPt. Quantification of (F) average sPts and (G) corresponding integral values. (H) Representative traces of normalized traces for specific maximum tetanic forces (sPo). Quantification of (I) average specific maximum tetanic forces and (J) corresponding integral values. N = 3 for Mdx;MyD88f/f and N = 4 for Mdx;MyD88scko. Error bars represent s.d. *P-value <0.05 from Mdx;MyD88f/f mice by unpaired t-test.
Ablation of MyD88 in satellite cells reduces grip strength and wet muscle weight in mdx mice
We first examined gross body weight and found it to be significantly reduced in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 2A). We next investigated whether a deficiency of MyD88 in satellite cells affects the muscle grip strength in mdx mice. Results showed that the average and maximum forelimb strengths normalized with body weight were significantly reduced in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 2B and C). Similarly, the average and maximum total four limb grip strength normalized with body weight were significantly lower in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 2D and E). After 24 h of measuring grip strength, the mice were euthanized and individual skeletal muscles and the heart were isolated and their wet weight was recorded. Our analysis showed that wet weight of diaphragm, tibialis anterior (TA), gastrocnemius (GA) and quadriceps muscle of Mdx;MyD88scko mice were significantly less compared to Mdx;MyD88f/f mice (Fig. 2F). In contrast, there was no significant difference in the wet weight of the heart between Mdx;MyD88f/f and Mdx;MyD88scko mice (Fig. 2G), suggesting no change in cardiac muscle. Western blot analysis showed that the protein levels of MyD88 are considerably reduced in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Supplementary Material, Fig. S2B). Furthermore, immunohistochemical analysis revealed that the levels of MyD88 were diminished in satellite cells of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Supplementary Material, Fig. S2C). Collectively, these results suggest that the deficiency of MyD88 in satellite cells leads to reduced muscle mass and strength in mdx mice.
Figure 2.
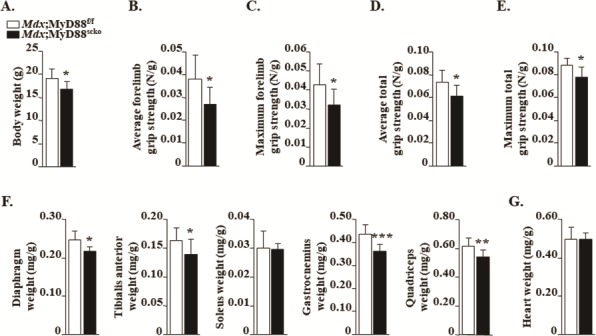
Effect of loss of MyD88 in satellite cells on grip strength and wet muscle weight in mdx mice. (A) Overall average body weight of 8-week-old male Mdx;MyD88f/f and Mdx;MyD88scko mice. (B) Average and (C) maximum forelimb grip strength of Mdx;MyD88f/f and Mdx;MyD88scko mice. (D) Average and (E) maximum total four-paw grip strength of Mdx;MyD88f/f and Mdx;MyD88scko mice. (F) Average wet weight of isolated diaphragm, TA, soleus, GA and quadriceps muscles. (G) Average wet weight of isolated heart. N = 8 for Mdx;MyD88f/f and N = 7 for Mdx;MyD88scko mice. Error bars represent s.d. *P-value < 0.05, **P-value < 0.01 and ***P-value < 0.001 from corresponding littermate Mdx;MyD88f/f mice by unpaired t-test.
Satellite cell-specific ablation of MyD88 exacerbates skeletal muscle histopathology in mdx mice
To understand the impact of satellite cell-specific ablation of MyD88 on the dystrophic muscle phenotype, we next generated transverse cryosections of hind limb muscles followed by performing hematoxylin and eosin (H&E) staining and morphometric analysis. Results showed that the myopathy was considerably exacerbated in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice. We found that the necrotic area (containing cellular infiltrates) in Mdx;MyD88f/f mice was partially filled with small, centronucleated (CNF) myofibers in skeletal muscle, a typical feature of mdx mice at this age. In contrast, necrotic area within skeletal muscle of Mdx;MyD88scko mice was devoid of small, CNF myofibers (Fig. 3A). Indeed, quantitative analysis showed that there was a drastic reduction in the percentage of CNF myofibers in the diaphragm, GA and TA muscles of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 3B–D). Our quantitative analysis also showed that the area under necrosis (i.e. devoid of myofibers) was significantly higher in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 3E). Although there was a trend toward a decrease in Mdx;MyD88scko mice, there was no significant difference in the average myofiber cross-sectional area (CSA) or minimal Feret’s diameter in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice (Fig. 3F and G). However, we found that the number of myofibers within skeletal muscle was significantly reduced in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 3H). To further assess muscle injury, we measured serum levels of creatine kinase (CK) in both groups. Consistent with muscle histopathology, we found that the serum levels of CK were significantly higher in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 3I).
Figure 3.
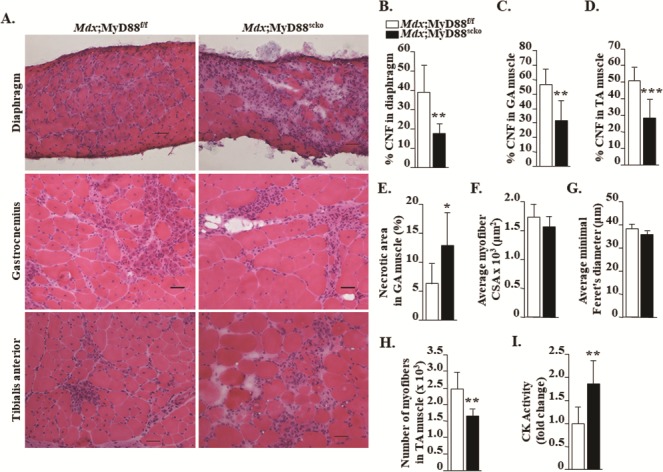
Satellite cell-specific ablation of MyD88 exacerbates skeletal muscle histopathology in mdx mice. (A) Representative photomicrographs of diaphragm, GA and TA muscle sections of 8-week-old Mdx;MyD88f/f and Mdx;MyD88scko mice. Quantification of percentage of CNF myofibers in (B) diaphragm, (C) GA and (D) TA muscle. (E) Percentage of necrotic area in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. Quantification of average myofiber (F) CSA and (G) minimal Feret’s diameter in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. N = 6 in each group. (H) Average number of myofibers in TA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. (I) Fold change in serum levels of CK activity in Mdx;MyD88f/f and Mdx;MyD88scko mice. Scale bar: 50 μm. N = 6 to 9 in each group. Error bars represent s.d. *P-value < 0.05, **P-value < 0.01 and ***P-value < 0.001 from corresponding littermate Mdx;MyD88f/f mice by unpaired t-test.
To understand whether the increased necrotic area was due to an increased rate of myofiber degeneration or due to insufficient regeneration, we measured the number of injured myofibers by performing immunostaining with Cy3-labelled IgG. Interestingly, while we found a significant increase in serum CK levels, the percentage of IgG-filled myofibers was comparable in the skeletal muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice (Supplementary Material, Fig. S4). It is notable that IgG+ myofibers represent the number of injured myofibers at the time of isolating muscle. Higher levels of CK could be attributed to multiple factors including a reduction in the rate of replenishment of injured myofibers in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice. Furthermore, it is possible that there is higher rate of muscle injury in other muscles of Mdx;MyD88scko mice that were not analyzed in our study. Collectively, our results suggest that muscle histopathology was significantly elevated in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice.
Ablation of MyD88 in satellite cells inhibits myofiber regeneration in mdx mice
Satellite cells are the major cell type that mediates muscle repair in response to acute injury (10–12). To specifically investigate the role of MyD88 in satellite cell function in the dystrophic microenvironment, we performed immunostaining for the embryonic isoform of myosin heavy chain (eMyHC), which is expressed only in newly formed myofibers. Our analysis showed that the number of eMyHC+ myofiber was drastically reduced in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Figs. 4A and B). Furthermore, the average CSA and minimal Feret’s diameter of eMyHC+ myofibers were significantly reduced in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 4C and D). We next measured mRNA and protein levels of markers of myogenesis in the skeletal muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. A significant reduction in mRNA levels of Myh3 (i.e. eMyHC) and myogenin, but not MyoD, was noticeable in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 4E). Furthermore, protein levels of eMyHC, MyoD and myogenin were also found to be markedly reduced in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 4F). Altogether, these results suggest that the ablation of MyD88 in satellite cells diminishes myofiber regeneration in dystrophic muscle of mdx mice.
Figure 4.
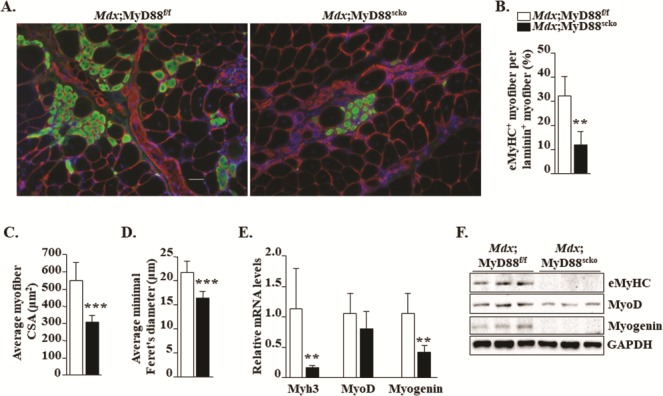
Ablation of MyD88 in satellite cells inhibits myofiber regeneration in mdx mice. (A) Representative photomicrographs of transverse GA muscle sections of Mdx;MyD88f/f and Mdx;MyD88scko mice immunostained for eMyHC (green color) and laminin (red color). Nuclei stained with DAPI. (B) Percentage of eMyHC+ myofiber per laminin+ myofiber. Quantification of eMyHC+ myofiber (C) CSA and (D) minimal Feret’s diameter in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. N = 6 or 7 in each group. (E) Relative mRNA levels of regeneration markers Myh3, MyoD and myogenin in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. (F) Representative immunoblots showing levels of eMyHC, MyoD and myogenin and an unrelated protein GAPDH in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. Scale bar: 50 μm. N = 5 mice in each group. Error bars represent s.d. *P-value < 0.05, **P-value < 0.01 and ***P-value < 0.001 from corresponding littermate Mdx;MyD88f/f mice by unpaired t-test.
Ablation of MyD88 reduces satellite cell number and their fusion with injured myofibers in mdx mice
We next investigated the cell-autonomous role of MyD88 in the regulation of satellite cell number in dystrophic muscle. We prepared transverse muscle sections of GA muscle and stained them for anti-Pax7 (a marker for satellite cells), anti-laminin (to mark the basal lamina) and DAPI (for labeling nuclei) followed by enumeration of Pax7+ cells within the basal lamina. Results showed that the frequency of Pax7+ cells per myofiber was significantly reduced in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 5A and B). Moreover, we also found that mRNA levels of Pax7 were significantly reduced in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 5C). We also used a fluorescence-activated cell sorting (FACS) approach to quantify the number of satellite cells in dystrophic muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. Consistent with our immunohistochemistry results, we found a drastic reduction in the proportion of α7-integrin+ cells (another marker of satellite cells) in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 5D and E). To understand whether MyD88 also has a role in the survival of satellite cells in dystrophic muscle, we performed a TUNEL assay on muscle sections. We did not find any TUNEL+ nuclei in GA muscle sections of Mdx;MyD88f/f and Mdx;MyD88scko mice (Supplementary Material, Fig. S5), suggesting that the inactivation of MyD88 does not induce apoptosis in satellite cells.
Figure 5.
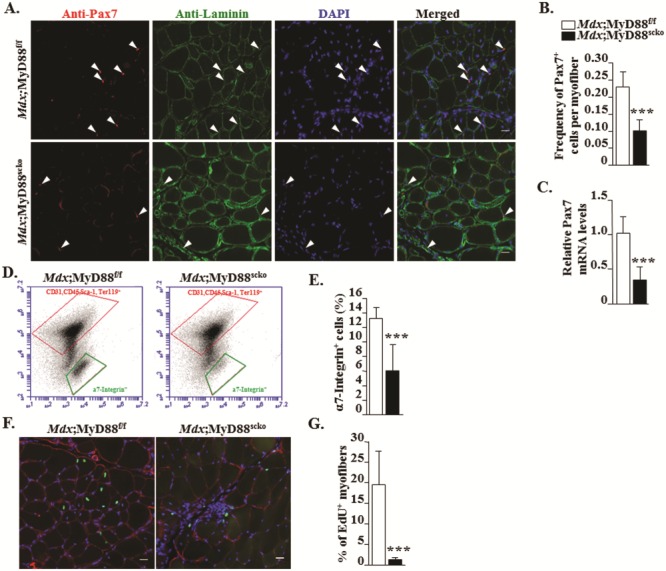
Ablation of MyD88 reduces satellite cell number and their fusion with injured myofibers in mdx mice. (A) Representative photomicrographs of transverse sections of GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice after immunostaining for Pax7 (red) and laminin (green). Nuclei were labelled with DAPI (blue). (B) Quantitative analysis showing frequency of Pax7+ cells per myofiber in Mdx;MyD88f/f and Mdx;MyD88scko mice. (C) Relative mRNA levels of Pax7 in GA muscle. (D) Representative FACS dot plots showing the percentage of satellite cells in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. Negative selection antibodies (CD31, CD45, Ter119) are in upper box, whereas positive selection antibody (α7-Integrin) is in lower box. (E) Quantification of percentage of α7-Integrin+ cells in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. N = 5 in each group. (F)Mdx;MyD88f/f and Mdx;MyD88scko mice were processed for the detection of EdU+ nuclei (green colored). The sections were also stained for laminin (red colored). Nuclei were counterstained with DAPI. (G) Percentage of EdU+ nuclei per myofiber. Scale bar: 20 μm. N = 5 for Mdx;MyD88f/f and N = 7 for Mdx;MyD88scko group. Error bars represent s.d. *P-value < 0.05, **P-value < 0.01 and ***P-value < 0.001 from corresponding littermate Mdx;MyD88f/f mice by unpaired t-test.
We also investigated whether the ablation of MyD88 affects the fusion of satellite cells with injured myofibers of mdx mice. For this experiment, 3-week-old Mdx;MyD88f/f and Mdx;MyD88scko mice were treated with tamoxifen for four consecutive days. Four weeks after the first tamoxifen injection, the mice were given an i.p. injection of 5-ethynyl-2’-deoxyuridine (EdU). The mice were euthanized a week later and the EdU+ nuclei within myofibers were visualized and enumerated. Results showed that the average number of intramyofiber EdU+ nuclei was significantly reduced in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 5F and G). Collectively, these results suggest that MyD88 is required for the proliferation and fusion of satellite cells, but not for their survival in dystrophic muscle of mdx mice.
Effect of satellite cell-specific ablation of MyD88 on interstitial fibrosis in dystrophic muscle
Progressive reduction in the ability of myofibers to regenerate leads to the development of interstitial fibrosis in skeletal muscle of DMD patients (1,6,7). Our preceding results showed that satellite cell-specific ablation of MyD88 diminishes skeletal muscle regeneration in mdx mice. To further understand the impact of the ablation of MyD88 in satellite cells, we compared levels of interstitial fibrosis in dystrophic muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. Specifically, transverse sections of diaphragm and GA muscle were stained with Masson’s trichrome staining and the area under fibrosis was quantified. Interestingly, there was a marked increase in fibrosis in both the diaphragm and GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 6A–C). Collagen I and III are the major collagens in the extracellular matrix of skeletal muscle (33). Consistent with the Masson’s trichrome staining results, our immunohistochemical analysis showed that there was a considerable increase in the levels of collagen I and III in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 6D). Moreover, mRNA levels of Col1a and Col3a1 were found to be significantly increased in GA muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 6E and F). These results suggest that the lack of MyD88 in satellite cells hastens the accumulation of fibrotic tissues, potentially due to reduced muscle regeneration.
Figure 6.
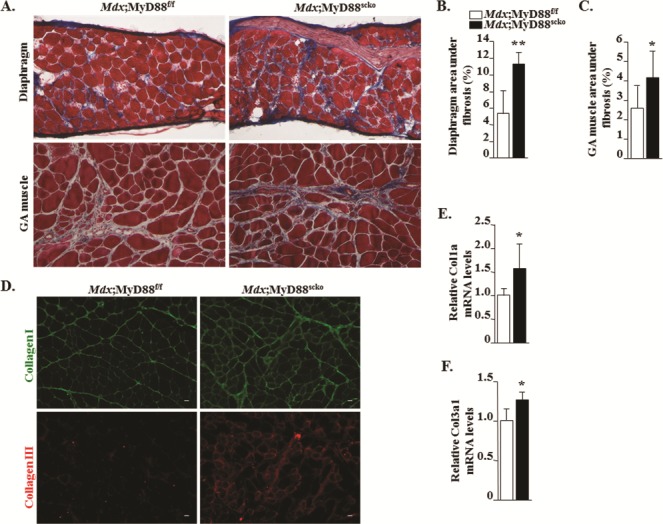
Effect of satellite cell-specific ablation of MyD88 on interstitial fibrosis in dystrophic muscle. (A) Representative Masson’s trichrome staining photomicrographs showing fibrosis (blue) in the diaphragm and GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. Quantification of percentage area under fibrosis in (B) diaphragm and (C) GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. (D) Representative photomicrographs of transverse sections of GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice after immunostaining for Collagen I (green) and Collagen III (red). Relative mRNA levels of (E)Col1a and (F)Col3a1. Scale bar: 20 μm. N = 5 or 6 in each group. Error bars represent s.d. *P-value < 0.05, **P-value < 0.01 and ***P-value < 0.001 from corresponding littermate Mdx;MyD88f/f mice by unpaired t-test.
Effect of satellite cell-specific deletion of MyD88 on inflammatory response and markers of autophagy in dystrophic muscle
Because of its critical role in TLR- and IL-1R–mediated signaling, MyD88 is considered as an important mediator of inflammation (26). However, satellite cells are a fraction of the cellular infiltrates in injured muscle and whether they play any role in the perpetuation of inflammation remains enigmatic. We next investigated whether the deletion of MyD88 in satellite cells has any effect on the number or type of macrophages, or the expression of pro- or anti-inflammatory molecules in dystrophic muscle of mdx mice. There are two subtypes of macrophages which have been suggested to play important roles in the regulation of myofiber necrosis and regeneration upon muscle injury (34). M1 macrophages are pro-inflammatory and dominate the necrotic phase of pathology in mdx mice contributing to muscle damage. In contrast, alternatively activated M2 macrophages prevail during the regenerative stage to facilitate myofiber repair. Interestingly, M2 macrophages have also been found to promote fibrogenesis in a number of tissues, including dystrophic muscle (35,36). By preparing mononuclear cell suspensions and performing FACS analysis, we examined the proportion of M1 and M2 macrophages in the F4/80+ cell population in dystrophic muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. While there was no significant difference in the proportion of M1 macrophages, there was a trend toward increased (although not statistically significant potentially due to small number in each group) proportion of M2 macrophages in dystrophic muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 7A–C). Our quantitative real time-PCR (qRT-PCR) analysis of cell surface markers also showed that there was no change in the mRNA levels of CD68 and Mac1, the markers for M1 macrophages (Fig. 7D). Interestingly, we found that the mRNA levels of CD163 and CD206, the markers of M2 macrophages, were significantly higher in dystrophic muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 7E). We also compared gene expression of certain cytokines and other mediators of inflammation. While there was no significant difference in mRNA levels of IL-1β, TNF-α, IL-4, IL-6, IL-10 and VCAM1, mRNA levels of ICAM1 and MMP-9 were found to be significantly increased in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 7F).
Figure 7.
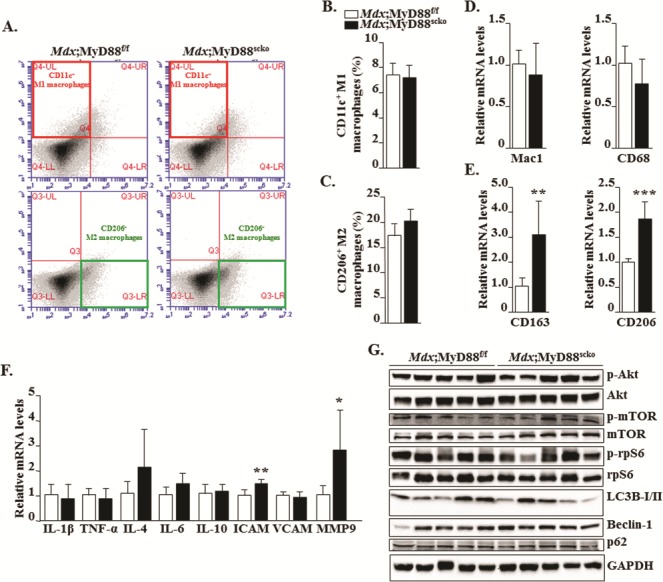
Effect of satellite cell-specific deletion of MyD88 on inflammatory response and autophagy in dystrophic muscle. (A) Representative FACS dot plots showing the percentage of M1 and M2 macrophages in GA muscle of 8-week-old Mdx;MyD88f/f and Mdx;MyD88scko mice. M1 macrophage positive selection antibody (CD11c+) is in the top left box and M2 macrophage positive selection antibody (CD206+) is in the bottom right box. Percentage of (B) CD11c+ M1 macrophages and (C) CD206+ M2 macrophages. Relative mRNA levels for (D) M1 macrophages (Mac1 and CD68) and (E) M2 macrophages (CD163 and CD206). (F) Relative mRNA levels for select cytokines and inflammatory mediators in skeletal muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. (G) Representative immunoblots showing proteins involved in the PI3K/Akt signaling pathway and autophagy in GA muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice. N = 5 mice in each group. Error bars represent s.d. *P-value < 0.05, **P-value < 0.01 and ***P-value < 0.001 from corresponding littermate Mdx;MyD88f/f mice by unpaired t-test.
The PI3K/Akt signaling pathway plays an important role in the regulation of skeletal muscle mass (37). Increased activation of this pathway also inhibits autophagy in dystrophic muscle of mdx mice leading to increased pathology (38). We investigated whether the deletion of MyD88 in satellite cells has any impact on the activation of the Akt/mTOR pathway in dystrophic muscle. There was no significant difference in the levels of phosphorylated Akt, mTOR or rpS6 proteins in skeletal muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice (Fig. 7G). Consistent with the levels of phosphorylated Akt, mTOR and rpS6 proteins, we did not find any significant difference in the markers of autophagy such as LC3B-I/II, Beclin-1 or p62 in dystrophic muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice (Fig. 7G).
Increased activation of Notch and Wnt signaling pathways in dystrophic muscle of Mdx;MyD88scko mice
Notch and Wnt are major signaling pathways that regulate multiple aspects of myogenesis (18,39,40). To gain further insight into how the deficiency of MyD88 in satellite cells affects disease progression in mdx mice, we examined the expression of the components of Notch and Wnt pathways. To study the activation of Notch signaling, we measured transcript levels of Notch ligands (Jagged1, Jagged2, DLL1 and DLL4), receptors (Notch1, Notch2 and Notch3) and target genes (Hes1, Hey1 and HeyL) by qRT-PCR assay. Interestingly, transcript levels of Jagged1, Notch1, Notch2, Notch3, Hes1 and HeyL were found to be significantly increased in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 8A–C). To study the activation of the Wnt pathway, we measured mRNA levels of Wnt ligands (Wnt3a, Wnt4, Wnt5a, Wnt7a and Wnt11), Wnt receptors (Frizzled-1, -2, -4 and -5) and Wnt target gene, Axin-2. Results showed that mRNA levels of all these molecules, except Frizzled-2, were significantly elevated in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 8D–F). Moreover, we found that protein levels of Wnt3a and Axin-2 were significantly higher in dystrophic muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 8G and H). Collectively, these results suggest that the ablation of MyD88 in satellite cells leads to aberrant activation of Notch and Wnt signaling pathways.
Figure 8.
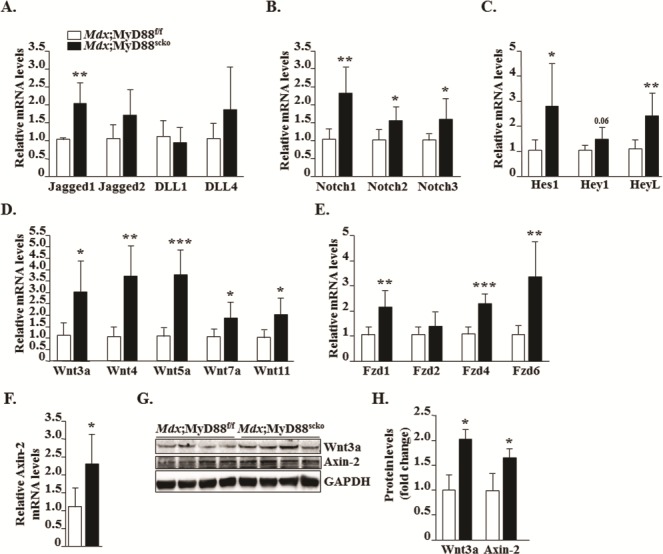
Increased expression of components of Notch and Wnt signalings in skeletal muscle of Mdx;MyD88scko mice. Relative mRNA levels of (A) Notch ligands, (B) Notch receptors and (C) Notch target genes in GA muscle of 8-week-old Mdx;MyD88f/f and Mdx;MyD88scko mice. Relative mRNA levels of indicated (D) Wnt ligands, (E) Wnt receptors and (F) Wnt target gene, Axin-2, in Mdx;MyD88f/f and Mdx;MyD88scko mice. (G) Representative immunoblots showing protein levels of Wnt3a and Axin-2 in Mdx;MyD88f/f and Mdx;MyD88scko mice. (H) Relative fold change in protein levels of Wnt3a and Axin-2 in Mdx;MyD88f/f and Mdx;MyD88scko mice. N = 5 mice. Error bars represent s.d. *P-value < 0.05, **P-value < 0.01 and ***P-value < 0.001 from corresponding littermate Mdx;MyD88f/f mice by unpaired t-test.
Discussion
Inflammation plays a major role in the pathogenesis of all diseases, including muscular dystrophy (1,41). Upon tissue injury, several endogenous mediators of inflammation engage TLRs to activate pro-inflammatory signaling pathways (42). It has been well established that MyD88 is a key component of TLR- and IL-1R–mediated signaling. Interestingly, recent studies suggest that MyD88 can also regulate cellular proliferation and differentiation in a cell-autonomous manner, independent of signaling from TLRs or IL-1β. For example, mutations in the MYD88 gene lead to the development of cancer in humans and mice (43,44). On similar lines, we have recently reported that MyD88 promotes myoblast fusion during post-natal growth, myofiber regeneration and overload-induced myofiber hypertrophy in mice (32). It has been reported that the genetic ablation of TLR4 or MyD88 improves skeletal muscle pathology in the models of muscular dystrophy (30,45–47). However, these studies were performed using mice in which MyD88 or TLR4 was globally inactivated. It is likely that the deletion of TLR4 or MyD88 in some cell types, especially those involved in the inflammatory immune response, attenuates the inflammatory response leading to reduced muscle damage and some improvement in pathology in dystrophic muscle. While satellite cells are the most important cell type for regenerative myogenesis, the satellite cell-specific role of MyD88 in dystrophic muscle remained unknown.
Our results demonstrate that the deletion of MyD88 in satellite cells exacerbates the muscle pathogenesis in the mdx model of DMD. Skeletal muscle of mdx mice starts undergoing necrosis at around 3 weeks followed by repeated cycles of regeneration and degeneration. Examination of muscle function after 5 weeks of inactivation of MyD88 in satellite cells demonstrated a significantly reduction in muscle contractile properties and a drastic reduction in grip strength (Figs. 1 and 2). At histological levels, we found that there was a significant increase in the area that is devoid of myofibers and there is an overall decrease in the number of myofibers in skeletal muscle of Mdx;MyD88scko mice (Figs. 3A, E and F). Interestingly, our analysis showed that there was no major difference in the number of IgG filled myofibers in skeletal muscle of Mdx;MyD88f/f and Mdx;MyD88scko mice (Supplementary Material, Fig. S4), suggesting that the lack of MyD88 in satellite cells may not have any major effect on the rate of myofiber degeneration rather it impairs the rate of myofiber regeneration upon initial injury. Indeed, we have found that the number of CNF myofibers (Fig. 3A–D) and the number of eMyHC+ myofibers (Fig. 4A and B) were markedly reduced in dystrophic muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice, suggesting that the satellite cell-specific deletion of MyD88 impairs myofiber regeneration.
We have recently shown that MyD88 does not affect the proliferation and differentiation of cultured myoblasts. Intriguingly, we found that MyD88 promotes fusion of cultured myoblasts after induction of the differentiation program (32). We also found that there was no significant difference in the number of satellite cells in 5d-injured TA muscle of control and myoblast-specific MyD88-KO mice, generated by crossing Myod1-Cre mice with MyD88f/f mice (32). While our previous studies highlighted the importance of MyD88 in myoblast fusion, the role of MyD88 in satellite cell function remained unanswered. Interestingly, we found that MyD88 is essential for satellite cell-mediated regenerative myogenesis. We found that the number of satellite cells and mRNA levels of Pax7 were significantly reduced in Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 5). Moreover, we noticed that myogenic differentiation markers, such as MyoD and myogenin, were also considerably reduced in the skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 4), further suggesting that MyD88 is essential for both the proliferation and differentiation of satellite cells. The role of MyD88 in satellite cells is consistent with previous reports suggesting that it plays an important role in proliferation and differentiation of several immune cell types (26). It is notable that myoblast-specific deletion of MyD88 has a relatively smaller effect on regeneration of injured myofibers in otherwise normal adult mice (32). In contrast, our results in this study demonstrate that deletion of MyD88 in satellite cells leads to a more dramatic inhibition in the regeneration of myofibers in mdx mice. This could be attributed to the fact that the mdx mice pathology involves chronic muscle injury and that in addition to the proliferation and differentiation of satellite cells, MyD88 also mediates fusion of myoblasts with injured myofibers. Indeed, our EdU incorporation assay also revealed that there was a significant reduction in the percentage of myofibers containing EdU+ nuclei in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 5F and G).
Our experiments also revealed that interstitial fibrosis was significantly increased in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 6). While the exact mechanisms remain unknown, it is known that a deficit or failure in muscle regeneration leads to the replacement of myofibers with fibrotic tissues especially in conditions of chronic muscle injury, such as muscular dystrophy (1,41). Since there is a drastic reduction in satellite cell number and markers of muscle regeneration (Figs. 4 and 5), it is possible that the increased level of fibrosis in skeletal muscle of Mdx;MyD88scko mice is a result of diminished myofiber regeneration. Alternatively, it is also possible that the deficiency of MyD88 in satellite cells leads to the dysregulation of some specific signaling pathways leading to increased fibrosis in dystrophic muscle. We observed that while the mRNA levels of pro- or anti-inflammatory cytokines remained comparable, there is a significant increase in the mRNA levels of MMP-9 in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice (Fig. 7F). Indeed, MMP-9 is one of the most important regulators of interstitial fibrosis in dystrophic muscle (33,48). We also found that there is a considerable increase in the markers of M2 macrophages in skeletal muscle of Mdx;MyD88scko mice (Fig. 7A, C and E). M2 macrophages are activated during the final stages of tissue regeneration, where they promote the repair process (34,49). Interestingly, M2 macrophages have also been linked to fibrosis in dystrophic muscle of mdx mice (35,36). Altogether, failed regeneration, increased levels of MMP-9 and increased abundance of M2 macrophages may be responsible for the increased fibrosis in skeletal muscle of Mdx;MyD88scko mice compared to Mdx;MyD88f/f mice.
An interesting observation of the present investigation is that while there is an impairment in muscle regeneration, there was a significant increase in the expression of the components of Notch and Wnt signaling pathways (Fig. 8). While Notch signaling is essential for the self-renewal and proliferation, it inhibits the differentiation of satellite cells into the myogenic lineage (39). Moreover, Notch3 signaling is also required for maintaining the quiescent satellite cell population (16,50). In contrast, the activation of Wnt signaling promotes satellite cell differentiation and fusion (18,40). Furthermore, elevated levels of the canonical Wnt pathway lead to fibrosis in dystrophic muscle (20). Previous studies have shown that temporal regulation of Notch and Wnt signaling is essential for regeneration of skeletal muscle upon injury (18). Even though we find increased activation of Notch signaling, the number of satellite cells was significantly reduced in dystrophic muscle of Mdx;MyD88scko compared to Mdx;MyD88f/f mice. It is possible that the activated Notch signaling may prevent the activation and subsequent differentiation of the remaining satellite cells into the myogenic lineage. However, there is also a possibility that the increased activation of Notch and Wnt signaling is a compensatory response due to a reduced number of satellite cells and diminished myofiber regeneration in Mdx;MyD88scko mice. It is noteworthy that we measured the expression levels of various components of Wnt and Notch signaling in whole muscle, which contains multiple cell types. Therefore, it is also possible that the increased expression of the components of the aforementioned pathways may be a result of their activation in other cell types as well in addition to the satellite cells.
It is now increasingly evidenced that in addition to controlling the inflammatory immune response, MyD88 has important functions in the regulation of proliferation and differentiation of several cell types including myogenic cells. We have previously demonstrated that MyD88 promotes skeletal muscle growth potentially through enhancing myoblast fusion. Indeed, overexpression of MyD88 improves transplantation and engraftment of exogenous myoblasts in injured skeletal muscle of WT mice (32). Our results in this study provide crucial evidence that MyD88-mediated signaling is essential for homeostasis and functioning of satellite stem cells in dystrophic muscle. However, it is noteworthy that the present study was performed at the age of 3–8 weeks, a time period when myofiber degeneration and regeneration are at peak in mdx mice. It remains to be investigated whether chronic inhibition of MyD88 in satellite cells will further worsen myopathy and progressive weight loss in mdx mice at more advanced ages. Future studies should also investigate the effects of satellite cell-specific inhibition of MyD88 in exercise-induced myofiber damage and repair in mdx mice.
Published microarray dataset deposited in the Gene Expression Omnibus (i.e. GSE1004, GSE3307 and GSE6011) demonstrates that the expression of MYD88 is variable among DMD patients compared to that of control subjects indicating that the gene expression of MYD88 may be deregulated in skeletal muscle and other cell types including satellite cells of DMD patients.
While more investigations are needed to understand the role of MyD88 in dystrophinopathies, our study provides initial evidence that enhancing the levels of MyD88 in muscle progenitor cells may be used as therapeutic approach to improve muscle regeneration and a mean to enhance the efficacy of stem cell-based therapy for muscular dystrophy.
Materials and Methods
Animals
Floxed MyD88 (strain: B6.129P2(SJL)-Myd88tm1Defr/J), Pax7-CreER (strain: B6;129-Pax7tm2.1(cre/ERT2)Fan/J) and mdx (strain: C57BL/10ScSn-Dmdmdx/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Floxed MyD88 (i.e. MyD88f/f) mice were crossed with Pax7-CreER mice to generate MyD88f/f;Pax7-CreER (i.e. MyD88scko) mice. MyD88scko mice were then crossed with mdx mice to generate Mdx;MyD88scko and littermate Mdx;MyD88f/f mice. The mice were bred for three generations in mdx background before using for experimentation. All genotypes were determined by PCR analysis from tail DNA. For inducible inactivation of MyD88, Mdx;MyD88f/f and Mdx;MyD88scko mice were given intraperitoneal (i.p.) injection of tamoxifen (10 mg/kg body weight) for 4 consecutive days. The mice were kept on tamoxifen-containing standard chow (Harlan Laboratories, Madison, WI) for the entire duration of the experiment. To study the contribution of myogenic cells in muscle regeneration, the mice were also given i.p. injection of EdU (100 μg per mouse) 10 days before euthanizing the mice. All experimental protocols with mice were approved in advance by the Institutional Biosafety Committee and the Institutional Animal Care and Use Committee at the University of Louisville (IACUC numbers: 13097 and 16663).
In vivo muscle functional assay
In vivo force measurements of the posterior lower leg muscles were conducted using the 1300A 3-in-1 Whole Animal System (Aurora Scientific, Aurora, Ontario, Canada) to evoke plantarflexion. Male mice were anesthetized with Avertin (2,2,2-Tribromoethanol; MilliporeSigma, St. Louis, MO, USA) and placed on the isothermal stage. Optimal muscle length that allows maximal isometric twitch force (Pt; a single muscular contraction and relaxation event) was determined by a sequence of twitch contractions at 150 Hz every second with small varying changes to muscle tension and current. After muscle optimization, an average sPt was generated from five stimulations at 150 Hz. To obtain maximum isometric tetanic contraction (Po), muscles were stimulated from 25 to 300 Hz in 25 Hz intervals resulting in 12 specific tetanic forces (sPo; a tetanic contraction occurring as a result of a specific stimulation frequency). There was a 30-s delay between the first two recordings and a 1-min delay between subsequent recordings to allow sufficient muscle recovery. Following the 300 Hz stimulation, a baseline recording was obtained. Then the muscles were fatigued at 150 Hz with one contraction per second for 180 s. Force recordings for obtained specific isometric twitch force sPt (mN/m) and tetanic force sPo (mN/m) were normalized by total mouse body weight. For all experiments, a pre-stimulus and post-stimulus baseline of 200 ms was recorded to establish a baseline recording. For all experiments, a 0.2 ms pulse width was used. Current was adjusted on an individual basis to evoke the maximum amount of force. Contractile events were recorded using the ASI 611A Dynamic Muscle Control software (Aurora Scientific) at a sampling rate of 2000 Hz. Force (mN/m) and corresponding integral values for sPt (mN/m/s) and Po (mN/m/s) were calculated using the accompanying ASI 611A Dynamic Muscle Analysis software (Aurora Scientific).
Grip strength measurements
To measure forelimb and total four-limb grip strength of mice, a digital grip-strength meter (Columbus Instruments, Columbus, OH, USA) was used. Before performing test, mice were acclimatized for 5 min. The mouse was allowed to grab the metal pull bar with the forepaws and in a separate experiment with or all four paws. The mouse tail was then gently pulled backward in the horizontal plane until it could no longer grasp the bar. The force at the time of release was recorded as the peak tension. Each mouse was tested five times with a 20–40 s break between tests. The average peak tension from five best attempts and maximum peak tension normalized against total body weight was defined as forelimb grip strength.
CK assay
The serum level of CK was determined using a commercially available kit (Stanbio Laboratory, TX, USA) following a protocol from the manufacturer.
Histology and morphometric analysis
We performed H&E staining or Masson’s trichrome staining on transverse muscle sections to visualize muscle structures or fibrosis, respectively, similar to as described (51,52). In brief, individual hind limb muscle was isolated, flash frozen in liquid nitrogen and sectioned using a microtome cryostat. For the assessment of gross morphology, 10 μm thick transverse sections of diaphragm, GA and TA muscles were stained with H&E dye. Stained sections were visualized on an inverted microscope (Nikon Eclipse TE 2000-U; Nikon, Melville, NY, USA) at room temperature (RT), a digital camera (Digital Sight DS-Fi1; Nikon) and NIS Elements BR 3.10 software (Nikon). The images were stored as TIFF files. Pictures of the whole muscle sections were captured and the percentage of CNF myofibers as well as total number of myofibers in each muscle was counted in the entire muscle section. Necrotic area in H&E-stained sections was determined by measuring percentage area filled with cellular infiltrate in whole muscle section. Average minimal Feret's diameter of eMyHC+ fibers was determined after measuring CSA for each fiber. The extent of fibrosis in muscle cryosections was determined using a Masson's trichrome staining kit following a protocol suggested by manufacturer (American MasterTech Scientific, Lodi, CA, USA). Morphometric analyses were quantified using Fiji software (U.S. National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry
For immunohistochemistry studies, muscle sections were blocked in 1% bovine serum albumin in phosphate-buffered saline (PBS) for 1 h and incubated with anti-Pax7 (1:5-1:10, Developmental Studies Hybridoma Bank (DSHB) Cat# PAX7, Iowa City, IA, USA) or anti-eMyHC (1:150, DSHB Cat# F1.652) along with anti-Laminin (1:150, MilliporeSigma Cat# L9393) or anti-MyD88 (1:40, R&D Systems, Minneapolis, MN, USA) in blocking solution at 4°C overnight under humidified conditions. The sections were washed briefly with PBS before incubation with Alexa Fluor® 488 (1:2000, Thermo Fisher Scientific Cat# A-11034, Rockford, IL, USA) and Alexa Fluor® 594 (1:2000, Thermo Fisher Scientific Cat# A-11037) or Alexa Fluor® 488 (1:2000, Thermo Fisher Scientific Catalog # A-1105) secondary antibody for 1 h at RT and then washed three times for 5 min with PBS. Damaged/permeabilized fibers in muscle cryosections were identified by immunostaining with Cy3-labelled goat anti-mouse IgG (1:3000, Thermo Fisher Scientific Cat# A10521). The slides were mounted using a non-fluorescing aqueous mounting medium (Polysciences, Warrington, PA, USA) and visualized at -0.4°C on a Nikon TiE 3000 Inverted Microscope (Nikon), a digital camera (DXM-1200C Coded Digital Camera) and Nikon NIS Elements AR software (Nikon). Images were stored as TIFF files and contrast levels were equally adjusted using Adobe Photoshop CS6 software (Adobe, San Jose, CA, USA).
Western blot
Quantitative estimation of various proteins in skeletal muscle tissue was done by western blot using a method as previously described (32). Briefly, skeletal muscle tissues were washed with PBS and homogenized in ice-cold lysis buffer [50-mM Tris pH 8.0, 200-mM NaCl, 50-mM NaF, 0.3% IGEPAL, 1-mM dithiothreitol, 1-mM benzamidine, 1-mM sodium orthovanadate and 100-μM phenylmethanesulfonyl fluoride] supplemented with Halt protease inhibitor cocktail (Thermo Fisher Scientific, Rockford, IL, USA). Approximately, 50–100 μg protein was resolved on each lane on 12% SDS-polyacrylamide gel, electrotransferred onto nitrocellulose membrane and probed using anti-MyD88 (1:500, R&D System, Minneapolis, MN, USA), anti-Wnt3a (1:500, SigmaMillipore), anti-Axin2 (1:5000, Abcam, Cambridge, MA, USA), anti-eMyHC (1:500, Developmental Studies Hybridoma Bank (DSHB) Cat# F1.652, Iowa City), anti-MyoD (1:500, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Myogenin (1:1000, Santa Cruz Biotechnology) and anti-GAPDH (1:2000, Cell Signaling Technology, Danvers, MA, USA). Signal detection was performed by an enhanced chemiluminescence detection reagent (Bio-Rad, Hercules, CA, USA). Approximate molecular masses were determined by comparison with the migration of prestained protein standards (Bio-Rad). Quantitative estimation of the bands’ intensity was performed using Fiji software (U.S. NIH).
RNA isolation and qRT-PCR assay
Total RNA isolation from muscle tissues and qRT-PCR was performed as previously described (53,54). Primers for qRT-PCR were designed using Vector NTI software and their sequence has been described in our previous publications (14,55).
FACS
The proportion of satellite cells and M1 and M2c macrophages in skeletal muscle tissues was analyzed by FACS as previously described (14,55). In brief, skeletal muscle tissues were digested with Collagenase II to prepare single cell suspensions. Approximately 2×106 cells were incubated in DMEM (supplemented with 2% fetal bovine serum and 25-mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) and dead cells (positive for propidium iodide staining) which were around ~1% were excluded from all FACS analysis. For satellite cell quantification from heterogeneous cell population, cells were immunostained with antibodies against CD45 (eBioscience Cat# 12-0451-83, San Diego, CA, USA), CD31 (BD Biosciences Cat# 553373, San Jose, CA, USA), Sca-1 (eBioscience Cat# 12-5981-82), Ter-119 (eBioscience Cat# 12-5921-82) for negative selection (all PE conjugated, eBiosciences) and with α7-Integrin (APC conjugated, Miltenyi Biotec) for positive selection.
Macrophages were quantified from cell population by selection of F4/80+ (PerCP Cy5.5-conjugated, eBiosciences) cells against negative selection by CD56/Sca-1 and Ter-119 (all PE-conjugated, eBiosciences). From F4/80+ cells, CD11c+ (APC-conjugated, eBiosciences) M1 and CD206+ (FITC-conjugated, BioLegend, San Diego, CA, USA) M2 macrophages were isolated. FACS analysis was performed on a C6 Accuri cytometer equipped with three lasers. The output data was processed and plots were prepared using FCS Express 4 RUO software (De Novo Software) and were exported to Adobe Photoshop CS6 software.
Statistical analysis
Results are expressed as mean ± standard deviation. Statistical analysis used unpaired Student's t-test (two-tailed) to compare quantitative data populations with normal distribution and equal variance. The α-level of significance was set at 0.05 for all comparison unless otherwise specified.
Supplementary Material
Data Availability
All relevant data related to this manuscript are available from the authors.
Authors’ Contributions
AK designed the work and wrote the manuscript. All authors edited the manuscript. YSG, ARS, KRB, GX and SMH performed the experiments.
Funding
This work was supported by NIH grants AR068313, AR059810 and AG029623 to AK and AR069985 to SMH. Figure S2A was produced with the assistance of Servier Medical Art.
References
- 1. Shin J., Tajrishi M.M., Ogura Y. and Kumar A. (2013) Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell Biol., 45, 2266–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Emery A.E.(1998) The muscular dystrophies. BMJ, 317, 991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kinane T.B., Mayer O.H., Duda P.W., Lowes L.P., Moody S.L. and Mendell J.R. (2018) Long-term pulmonary function in Duchenne muscular dystrophy: comparison of eteplirsen-treated patients to natural history. J. Neuromuscul. Dis., 5, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D.M., Duval B., Aoyagi A.. et al. (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol., 71, 304–313. [DOI] [PubMed] [Google Scholar]
- 5. Hoffman E.P., Brown R.H. Jr. and Kunkel L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. [DOI] [PubMed] [Google Scholar]
- 6. Blake D.J., Weir A., Newey S.E. and Davies K.E. (2002) Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiological Reviews, 82, 291–329. [DOI] [PubMed] [Google Scholar]
- 7. Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M. and Sweeney H.L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA, 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang N.C., Chevalier F.P. and Rudnicki M.A. (2016) Satellite cells in muscular dystrophy - lost in polarity. Trends Mol. Med., 22, 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Negroni E., Bigot A., Butler-Browne G.S., Trollet C. and Mouly V. (2016) Cellular therapies for muscular dystrophies: frustrations and clinical successes. Hum. Gene Ther., 27, 117–126. [DOI] [PubMed] [Google Scholar]
- 10. Yin H., Price F. and Rudnicki M.A. (2013) Satellite cells and the muscle stem cell niche. Physiological Reviews, 93, 23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bentzinger C.F., Wang Y.X. and Rudnicki M.A. (2012) Building muscle: molecular regulation of myogenesis. Cold Spring Harb. Perspect. Biol., 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buckingham M., Bajard L., Chang T., Daubas P., Hadchouel J., Meilhac S., Montarras D., Rocancourt D. and Relaix F. (2003) The formation of skeletal muscle: from somite to limb. J. Anat., 202, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuroda K., Tani S., Tamura K., Minoguchi S., Kurooka H. and Honjo T. (1999) Delta-induced notch signaling mediated by RBP-J inhibits MyoD expression and myogenesis. J. Biol. Chem., 274, 7238–7244. [DOI] [PubMed] [Google Scholar]
- 14. Hindi S.M., Paul P.K., Dahiya S., Mishra V., Bhatnagar S., Kuang S., Choi Y. and Kumar A. (2012) Reciprocal interaction between TRAF6 and notch signaling regulates adult myofiber regeneration upon injury. Mol. Cell. Biol., 32, 4833–4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kuang S., Kuroda K., Le Grand F. and Rudnicki M.A. (2007) Asymmetric self-renewal and commitment of satellite stem cells in muscle. Cell, 129, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bjornson C.R., Cheung T.H., Liu L., Tripathi P.V., Steeper K.M. and Rando T.A. (2012) Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem Cells, 30, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mourikis P., Sambasivan R., Castel D., Rocheteau P., Bizzarro V. and Tajbakhsh S. (2012) A critical requirement for notch signaling in maintenance of the quiescent skeletal muscle stem cell state. Stem Cells, 30, 243–252. [DOI] [PubMed] [Google Scholar]
- 18. Brack A.S., Conboy I.M., Conboy M.J., Shen J. and Rando T.A. (2008) A temporal switch from notch to Wnt signaling in muscle stem cells is necessary for normal adult myogenesis. Cell Stem Cell, 2, 50–59. [DOI] [PubMed] [Google Scholar]
- 19. Hindi S.M., Tajrishi M.M. and Kumar A. (2013) Signaling mechanisms in mammalian myoblast fusion. Sci. Signal., 6, re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Biressi S., Miyabara E.H., Gopinath S.D., Carlig P.M. and Rando T.A. (2014) A Wnt-TGFbeta2 axis induces a fibrogenic program in muscle stem cells from dystrophic mice. Sci. Transl. Med., 6, 267ra176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Abou-Khalil R., Le Grand F., Pallafacchina G., Valable S., Authier F.J., Rudnicki M.A., Gherardi R.K., Germain S., Chretien F., Sotiropoulos A.. et al. (2009) Autocrine and paracrine angiopoietin 1/Tie-2 signaling promotes muscle satellite cell self-renewal. Cell Stem Cell, 5, 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hindi S.M. and Kumar A. (2016) TRAF6 regulates satellite stem cell self-renewal and function during regenerative myogenesis. J. Clin. Invest., 126, 151–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shi H., Verma M., Zhang L., Dong C., Flavell R.A. and Bennett A.M. (2013) Improved regenerative myogenesis and muscular dystrophy in mice lacking Mkp5. J. Clin. Invest., 123, 2064–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Janeway C.A. Jr. and Medzhitov R. (2002) Innate immune recognition. Annu. Rev. Immunol., 20, 197–216. [DOI] [PubMed] [Google Scholar]
- 25. Takeuchi O. and Akira S. (2010) Pattern recognition receptors and inflammation. Cell, 140, 805–820. [DOI] [PubMed] [Google Scholar]
- 26. Akira S. and Takeda K. (2004) Toll-like receptor signalling. Nat. Rev. Immunol., 4, 499–511. [DOI] [PubMed] [Google Scholar]
- 27. Kawai T. and Akira S. (2007) Signaling to NF-kappaB by toll-like receptors. Trends Mol. Med., 13, 460–469. [DOI] [PubMed] [Google Scholar]
- 28. Zhang S., Li C.Z., Yan H., Qiu W., Chen Y.G., Wang P.H., Weng S.P. and He J.G. (2012) Identification and function of myeloid differentiation factor 88 (MyD88) in Litopenaeus vannamei. PLoS One, 7, e47038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Janssens S. and Beyaert R. (2002) A universal role for MyD88 in TLR/IL-1R-mediated signaling. Trends Biochem. Sci., 27, 474–482. [DOI] [PubMed] [Google Scholar]
- 30. Henriques-Pons A., Yu Q., Rayavarapu S., Cohen T.V., Ampong B., Cha H.J., Jahnke V., Van der Meulen J., Wang D., Jiang W.. et al. (2014) Role of Toll-like receptors in the pathogenesis of dystrophin-deficient skeletal and heart muscle. Hum. Mol. Genet., 23, 2604–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Giordano C., Mojumdar K., Liang F., Lemaire C., Li T., Richardson J., Divangahi M., Qureshi S. and Petrof B.J. (2015) Toll-like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum. Mol. Genet., 24, 2147–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hindi S.M., Shin J., Gallot Y.S., Straughn A.R., Simionescu-Bankston A., Hindi L., Xiong G., Friedland R.P. and Kumar A. (2017) MyD88 promotes myoblast fusion in a cell-autonomous manner. Nat. Commun., 8, 1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li H., Mittal A., Makonchuk D.Y., Bhatnagar S. and Kumar A. (2009) Matrix metalloproteinase-9 inhibition ameliorates pathogenesis and improves skeletal muscle regeneration in muscular dystrophy. Hum. Mol. Genet., 18, 2584–2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tidball J.G., Dorshkind K. and Wehling-Henricks M. (2014) Shared signaling systems in myeloid cell-mediated muscle regeneration. Development, 141, 1184–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vidal B., Serrano A.L., Tjwa M., Suelves M., Ardite E., De Mori R., Baeza-Raja B., Martinez de Lagran M., Lafuste P., Ruiz-Bonilla V.. et al. (2008) Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes. Dev., 22, 1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Villalta S.A., Nguyen H.X., Deng B., Gotoh T. and Tidball J.G. (2009) Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet., 18, 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Egerman M.A. and Glass D.J. (2014) Signaling pathways controlling skeletal muscle mass. Crit. Rev. Biochem. Mol. Biol., 49, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. De Palma C., Morisi F., Cheli S., Pambianco S., Cappello V., Vezzoli M., Rovere-Querini P., Moggio M., Ripolone M., Francolini M.. et al. (2012) Autophagy as a new therapeutic target in Duchenne muscular dystrophy. Cell Death Dis., 3, e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mourikis P. and Tajbakhsh S. (2014) Distinct contextual roles for Notch signalling in skeletal muscle stem cells. BMC Dev. Biol., 14, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maltzahn J., Chang N.C., Bentzinger C.F. and Rudnicki M.A. (2012) Wnt signaling in myogenesis. Trends Cell Biol., 22, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bhatnagar S. and Kumar A. (2010) Therapeutic targeting of signaling pathways in muscular dystrophy. J. Mol. Med. (Berl), 88, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kawai T. and Akira S. (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol., 11, 373–384. [DOI] [PubMed] [Google Scholar]
- 43. Salcedo R., Cataisson C., Hasan U., Yuspa S.H. and Trinchieri G. (2013) MyD88 and its divergent toll in carcinogenesis. Trends Immunol., 34, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang J.Q., Jeelall Y.S., Ferguson L.L. and Horikawa K. (2014) Toll-Like receptors and cancer: MYD88 mutation and inflammation. Front Immunol., 5, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sachdev U., Cui X., Xu J., Xu J. and Tzeng E. (2014) MyD88 and TRIF mediate divergent inflammatory and regenerative responses to skeletal muscle ischemia. Physiological Reports, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Uaesoontrachoon K., Cha H.J., Ampong B., Sali A., Vandermeulen J., Wei B., Creeden B., Huynh T., Quinn J., Tatem K.. et al. (2013) The effects of MyD88 deficiency on disease phenotype in dysferlin-deficient A/J mice: role of endogenous TLR ligands. J. Pathol., 231, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hindi S.M. and Kumar A. (2016) Toll-like receptor signalling in regenerative myogenesis: friend and foe. J. Pathol., 239, 125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kumar A., Bhatnagar S. and Kumar A. (2010) Matrix metalloproteinase inhibitor batimastat alleviates pathology and improves skeletal muscle function in dystrophin-deficient mdx mice. Am. J. Pathol., 177, 248–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kharraz Y., Guerra J., Mann C.J., Serrano A.L. and Munoz-Canoves P. (2013) Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm., 2013, 491497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kitamoto T. and Hanaoka K. (2010) Notch3 null mutation in mice causes muscle hyperplasia by repetitive muscle regeneration. Stem Cells, 28, 2205–2216. [DOI] [PubMed] [Google Scholar]
- 51. Hindi S.M., Sato S., Xiong G., Bohnert K.R., Gibb A.A., Gallot Y.S., McMillan J.D., Hill B.G., Uchida S. and Kumar A. (2018) TAK1 regulates skeletal muscle mass and mitochondrial function. JCI Insight, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Paul P.K., Gupta S.K., Bhatnagar S., Panguluri S.K., Darnay B.G., Choi Y. and Kumar A. (2010) Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol., 191, 1395–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mittal A., Bhatnagar S., Kumar A., Lach-Trifilieff E., Wauters S., Li H., Makonchuk D.Y., Glass D.J. and Kumar A. (2010) The TWEAK-Fn14 system is a critical regulator of denervation-induced skeletal muscle atrophy in mice. J. Cell Biol., 188, 833–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paul P.K., Bhatnagar S., Mishra V., Srivastava S., Darnay B.G., Choi Y. and Kumar A. (2012) The E3 ubiquitin ligase TRAF6 intercedes in starvation-induced skeletal muscle atrophy through multiple mechanisms. Mol. Cell. Biol., 32, 1248–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hindi S.M., Shin J., Ogura Y., Li H. and Kumar A. (2013) Matrix metalloproteinase-9 inhibition improves proliferation and engraftment of myogenic cells in dystrophic muscle of mdx mice. PLoS One, 8, e72121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.