

Abstract
Background
Although the peroxisome proliferator-activated receptor-γ (PPARγ) agonist rosiglitazone has significant anti-inflammatory properties, no scientific studies have provided new insights in its pharmacological properties with respect to acute respiratory distress syndrome (ARDS). The present investigation aimed to evaluate whether rosiglitazone can reduce apoptosis and inflammation in a lipopolysaccharide (LPS)-induced acute respiratory distress syndrome in vitro model.
Material/Methods
Human umbilical vein endothelial cells (HUVECs) were treated with 1 μg/ml LPS in the absence or presence of 10 μM rosiglitazone for 24 h. Cell viability was measured by MTT assay. Flow cytometry was used to examine the cell apoptosis and ROS production in HUVECs response to LPS and rosiglitazone. The levels of pro-inflammatory cytokine factors, including TNF-α, IL-6, CXCL12, and CXCR4, were measured by ELISA, real-time PCR, and Western blot assay, respectively. The expression of PPARγ, Bcl-2, and Bax and the activity of JAK2 and STAT3 were also investigated by Western blot assay.
Results
We found that rosiglitazone significantly inhibited LPS-induced cell apoptosis, ROS production, and inflammation in HUVECs. Furthermore, we found a significant reduction of JAK2/STAT3 activation and the Bax/Bcl-2 ratio in LPS-induced HUVECs response to rosiglitazone treatment.
Conclusions
Treatment with rosiglitazone can reduce apoptosis and inflammation in HUVECs induced by LPS.
MeSH Keywords: Apoptosis; Inflammation; Janus Kinase 2; Respiratory Distress Syndrome, Adult; STAT3 Transcription Factor
Background
Acute respiratory distress syndrome (ARDS) is an acute respiratory failure syndrome caused by severe infection, shock, trauma, and other conditions, characterized by progressive respiratory distress and refractory hypoxia in clinical patients [1,2]. Diffuse pulmonary interstitial and alveolar edema caused by alveolar capillary endothelial cells and alveolar epithelial cells are the main pathological changes of ARDS [3]. The inflammatory cytokines networks, as well as the interaction of inflammation, coagulation, and fibrinolysis pathways, are involved in the development of ARDS [4,5]. Excessive inflammatory reaction caused by alveolar epithelial cells and capillary endothelial cell injury is the main mechanism of ARDS pathogenesis [6].
A series of systemic inflammations caused by infection is the main cause of ARDS, among which the endotoxemia caused by Gram-negative bacterial infection accounts for the most of the infection factors in ARDS. Lipopolysaccharide (LPS) is the main component of endotoxin [7]; it is a strong injury factor, which can activate macrophages after entering the body and promote the production of TNF-α, IL-6, IL-1β, and IL-8 [8]. Persistent elevation of pro-inflammatory cytokines in ARDS is associated with poor patient outcomes [9]. Recently, the critical role of lung cell apoptosis has been evaluated in ARDS. In microenvironments in which frequent apoptosis occurs in alveolar epithelial cells and capillary endothelial cells, decreasing apoptosis of alveolar wall cells might be of benefit for the suppression of lung tissue damage and could be a key element in the treatment of patients with ARDS [10,11].
Peroxisome proliferator-activated receptor-γ (PPARγ) is a member of the nuclear hormone receptor superfamily and rosiglitazone is its specific agonist. Research shows that PPARγ receptor agonists (rosiglitazone) can reduce the inflammatory response of sepsis, prevent cell damage, and protect the body [12,13]. The main mechanism of PPARs protection in sepsis is by protecting mitochondrial function in the metabolism of ATP from damage through 3 subtypes (PPAR-α, PPAR-β, and PPAR-γ), inhibiting inflammatory reaction [14,15]. Liu et al. demonstrated an anti-inflammatory role for rosiglitazone during lung injury when animals were pretreated with rosiglitazone prior to the induction of lung injury [16]. This anti-inflammatory and protective role for rosiglitazone was also shown in a model of pancreatitis-associated lung injury [17].
In order to evaluate the effect of the PPAR-γ agonist rosiglitazone in preventing lung cell damage, we generated LPS-induced ARDS in HUVECs and analyzed cell patterns, including apoptosis, inflammation, and signaling transduction. The results suggest it is a novel therapeutic target of PPAR-γ to treat ARDS in human.
Material and Methods
Cell culture and treatment
Human umbilical vein endothelial cells (HUVECs) were purchased from Sanko Junyaku Co. (Tokyo, Japan) and grown at 37°C in 5% CO2 in DMEM with 10% FBS, penicillin (100 U/ml), and streptomycin (100 U) supplement. HUVECs (5×103 cell/well) were incubated at 37°C in a 5% CO2 incubator overnight in 96-well plates. Then, cells were treated with 1 μg/ml LPS in the absence or presence of PPARγ agonist rosiglitazone (10 μM; Selleck, Houston, TX, USA) for 24 h.
Cell viability assay
Cell viability was determined using a colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, as described previously [18]. HUVECs were treated with 1 μg/ml LPS in the absence or presence of rosiglitazone (10 μM) for 0, 24, 48, or 72 h and cultured in new medium containing 0.5 mg/ml MTT for a further 3 h. The blue formazan products in HUVECs were dissolved in DMSO and spectrophotometrically measured at a wavelength of 550 nm.
Flow cytometry assay
HUVECs were treated with 1 μg/ml LPS in the absence or presence of rosiglitazone (10 μM) for 24 h and collected for cell apoptosis and reactive oxygen species (ROS) assay. Annexin-V fluorescein isothiocyanate (FITC)/propidium iodide (PI) double-staining assays (Biovision, Inc, Mountain View, CA, USA) were performed, according to the manufacturer’s protocol. Briefly, at 24 h after treatment, the cells were collected and resuspended in 500 μl binding buffer containing 5 μl annexin-V/FITC and 5 μl PI, and subsequently incubated for 5 min in the dark at room temperature. Analysis was immediately performed using a flow cytometer (BD Biosciences, San Diego, CA, USA). After the cells were treated as indicated, 10 μM DCFH-DA was added according to the manufacturer’s protocol, and fluorescence intensity indicating ROS content was measured using flow cytometry.
ELISA
The experimental procedure was performed using an ELISA kit (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s instructions. HUVECs were treated with 1 μg/ml LPS in the absence or presence of rosiglitazone (10 μM) for 24 h. Cultured supernatants were collected and subjected to ELISA for measurement of TNF-α, IL-6, CXCL12, and CXCR4.
Real-time PCR assay
Total RNA was prepared from the tissue samples or breast cancer cell lines by using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). First-strand cDNA was synthesized from total RNA with the M-MLV reverse transcriptase (Promega, Madison, WI, USA) following the manufacturer’s manual. Quantitative real-time PCR was performed using the SYBR Green mix (Thermo Fisher Scientific) on the ABI Prism 7300 sequence detection system (Applied Biosystems, Foster City, CA, USA). Primer sequences used were as follows:
CXCL12, 5′-TTCCCAATTCACATCTAACC-3′
and 5′-TCACATAGCACATTGTTCTC-3′;
CXCR4, 5′-ACAGTCAGAGGCCAAGGAAG-3′
and 5′-GGAACACAACCACCCACAAG-3′;
GAPDH, 5′-CACCCACTCCTCCACCTTTG-3′
and 5′-CCACCACCCTGTTGCTGTAG-3′.
Relative quantification of the signals was performed by normalizing the signals of different genes with that of GAPDH. The gene expression was calculated using the 2−ΔΔCT method.
Western blotting
After being fully lysed, cells were centrifuged at 12 000 rpm for 15 min at 4°C and the supernatant was collected. BCA protein quantification kit (BCA, thermo, Shanghai) was used to quantify the protein contents. Then, cell samples (15 μl) were run on 12% SDS-PAGE gel and transferred to a nitrocellulose filter membrane (Millipore, Shanghai, China) electrophoretically. Blots were blocked with 5% skim milk at room temperature for 1 h, followed by incubation with primary antibodies against CXCL12 (cat. no., ab155090; dilution, 1: 8000; Abcam, Cambridge, MA, USA), CXCR4 (cat. no., ab124824; dilution, 1: 100; Abcam), PPARγ (cat. no., ab191407; dilution, 1: 1000; Abcam), Bcl-2 (cat. no., Sc-492; dilution, 1: 300; Santa Cruz Biotechnology, Dallas, TX, USA), Bax (cat. no., Sc-493; dilution, 1: 300; Santa Cruz Biotechnology), p-JAK2 (cat. no., ab108596; dilution, 1: 500; Abcam), JAK2 (cat. no., ab195055; dilution, 1: 1000; Abcam), p-STAT3 (cat. no., ab30647; dilution, 1: 1000; Abcam), STAT3 (cat. no., ab68153; dilution, 1: 2000; Abcam), and GAPDH (cat. no. 5174; dilution, 1: 2000; Cell Signaling Technology, Inc., Danvers, MA, USA). After incubation with HRP-linked secondary antibodies (cat. no. A0208 and A0216; 1: 1000; Beyotime Institute of Biotechnology, Haimen, China) for 1 h at 37°C, blots were detected using the Clarity Western ECL kit (Bio-Rad Laboratories Inc., Richmond, CA, USA). Band intensities were quantified by using ImageJ software.
Statistical analysis
All analyses were done by t test, unless stated otherwise. Data are presented as mean ±S.D. Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA). Differences were considered significant when P value was less than 0.05.
Results
Cytotoxicity of LPS to HUVECs
Cell viability was assayed to evaluate the cytotoxicity of LPS to HUVECs. Treatment of HUVECs with 1 μg/ml LPS for 24, 48, and 72 h significantly inhibited the cell viability by 15.7%, 31.2%, and 49.3%, respectively (P<0.01; Figure 1). However, rosiglitazone treatment at a concentration of 10 μM, in addition to LPS stimulation for 24, 48, and 72 h, markedly increased cell viability of HUVECs by 4.7%, 22.7%, and 44.1%, respectively (P<0.05, P<0.01; Figure 1). These results suggest that rosiglitazone reduces LPS-induced decrease in cell viability of HUVECs.
Figure 1.
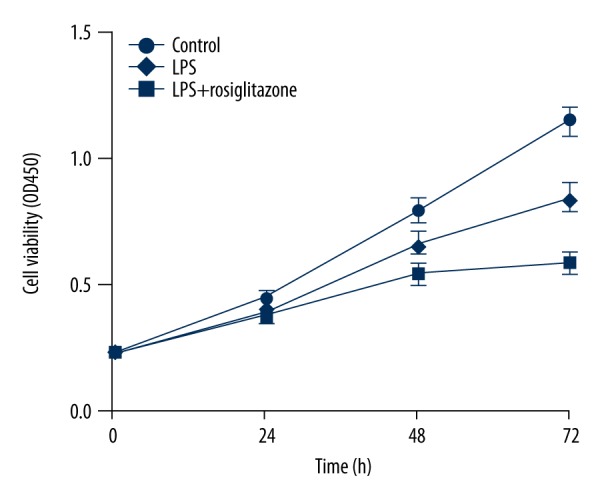
Rosiglitazone inhibited LPS-induced cell viability reduction in HUVECs. HUVECs were treated with 1 μg/ml LPS in the absence or presence of rosiglitazone (10 μM) for 0, 24, 48, or 72 h. Cell viability was measured by MTT assay.
Induction of apoptosis and ROS production by LPS in HUVECs
To determine the effects of LPS on cell apoptosis and ROS release, flow cytometry analysis was carried out. Exposure to 1 μg/ml LPS for 24 h obviously increased the number of apoptotic cells in HUVECs by 7.4-fold, and 10 μM rosiglitazone treatment attenuated the increase by 62.2% (P<0.01; Figure 2A, 2B). Exposure to 1 μg/ml LPS for 24 h obviously increased the content of ROS in HUVECs by 3.5-fold, and 10-μM rosiglitazone treatment attenuated the increase by 45.9% (P<0.01; Figure 2C, 2D). These results indicate that rosiglitazone reduces LPS-induced increases in cell apoptosis and ROS production of HUVECs.
Figure 2.

Rosiglitazone inhibited LPS-induced cell apoptosis and ROS production in HUVECs. HUVECs were treated with 1 μg/ml LPS in the absence or presence of rosiglitazone (10 μM) for 24 h. Cell apoptosis (A, B) and ROS production (C, D) were measured by flow cytometry. ** P<0.01 compared with control; ## P<0.01 compared with LPS.
Induction of pro-inflammatory cytokine secretion by LPS in HUVECs
To determine the effects of LPS on inflammation, the contents of pro-inflammatory cytokines, including TNF-α, IL-6, CXCL12, and CXCR4, were analyzed by ELISA, real-time PCR, and Western blotting. ELISA showed that the concentration of TNF-α, IL-6, CXCL12, and CXCR4 was significantly increased by 2.5-, 1.9-, 1.5-, and 1.8-fold, respectively, in HUVECs in response to 1 μg/ml LPS for 24 h, and 10 μM rosiglitazone treatment attenuated the increase of TNF-α, IL-6, CXCL12, and CXCR4 concentration by 34.9%, 27.0%, 40.1%, and 41.6%, respectively (P<0.01; Figure 3A–3D). Using real-time PCR and Western blotting assay, similar results were also obtained (P<0.01; Figure 3E, 3F). These results demonstrate that rosiglitazone reduces LPS-induced increases in TNF-α, IL-6, CXCL12, and CXCR4 content in HUVECs.
Figure 3.

Rosiglitazone inhibited LPS-induced pro-inflammatory cytokine levels in HUVECs. HUVECs were treated with 1 μg/ml LPS in the absence or presence of rosiglitazone (10 μM) for 24 h. Contents of pro-inflammatory cytokine, including TNF-α (A), IL-6 (B), CXCL12 (C), and CXCR4 (D), were measured by ELISA. Expression of CXCL12 and CXCR4 was also measured by real-time PCR (E) and Western blot assay (F). ** P<0.01 compared with control; ## P<0.01 compared with LPS.
Induction of JAK2/STAT3 activation by LPS in HUVECs
Effects of LPS on the phosphorylation of JAK2 and STAT3 were determined to evaluate the signal-transducing mechanisms of apoptosis and inflammation. Exposure of HUVECs to 1 μg/ml LPS for 3 h obviously increased the levels of phosphorylated JAK2 and STAT3 (Figure 4A). Levels of phosphorylated JAK2 and STAT3 in HUVECs were increased by 4.9- and 2.8-fold, respectively, and 10-μM rosiglitazone treatment attenuated the increase by 34.2% and 38.9%, respectively (P<0.01). These results suggest that rosiglitazone reduces LPS-induced activation of JAK2/STAT3 in HUVECs.
Figure 4.

Rosiglitazone inhibited LPS-induced JAK2/STAT3 activation and the expression of Bax/Bcl-2 in HUVECs. HUVECs were treated with 1 μg/ml LPS in the absence or presence of rosiglitazone (10 μM) for 3 or 24 h. Protein expression of p-JAK2, JAK2, p-STAT3, and STAT3 was measured by Western blot assay (A). Protein expression of PPARγ, Bcl-2, and Bax was also measured by Western blot assay (B). ** P<0.01 compared with control; ## P<0.01 compared with LPS.
Reduction of PPARγ and Bcl-2/Bax ratio by LPS in HUVECs
To evaluate the downstream events of LPS-involved JAK2/STAT3 activation, the expression of Bcl-2, Bax, and PPARγ was immunodetected (Figure 4B). Exposure of HUVECs to 1 μg/ml LPS for 24 h decreased the levels of PPARγ and Bcl-2 by 43.8% and 55.1%, respectively, but increased the level of Bax by 4.3-fold, and 10-μM rosiglitazone treatment increased the levels of PPARγ and Bcl-2 by 1.4- and 0.45-fold, respectively, and decreased the level of Bax by 40.4% (P<0.01). These results indicate that rosiglitazone inhibits LPS-induced reduction of PPARγ and Bcl-2/Bax ratio in HUVECs.
Discussion
The main pulmonary issue in ARDS is failure of the semipermeable endothelial barrier between blood and interstitial spaces, leading to increased endothelial permeability with interstitial and later alveolar edema [19]. Microvascular endothelial cells play an important role in the disruption of the blood vessel barrier, leading to the release of pro-inflammatory cytokines [20]. Similar to results of a previous study [21], HUVECs, which served as a model for the macrovasculature, were treated with LPS as a suitable in vitro ARDS model. The present study investigated the mechanism by which the PPARγ agonist rosiglitazone suppresses damage to HUVECs. Rosiglitazone treatment protected HUVECs from LPS-induced cell apoptosis, ROS production, and inflammatory cytokine releases, as well as signaling transduction.
Increased cell viability, decreased cell apoptosis, and ROS production in HUVECs were demonstrated by rosiglitazone treatment in addition to stimulation with LPS. The present results of the anti-apoptotic and inhibitory effects of rosiglitazone on ROS production are in line with previous studies. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ -induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production [22]. Rosiglitazone promotes TNF-related apoptosis-inducing ligand-induced apoptosis by ROS-mediated upregulation of death receptor 5 and downregulation of c-FLIP [23]. Rosiglitazone and PPARγ overexpression protect mitochondrial membrane potential and prevent apoptosis by upregulating anti-apoptotic Bcl-2 family proteins [24]. Similarly, the present study demonstrated that treatment of HUVECs with rosiglitazone significantly inhibited LPS-induced increase in the ratio of Bax/Bcl-2 protein expression. LPS can potentially be human pathogens. Sharifi et al. [25], studying the effect of LPS on cytotoxicity and apoptosis in PC12 neuronal cells, showed that LPS can cause PC12 cell death, in which apoptosis plays an important role, possibly by the mitochondrial pathway through higher expression of Bax and caspase 3 protein, while the expression of Bcl-2 protein was not changed significantly.
Moreover, several studies have demonstrated the anti-inflammatory potential of rosiglitazone in vitro [26] and in vivo [27]. The clinical prognosis of ARDS correlates with the extent of the intra-alveolar inflammation that survive ARDS demonstrate reduced levels of inflammatory cytokines compared to nonsurvivors [28]. In the present study, we found that rosiglitazone significantly decreased the levels of TNF-α, IL-6, CXCL12, and CXCR4 in LPS-induced HUVECs, consistent with our findings that pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 were found arise in the early phase of inflammation and play a crucial role in ARDS [29]. The chemokine CXCL12 binds to the chemokine receptor CXCR4 and the CXCL12-CXCR4 interaction may contribute to augmentation of inflammation [30] and is associated with the pathological development of acute inflammation after pulmonary fibrosis [31].
The JAK (Janus kinase)/STAT (signal transducer and activator of transcription) cascade is an essential inflammatory signaling pathway that mediates the immune responses. Previous studies demonstrated that JAK2/STAT3 activation mediated IL-6 and TGF-β1 expression in pancreatic inflammation [32] and inhibition of IL-6 and TNF-α-induced JAK2/STAT3 activation protects the lungs against severe acute pancreatitis-associated acute lung injury [33]. STAT3 phosphorylation was increased by CXCL12 stimulation, which involved interactions between CXCR4 and JAK2 in small cell lung cancer cells [34]. In addition, inhibition of JAK2/STAT3 pathways attenuated lung damage through decreasing apoptotic cells in limb I/R-induced acute lung injury [35]. In the present study, we also found that LPS significantly induced JAK2/STAT3 activation and rosiglitazone inhibited LPS-induced JAK2/STAT3 activation in HUVECs.
Conclusions
In conclusion, the present study demonstrated that in vitro PPARγ was involved in the pathogenesis of ARDS. Downregulation of the LPS-mediated JAK2/STAT3 signaling cascade attenuated cell apoptosis and inflammatory responses. Further elucidation of the molecular mechanisms underlying ARDS will be crucial for the development of novel treatments in the future.
Footnotes
Conflict of interest
None.
Source of support: This work was funded by the Major Research Project Fund from Wuxi Municipal Health and Family Planning Commission (Z201601)
References
- 1.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–40. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Randolph AG. Management of acute lung injury and acute respiratory distress syndrome in children. Crit Care Med. 2009;37:2448–54. doi: 10.1097/CCM.0b013e3181aee5dd. [DOI] [PubMed] [Google Scholar]
- 3.Copetti R, Soldati G, Copetti P. Chest sonography: A useful tool to differentiate acute cardiogenic pulmonary edema from acute respiratory distress syndrome. Cardiovasc Ultrasound. 2008;6:16. doi: 10.1186/1476-7120-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ware LB, Matthay MA, Parsons PE, et al. Pathogenetic and prognostic significance of altered coagulation and fibrinolysis in acute lung injury/acute respiratory distress syndrome. Crit Care Med. 2007;35:1821–28. doi: 10.1097/01.CCM.0000221922.08878.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brigham KL. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Curr Infect Dis Rep. 2005;7:327–28. doi: 10.1007/s11908-005-0004-2. [DOI] [PubMed] [Google Scholar]
- 6.Song L, Zhu Y, Jin M, Zang B. Hydroxysafflor yellow a inhibits lipopolysaccharide-induced inflammatory signal transduction in human alveolar epithelial A549 cells. Fitoterapia. 2013;84:107–14. doi: 10.1016/j.fitote.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 7.Dunn DL. Role of endotoxin and host cytokines in septic shock. Chest. 1991;100:164S–68S. doi: 10.1378/chest.100.3_supplement.164s. [DOI] [PubMed] [Google Scholar]
- 8.Zhang D, Chen L, Li S, et al. Lipopolysaccharide (LPS) of Porphyromonas gingivalis induces IL-1β, TNF-α and IL-6 production by THP-1 cells in a way different from that of Escherichia coli LPS. Innate Immun. 2008;14:99–107. doi: 10.1177/1753425907088244. [DOI] [PubMed] [Google Scholar]
- 9.Meduri GU, Headley S, Kohler G, et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS: Plasma IL-1β and IL-6 levels are consistent and efficient predictors of outcome over time. Chest. 1995;107:1062–73. doi: 10.1378/chest.107.4.1062. [DOI] [PubMed] [Google Scholar]
- 10.Martin TR, Hagimoto N, Nakamura M, Matute-Bello G. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc. 2005;2:214–20. doi: 10.1513/pats.200504-031AC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L899–914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- 12.Lee S, Kim W, Kang KP, et al. Agonist of peroxisome proliferator-activated receptor-γ, rosiglitazone, reduces renal injury and dysfunction in a murine sepsis model. Nephrol Dial Transplant. 2005;20:1057–65. doi: 10.1093/ndt/gfh705. [DOI] [PubMed] [Google Scholar]
- 13.Sha L, YuJie G, WeiQing J, HongGuang B. Effects of rosiglitazone on outcome in mice with sepsis. Prog Mod Biomed. 2010;10:4252–54. 4294. [Google Scholar]
- 14.Cree MG, Zwetsloot JJ, Herndon DN, et al. Insulin sensitivity and mitochondrial function are improved in children with burn injury during a randomized controlled trial of fenofibrate. Ann Surg. 2007;245:214–21. doi: 10.1097/01.sla.0000250409.51289.ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosmann M, Ward PA. The inflammatory response in sepsis. Trends Immunol. 2013;34:129–36. doi: 10.1016/j.it.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D, Zeng B, Zhang S, Yao S. Rosiglitazone, an agonist of peroxisome proliferator-activated receptor γ, reduces pulmonary inflammatory response in a rat model of endotoxemia. Inflamm Res. 2005;54:464–70. doi: 10.1007/s00011-005-1379-0. [DOI] [PubMed] [Google Scholar]
- 17.Chen C, Xu S, Wang WX, et al. Rosiglitazone attenuates the severity of sodium taurocholate-induced acute pancreatitis and pancreatitis-associated lung injury. Arch Med Res. 2009;40:79–88. doi: 10.1016/j.arcmed.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 18.Cherng YG, Chang HC, Lin YL, et al. Apoptotic insults to human chondrocytes induced by sodium nitroprusside are involved in sequential events, including cytoskeletal remodeling, phosphorylation of mitogen-activated protein kinase kinase kinase-1/c-Jun N-terminal kinase, and Bax-Mitochondria-Mediated caspase activation. J Orthop Res. 2008;26:1018–26. doi: 10.1002/jor.20578. [DOI] [PubMed] [Google Scholar]
- 19.Herwig MC, Tsokos M, Hermanns MI, et al. Vascular endothelial cadherin expression in lung specimens of patients with sepsis-induced acute respiratory distress syndrome and endothelial cell cultures. Pathobiology. 2013;80:245–51. doi: 10.1159/000347062. [DOI] [PubMed] [Google Scholar]
- 20.Bogatcheva NV, Zemskova MA, Kovalenkov Y, et al. Molecular mechanisms mediating protective effect of cAMP on lipopolysaccharide (LPS)-induced human lung microvascular endothelial cells (HLMVEC) hyperpermeability. J Cell Physiol. 2009;221:750–59. doi: 10.1002/jcp.21913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Zhang L, Jin Z, et al. Anti-apoptotic PTD-FNK protein suppresses lipopolysaccharide-induced acute lung injury in rats. Exp Mol Pathol. 2007;83:377–84. doi: 10.1016/j.yexmp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Jung TW, Lee JY, Shim WS, et al. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J Neurol Sci. 2007;253:53–60. doi: 10.1016/j.jns.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 23.Kim YH, Jung EM, Lee TJ, et al. Rosiglitazone promotes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by reactive oxygen species-mediated up-regulation of death receptor 5 and down-regulation of c-FLIP. Free Radic Biol Med. 2008;44:1055–68. doi: 10.1016/j.freeradbiomed.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 24.Wu JH, Lin TA, Wu KK. Rosiglitazone and PPAR-gamma overexpression protect mitochondrial membrane potential and prevent apoptosis by upregulating anti-poptotic Bcl-2 family proteins. J Cell Physiol. 2009;220:58–71. doi: 10.1002/jcp.21730. [DOI] [PubMed] [Google Scholar]
- 25.Sharifi AM, Hoda FE, Noor AM. Studying the effect of LPS on cytotoxicity and apoptosis in PC12 neuronal cells: Role of Bax, Bcl-2, and Caspase-3 protein expression. Toxicol Mech Methods. 2010;20:316–20. doi: 10.3109/15376516.2010.486420. [DOI] [PubMed] [Google Scholar]
- 26.Zhang LL, Gao CY, Fang CQ, et al. PPARγ attenuates intimal hyperplasia by inhibiting TLR4-mediated inflammation in vascular smooth muscle cells. Cardiovasc Res. 2011;92:484–93. doi: 10.1093/cvr/cvr238. [DOI] [PubMed] [Google Scholar]
- 27.Fidan E, Ersoz HO, Yilmaz M, et al. The effects of rosiglitazone and metformin on inflammation and endothelial dysfunction in patients with type 2 diabetes mellitus. Acta Diabetol. 2011;48:297–302. doi: 10.1007/s00592-011-0276-y. [DOI] [PubMed] [Google Scholar]
- 28.Minamino T, Komuro I. Regeneration of the endothelium as a novel therapeutic strategy for acute lung injury. J Clin Invest. 2006;116:2316–19. doi: 10.1172/JCI29637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giebelen IA, van Westerloo DJ, Larosa GJ, et al. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock. 2007;28:700–3. doi: 10.1097/shk.0b013e318054dd89. [DOI] [PubMed] [Google Scholar]
- 30.Dotan I, Werner L, Vigodman S, et al. CXCL12 is a constitutive and inflammatory chemokine in the intestinal immune system. Inflamm Bowel Dis. 2010;16:583–92. doi: 10.1002/ibd.21106. [DOI] [PubMed] [Google Scholar]
- 31.Shu HK, Yoon Y, Hong S, et al. Inhibition of the CXCL12/CXCR4-axis as preventive therapy for radiation-induced pulmonary fibrosis. PLoS One. 2013;8:750–52. doi: 10.1371/journal.pone.0079768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ji HY, Kim KH, Kim H. SOCS 3 and PPAR-γ ligands inhibit the expression of IL-6 and TGF-β1 by regulating JAK2/STAT3 signaling in pancreas. Int J Biochem Cell Biol. 2008;40:677–88. doi: 10.1016/j.biocel.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 33.Han X, Wang Y, Chen H, et al. Enhancement of ICAM-1 via the JAK2/STAT3 signaling pathway in a rat model of severe acute pancreatitis-associated lung injury. Exp Ther Med. 2016;11:788–96. doi: 10.3892/etm.2016.2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pfeiffer M, Hartmann TN, Leick M, et al. Alternative implication of CXCR4 in JAK2/STAT3 activation in small cell lung cancer. Br J Cancer. 2009;100:1949–56. doi: 10.1038/sj.bjc.6605068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhao YR, Wang D, Liu Y, et al. The PI3K/Akt, p38MAPK, and JAK2/STAT3 signaling pathways mediate the protection of SO2 against acute lung injury induced by limb ischemia/reperfusion in rats. J Physiol Sci. 2016;66:1–11. doi: 10.1007/s12576-015-0418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]