

Abstract
Photoreceptor dysplasia, characterized by formation of folds and (pseudo-)rosettes in the outer retina, is associated with loss of functional nuclear receptor subfamily 2 group E member 3 (NR2E3) and neural retina leucine-zipper (NRL) in both humans and mice. A sensitized chemical mutagenesis study to identify genetic modifiers that suppress photoreceptor dysplasia in Nr2e3rd7mutant mice identified line Tvrm222, which exhibits a normal fundus appearance in the presence of the rd7 mutation. The Tvrm222 modifier of Nr2e3rd7/rd7 was localized to Chromosome 6 and identified as a missense mutation in the FERM domain containing 4B (Frmd4b) gene. The variant is predicted to cause the substitution of a serine residue 938 with proline (S938P). The Frmd4bTvrm222 allele was also found to suppress outer nuclear layer (ONL) rosettes in Nrl–/– mice. Fragmentation of the external limiting membrane (ELM), normally observed in rd7 and Nrl–/–mouse retinas, was absent in the presence of the Frmd4bTvrm222 allele. FRMD4B, a binding partner of cytohesin 3, is proposed to participate in cell junction remodeling. Its biological function in photoreceptor dysplasia has not been previously examined. In vitro experiments showed that the FRMD4B938P variant fails to be efficiently recruited to the cell surface upon insulin stimulation. In addition, we found a reduction in protein kinase B phosphorylation and increased levels of cell junction proteins, Catenin beta 1 and tight junction protein 1, associated with the cell membrane in Tvrm222 retinas. Taken together, this study reveals a critical role of FRMD4B in maintaining ELM integrity and in rescuing morphological abnormalities of the ONL in photoreceptor dysplasia.
Introduction
Neuroretinal dysplasia is a congenital maldevelopment of the retina, characterized by undulation of neuroretinal layers with formation of folds and rosettes and gliosis (1,2). The dysplastic lesions can result from environmental insults and/or occur in genetic diseases such as Meckel, Joubert and enhanced S-cone syndromes (ESCS) in human populations (3–5).
Neural retina leucine-zipper (NRL) and nuclear receptor subfamily 2 group E member 3 (NR2E3) are two critical components of the hierarchical transcriptional pathway necessary for proper photoreceptor development. It has been reported that NRL dictates photoreceptor precursors to commit to rod photoreceptors by inducing expression of rod-specific genes (6). NRL deficiency causes an increased number of cone-like cells at the expense of rods because of a skewed differentiation of immature precursor cells to cone cells in mouse retina (7,8). NR2E3 on the other hand, is transcriptionally regulated by NRL and can divert cell fate commitment to the rod lineage by suppressing cone cell genes (9). Lack of NR2E3 results in excessive blue cone photoreceptors, which arise from photoreceptor precursors (10). In mouse retinas, loss of functional NRL or NR2E3 causes dysplastic lesions in Nrl–/– and homozygous retinal degeneration (rd) 7 mutant mice, respectively (7,11). Clinically, patients harboring either NRL or NR2E3 mutations manifest early-onset night blindness, impaired visual acuity and enhanced sensitivity to short-wavelength light that is often associated with ESCS (12,13). No treatment is currently available (14,15). Thus, a clearer understanding of the pathogenic mechanisms underlying the disease is vital for developing potential therapeutic interventions.
The retina originates from the neuroepithelium and develops into a tissue with laminar structure. Tissue assembly and maintenance of the outer retina largely depends on proper differentiation of photoreceptors and establishment of cell–cell contacts with their neighboring Müller glial cells (16,17). These cell contacts form the external limiting membrane (ELM), a belt-like structure composed of cell junctions between Müller glia and the inner segments (ISs) of photoreceptors, which is critical for maintaining structural integrity of the photoreceptor layer and selective diffusion into the inner retina (18). Previous studies have shown that a defective ELM is often associated with dysplastic folds and pseudo-rosettes in Crumbs 1 (Crb1) rd8 homozygous (19) and Nrl deficient mice (20).
In the present study, we characterized a mouse model, Tvrm222, identified in a sensitized N-ethyl-N-nitrosourea (ENU) mutagenesis study of rd7, in which the aberrant morphology associated with photoreceptor dysplasia was largely ameliorated. The molecular basis of Tvrm222 was identified as a missense variant in the FERM domain containing 4B (Frmd4b) gene, which encodes FRMD4B, a binding partner of Cytohesin 3 (CYTH3/GRP1: general receptor of phosphoinositides 1) (21). This variant is predicted to cause a substitution of serine 938 by proline (S938P) in the protein. The Frmd4bTvrm222 allele is also capable of suppressing photoreceptor dysplasia in Nrl–/– mice, suggesting a broader impact of FRMD4B on modifying dysplastic retinal lesions. In both models, suppression of photoreceptor dysplasia was accompanied by a normalization of ELM structure. In concert with the observed rescue of ELM fragmentation, we documented augmentation of cell surface-associated zonula adherens (ZA) and zonula occludens (ZO) proteins in the Tvrm222 mouse eyes, suggesting that Frmd4bTvrm222 allele facilitates maintenance of ELM integrity. At the molecular level, in vitro studies show that the FRMD4B938P variant displays reduced cell surface targeting upon insulin stimulation, compared with its wild-type counterpart. In addition, we noted a reduced phosphorylation of protein kinase B (AKT) in the retina of mice homozygous for Frmd4bTvrm222 allele.
Collectively, our data support a role of FRMD4B as a key player in maintaining the ELM integrity and protecting retinal architecture against formation of dysplastic lesions.
Results
Tvrm222, generated by ENU mutagenesis, suppresses photoreceptor dysplasia in rd7/rd7 mice
Homozygous B6.Cg-Nr2e3rd7/J mice display a pan-retinal fundus spotting phenotype. However, Nr2e3rd7/rd7 offspring on mixed genetic backgrounds often exhibit a variable degree of spotting (22), suggesting the existence of genetic modifiers. To identify such rd7 modifiers, we conducted a sensitized ENU mutagenesis screen using B6.Cg-Nr2e3rd7/J mutant mice, and have screened and established a number of mouse lines with altered retinal phenotypes. Mice from one of these lines, Tvrm222, displayed a significant reduction of retinal spots compared with B6.Cg-Nr2e3rd7/J homozygotes (Fig. 1A–C, G; note: all of the Tvrm222 mice referred to in this study were homozygous for both rd7 and modifier alleles, unless otherwise indicated). Consistent with fewer retinal spots in fundus images, retinal folds as observed by light microscopy were significantly reduced in Tvrm222 eyes (Fig. 1F), compared with rd7 homozygotes in which retinal dysplastic lesions are abundant (Fig. 1E). The retinal folds are widespread in Nr2e3rd7/rd7 mouse retinas at 2 weeks of age compared with that found in Tvrm222 retinas (Fig. 1H–J) and also at 6 months of age (Fig. 1K–M), suggesting that the suppression in Tvrm222 retinas begins during development rather than as a consequence of cell death occurring as the mutant ages (23).
Figure 1.

Tvrm222 mice show suppression of rd7-associated photoreceptor dysplasia. Fundus images (A–C; quantification of the numbers of dysplastic lesions observed in fundus images of the central retina are shown in G) and posterior eye sections stained with H&E (D–F) from 1-month-old WT, Nr2e3rd7/rd7, and Nr2e3rd7/rd7; Frmd4bTvrm222/Tvrm222 (Tvrm222) mice. Note spotting phenotype in the rd7 fundus image (B) and the corresponding retinal folds or lesions shown in histological sections (E). Scale bar=100μm. For quantification in (G), the results are mean±SD, n = 5 (WT), 6 (rd7) or 13 (Tvrm222); **P < 0.0001 (one-way ANOVA, post-hoc Tukey’s test). (H-M) Representative retinal sections from 2-week-old (H–J) and 6-month-old (K–M) WT, Nr2e3rd7/rd7 and Tvrm222 mice are shown by H&E staining. Note the undulation of the retinal layers in the Nr2e3rd7/rd7 mice and the absence of retinal folds in sections from Tvrm222 mice at both ages surveyed. Scale bar=20μm. IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium.
Human ESCS patients show early-onset night blindness and enhanced cone response (13,24). Therefore, in addition to the morphological characterizations, outer retinal function in Tvrm222 mice was assessed by electroretinography (ERG) at 1.5 months. However, no marked photopic (10 cd s/m2) or scotopic (0.25 cd s/m2) ERG modifications were recorded in Tvrm222 mice relative to Nr2e3rd7/rd7 (Supplementary Material, Fig. S1A–D) and wild-type mice. Since RD has been documented in ESCS patients, we additionally measured the thickness of the outer nuclear layer (ONL). The results revealed a progressive degeneration found in both Nr2e3rd7/rd7 and Tvrm222 mice with age, in comparison to the wild-type mice (Supplementary Material, Fig. S1E and F).
The Tvrm222 variant does not affect cell proliferation and gliosis in Nr2e3rd7/rd7 retina
NR2E3, as a transcription factor, promotes rod cell commitment by inhibiting cone-specific genes. Previous studies have indicated that loss of NR2E3 leads to an increased number of blue cones in mouse retinas, which has been suggested to contribute to formation of retinal folds in Nr2e3rd7/rd7 mice (11,25). We, therefore, tested whether the Tvrm222 variant suppresses photoreceptor dysplasia by inhibiting aberrant cell proliferation. Immunofluorescence staining for phospho-histone 3 (PH3), a cell proliferation marker, showed a significant increase in the number of PH3-positive cells in Nr2e3rd7/rd7 retinas compared with the wild-type (Fig. 2A–D). However, the retinas of rd7 homozygotes carrying the Tvrm222 locus in homozygosity as well also displayed increased numbers of PH3-positive cells compared with controls, similar to that observed in the Nr2e3rd7/rd7 retinas (Fig. 2E and F). No significant differences in the number of PH3-positive cells between the Nr2e3rd7/rd7 and Tvrm222 mice were observed (Fig. 2G), suggesting that the modifying effect of the Tvrm222 allele does not affect cell proliferation. This finding was further validated by staining wild-type (WT), Nr2e3rd7/rd7 and Tvrm222 retinas for blue opsin. Compared with WT, a mild increase of blue opsin was observed in Nr2e3rd7/rd7retinas, which was not reduced in Tvrm222 (Supplementary Material, Fig. S2A–D). This suggests that neither excessive cell number nor blue cone photoreceptors play a major role in the Tvrm222 modification of rd7-associated photoreceptor dysplasia.
Figure 2.

Neither proliferation of retinal cells nor Müller gliosis is associated with the phenotypic modification by Tvrm222. Cell proliferation in 5-day-old (P5) retina from WT (A, B), Nr2e3rd7/rd7 (C, D) and Tvrm222 (E, F) mice was assessed by immunofluorescence staining using an antibody against phosphorylated histone (PH) 3. Stained cells were counted and analyzed by one-way ANOVA with Tukey’s test for multiple comparisons (G). The results are mean±SD. n = 3 for each mouse strain; **P < 0.01. Scale bar=200µm (×10) or 20 µm (×63). (H–M) PNA and ARR3 shown by immunofluorescence staining using antibodies against PNA (green) and ARR3 (red). Note the localization of cone cell bodies (arrows). Genotypes of mice used are as described above. Scale bar=50 µm (×20) or 20 µm (×63). (N) Representative western blot probed with anti-ARR3. GAPDH was used as the loading control. (O–T) Eye sections from 2-week-old mice of different genotypes were stained with an antibody against SRY Box 9 (SOX9), a Müller cell marker. Note the localization of somata of Müller glia (arrows). Scale bar=50 µm (×20) or 20 µm (×63). (U) Representative western blot probed with anti-SOX9. GAPDH was used as the loading control. (V–Y) Glial activation was assessed by staining of retinas from 2-week-old mice (genotypes as described above) with a GFAP antibody. Scale bar=20 µm. IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer.
Loss of NR2E3 leads to abnormal photoreceptor development and morphology. Labeling of cone photoreceptors with arrestin 3 (ARR3, a.k.a. cone arrestin), a marker for cone cell body and peanut agglutinin (PNA), which stains plasma membrane near cone IS and outer segment (OS), showed abundant mislocalization of cone photoreceptors in the ONL in both Nr2e3rd7/rd7 and Tvrm222 retinas, in contrast to those in WT retinas where cones are predominantly localized near the apical terminus of the ONL (Fig. 2H–M). Furthermore, a large number of ovoid-shaped cone IS/OS in both Nr2e3rd7/rd7 and Tvrm222 retinas was observed, compared with the elongated cone IS/OS in WT retinas (Supplementary Material, Fig. S2E–G). In addition, the expression level of ARR3 was not significantly different between the Nr2e3rd7/rd7 and Tvrm222 mice (Fig. 2N). Collectively, these results demonstrate that the Tvrm222 modifier does not correct the developmental abnormalities of photoreceptors seen in Nr2e3rd7/rd7 retinas.
Müller cell gliosis (26), another hallmark of photoreceptor dysplasia, was also examined. Anti-SRY-Box 9 (SOX9), an adult Müller cell marker, glutamine synthetase (GS), a protein that is exclusively expressed by Müller glia (27) and anti-glial fibrillary acidic protein (GFAP), a marker for activated glial cells were used to detect potential changes in the amount of Müller glia and their activation status. No difference was noticed in terms of localization of somata of Müller cells in the inner nuclear layer (INL) among the three different genotypes (Fig. 2O–T), as well as in the expression level of SOX9 assayed by western blot (Fig. 2U). This observation was further validated by immunofluorescence staining and western blot assays for GS (Supplementary Material, Fig. S2H–K).
We subsequently examined Müller gliosis in response to the dysplastic lesions. GFAP staining was prominent in Nr2e3rd7/rd7 retina relative to that found in WT retina (Fig. 2V and W), indicating presence of gliotic Müller cells. GFAP staining in Tvrm222 mouse retinas was markedly absent compared with that observed in Nr2e3rd7/rd7 eyes (Fig. 2X). However, residual staining was still detectable around the small number of remaining retinal folds (Fig. 2Y). This strongly suggests that activation of Müller glia observed in Nr2e3rd7/rd7 retinas is a response to the presence of dysplastic lesions and lack of Müller gliosis in Tvrm222 retinas is the consequence, rather than the overall cause of the absence of rd7-induced dysplasia in Tvrm222 mutants.
Defective ELM associated with Nr2e3rd7/rd7 photoreceptor dysplasia is rescued by the Tvrm222 variant
Since ELM fragmentation has been identified as a prominent feature associated with dysplastic lesions in mouse retinas, including Nrl–/– mice and Crb1rd8/rd8 mice (19,20), this prompted us to determine if Nr2e3rd7/rd7 showed a similar ELM fragmentation and if so, whether the Tvrm222 allele had an impact on the ELM phenotype. By comparing the ELM in the Nr2e3rd7/rd7 retina with that of Tvrm222 mutants by immunofluorescence staining using antibodies against Catenin beta 1 (CTNNB1) (β-catenin) and tight junction protein 1 (TJP1) (formerly ZO-1), we noted that the ELM in Nr2e3rd7/rd7 retinas appeared diffuse or fragmented compared with the compact and continuous staining of the ELM observed in the WT retinas (Fig. 3A, B, D and E). In striking contrast to the ELM in Nr2e3rd7/rd7 retinas, ELM integrity was preserved in Tvrm222 retinas (Fig. 3C and F).
Figure 3.

Suppression of photoreceptor dysplasia in Tvrm222 mice is associated with normalization of retinal lamination, and intact cell junctions at the ELM. (A–F) Retinal sections from 1-month-old WT, Nr2e3rd7/rd7 and Tvrm222 mice were subject to immunofluorescence staining using antibodies against CTNNB1 (β-catenin) (A–C) and TJP1 (ZO-1) (D–F), respectively. Scale bar=20 μm. The fragmentation of the ELM is marked by arrows. Ultra-structural assessment of the retinal architecture from 1-month-old WT (G, J), Nr2e3rd7/rd7 (H, K) and Tvrm222 (I, L) mice. The ELM is marked by yellow arrows. Scale bar=5 μm (×3000) or 2 μm (×10 000). IPL, inner plexiform layer; INL, inner nuclear layer; ONL, outer nuclear layer; IS, inner segment; OS, outer segment.
Ultra-structural assessment of the photoreceptor morphology was performed. Mouse eyes were collected at 1 month of age when retinal folds are abundant. Unlike the distinct ELM structure that forms a visible linear profile in the WT retinas (Fig. 3G and J), junctions comprising the ELM were missing and the IS/OS were distorted in Nr2e3rd7/rd7 retinas (Fig. 3H). Images at higher magnification further revealed diffuse junctional plaques, and aberrant photoreceptor organization with disordered IS and OS in the homozygous rd7 retinas, which was particularly striking at sites where dysplastic whorls were present (Fig. 3K). In contrast, the ELM structure appeared intact in the Tvrm222 retinas (Fig. 3I) and IS/OS morphology was preserved in these retinas without dysplastic folds (Fig. 3L). Collectively, our results demonstrate that disruption of ELM integrity is closely associated with photoreceptor dysplasia. More importantly, we found that the photoreceptor dysplasia was prevented, with intact ELM and distinct IS/OS in mice harboring the Tvrm222 allele.
Identification of an Frmd4b variant as the genetic suppressor of rd7
In order to determine the nature of the genetic suppression of rd7 by Tvrm222, we conducted genetic mapping and exome sequence analyses of Tvrm222 mice. An initial mapping position was obtained by quantitative trait locus (QTL) analysis of a cohort of 51 animals from a backcross of (C57BL/6N ×C57BL/6J.Cg-rd7/rd7; Tvrm222/Tvrm222) F1 × C57BL/6J.Cg-rd7/rd7 phenotyped by indirect ophthalmoscopy. A significant QTL was found on Chromosome 6 at 34.671 cM [LOD =2.537, P(F) =0.00063]. The location on Chromosome 6 was confirmed in a second cross of (DBA/2J x C57BL/6J.Cg-rd7/rd7; Tvrm222/Tvrm222) F1 ×C57BL/6J.Cg-rd7/rd7), in which 21 animals homozygous for rd7 were phenotyped. Marker D6Mit29 co-segregated with the suppressed phenotype. Whole exome sequencing of C57BL/6J.Cg-rd7/rd7; Tvrm222/Tvrm222 genomic DNA was carried out. After filtering the sequencing results for novel and significant mutations and re-sequencing to confirm the nucleotide changes, only a thymine to cytosine transversion in the Frmd4b gene co-segregated with Tvrm222 (Supplementary Material, Table S1). This variant is predicted to lead to a substitution of a serine residue 938 by proline (S938P) (Fig. 4A). The FRMD4BS938P variation is located in a threonine/serine rich domain near the carboxyl terminus of the protein. Cross-species alignment of mouse FRMD4B indicates that the residue corresponding to S938 is normally a proline in human (P991) and rat (P992) reference sequences (Fig. 4B).
Figure 4.

An Frmd4b variant was identified as the modifier that rescues rd7-associated photoreceptor dysplasia. (A) Sequencing traces around the Tvrm222 variation in the Frmd4b gene. The arrow indicates the transversion of thymine to cytosine (B) Alignment of human, rat and mouse FRMD4B sequences around the Tvrm222 variation. Note the proline (marked by an arrow) in human and rat reference sequences is substituted by a serine in mice. The Tvrm222 variation causes a replacement of the serine with proline.
Because a suitable FRMD4B antibody for western or immunostaining analysis was not available, tissue expression of Frmd4b at the transcript level was examined. Frmd4b expression was found in abundance in whole eye, retina, retinal pigment epithelium, brain, lung and colon (Supplementary Material, Fig. S3A).
In addition, we tested whether the Frmd4bTvrm222 variant alone caused any obvious retinal phenotypes in the absence of the Nr2e3rd7/rd7 mutation, by segregating the Frmd4bTvrm222 allele from the rd7 mutation through outcrossing with WT C57BL/6J mice. Based on histological examinations, no retinal phenotype was observed in either young (2 weeks) or adult (6 months) mouse retinas of Frmd4bTvrm222/Tvrm222 homozygotes in the absence of the Nr2e3rd7/rd7 mutation (Supplementary Material, Fig. S3B–G).
Frmd4bTvrm222 suppresses photoreceptor dysplasia in Nrl deficient mice
In order to examine the modifying effects of Frmd4bTvrm222 on other models in which photoreceptor dysplasia is observed, we crossed the model, after segregation from rd7, with Nrl deficient mice (Fig. 5A and C). We found that the Frmd4bTvrm222 allele was also able to suppress the photoreceptor dysplasia normally observed in the Nrl–/– mice (Fig. 5B and D). Notably, the Frmd4bTvrm222 variant was also able to suppress ELM fragmentation because of the Nrl deficiency indicated by continuous CTNNB1 and TJP1 staining (Fig. 5E–H).
Figure 5.
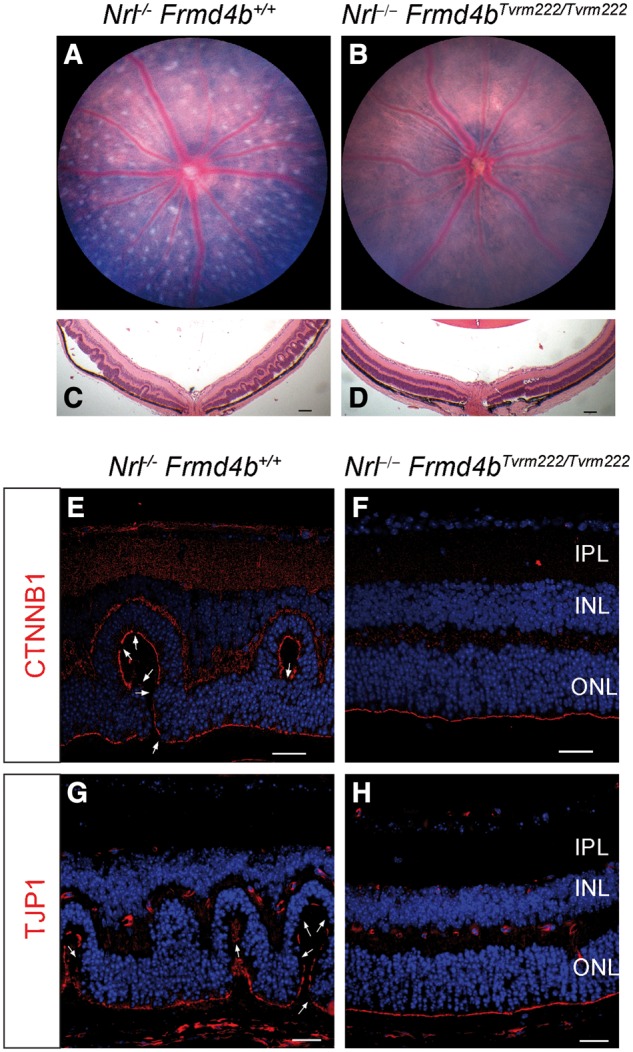
The Frmd4bTvrm222 allele suppresses photoreceptor dysplasia caused by ablation of Nrl. The fundus images of 1-month-old Nrl–/–(A) and Nrl–/–Frmd4bTvrm222/Tvrm222 (B) mice. Note that the Nrl–/– retina displays a spotting phenotype while the Nrl–/–Frmd4bTvrm222/Tvrm222 retina shows a remarkable reduction of the retinal spotting. H&E stained retinal sections from Nrl–/– (C) and Nrl–/–Frmd4bTvrm222/Tvrm222 (D) mice. Note a significant reduction in the number of dysplastic lesions in Nrl−/− mice harboring the Frmd4bTvrm222 allele. Scale bar=100μm. Immunofluorescence staining of Nrl–/– retinal sections with CTNNB1 and TJP1 show fragmentation of the ELM noted by the arrows (E, G). Such breaks were rarely observed in Nrl–/–Frmd4bTvrm222/Tvrm222 retinas (F, H). Scale bar=20 μm.
Membrane recruitment of FRMD4B is altered by the 938P variant
Previously, it has been reported that the majority of FRMD4B molecules exist as a complex with CYTH3. This complex is sequestered in the cytoplasm by serum starvation and is recruited to the plasma membrane upon stimulation with insulin (21). To probe the potential consequence of the S938P variation in FRMD4B, COS7 cells were transiently transfected with vectors expressing epitope-tagged CYTH3 and either WT FRMD4B or the 938P variant. Transfected cells were subjected to serum starvation before insulin supplementation. With serum starvation only, both the WT and the 938P variant were diffusely localized in the cytoplasm (Fig. 6A–D). After insulin treatment, WT FRMD4B translocated to the cell surface where it co-localized with the cell surface indicated by staining for sub-cortical actin fibers (Fig. 6E and F), consistent with previous findings (21). In contrast, a large proportion of the FRMD4B938P variant remained cytosolic even with insulin treatment (Fig. 6G and H). To confirm this observation, we also performed subcellular fractionation using protein extracts from HEK-293T cells transiently transfected with vectors expressing epitope-tagged CYTH3 and either WT FRMD4B or the 938P variant. Little difference in cell surface targeting between the WT FRMD4B and the 938P variant was observed without insulin treatment (Fig. 6I). However, more WT FRMD4B was partitioned to the membrane fraction compared with FRMD4B938P after insulin supplementation (Fig. 6J).
Figure 6.

Membrane recruitment of FRMD4B938P upon insulin stimulation is reduced in vitro (A–H). COS7 cells were co-transfected with 3×FLAG-CYTH3 and either 3×myc-FRMD4Bwt (A, B; E, F) or 3×myc-FRMD4B938P (C, D; G, H), and subject to serum starvation (without insulin; A–D) or serum starvation followed by treatment with 100 nM insulin (E–H). 3×myc-FRMD4B was detected by immunofluorescence using antibody against the myc tag (green). Counterstaining with phalloidin (red) shows sub-plasma membrane actin cytoskeleton. Note the cell surface associated FRMD4Bwt (arrows). Scale bar=20 μm. (I, J) Subcellular fractionation profiles of HEK-293T cells transiently co-transfected with 3×FLAG-CYTH3 and either 3×myc-FRMD4Bwt or 3×myc-FRMD4B938P, and untreated (I) or treated (J) with insulin after serum starvation. Na+/K+-ATPase (ATP1A1) was used as the loading control for membrane fractions. PNS, post-nuclear supernatant; S100, supernatant after 100 000×g centrifugation; P100, pellet after 100 000×g centrifugation.
Since it has been shown that the majority of intracellular FRMD4B binds to CYTH3, we further tested whether membrane recruitment of FRMD4B after insulin stimulation is dependent on CYTH3 and is affected by the S938P variation. We found that, in the absence of CYTH3, both exogenous WT FRMD4B and the FRMD4B938P variant were predominantly aggregated in the cytoplasm upon insulin stimulation (Supplementary Material, Fig. S4A–D), suggesting a dependence of FRMD4B on CYTH3 for membrane recruitment regardless of the variant (28). Therefore, we next asked whether the FRMD4B938P variant protein has a lower binding affinity for CYTH3 compared with WT FRMD4B, which could in turn lead to the reduced membrane recruitment of FRMD4B938P. By carrying out co-immunoprecipitation studies using epitope-tagged CYTH3 and FRMD4B expressed in HEK-293T cells, we found that the binding affinity between FRMD4B and CYTH3 was not affected by the FRMD4B938P variant with or without insulin stimulation (Fig. 7A and B), indicating that this variant does not affect the physical interaction with CYTH3. Hence, the reduced membrane recruitment of FRMD4B938P was not because of altered binding affinity between CYTH3 and FRMD4B.
Figure 7.

The S938P variation does not affect binding affinity between FRMD4B and CYTH3 in vitro (A, B). Co-immunoprecipitation of CYTH3 and FRMD4B with protein extracts prepared from HEK-293T cells. Cells were co-transfected with 3×FLAG-CYTH3 and myc-tagged HSP90B1 (GRP94) as a negative control (for non-specific MYC interactions), and 3×myc-FRMD4Bwt or 3×myc-FRMD4B938P. Cells were either untreated (A) or treated (B) with 100 nM insulin. Co-immunoprecipitation was performed on post-nuclear supernatant (PNS) using anti-FLAG M2 antibody conjugated agarose beads. UB, unbound; BD, bound.
The Frmd4bTvrm222 variant leads to reduced AKT phosphorylation and increased cell junction proteins in Tvrm222 retinas
Our identification of Tvrm222 as an allele of Frmd4b suggested two molecular phenotypes associated with development and cell junction. Because membrane recruitment of CYTH3–FRMD4B is triggered by insulin, a phosphoinositide 3-kinase (PI3K)-AKT pathway agonist, and both CYTH3 and AKT interact with phosphatidylinositol (3–5)-trisphosphate (PIP3) (29,30), a key component of the PI3K-AKT pathway, we examined whether the PI3K-AKT pathway is affected by altered CYTH3–FRMD4B membrane recruitment. Western blot analysis of post-nuclear protein extract from retina showed that the level of phosphorylated AKT (Ser473) in cytosol was significantly reduced in both Tvrm222 and WT mice homozygous for Frmd4bTvrm222 allele compared with their WT and rd7 counterparts (Fig. 8A and B). It indicates a hypoactive state of AKT in mice homozygous for Frmd4bTvrm222 allele and suggests a potential implication of AKT pathway in mediating the Tvrm222 modification of rd7-associated photoreceptor dysplasia.
Figure 8.

Phosphorylation of AKT is reduced and membrane-associated CTNNB1 and TJP1 are increased in Tvrm222 mouse retinas. (A, B) Post-nuclear protein extracts from mouse eyecups were analyzed by western blot, probed with the antibody against phosphorylated AKT (Ser473). Pan-AKT was used as the loading control. The western blotting results were quantitated as mean±SD, n = 5 for WT, Nr2e3rd7/rd7 and Tvrm222, n = 4 for Frmd4bTvrm222/Tvrm222; **P = 0.0004, *P = 0.0142 (one-way ANOVA, post hoc Tukey’s test). (C–F) Membrane fractionation of mouse eyecups with subsequent western blot analyses of each fraction probed with antibodies against CTNNB1 (C) and TJP1 (E). ATP1A1 was used as the loading control for membrane fractions. Note there is a statistically significant increase in membrane-associated (P100) CTNNB1 and TJP1 in Tvrm222 mouse retinas relative to those in either WT or Nr2e3rd7/rd7 mice. Membrane-associated CTNNB1 and TJP1 were quantified by ImageJ (D, F). The levels of membrane-associated CTNNB1 and TJP1 were normalized to corresponding ATP1A1 levels. Results from four independent experiments were used in the quantification. Statistical analysis was performed by one-way ANOVA with Tukey’s test for multiple comparisons. The results are mean±SD, *P < 0.05, **P < 0.01. PNS, post-nuclear supernatant; S100, supernatant after 100 000×g centrifugation; P100, pellet after 100 000×g centrifugation.
Since the ELM appears intact in the Tvrm222 retina, we examined whether membrane-associated cell junction components were affected in Tvrm222 retinas compared with either WT or Nr2e3rd7/rd7 retinas. Subcellular fractionation revealed that CTNNB1 (Fig. 8C and D) and TJP1 (Fig. 8E and F) were present in greater abundance in the membrane fraction in the Tvrm222 mouse retinas, compared with that in both WT and Nr2e3rd7/rd7 retinas. This suggests that stabilization of cell junction at the ELM is associated with the rescue of rd7-associated photoreceptor dysplasia by the Frmd4bTvrm222 variant.
Collectively, our results revealed that FRMD4B membrane recruitment was affected because of the presence of 938P variant, accompanied by reduced phosphorylation of AKT and increased levels of cell junctional proteins at the cell surface.
Discussion
Maintaining structural integrity and stability of the retina relies on proper development of retinal precursor cells and the establishment of junctions between the differentiating photoreceptors and their neighboring Müller glial cells (31,32). The continuous intermediate junctions localized between photoreceptor ISs and the apical processes of Müller cells comprise the ELM. Disruption of cell junctions of the ELM has been noted in multiple retinopathies (19,33–35).
In this study, we first discovered that ELM fragmentation is also closely associated with photoreceptor dysplasia in rd7 homozygotes. More importantly, we identified a novel genetic modifier that suppresses rd7-associated dysplastic lesions, such as folds and rosettes in the ONL and maintains an intact ELM. Consistently then, the Tvrm222 modifier, after segregation from rd7, was also able to prevent the ELM fragmentation and photoreceptor dysplasia phenotype caused by Nrl deficiency.
The modifier allele was identified as a variant of FRMD4B, a protein recently identified as a key scaffolding protein involved in cell junction remodeling, cytoskeletal dynamics and epithelial polarization (36). The S938P variation of FRMD4B is located in the region spanning amino acids 542–972, which was suggested to bind the Par-3 family cell polarity regulator (PARD3), and is critical for the localization of FRMD4B to adherens junctions (36). Given that this variation alters an amino acid, it may affect the structure of the PARD3-binding region of FRMD4B, and subsequently influence the binding of FRMD4B938P to PARD3, and therefore, cell surface targeting of FRMD4B. Consistently, we found that membrane recruitment of this variant, FRMD4B938P, was reduced after insulin stimulation in vitro compared with its WT counterpart.
In light of the spatial proximity of the CYTH3–FRMD4B complex with AKT, as both interact with PIP3 at the plasma membrane, it is natural to hypothesize a potential influence of membrane recruitment of CYTH3–FRMD4B on AKT phosphorylation (Fig. 9). Our data showed that AKT phosphorylation is reduced in the mice homozygous for the Frmd4bTvrm222 allele. AKT is a core component for incorporating a variety of cellular signals from extracellular cues, and profoundly influences growth and development of cells (37,38). It is worth noting that constitutive activation of AKT is documented to play a major role in promoting cell motility, destabilizing cell junctions and losing apical-basal polarity (39,40). Down regulating the phosphorylation of AKT could potentially stabilize cell adhesions and rescue the fragmented ELM in dysplastic mouse retinas.
Figure 9.

Hypothetical model of potential interactions of the CYTH3–FRMD4B complex with AKT and ARF6 pathways for modifying dysplastic lesions in retina based on information from the cited literature and our finding in this study. A reduced cell surface targeting of FRMD4B938P variant was noted in this study, which potentially generates a variety of biological consequences by mediating the functions of ARF6 and/or AKT pathways. AKT, protein kinase B; ARF6, ADP ribosylation factor 6; CYTH3, cytohesin 3; PAR, Par3-Par6-aPKC; IRS, insulin receptor substrate; RTK, receptor tyrosine kinase; P, phosphate group; PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3–5)-trisphosphate.
FRMD4B was also reported to directly participate in cell junction remodeling, cytoskeletal dynamics and epithelial polarization by linking the PAR complex and ADP-ribosylation factor 6 (ARF6). Physical binding of FRMD4B to PARD3 was suggested to be a critical prerequisite for the biological function of CYTH3 in regulating the GDP-GTP exchange of ARF6 (41–43). Thus, it is reasonable to speculate that the PAR–ARF6 complex might be an alternative pathway, through which the change in membrane recruitment of CYTH3–FRMD4B938P influences cell junction dynamics in the retina to modify photoreceptor dysplasia (Fig. 9).
Although representing distinct pathways, AKT and ARF6 are likely to regulate the growth and development of photoreceptor cells in a coordinated fashion. FRMD4B is an appealing candidate to integrate and propagate these molecular signals to regulate cell growth and development, as well as to prevent developmental abnormalities (e.g. defective cell–cell contact) of dysplastic lesions in mouse retinas.
Materials and Methods
Mice and phenotyping
Procedures involving mice were approved by The Jackson Laboratory’s Institutional Animal Care and Use Committee. For mutagenesis, rd7 (Nr2e3rd7/rd7) mice on the C57BL/6J background (B6.Cg-Nr2e3rd7/J) were injected with a solution of N-ethyl-N-nitrosourea (70 mg/kg) intraperitoneally at 10–12 weeks of age (G0). Subsequently, G0 mice were mated to untreated rd7 mice to produce G1 offspring. Backcrossing to untreated rd7 mice generated G2 female mice, which were mated to their G1 sire. Several mouse lines, including Tvrm222 (B6.Cg-Frmd4b<Tvrm222> Nr2e3<rd7>/Pjn, JR#30623) were identified by indirect ophthalmoscopy as having distinctive fundus appearances compared with Nr2e3rd7/rd7 mice. The Nrl mutant strain (B6.129-Nrltm1Asw/J, JR#021152) was obtained from The Jackson Laboratory’s strain repository.
Fundus examinations were carried out using a Heine Omega 500 indirect ophthalmoscope with a 60-diopter aspheric lens after dilation of pupils with 1% atropine sulfate. Fundus images were captured using Micron III and IV cameras (Phoenix Research Laboratories). Outer neuroretinal function was tested by ERG. B6.Cg-Nr2e3rd7/J homozygotes, Tvrm222 and C57BL/6J WT mice were examined at 1.5 months of age. ERGs were recorded as previously described (44). Briefly, the mice were dark-adapted overnight and anesthetized by an intraperitoneal injection with ketamine (80 mg/kg)/xylazine (16 mg/kg). The corneas were anesthetized (1% proparacaine HCl) and pupils dilated (1 drop mydriacyl, 2.5% phenylephrine HCl, 1% cyclophentolate). The mice were placed on a temperature-regulated heating pad during the recording session. The amplitude of the a-wave was measured 10 ms after flash onset from the pre-stimulus baseline. The amplitude of the b-wave was measured from the trough of the a-wave to the peak of the b-wave.
Mapping and sequencing
A genome-wide scan was conducted on DNA from 51 offspring of a (C57BL/6NJ ×B6.Cg-Tvrm222/Tvrm222; rd7/rd7) F1 × B6.Cg-rd7/rd7 backcross using 143 validated single-nucleotide polymorphism SNP markers evenly spaced across the whole genome that distinguish between the C57BL/6J and C57BL/6NJ genetic backgrounds. The map position was narrowed down by testing multiple markers in recombinant animals to a region between the markers rs259773143 and rs30672468 on Chromosome 6. High-quality genomic DNA was prepared from B6.Cg-Tvrm222/Tvrm222; rd7/rd7 mice for exome capture and sequencing with an Illumina HiSeq2500. High throughput sequencing (HTS) data were analyzed using a local JAX Galaxy interface pipeline (45–47). Sequence reads were quality assessed using FastQC v0.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and aligned to the mouse reference genome (mm10) from UCSC, released December 2011, using BWA v1.2.3 (48). PCR duplicates were removed using SAMtools rmdup v.1.0.0 (49). SNPs and indels were called using SAMtools mpileup v1.0.0 (49) and genomic and functional annotations were assigned to the variants using SnpEff v0.9 (50).
The PCR primers used for: genotyping Tvrm222 mice; sequencing candidate variants within the minimal region on Chromosome 6 revealed by HTS, including Tacr1, Dctn1, Grcc10 and Klra6; and assessing the expression of Tvrm222 transcripts in different tissues are presented in Supplementary Material, Table S2.
Plasmid constructs
The Frmd4b open reading frame was amplified from cDNA and was inserted into pCMV-3Tag-2B (Agilent Technologies), which was cleaved by BamHI/HindIII, using cold fusion recombination cloning (System Biosciences). The Frmd4bTvrm222 sequence was directly site mutagenized (Agilent Technologies) from pCMV-3Tag-2B-Frmd4bwt. The Cyth3 coding sequence was amplified from the cDNA. The PCR product was cleaved with EcoRI and HindIII and inserted into pCMV-3Tag-1A (Agilent Technologies) by ligation. All the primers used for amplifying plasmid DNA are listed in Supplementary Material, Table S2.
Cell culture
Both HEK-293T and COS7 cells were cultured in Dulbecco’s modified eagle medium (DMEM, high glucose, ThermoFisher Scientific) supplemented with 10% fetal bovine serum. Transient transfection was performed using TransIT-2020 reagent (Mirus Bio) according to the manufacturer’s instruction. The cells were starved by culturing in serum-free DMEM overnight (>18 h) before subsequent supplementation with insulin (100 nM) as previously described (21,51,52). The cells treated with serum-free DMEM served as controls.
Histology, immunohistochemistry and immunocytochemistry
For histology, whole eyes, enucleated after carbon dioxide (CO2) euthanasia, were fixed with methanol:acetic acid (3:1) for at least 5 h prior to embedding in paraffin. Sections were de-paraffinized and stained with hematoxylin and eosin (H&E). For transmission electron microscopy (TEM), mice were subjected to cardiac perfusion with phosphate-buffered 2.5% glutaraldehyde/2% paraformaldehyde (PFA) solution after CO2 asphyxiation. The enucleated whole eyes were immersed in the same fixative for 30 min at 4°C. Subsequently, after removal of the anterior segment, the eyecup was immersed in the same fixative diluted with PBS (1:10) overnight at 4°C prior to further processing for TEM examinations by advanced microscopy techniques camera system as previously described (44). For immunohistochemistry, enucleated whole eyes were fixed with 4% PFA and embedded in paraffin. Sections were subject to antigen retrieval using heated 10 mM citrate buffer and incubated with antibodies as previously described (53). Cultured cells were fixed with 2% PFA for ∼10 min after incubation with insulin (diluted with DMEM). Subsequently, the fixed cells were subject to antigen retrieval with 0.1% sodium dodecyl sulfate (SDS) for 5 min. The cells were then blocked with 5% normal donkey serum and incubated with antibodies overnight at 4°C. Immunostained samples were examined with a Leica SP5 confocal microscope.
Protein preparation and western blot
Eyecups were homogenized in modified radioimmunoprecipitation assay buffer (Tris, pH 7.4, 65 mM; NP40: 1%; NaCl: 0.9%; Na-deoxycholate: 0.25%; EDTA: 1 mM) with protease (Sigma-Aldrich, P8340) and phosphatase inhibitors (20 mM sodium fluoride, 100 mM sodium pyrophosphate, 100 mM sodium orthovanadate, 100 mM ammonium molybdate and phenylmethanesulfonyl fluoride, Sigma-Aldrich, P7626), using a motorized dounce homogenizer. Protein extracts were denatured with SDS and separated by SDS-polyacrylamide gel electrophoresis using BIO-RAD TGX pre-cast gels. Proteins were then transferred to nitrocellulose for western blot analysis.
For subcellular fractionation, eyecups from two individual mice of the same genotype were pooled and homogenized in HEPES buffer (NaCl: 115 mM; CaCl2: 1.2 mM; MgCl2: 1.2 mM; K2HPO4: 2.4 mM; HEPES: 20 mM, pH 7.5) with an EDTA-free protease inhibitor (ROCHE, 11836170001). HEK-293T cells were harvested from 60 mm petri dishes after transfection. The cells were lysed and homogenized using a similar procedure and buffer for collecting tissue samples. Protein extracts were centrifuged at 13 400×g for 15 min at 4°C. The pellet, including the nucleus and heavy membranes, was passed through a fine needle, followed by two washes with HEPES buffer. The collected supernatant labeled post-nuclear supernatant (PNS), contained light membranes including the plasma membrane. A portion of the PNS was subject to ultracentrifugation at 100 000 × g for 45 min to separate the membrane (P100) and the cytosolic proteins (S100).
For immunoprecipitation, HEK-293T cells were plated on 60 mm petri dishes for transfection. Cells were lysed with lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mM EDTA and 1% Triton X-100) containing protease inhibitors and incubated with FLAG-agarose beads (Sigma-Aldrich FLAG® Immunoprecipitation Kit, FLAGIPT1) according to the manufacturer’s protocol.
Antibodies
Antibodies were used as follow: rabbit anti-cone arrestin (EMD Millipore, AB15282), goat anti-β-catenin (Santa Cruz; sc-1496), mouse anti-GS (Chemicon; MAB302), rabbit anti-blue-opsin (Chemicon; AB5407), biotin conjugated anti-PNA (Vector Laboratories; BA-0074), rabbit anti-SOX9 (EMD Millipore, AB5535) and rabbit anti-ZO-1 (Abcam; ab59720) were used in 1:200 dilution for immunofluorescence staining and 1:1000 for western blots (SOX9 was used in 1:500 for western blot). Rabbit anti-PH3 (Abcam, ab47297) and rabbit anti-GFAP (Dako, Z0334) were used in 1:200 dilution for immunofluorescence. Mouse monoclonal antibodies against FLAG-tag (Sigma-Aldrich; F1804) and MYC-tag (Cell Signaling; #2276) were diluted in 1:500 for immunocytochemistry and 1:2000 for western blot analysis. Rabbit anti-phospho AKT (Ser473, Cell Signaling; #4060) was diluted in 1:1000 for western blot analysis. Rabbit anti-GAPDH (Cell Signaling; #2118) and mouse anti-ATPase Na+/K+ transporting subunit alpha 1 (ATP1A1) (Novus Biologicals; NB300-146) were used in 1:1000 dilution for western blot analysis.
Image analyses and statistics
The ImageJ/Fiji program was used for processing the images and quantifying the immunoblotting results. Data were analyzed by two-way ANOVA with post hoc Tukey’s test or one-way ANOVA with post hoc Tukey’s test for multiple comparisons (for comparison of three groups of samples). Each group has at least three biological replicates. Results are reported as mean ±SD. P < 0.05 was reported as statistical significance.
Authors’ Contributions
This project was conceived and supervised by L.Z. and P.M.N., while Y.K., L.Z., J.K.N. and P.M.N. designed experiments; Y.K. and L.Z. conducted experiments, analyzed the data and assembled the manuscript; J.R.C., W.L.H. and L.S. conducted experiments; J.K.N. and P.M.N. analyzed the data, assembled and edited the manuscript.
Supplementary Material
Acknowledgements
We thank The Jackson Laboratory Scientific Services including Sequencing, Histology and Imaging Sciences for their expert support.
Conflict of Interest statement. None declared.
Funding
This work was supported by NIH grants EY028561 (J.K.N.); EY016501 and EY11996 (P.M.N.) and an International Retinal Research Foundation (IRRF) grant (L.Z.) on Genetic Modifiers of enhanced S-cone Syndrome. The Jackson Laboratory Scientific Services are supported by NIH Grant CA034196.
References
- 1. Wilkie D.A. (2011) In Equine Ophthalmology, 2nd edn.W.B. Saunders, St Louis, pp. 367–396. [Google Scholar]
- 2. Wilcock B.P., Njaa B.L. (2016) In Jubb, Kennedy & Palmer's Pathology of Domestic Animals, Vol. 1, 6th edn W.B. Saunders, St Louis, pp. 407–508.e402. [Google Scholar]
- 3. Lahav M., Albert D.M., Wyand S. (1973) Clinical and histopathologic classification of retinal dysplasia. Am. J. Ophthalmol., 75, 648–667. [DOI] [PubMed] [Google Scholar]
- 4. Whiteley H.E. (1991) Dysplastic canine retinal morphogenesis. Invest. Ophthalmol. Vis. Sci., 32, 1492–1498. [PubMed] [Google Scholar]
- 5. Albert D.M., Lahav M., Colby E.D., Shadduck J.A., Sang D.N. (1977) Retinal neoplasia and dysplasia. I. Induction by feline leukemia virus. Invest Ophthalmol. Vis. Sci., 16, 325–337. [PubMed] [Google Scholar]
- 6. Rehemtulla A., Warwar R., Kumar R., Ji X., Zack D.J., Swaroop A. (1996) The basic motif-leucine zipper transcription factor Nrl can positively regulate rhodopsin gene expression. Proc. Natl. Acad. Sci. U S A, 93, 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mears A.J., Kondo M., Swain P.K., Takada Y., Bush R.A., Saunders T.L., Sieving P.A., Swaroop A. (2001) Nrl is required for rod photoreceptor development. Nat. Genet., 29, 447–452. [DOI] [PubMed] [Google Scholar]
- 8. Daniele L.L., Lillo C., Lyubarsky A.L., Nikonov S.S., Philp N., Mears A.J., Swaroop A., Williams D.S., Pugh E.N. Jr (2005) Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Invest. Ophthalmol. Vis. Sci., 46, 2156–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen J., Rattner A., Nathans J. (2005) The rod photoreceptor-specific nuclear receptor Nr2e3 represses transcription of multiple cone-specific genes. J. Neurosci., 25, 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cheng H., Khan N.W., Roger J.E., Swaroop A. (2011) Excess cones in the retinal degeneration rd7 mouse, caused by the loss of function of orphan nuclear receptor Nr2e3, originate from early-born photoreceptor precursors. Hum. Mol. Genet., 20, 4102–4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Haider N.B., Naggert J.K., Nishina P.M. (2001) Excess cone cell proliferation due to lack of a functional NR2E3 causes retinal dysplasia and degeneration in rd7/rd7 mice. Hum. Mol. Genet., 10, 1619–1626. [DOI] [PubMed] [Google Scholar]
- 12. Wright A.F., Reddick A.C., Schwartz S.B., Ferguson J.S., Aleman T.S., Kellner U., Jurklies B., Schuster A., Zrenner E., Wissinger B.. et al. (2004) Mutation analysis of NR2E3 and NRL genes in enhanced S Cone syndrome. Hum. Mutat., 24, 439.. [DOI] [PubMed] [Google Scholar]
- 13. Haider N.B., Jacobson S.G., Cideciyan A.V., Swiderski R., Streb L.M., Searby C., Beck G., Hockey R., Hanna D.B., Gorman S.. et al. (2000) Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat. Genet., 24, 127–131. [DOI] [PubMed] [Google Scholar]
- 14. Iannaccone A., Fung K.H., Eyestone M.E., Stone E.M. (2009) Treatment of adult-onset acute macular retinoschisis in enhanced s-cone syndrome with oral acetazolamide. Am. J. Ophthalmol., 147, 307–312 e302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milam A.H., Rose L., Cideciyan A.V., Barakat M.R., Tang W.X., Gupta N., Aleman T.S., Wright A.F., Stone E.M., Sheffield V.C.. et al. (2002) The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc. Natl. Acad. Sci. U S A, 99, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swaroop A., Kim D., Forrest D. (2010) Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat. Rev. Neurosci., 11, 563–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heynen S.R., Meneau I., Caprara C., Samardzija M., Imsand C., Levine E.M., Grimm C. (2013) CDC42 is required for tissue lamination and cell survival in the mouse retina. PLoS One, 8, e53806.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. West E.L., Pearson R.A., Tschernutter M., Sowden J.C., MacLaren R.E., Ali R.R. (2008) Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors. Exp. Eye Res., 86, 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mehalow A.K., Kameya S., Smith R.S., Hawes N.L., Denegre J.M., Young J.A., Bechtold L., Haider N.B., Tepass U., Heckenlively J.R.. et al. (2003) CRB1 is essential for external limiting membrane integrity and photoreceptor morphogenesis in the mammalian retina. Hum. Mol. Genet., 12, 2179–2189. [DOI] [PubMed] [Google Scholar]
- 20. Stuck M.W., Conley S.M., Naash M.I. (2012) Defects in the outer limiting membrane are associated with rosette development in the Nrl-/- retina. PLoS One, 7, e32484.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Klarlund J.K., Holik J., Chawla A., Park J.G., Buxton J., Czech M.P. (2001) Signaling complexes of the FERM domain-containing protein GRSP1 bound to ARF exchange factor GRP1. J. Biol. Chem., 276, 40065–40070. [DOI] [PubMed] [Google Scholar]
- 22. Haider N.B., Zhang W., Hurd R., Ikeda A., Nystuen A.M., Naggert J.K., Nishina P.M. (2008) Mapping of genetic modifiers of Nr2e3 rd7/rd7 that suppress retinal degeneration and restore blue cone cells to normal quantity. Mamm. Genome, 19, 145–154. [DOI] [PubMed] [Google Scholar]
- 23. Chen J., Nathans J. (2007) Genetic ablation of cone photoreceptors eliminates retinal folds in the retinal degeneration 7 (rd7) mouse. Invest. Ophthalmol. Vis. Sci., 48, 2799–2805. [DOI] [PubMed] [Google Scholar]
- 24. Schorderet D.F., Escher P. (2009) NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum. Mutat., 30, 1475–1485. [DOI] [PubMed] [Google Scholar]
- 25. Webber A.L., Hodor P., Thut C.J., Vogt T.F., Zhang T., Holder D.J., Petrukhin K. (2008) Dual role of Nr2e3 in photoreceptor development and maintenance. Exp. Eye Res., 87, 35–48. [DOI] [PubMed] [Google Scholar]
- 26. Dyer M.A., Cepko C.L. (2000) Control of Muller glial cell proliferation and activation following retinal injury. Nat. Neurosci., 3, 873–880. [DOI] [PubMed] [Google Scholar]
- 27. Germer A., Jahnke C., Mack A., Enzmann V., Reichenbach A. (1997) Modification of glutamine synthetase expression by mammalian Muller (glial) cells in retinal organ cultures. Neuroreport, 8, 3067–3072. [DOI] [PubMed] [Google Scholar]
- 28. Mansour M., Lee S.Y., Pohajdak B. (2002) The N-terminal coiled coil domain of the cytohesin/ARNO family of guanine nucleotide exchange factors interacts with the scaffolding protein CASP. J. Biol. Chem., 277, 32302–32309. [DOI] [PubMed] [Google Scholar]
- 29. Czech M.P. (2000) PIP2 and PIP3: complex roles at the cell surface. Cell, 100, 603–606. [DOI] [PubMed] [Google Scholar]
- 30. Liao Y., Hung M.C. (2010) Physiological regulation of Akt activity and stability. Am. J. Transl. Res., 2, 19–42. [PMC free article] [PubMed] [Google Scholar]
- 31. Morrow E.M., Furukawa T., Cepko C.L. (1998) Vertebrate photoreceptor cell development and disease. Trends Cell Biol., 8, 353–358. [DOI] [PubMed] [Google Scholar]
- 32. Shen W., Fruttiger M., Zhu L., Chung S.H., Barnett N.L., Kirk J.K., Lee S., Coorey N.J., Killingsworth M., Sherman L.S.. et al. (2012) Conditional Mullercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J. Neurosci., 32, 15715–15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rich K.A., Figueroa S.L., Zhan Y., Blanks J.C. (1995) Effects of Muller cell disruption on mouse photoreceptor cell development. Exp. Eye Res., 61, 235–248. [DOI] [PubMed] [Google Scholar]
- 34. Omri S., Omri B., Savoldelli M., Jonet L., Thillaye-Goldenberg B., Thuret G., Gain P., Jeanny J.C., Crisanti P., Behar-Cohen F. (2010) The outer limiting membrane (OLM) revisited: clinical implications. Clin. Ophthalmol., 4, 183–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ito S., Miyamoto N., Ishida K., Kurimoto Y. (2013) Association between external limiting membrane status and visual acuity in diabetic macular oedema. Br. J. Ophthalmol., 97, 228–232. [DOI] [PubMed] [Google Scholar]
- 36. Ikenouchi J., Umeda M. (2010) FRMD4A regulates epithelial polarity by connecting Arf6 activation with the PAR complex. Proc. Natl. Acad. Sci. U S A, 107, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vivanco I., Sawyers C.L. (2002) The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer, 2, 489–501. [DOI] [PubMed] [Google Scholar]
- 38. Manning B.D., Toker A. (2017) AKT/PKB signaling: navigating the network. Cell, 169, 381–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Grille S.J., Bellacosa A., Upson J., Klein-Szanto A.J., van Roy F., Lee-Kwon W., Donowitz M., Tsichlis P.N., Larue L. (2003) The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res., 63, 2172–2178. [PubMed] [Google Scholar]
- 40. Larue L., Bellacosa A. (2005) Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene, 24, 7443–7454. [DOI] [PubMed] [Google Scholar]
- 41. Palacios F., Price L., Schweitzer J., Collard J.G., D'Souza-Schorey C. (2001) An essential role for ARF6-regulated membrane traffic in adherens junction turnover and epithelial cell migration. EMBO J., 20, 4973–4986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen P.W., Jian X., Yoon H.Y., Randazzo P.A. (2013) ARAP2 signals through Arf6 and Rac1 to control focal adhesion morphology. J. Biol. Chem., 288, 5849–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sabe H. (2003) Requirement for Arf6 in cell adhesion, migration, and cancer cell invasion. J. Biochem., 134, 485–489. [DOI] [PubMed] [Google Scholar]
- 44. Collin G.B., Hubmacher D., Charette J.R., Hicks W.L., Stone L., Yu M., Naggert J.K., Krebs M.P., Peachey N.S., Apte S.S.. et al. (2015) Disruption of murine Adamtsl4 results in zonular fiber detachment from the lens and in retinal pigment epithelium dedifferentiation. Hum Mol Genet, 24, 6958–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M., Nekrutenko A., Taylor J. (2010) Galaxy: a web-based genome analysis tool for experimentalists. Curr. Protoc. Mol. Biol., 19, Chapter 19, Unit 19 10 11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Giardine B., Riemer C., Hardison R.C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J.. et al. (2005) Galaxy: a platform for interactive large-scale genome analysis. Genome Res., 15, 1451–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Goecks J., Nekrutenko A., Taylor J., Galaxy Team T. (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol., 11, R86.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. (2012) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: sNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li J., Malaby A.W., Famulok M., Sabe H., Lambright D.G., Hsu V.W. (2012) Grp1 plays a key role in linking insulin signaling to glut4 recycling. Dev. Cell, 22, 1286–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vander Haar E., Lee S.I., Bandhakavi S., Griffin T.J., Kim D.H. (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat. Cell Biol., 9, 316–323. [DOI] [PubMed] [Google Scholar]
- 53. Shi S.R., Shi Y., Taylor C.R. (2011) Antigen retrieval immunohistochemistry: review and future prospects in research and diagnosis over two decades. J. Histochem. Cytochem., 59, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.