

Abstract
Background
Hyperactivation of phosphoinositide 3-kinase (PI3K) signaling is common in cancers, but the precise role of the pathway in glioma biology remains to be determined. Some understanding of PI3K signaling mechanisms in brain cancer comes from studies on neural stem/progenitor cells (NSPCs), where signals transmitted via the PI3K pathway cooperate with other intracellular pathways and downstream transcription factors to regulate critical cell functions.
Methods
To investigate the role of the PI3K pathway in glioma initiation and development, we generated a mouse model targeting the inducible expression of a PIK3CAH1047A oncogenic mutant and deletion of the PI3K negative regulator, phosphatase and tensin homolog (PTEN), to NSPCs.
Results
Expression of a Pik3caH1047A was sufficient to generate tumors with oligodendroglial features, but simultaneous loss of PTEN was required for the development of invasive, high-grade glioma. Pik3caH1047A–PTEN mutant NSPCs exhibited enhanced neurosphere formation which correlated with increased Wnt signaling, while loss of cAMP response element binding protein (CREB) in Pik3caH1047A–Pten mutant tumors led to longer symptom-free survival in mice.
Conclusion
Taken together, our findings present a novel mouse model for glioma demonstrating that the PI3K pathway is important for initiation of tumorigenesis and that disruption of downstream CREB signaling attenuates tumor expansion.
Keywords: CREB, mouse model, neural stem cells, PIK3CA, PTEN
Importance of the study
This study demonstrates that PI3K mutations in NSPCs can initiate glioma-like tumors. Mutant NSPCs exhibited increased PI3K signaling but also showed enhanced CREB transcription factor activation, which promotes cell proliferation, as well as enhanced Wnt signaling, which promotes self-renewal. Tumors were switched to a less malignant state by simultaneous deletion of CREB, raising the possibility that CREB may be an important anticancer drug target.
Genetically engineered mouse models have demonstrated that various genes can initiate glioma development, with the first models expressing constitutively activated mutant growth factor receptors, including epidermal growth factor receptor (EGFR), or mutated signaling molecules, including Ras and Akt.1,2 Genes enabling the initiation of high-grade gliomas (HGGs) include combinations of NF1, TP53, PTEN, Akt, Ras, and INK4a/ARF.3–6 Models targeting glial cells,1,4,7 oligodendrocyte precursor cells, astrocytes, or neurons also initiate HGG.8,9 The role of brain tumor stem cells (BTSCs) has been demonstrated by targeting genetic mutations/deletions in neural stem/progenitor cells (NSPCs),2,3,5,6,9–11 highlighted by a study demonstrating that ablation of BTSCs inhibits tumor growth.12
The signaling pathways driving HGG overlap with those activated in other cancers and involve complex interactions between oncogenes and tumor suppressors, many of which regulate the phosphoinositide 3-kinase (PI3K) and mitogen-activated protein kinase (MAPK) pathways. These pathways trigger a cascade of downstream kinases leading to the activation of transcription factors, including the cAMP response element binding protein (CREB), which is upregulated in glioblastoma (GBM) and has a role in NSPC and GBM cell proliferation.13,14 Emerging evidence suggests that oncogenic PI3K and MAPK signals converge with Wnt signaling to regulate cancer cell growth and proliferation15 and that in GBM, Wnt signaling has a role in cancer stem cell maintenance.16
When one considers the upstream components of the PI3K pathway, including EGFR, the PI3K catalytic and regulatory subunits, as well as the pathway’s major negative regulator, phosphatase and tensin homolog (PTEN), 63% of HGGs exhibit an alteration in at least one of these genes.17 A major catalytic subunit of PI3K, encoded by the phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha gene (PIK3CA), is among the 3 top oncogenic drivers in GBM; the other drivers are EGFR and TP53.18 Indeed, PIK3CA mutations are reported in up to 17% of low- and high-grade pediatric and adult brain cancers, with higher-grade and treatment-resistant tumors exhibiting higher mutation rates.17,19 Full activation of the PI3K pathway not only requires enhanced catalytic activity to drive key downstream kinases such as Akt but also requires the inactivation of the phosphatase activity of PTEN.20,21 Deletion of PTEN in NSPCs leads to enhanced proliferative activity in the stem cell niche but no tumor development.22,23Pten deletion in combination with p53 and/or Rb1 loss results in the development of astrocytic high-grade tumors.4 However, the effects of pro-oncogenic mutations involving the PI3K catalytic (p110α) and regulatory (p85) subunits have not been investigated.
To better understand the role of the PI3K pathway, we targeted the PI3K pathway in mouse NSPCs by conditional activation of an oncogenic mutation in Pik3ca, the gene encoding the PI3K p110α catalytic subunit,24 in combination with PTEN deletion and show that these mutations are sufficient to initiate tumor growth. To investigate the downstream transcriptional programs regulated by the PI3K pathway, we deleted CREB and show that CREB loss slows tumor growth and leads to a less aggressive phenotype.
Materials and Methods
Ethics Statement
Mouse experiments were carried out with the approval of The University of Melbourne, School of Biomedical Sciences (AEC No 1112336.1) and Peter MacCallum Cancer Center (AEEC No. E406) Animal Ethics Committees.
Mouse Breeding
Mice heterozygous for a latent Cre recombinase (Cre)-inducible knock-in of the Pik3caH1047R mutation (Pik3caH1047R-lox)24 and/or 2 Cre-inducible Pten deletion alleles (PtenloxP/loxP)23 were crossed with mice expressing a single (heterozygous) Nestin-CreERT2 transgene.25 Further F1 crosses generated mice which were heterozygous for mutant Pik3caH1047R (Pik3caH1047R-lox–Nestin-CreERT2) and homozygous Pten deletion (PtenloxP/loxP–Nestin-CreERT2) (Pten∆). Mice were housed and genotyped as previously described.24,26 See Supplementary Methods for details.
In Vivo Tumor Induction
Control (Pik3caH1047R-lox–Ptenlox/lox), double mutant (Pik3caH1047R-lox–Ptenlox/lox–Nestin-CreERT2), or triple mutant (Pik3caH1047R-lox–Ptenlox/lox–Creblox/lox–Nestin-CreERT2) mice were treated with tamoxifen at either 1 day or 6–8 weeks of age, as described previously.27
Histology and protein analysis
Brain tissue was fixed by intracardiac perfusion with 4% paraformaldehyde, and immunohistochemistry was performed as described previously.26 For western blotting, cells were lysed in radioimmunoprecipitation assay buffer. Antibodies are detailed in the Supplementary Methods.
NSPC Culture
Refer to the Supplementary Methods for isolation of NSPC protocol. For experiments involving NSPCs, mutant NSPCs were generated by transient (24 h) treatment with 0.02 mg/mL 4-hydroxy-tamoxifen (4-OHT) in ethanol to induce Cre-mediated recombination of floxed alleles, while control cells were derived from the same genotype but treated with vehicle only (ethanol). 4-OHT was removed after 24 h and never included in the medium in further experiments.
Cell Cycle Analysis
Cells in phosphate buffered saline (PBS) were fixed in cold 100% ethanol. Cells were washed 3 times with PBS and incubated with 4′,6′-diamidino-2-phenylindole (DAPI) (5 μg/mL; Thermo Fisher Scientific) for 15 min on ice. Cells were then washed, suspended in PBS, and analyzed by fluorescence activated cell sorting (LSR Fortessa 405 nm). Cells were gated with forward scatter width versus DAPI (405 nm) to exclude doublets and debris. A histogram of DAPI versus count was generated and analyzed using FlowLogic v6 software to identify the population (%) of cells in each of the cell cycle phases.
Proliferation Assays
Resazurin solution (Sigma) was diluted in media and added to wells, in 96-well plates, to obtain a 10% v/v solution before incubation for 3 h at 37°C. Plates were analyzed using the EnSpire Plate Reader (PerkinElmer).
Migration Assay
Single neurospheres in Dulbecco’s modified Eagle’s medium F12 with growth factors and supplements (see NSPC Culture method, above) were placed into a 96-well plate (1 sphere/well) for 48 h and assayed using the Trevigen 96-well 3D spheroid basement membrane extracts cell invasion assay kit (Bio Scientific). Neurospheres were incubated either with (test) or without (control) addition of invasion matrix for 48 h. Spheres were photographed and invasion area was calculated by measuring the area of spread and subtracting the control neurosphere invasion area using ImageJ (http://rsb.info.nih.gov/ij/index.html).
Extreme Limiting Dilution Analysis
Neurospheres were dissociated and plated in suspension media at 20, 10, 5, and 1 cell per well with 24 replicates per cell density. Wells were photographed after 7 days and the number of wells with one or more spheres greater than ~20 µm diameter was scored. Data were analyzed using online analysis28 (http://bioinf.wehi.edu.au/software/elda/).
Statistical analysis
Student’s t-test was used and results presented as mean ± SD or SEM, as indicated in relevant sections. P < 0.05 (*) was considered significant.
Results
Activation of Pik3caH1047R Expression in NSPCs Is Sufficient for Tumor Initiation but Simultaneous Deletion of Pten Is Necessary for the Development of Invasive Tumors
The data from all mice analyzed are summarized in Table 1. Control Pik3caH1047R-lox–PtenloxP/loxP mice without the Nestin-CreERT2 transgene but treated with tamoxifen did not develop neurological symptoms or show evidence of tumor growth over the experimental time window of >200 days (Fig. 1). Single mutant PtenΔ mice showed no overt abnormalities but did exhibit increased subventricular zone (SVZ) cellularity and proliferation compared with controls (Fig. 1A), as reported previously.23 Examination of single mutant Pik3caH1047R mouse brains between 42 and 222 days post tamoxifen administration revealed the presence of tumors in/adjacent to the lateral ventricles (LVs) (Fig. 1A). Tumors from 4 Pik3caH1047R mice exhibited numerous cells with perinuclear halos or “fried egg” cells with oligodendrocyte transcription factor (Olig2) expression; features associated with oligodendroglioma (Fig. 1A and Supplementary Figure S1).
Table 1.
Mice used in this study and the treatment and outcome of the analysis
| Mutation(s)* | Tamoxifen Treated Mice | Tamoxifen at Age (days) | Endpoint Post- tamoxifen (days) | Tumors | Histopathology |
|---|---|---|---|---|---|
| Pik3ca H1047R ‒nesCre | 7 | 42–56 | >222 | Yes (100% from 4 examined) | Low grade, oligodendroglioma |
| Pten Δ ‒nesCre | 3 | 42–56 | >214 | No (3 examined) | — |
| Pik3ca H1047R ‒Pten Δ ‒nesCre | 30 | 42–56 | 55 to 90 | Yes (100% from 20 examined) | High-grade glioma |
| Pik3ca H1047R ‒Pten Δ ‒nesCre | 3 | 1 | 28 to 43 | Yes (100% from 3 examined) | High-grade glioma |
| *Pik3ca H1047R ‒Pten Δ ‒nesCre | 0 | — | >200 | No (2 examined) | — |
| Pik3ca H1047R ‒Pten Δ ‒CREB Δ ‒nesCre | 14 | 42–56 | 71 to >167 | Yes (100% from 6 examined) | Glioma with large cells |
| *Pik3ca H1047R ‒Pten Δ | 2 | 42–56 | >214 | No (2 examined) | — |
*Pik3caH1047R‒PtenΔ mice without the Cre transgene which were treated with tamoxifen or Pik3caH1047R‒PtenΔ‒nesCre mice not tamoxifen treated showed no signs of disease or neurological symptoms, nor evidence of brain tumors.
Fig. 1.

Pik3ca H1047R tumors exhibit oligodendroglial features while combined Pik3caH1047R expression and Pten-deleted tumors exhibit extensive growth. (A) Hematoxylin and eosin (HE) staining of brains from wild-type control mice (tamoxifen-treated Pik3caH1047R‒Ptenlox/lox without Nestin-CreERT2) showing the SVZ layer (arrow) harboring NSPCs. Single mutant homozygous PtenΔ mice showed thickening of the SVZ layer (arrow). Single mutant heterozygous Pik3caH1047R tumor (all brains were dissected at 181 days post-tamoxifen), within the dotted outline, showing cells with characteristic perinuclear halos (arrow). Scale bar is 250 μm for the 2 left images and 100 μm for Pik3caH1047R. (B) Body weight of a representative adult mouse (6 wk of age) which received tamoxifen (Tam) at day 0. (C) Kaplan–Meier survival analysis shows that only tamoxifen-treated double mutant Pik3caH1047R‒PtenΔ adult (n = 10) and newborn (P1) mouse (n = 3) cohorts compared with WT controls (n = 12). (D) HE staining of a double mutant Pik3caH1047R‒PtenΔ brain (all brains were dissected at 55 or 62 days post-tamoxifen) show multiple tumors (*) growing out from the neurogenic zone (arrows) into the lateral ventricles (LV) (upper panel). Scale bar is 200 μm. Anterior brain sections (lower panels) demonstrate almost complete occlusion of the ventricular space by the tumor tissue. Scale bar is 1 mm.
Tamoxifen treatment of Pik3caH1047R-lox–PtenloxP/loxP–Nestin-CreERT2 (referred to as Pik3caH1047R–PtenΔ) 6- to 8-week-old mice resulted in a completely penetrant (100%; 30/30 mice) neurological phenotype 55–90 days after tamoxifen administration. An early sign of ill health was a decline in body weight (Fig. 1B), followed by the appearance of progressively more severe neurological symptoms. The first neurological manifestation was ataxia, followed by sporadic seizures and a gradual increase in seizure frequency and length. The experimental endpoint was when weight loss was more than 15% of the start weight and/or when seizures occurred more than 3 times per day, and when a seizure lasted more than 1 min. Double mutant mice reached the experimental endpoint between 55 and 90 days (Fig. 1C). Single mutant Pik3caH1047R expression or Pten mice showed no symptoms or reduced survival over 200 days (Fig. 1B, C and Table 1). When Pik3caH1047R–PtenΔ mutations were activated in 1-day-old newborn mice (P1) via tamoxifen treatment of the mother and transmission of the tamoxifen to pups via the mother’s milk, the pups began exhibiting severe neurological symptoms between 28 and 43 days (Fig. 1C); faster than in adult mice treated with tamoxifen but with morphologically similar tumors to adult mice.
Adult Pik3caH1047R–PtenΔ brains exhibited hypercellularity of the SVZ, with multiple tumor nodules protruding into the LV (Fig. 1D, upper panel) and tumors filling the anterior LV (Fig. 1D lower panels). Hypercellular clusters were present along the SVZ with tumor cell migration into tumor nodules (Fig. 2A, B). Tumor cells invaded the brain parenchyma and white matter tracts, including the corpus callosum (Fig. 2C). Apoptotic cells and mitoses were present, lying among atypical, irregularly arranged tumor cells and blood vessels (Fig. 2D and Supplementary Figure S2A). Rosette-like cell arrangements were noted in one tumor (Supplementary Figure S2B).
Fig. 2.

Mutant Pik3caH1047R‒PtenΔ NSPC initiated tumors exhibit features consistent with malignant glioma. (A) A prominent tumor nodule (*) and hyperplastic germinal zones (arrows) (A, B). Tumor cell invasion (arrows) into the brain parenchyma and corpus callosum (demarcated by the dotted lines) (C); (D) tumor vascularization (arrows). Expression of astrocytoma markers GFAP (E) and Tuj1 (F). (G) Elevated expression of pAkt(Ser473), pRpS6, pMAPK (pERK1/2), and pCREB. The dotted lines represent the SVZ‒tumor interface, with tumors lying above the dotted line. Scale bars are: 200 μm for (A), (E), (F); 100 μm for (B); 500 μm for (C); 20 μm for (D) (all brains were dissected at 55 or 62 days post-tamoxifen).
Pik3ca H1047R –Pten Δ tumors expressed glial fibrillary acidic protein (GFAP) (Fig. 2E) and β-III-tubulin (Tuj1) (Fig. 2F): evidence of glial and neuronal differentiation. Phosphorylated (p)Akt and pRps6 were expressed in tumor cells (Fig. 2G), demonstrating PI3K pathway activation. Activation of phospho‒extracellular signal-regulated kinase (ERK)1/2 (pERK1/2 [pMAPK]) and phospho-CREB (pCREB) expression showed that other oncogenic signaling and transcriptional pathways were coactivated (Fig. 2G). Ki67 and nestin expression demonstrated that tumors also harbored proliferating, immature cells, with some Olig2-positive cells observed (Supplementary Figure S3). Overall, Pik3caH1047R–PtenΔ tumors exhibited heterogeneous features, consistent with high-grade (grade III) astrocytic tumors.
Mutant NSPCs Exhibit Enhanced PI3K Signaling and Subtle Shift in Cell Cycle
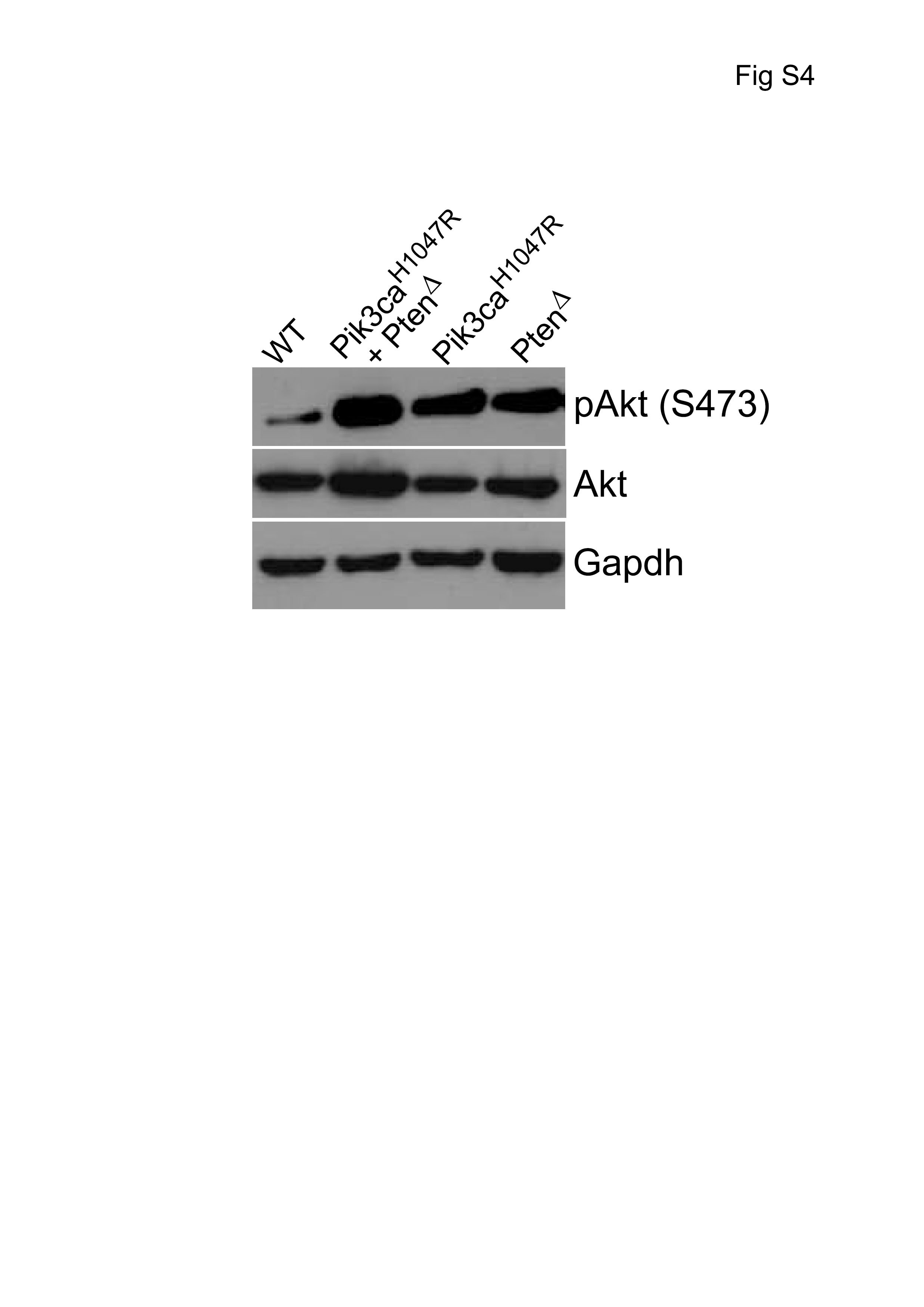
To investigate the cellular characteristics of mutant cells, NSPCs were isolated from the SVZ of 4-week-old mice carrying the Nestin-Cre transgene and latent Pik3caH1047R, Ptenlox/lox, and double mutant Pik3caH1047R–Ptenlox/lox alleles. NSPCs were propagated as neurospheres, and mutations were activated using 4-OHT. Comparison of PI3K pathway activation by expression analysis of pAkt(Ser473) in wild type (WT), Pik3caH1047R–PtenΔ, Pik3caH1047R, and PtenΔ NSPCs showed that PI3K activation was strongest in double mutant Pik3caH1047R–PtenΔ NSPCs (Supplementary Figure S4). Short-term proliferation assays revealed no significant differences between genotypes. Cell cycle phase analysis revealed subtle differences in S-phase, where Pik3caH1047R–PtenΔ and PtenΔ NSPCs showed more cells in S-phase (DNA-replication phase) than both Pik3caH1047R and WT control cells (Supplementary Figure S5).
Incomplete Recombination Directed by the Nestin-CreERT2 Transgene
PCR analysis of the Pten allele in NSPCs carrying the loxP alleles and the Nestin-CreERT2 transgene to determine DNA recombination efficiency following 4-OHT treatment showed that NSPC cultures harbored a mixture of mutant and nonmutant cells (Supplementary Figure S6), irrespective of the presence or absence of further floxed alleles. Given reports showing that Nestin promoters may drive inefficient Cre-mediated recombination of some floxed alleles in NSPCs,29 we surmised that the Nestin-CreERT2 transgene led to incomplete recombination of the Ptenlox/lox allele in 4-OHT treated cells. In vivo, mosaic expression of the oncogenic mutations is sufficient for tumor initiation, since even a small population of mutant founder/initiating tumor cells can proliferate to give rise to the larger tumor mass and disease. Considering this, further in vitro experiments were performed using NSPCs derived from the SVZ of Pik3caH1047R–Ptenlox/lox–UBC-CreERT2 mice generated previously,30 where CreERT2 expression under the control of the Ubiquitin promoter results in complete recombination in all cells exposed to 4-OHT, demonstrated by PCR (Supplementary Figure S6) and loss of PTEN protein (Supplementary Figure S7).
Pik3ca H1047R –Pten Δ Mutant NSPCs Exhibit Abnormal Morphology and Enhanced Migration
Pik3ca H1047R –Pten Δ neurospheres formed loosely aggregated rough-edged spheres (Fig. 3A) compared with parental WT neurospheres. Moreover, mutant NSPCs had more filopodia compared with controls (Fig. 3A, B), a feature associated with a more invasive, malignant phenotype in GBM.31 Incorporation of the thymidine analog, 5-ethynyl-2ʹ-deoxyuridine (EdU) and flow cytometry analysis showed that Pik3caH1047R–PtenΔ cells had a higher proliferation rate compared with WT cells (Fig. 3C). Mutant cells showed enhanced pAkt expression and increased expression of cyclins B1 and D1 (Fig. 3D).
Fig. 3.

Pik3ca H1047R -Pten Δ mutations enhance the growth and migratory capacity of NSPCs. (A) WT neurospheres exhibit smooth circumscribed borders, while mutant Pik3caH1047R‒PtenΔ mutant neurospheres exhibit irregular edges. The inset, above right, highlights part of a neurosphere with filopodia. Scale bar for the main images is 20 μm. (B) The number of filopodia per cell is higher in mutant neurospheres (mean ± SD, n = 5; ***P < 0.001, Student’s t-test). (C) Pik3caH1047R‒PtenΔ NSPCs exhibited enhanced proliferation, measured by EdU incorporation over 24 h. ****P < 0.00001. (D) Western blot shows that Pik3caH1047R‒PtenΔ NSPCs express increased pAkt, cyclin B1, and cyclin D1, with or without EGF and basic fibroblast growth factor in the neurosphere medium. (E) Mutant NSPCs exhibit a higher migratory capacity compared with control NSPCs. The lower panels are “threshold” converted images (of the central [+ matrix] images) to enhance the contrast to show the extent of cell migration. Scale bar on top left panel of (c) is 50 μm and applies to all images in the panel. (F) Quantitation of migration is expressed as area of spread (mean ± SD, n = 5; ***P < 0.001, Student’s t-test).
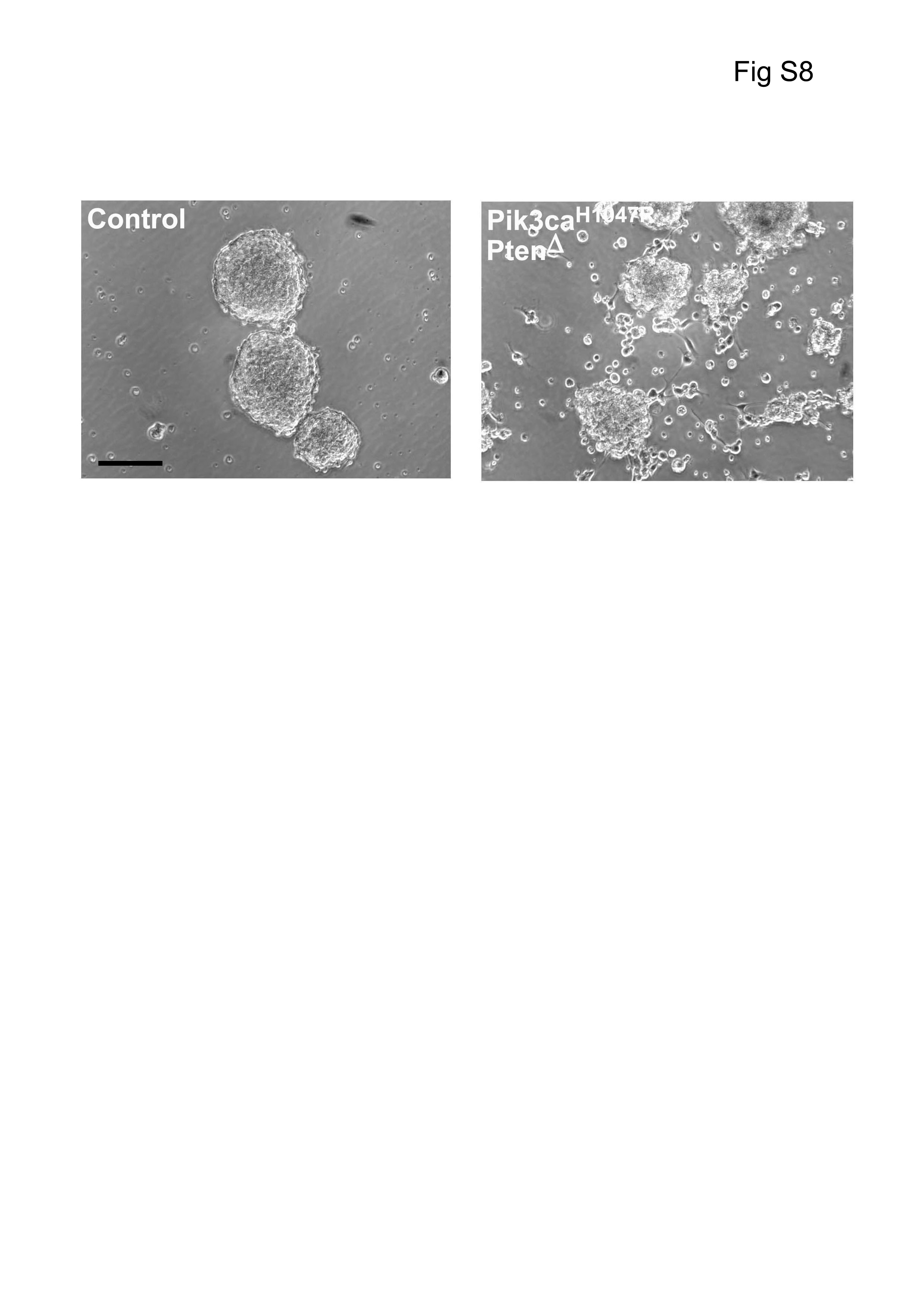
Seeding single neurospheres into a gel matrix showed that Pik3caH1047R–PtenΔ NSPCs migrated to cover more than 3 times the area compared with WT NSPCs (Fig. 3E, F), demonstrating increased migratory capacity. Moreover, seeding cells at high density for 72 h showed that mutant NSPCs detached and migrated away from spheres, unlike control NSPCs at the same density (Supplementary Figure S8).
Enhanced Sphere-Forming Capacity, Persistent Nestin Expression, and Wnt Pathway Activation in Pik3caH1047R–PtenΔ Mutant NSPCs
To determine the long-term proliferation differences between mutant and control NSPCs, cumulative growth was measured over 8 passages (8 wk). Pik3caH1047R–PtenΔ cells showed significantly higher cell numbers from week 2 onward and almost 4 times more cells by 8 weeks (Fig. 4A). To investigate this difference, we examined the neurosphere-forming efficiency of NSPCs using extreme limiting dilution analysis (ELDA).28Pik3caH1047R–PtenΔ cells (1 neurosphere forming unit [NSFU])/1.55 cells seeded) formed spheres more efficiently than control cells (1 NSFU/2.01 cells seeded) (Fig. 4B). To test the neurosphere-forming stability of the NSPCs, we subjected the cells to alternating rounds of growth in neurosphere medium, serum-containing medium (to differentiate the cells), and a return to neurosphere medium (Fig. 4C). Flow cytometry analysis demonstrated that the immature marker, nestin, was still expressed in 70% of Pik3caH1047R, PtenΔ differentiated cells, compared with 52% of WT after 7 days (Fig. 4D). Returning differentiated cells to serum-free neurosphere conditions revealed a significantly higher sphere-forming capacity in mutant cells (1 NSFU/15.4 cells seeded) compared with control cells (1 NSFU/36.0 cells seeded) (Fig. 4E). Independent assessment of immature and differentiation markers using nestin+, GFAP+, and Tuj1+ cells grown on laminin for 5 days showed enhanced nestin+ cell stability in mutant NSPCs, under differentiating conditions (Supplementary Fig. S9).
Fig. 4.

Pik3ca H1047R ‒Pten Δ mutant NSPCs exhibit enhanced and stable sphere-forming potential under conditions promoting differentiation. (A) Cumulative cell number of control and Pik3caH1047R‒PtenΔ NSPCs over 8 passages (1000 cells/mL seeded/1 passage/7 days). Cell counts were performed in triplicate from separate wells. (B) NSPCs seeded at low densities showed a selective advantage in forming a neurosphere compared with control cells; n = 3, Student’s t-test, **P < 0.01. (C) The protocol used to test the maintenance of nestin expression and neurosphere-forming capacity following differentiation. (D) Nestin expression by flow cytometry after differentiation for 7 days. (E) Sphere-forming efficiency determined by extreme limiting dilution analysis (ELDA) after cells were returned to neurosphere medium; n = 3, ****P < 0.0001. See Materials and Methods for details.
Phosphoprotein measurement showed an upregulation of phosphorylation of various PI3K/Akt pathway–associated factors in mutant NSPCs compared with WT cells (Fig. 5A). To investigate the biological basis of the enhanced neurosphere-forming capacity in Pik3caH1047R–PtenΔ NSPCs, we postulated that Wnt signaling was upregulated in mutant cells, due to the increase in glycogen synthase kinase 3 (GSK3)β phosphorylation and Wnt signaling. Analysis of NSPCs showed that β-catenin was expressed at higher levels in mutant nuclei compared with WT cells (Fig. 5B), indicating activation of Wnt signaling. Messenger RNA expression of Wnt pathway stem-cell factors and targets was higher in mutant NSPCs compared with controls (Fig. 5C). CHIR99021 (“CHIR”), a GSK3 inhibitor and Wnt pathway activator, increased neurosphere-forming potential of mutant NSPCs, which was 1:1 (spheres formed per cells seeded) for CHIR-treated NSPCs compared with 1:2.6 for vehicle-treated cells (Fig. 5D). Upstream inhibition of the Wnt pathway using IWP-2 (inhibitor of Wnt processing) did not affect sphere-forming capacity compared with vehicle-treated mutant NSPCs.
Fig. 5.

Pik3ca H1047R ‒Pten Δ mutant NSPCs exhibit enhanced PI3K downstream activity, MAPK activation, Wnt pathway activation, and Wnt pathway‒dependent self-renewal capacity. (A) A PathScan Akt Signaling Antibody Array showing the phosphoprotein expression levels of PI3K/Akt-dependent factors in lysates from Pik3caH1047R‒PtenΔ mutant and WT NSPCs. (B) Western blot showing increased nuclear β-catenin expression in Pik3caH1047R‒PtenΔ mutant NSPCs. Nuclear proteins, CREB, and histone-H3 were used as loading controls (C) Quantitative real-time PCR analysis of Wnt pathway factors showing upregulation of the Wnt signaling in Pik3caH1047R‒PtenΔ mutant cells. (D) Extreme limiting dilution analysis of NSPCs showing that Wnt pathway activation by pharmacological inhibition of GSK3β using CHIR99021 (2 µM) increases neurosphere-forming capacity of mutant NSPCs, while Wnt inhibition using IWP-2 (5 µM) reduces neurosphere forming capacity to WT control levels. All error bars are SEM from n = 3. *P < 0.05 or as indicated in Fig. 5D.
Creb1 Deletion Ameliorates Malignancy of Pik3caH1047R–PtenΔ Tumors
Pik3ca H1047R–PtenΔ tumors expressed high levels of pCREB (Fig. 2G), and this expression overlapped with nestin and GFAP expressing SVZ cells (Fig. 6A). We previously showed that mouse SVZ cells and human glioma express high levels of activated CREB (pCREB).14,32 To determine the contribution of CREB in brain tumor growth, we used Creblox/lox mice26 to generate triple mutant Pik3caH1047R-lox–Ptenlox/lox–Creblox/lox–Nestin-CreERT2 mice, resulting in deletion of CREB, in addition to the Pik3ca and Pten mutations to generate Pik3caH1047R–PtenΔ-CrebΔ mice.
Fig. 6.

Creb deletion in Pik3caH1047R‒PtenΔ NSPCs increases survival by slowing tumor growth. (A) Immunofluorescence analysis of Pik3caH1047R‒PtenΔ tumors for expression of pCREB, the neural stem cell marker, nestin, and glial marker, GFAP. The SVZ‒tumor boundary is demarcated by the dotted lines, with tumors lying below the line. Scale bars = 100 µm. (B) Hematoxylin and eosin staining showing cellularity differences in tumors derived from double mutant (Pik3caH1047R‒PtenΔ) (DM) and triple mutant (Pik3caH1047R‒PtenΔ‒CrebΔ) mice. Triple mutant tumors demonstrated the presence of large cells (arrow). The SVZ nontumor‒tumor boundary is demarcated by the dotted lines; T = tumor, N = nontumor. Scale bars = 100 µm. (C) Quantitative analysis of tumor cellularity between double mutant and triple mutant (mean ± SD, n = 3; **P < 0.01, Student’s t-test). (D) Kaplan–Meier survival analysis of double mutant mice (n = 10), triple mutant CrebΔ mice (n = 11), and WT mice (all brains were dissected at 167 days post-tamoxifen).
CREB-deleted brain tumors exhibited the presence of large cells (Fig. 6B and Supplementary Figure S10) with little or no invasion into nontumor brain parenchyma (Supplementary Figure S10A). CREB-deleted tumors showed a reduction in cell density compared with double mutant tumors (Figure 6B, C and Supplementary Figure S10), with 65% of the cell density of Pik3caH1047R–PtenΔ tumors. Moreover, CREB mutant mice were symptom free for longer (median 125 days) compared with double mutant Pik3caH1047R–PtenΔ mice (median 70 days) (Fig. 6D).
NSPCs were isolated from Pik3caH1047R-lox–Ptenlox/lox–Creblox/lox–Nestin-CreERT2 mice and treated with 4-OHT, which led to efficient recombination of the Creblox/lox allele (Supplementary Figure S6), unlike the Ptenlox/lox allele. Assays showed that proliferation was reduced in CREB-deleted NSPCs compared with Pik3caH1047R–PtenΔ cells (Supplementary Figure S11B). Cell cycle analysis showed that CREB mutant NSPCs had fewer (5.9%) S-phase cells and more (80.2%) G0/G1 phase cells compared with Pik3caH1047R–PtenΔ (12.8% S phase; 70% G0/G1 phase) NSPCs (Supplementary Figure S11C). Sphere-forming capacity of CrebΔ NSPCs did not differ compared with Pik3caH1047R–PtenΔ NSPCs (not shown).
Discussion
To our knowledge, no studies have demonstrated a direct involvement of PI3K subunit mutations in glioma in vivo. Large-scale sequencing data suggest that PIK3CA is a key driver of malignant astrocytic brain cancer.18 Our study demonstrates that expression of constitutive active Pik3caH1047R can initiate slow-growing oligodendroglial-like tumors when targeted to NSPCs. In patients, PIK3CAH1047R mutations are seen in both low- and high-grade gliomas, including anaplastic oligodendroglioma and glioblastoma.17,19 Homozygous loss of Pten led to increased SVZ cellularity but not tumor development, similar to the phenotype previously reported using Ptenlox/lox–Nestin-Cre mice.23,33 Coexistent mutation of both PIK3CA and deletion of PTEN is rare in glioma patients but the combined genetic aberrations are not absolutely redundant.34 Inactivation of PTEN and constitutive activation of PIK3CA, which led to maximal PI3K pathway activation (Fig. 3D),35 is presumably required to drive tumorigenesis, highlighting the importance of cooperative cancer-driving mutations which lead to robust PI3K pathway activation. Further, the Pik3ca mutation only affects p110α, while PTEN will affect both the p110α and other p110 subunits, including p110β, which has a role in glioma cell function,36,37 further enhancing PI3K signaling.
Pik3ca H1047R–PtenΔ NSPCs exhibited enhanced migratory properties and sphere-forming stem cell–associated characteristics, consistent with previous studies demonstrating that PI3K pathway activation promotes GBM cell migration,38 neurosphere formation, and tumorigenicity of glioma stem cells.39Pik3caH1047R–PtenΔ NSPCs maintained nestin expression and sphere-forming capacity even after forced differentiation, suggesting that Pik3caH1047R–PtenΔ mutations stabilize stem cell properties, and, in a clinical setting, these mutations may impart stable, therapy-resistant characteristics with enhanced capacity for tumor recurrence.
Phospho-GSK3β expression was increased in mutant NSPCs, suggesting hyperactivation of the Wnt pathway.40 Wnt and PI3K signaling are commonly coactivated in stem cells, consistent with enhanced neurosphere-forming capacity of Pik3caH1047R–PtenΔ mutant NSPCs. Wnt signaling is critical for maintaining efficient self-renewal of NSPCs41,42 and glioma stem cell self-renewal.43,44 The clinical importance of Wnt signaling in gliomas is highlighted by the correlation of Wnt factors with independent markers of poor prognosis.45
We and others have shown that the kinase-inducible transcription factor CREB and its activated form, pCREB, are expressed in GBM14,46 and that CREB has a role in glioma cell proliferation.14 Aside from the expected upregulation of the PI3K pathway factors in Pik3caH1047R–PtenΔ tumors, the MAPK pathway was also upregulated, consistent with observations that CREB sits at a transcriptional convergence point of the PI3K and MAPK pathways.14 CREB deletion in Pik3caH1047R–PtenΔ NSPCs showed that mutant Pik3caH1047R–PtenΔ–CrebΔ mice were symptom free for a longer time compared with double mutant Pik3caH1047R–PtenΔ mice. CREB loss resulted in reduced tumor cellularity and larger tumor cells, suggestive of a less malignant, giant-cell GBM, which has a better prognosis compared with classical GBM.47 Moreover, no invasion into white matter tracts was observed in triple mutant brains. This is the first in vivo data demonstrating that CREB deletion in tumor cells suppresses tumor growth, consistent with studies correlating aberrant CREB expression and activation with poor prognosis in many neoplasms, including lung48 and breast.49
Overall, our data lead us to propose a model in which the PI3K pathway is critical and sufficient for reprogramming normal NSPCs into tumor-initiating cells. Additionally, we show that mutation of the catalytic PI3K subunit, p110α, leads to transformation of NSPCs into low-grade oligodendroglial-like tumors but that simultaneous PTEN loss enhances oncogenesis, resulting in fast, aggressive tumor growth. As the PI3K mutant tumor cells mature, a CREB-dependent transcriptome is activated, which supports tumor growth. MAPK signaling is also coactivated, further enhancing CREB activation. The Wnt pathway is also hyperactivated and contributes to promoting NSPC self-renewal. Deleting CREB in tumor cells alters the tumor cell phenotype, slows tumor cell growth, and extends symptom-free survival. Recently developed experimental compounds which target CREB can inhibit tumor cell proliferation in vitro and in vivo.50 Co-targeting archetypal oncogenic pathways, including PI3K, MAPK, and Wnt, as well as transcription factors such as CREB, may prove to be an effective therapeutic approach for difficult to treat cancers, including HGGs.
Supplementary Material
Supplementary material is available at Neuro-Oncology online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by the Department of Pathology, CASS Foundation grant 6236. W.A.P. was supported in part by project grant 1080491 from the National Health and Medical Research Council (NHMRC) of Australia.
Acknowledgments
We thank Rob Ramsay, Andrew Allen, Daniel Gough, Jason Cain, and Ryan Hutchinson for helpful scientific discussions; Tina Isaakidis for proofreading, Sarah Louise Taverner for dedicated animal care and monitoring throughout this project, as well as Teresa Drever, Jessica Sturrock, Michelle Williams, and Marica Kesar for running and managing an excellent animal facility; Daniel Blashki and Vanta Jameson for flow cytometry assistance.
Conflict of interest statement. The authors confirm that they have no conflict of interest regarding the content of the present article.
References
- 1. Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12(23):3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25(1):55–57. [DOI] [PubMed] [Google Scholar]
- 3. Alcantara Llaguno S, Chen J, Kwon CH, et al. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15(1):45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chow LM, Endersby R, Zhu X, et al. Cooperativity within and among Pten, p53, and Rb pathways induces high-grade astrocytoma in adult brain. Cancer Cell. 2011;19(3):305–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ozawa T, Riester M, Cheng YK, et al. Most human non-GCIMP glioblastoma subtypes evolve from a common proneural-like precursor glioma. Cancer Cell. 2014;26(2):288–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Alcantara Llaguno SR, Wang Z, Sun D, et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell. 2015;28(4):429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Danks RA, Orian JM, Gonzales MF, et al. Transformation of astrocytes in transgenic mice expressing SV40 T antigen under the transcriptional control of the glial fibrillary acidic protein promoter. Cancer Res. 1995;55(19):4302–4310. [PubMed] [Google Scholar]
- 8. Friedmann-Morvinski D, Bushong EA, Ke E, et al. Dedifferentiation of neurons and astrocytes by oncogenes can induce gliomas in mice. Science. 2012;338(6110):1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu C, Sage JC, Miller MR, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146(2):209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jacques TS, Swales A, Brzozowski MJ, et al. Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. EMBO J. 2010;29(1):222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robinson JP, VanBrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene. 2010;29(3):335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu Z, Khan MA, Weiler M, et al. Targeting self-renewal in high-grade brain tumors leads to loss of brain tumor stem cells and prolonged survival. Cell Stem Cell. 2014;15(2):185–198. [DOI] [PubMed] [Google Scholar]
- 13. Peltier J, O’Neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol. 2007;67(10):1348–1361. [DOI] [PubMed] [Google Scholar]
- 14. Daniel P, Filiz G, Brown DV, et al. Selective CREB-dependent cyclin expression mediated by the PI3K and MAPK pathways supports glioma cell proliferation. Oncogenesis. 2014;3:e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Padala RR, Karnawat R, Viswanathan SB, Thakkar AV, Das AB. Cancerous perturbations within the ERK, PI3K/Akt, and Wnt/β-catenin signaling network constitutively activate inter-pathway positive feedback loops. Mol Biosyst. 2017;13(5):830–840. [DOI] [PubMed] [Google Scholar]
- 16. Rheinbay E, Suvà ML, Gillespie SM, et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013;3(5):1567–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kita D, Yonekawa Y, Weller M, Ohgaki H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 2007;113(3):295–302. [DOI] [PubMed] [Google Scholar]
- 18. Tamborero D, Gonzalez-Perez A, Perez-Llamas C, et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep. 2013;3:2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Broderick DK, Di C, Parrett TJ, et al. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64(15):5048–5050. [DOI] [PubMed] [Google Scholar]
- 20. Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Di Cristofano A, Pandolfi PP. The multiple roles of PTEN in tumor suppression. Cell. 2000;100(4):387–390. [DOI] [PubMed] [Google Scholar]
- 22. Gregorian C, Nakashima J, Le Belle J, et al. Pten deletion in adult neural stem/progenitor cells enhances constitutive neurogenesis. J Neurosci. 2009;29(6):1874–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Groszer M, Erickson R, Scripture-Adams DD, et al. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294(5549):2186–2189. [DOI] [PubMed] [Google Scholar]
- 24. Kinross KM, Montgomery KG, Kleinschmidt M, et al. An activating Pik3ca mutation coupled with Pten loss is sufficient to initiate ovarian tumorigenesis in mice. J Clin Invest. 2012;122(2):553–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Imayoshi I, Ohtsuka T, Metzger D, Chambon P, Kageyama R. Temporal regulation of Cre recombinase activity in neural stem cells. Genesis. 2006;44(5):233–238. [DOI] [PubMed] [Google Scholar]
- 26. Mantamadiotis T, Lemberger T, Bleckmann SC, et al. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet. 2002;31(1):47–54. [DOI] [PubMed] [Google Scholar]
- 27. Casanova E, Fehsenfeld S, Lemberger T, Shimshek DR, Sprengel R, Mantamadiotis T. ER-based double iCre fusion protein allows partial recombination in forebrain. Genesis. 2002;34(3):208–214. [DOI] [PubMed] [Google Scholar]
- 28. Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347(1–2):70–78. [DOI] [PubMed] [Google Scholar]
- 29. Liang H, Hippenmeyer S, Ghashghaei HT. A Nestin-cre transgenic mouse is insufficient for recombination in early embryonic neural progenitors. Biol Open. 2012;1(12):1200–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kinross KM, Montgomery KG, Mangiafico SP, et al. Ubiquitous expression of the Pik3caH1047R mutation promotes hypoglycemia, hypoinsulinemia, and organomegaly. FASEB J. 2015;29(4):1426–1434. [DOI] [PubMed] [Google Scholar]
- 31. Memmel S, Sukhorukov VL, Höring M, et al. Cell surface area and membrane folding in glioblastoma cell lines differing in PTEN and p53 status. PLoS One. 2014;9(1):e87052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dworkin S, Malaterre J, Hollande F, Darcy PK, Ramsay RG, Mantamadiotis T. cAMP response element binding protein is required for mouse neural progenitor cell survival and expansion. Stem Cells. 2009;27(6):1347–1357. [DOI] [PubMed] [Google Scholar]
- 33. Amiri A, Cho W, Zhou J, et al. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J Neurosci. 2012;32(17):5880–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hartmann C, Bartels G, Gehlhaar C, Holtkamp N, von Deimling A. PIK3CA mutations in glioblastoma multiforme. Acta Neuropathol. 2005;109(6):639–642. [DOI] [PubMed] [Google Scholar]
- 35. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–13378. [DOI] [PubMed] [Google Scholar]
- 36. Chen H, Mei L, Zhou L, et al. PTEN restoration and PIK3CB knockdown synergistically suppress glioblastoma growth in vitro and in xenografts. J Neurooncol. 2011;104(1):155–167. [DOI] [PubMed] [Google Scholar]
- 37. Pridham KJ, Le L, Guo S, et al. PIK3CB/p110beta is a selective survival factor for glioblastoma. Neuro Oncol. 2017;20(4):494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kubiatowski T, Jang T, Lachyankar MB, et al. Association of increased phosphatidylinositol 3-kinase signaling with increased invasiveness and gelatinase activity in malignant gliomas. J Neurosurg. 2001;95(3):480–488. [DOI] [PubMed] [Google Scholar]
- 39. Wei Y, Jiang Y, Zou F, et al. Activation of PI3K/Akt pathway by CD133-p85 interaction promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad Sci U S A. 2013;110(17):6829–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McManus EJ, Sakamoto K, Armit LJ, et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24(8):1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kalani MY, Cheshier SH, Cord BJ, et al. Wnt-mediated self-renewal of neural stem/progenitor cells. Proc Natl Acad Sci U S A. 2008;105(44):16970–16975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wexler EM, Paucer A, Kornblum HI, Palmer TD, Plamer TD, Geschwind DH. Endogenous Wnt signaling maintains neural progenitor cell potency. Stem Cells. 2009;27(5):1130–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pulvirenti T, Van Der Heijden M, Droms LA, Huse JT, Tabar V, Hall A. Dishevelled 2 signaling promotes self-renewal and tumorigenicity in human gliomas. Cancer Res. 2011;71(23):7280–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang N, Wei P, Gong A, et al. FoxM1 promotes β-catenin nuclear localization and controls Wnt target-gene expression and glioma tumorigenesis. Cancer Cell. 2011;20(4):427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu C, Tu Y, Sun X, et al. Wnt/beta-catenin pathway in human glioma: expression pattern and clinical/prognostic correlations. Clin Exp Med. 2011;11(2):105–112. [DOI] [PubMed] [Google Scholar]
- 46. Barresi V, Mondello S, Branca G, Rajan TS, Vitarelli E, Tuccari G. p-CREB expression in human gliomas: potential use in the differential diagnosis between astrocytoma and oligodendroglioma. Hum Pathol. 2015;46(2):231–238. [DOI] [PubMed] [Google Scholar]
- 47. Kozak KR, Moody JS. Giant cell glioblastoma: a glioblastoma subtype with distinct epidemiology and superior prognosis. Neuro Oncol. 2009;11(6):833–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu W, Wu Y, Wang L, et al. Protein signature for non-small cell lung cancer prognosis. Am J Cancer Res. 2014;4(3):256–269. [PMC free article] [PubMed] [Google Scholar]
- 49. Chhabra A, Fernando H, Watkins G, Mansel RE, Jiang WG. Expression of transcription factor CREB1 in human breast cancer and its correlation with prognosis. Oncol Rep. 2007;18(4):953–958. [PubMed] [Google Scholar]
- 50. Xie F, Li BX, Kassenbrock A, et al. Identification of a potent inhibitor of CREB-mediated gene transcription with efficacious in vivo anticancer activity. J Med Chem. 2015;58(12):5075–5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.