

ABSTRACT
International experts gathered at the Mayo Clinic (Rochester MN, USA) on February 27th-28th, 2017 for a meeting entitled ‘Basic and Translational Facets of the Epigenetics of GI Diseases’. This workshop summarized recent advances on the role of epigenetics in the pathobiology of gastrointestinal (GI) diseases. Highlights of the meeting included recent advances on the involvement of different epigenetic mechanisms in malignant and nonmalignant GI disorders and the epigenetic heterogeneity exhibited in these diseases. The translational value of epigenetic drugs, as well as the current and future use of epigenetic changes (i.e., DNA methylation patterns) as biomarkers for early detection tools or disease stratification were also important topics of discussion.
KEYWORDS: Gastrointestinal diseases, epigenetics, chromatin, miRNA, DNA methylation, biomarkers, therapeutics
The field of epigenetics centers on molecular events that regulate stable alterations in gene expression (as well as other nuclear functions) that are not due to changes in the DNA sequence (i.e., mutations). Epigenetic mechanisms include: i) changes in DNA methylation; ii) regulation of chromatin dynamics and structure (e.g., through alterations in histone modifications or nucleosome positioning) and; iii) changes in expression and activity of noncoding RNAs (Figure 1). The consideration of epigenetic mechanisms and their alterations at a genome-wide scale is called epigenomics. In this meeting report we provide a summary of recent advances and translational implications of the aforementioned epigenetic mechanisms for the pathogenesis, diagnosis, and treatment of GI diseases.
Figure 1.
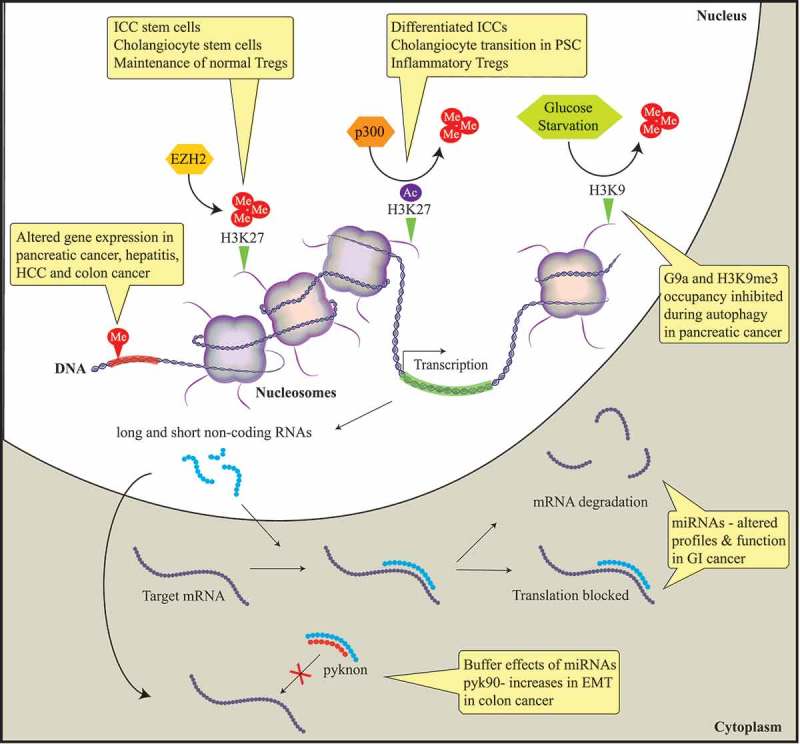
Diagram describing the main epigenetic mechanisms identified to date that have a role in the pathobiology of benign and malignant gastrointestinal diseases.
In this meeting report, we summarize and discuss presentations by international experts in the field of DNA methylation, histone-based epigenetics, and microRNAs. Epigenetic mechanisms that induce stable alterations to gene expression without alteration of the DNA sequence result in aberrant gene expression. Histone methylation alters gene expression in pancreatic cancer, hepatitis, hepatocellular cancer (HCC), and colon cancer. EZH2, a histone methyltransferase, induces the epigenetic gene silencing mark H3K27me3. This epigenetic mark mobilizes the progenitor interstitial cells of Cajal (ICC) to transition to mature ICC in gastrointestinal (GI) motility disorders. In addition, this mark maintains normal cholangiocytes and Regulatory T cells (T-regs) through FN1 and FOXP3 target silencing, respectively. The histone acetylase complex p300 replaces H3K27 methylation with acetylation resulting in FN1 transcription, promoting cholangiocytes to transition to primary sclerosing cholangitis (PSC) in liver disease. Autophagy in pancreatic cancer is associated with a depression of gene expression caused by the dissociation of the histone methyltransferase G9a and H3K9me3 from gene promoters. Non-protein-coding RNAs are associated with the initiation and progression of GI diseases through negatively regulating protein expression by binding to the promoter regions of targeted mRNAs leading to translational repression or mRNA degradation. Pyknons are lncRNAs that buffer the effects of miRNAs; pyk90 plays a role in epithelial-to-mesenchymal transition (EMT) in colon cancer via a targeted decreased expression of E-cadherin.
Epigenetic dynamics in non-neoplatic diseases
Non-neoplastic diseases affecting the GI system include diabetes, pancreatitis, hepatitis, cholangiopathies, and inflammatory bowel disease (IBD). Genetic mutations have been identified as causing or enhancing susceptibility for each of these diseases; however, such mutations are observed in the minority of patients, suggesting that alternative factors regulate disease incidence. In addition, these diseases have highly variable presentation between patients, and this variability is not fully explained by inherited genetic factors [1–10]. Thus, aspects of these diseases are likely to be regulated by epigenetic mechanisms.
Dr. Tamas Ordog described progress in understanding the epigenetic regulation of the interstitial cells of Cajal (ICC), which are electrical pacemaker and neuromodulator cells of the GI neuromuscular system. Alterations in ICC number or function contribute to dysregulation of gastric emptying in diabetes [11–13]. ICC differentiation and function depend on expression of the receptor tyrosine kinase, Kit. Gene expression of Kit was found to be repressed by trimethylation of histone 3, lysine residue 27 (H3K27me3) at its promoter in both ICC stem cells (ICC-SC) and dedifferentiated ICC. In differentiating ICC, H3K27me3 is replaced by the activating mark, H3K27 acetylation (ac) in a super-enhancer region regulating Kit expression. Kit expression and the differentiation of ICC was shown to be negatively regulated by Enhancer of Zeste Homolog 2 (EZH2), the histone methyltransferase that writes the H3K27me3 mark on chromatin. EZH2 inhibition mobilizes ICC progenitors to transition into mature ICC in GI motility disorders where ICC loss has been observed. However, prolonged EZH2 inhibition leads to ICC-SC exhaustion and, eventually, to ICC-SC depletion. These findings indicate that appropriate EZH2 dosage is necessary for ICC homeostasis. In diabetic mice, ICC-stem cell (SC) failure was the main cause of ICC depletion. Additional studies showed that succinate, a mitochondrial metabolite that accumulates in diabetes, reduced Kit expression by stimulating promoter occupancy by H3K27me3. Kit+ ICC were reduced in mice with conditional deletion of Sdhc encoding subunit C of the succinate-oxidizing enzyme, succinate dehydrogenase [12]. Together, these results reveal a metabolic-epigenetic regulation of phenotypic transitions in the GI neuromuscular system that may be pathogenetically significant.
Dr. Robert Huebert presented work on the epigenetic regulation of the pathophysiology of primary sclerosing cholangitis (PSC), a fibro-obliterative cholangiopathy that leads to end-stage liver disease. It was noted that although the disease has complex and multifactorial causes, signal integration at the level of the epigenome may make the disease amenable to epigenetic pharmacology. Dr. Huebert highlighted recently published work indicating that signaling via the hedgehog/GLI pathway in cholangiocytes overcomes an EZH2-dependent epigenetic barrier to promote cholangiocyte expansion [14]. His presentation also showed that in normal cholangiocytes, fibronectin (FN1) transcription is homeostatically silenced by EZH2 and, in PSC, the p300 histone acetylase complex replaces H3K27 methylation with H3K27ac to initiate FN1 transcription. It was further demonstrated that cholangiocyte differentiation from induced pluripotent stem cells (iPSC) required similar epigenetic alterations. These findings are being used as a basis to test therapeutic approaches for biliary diseases based on targeting the newly discovered pathways with epigenetic drugs.
Recent progress in understanding the role of epigenetics in IBD was presented by Dr. William Faubion. Regulatory T cells (Tregs) play important roles in autoimmune diseases such as IBD. These cells are defined by the expression of the lineage-specific expression of the transcription factor Forkhead Box P3 (FOXP3) [15]. Dr. Faubion’s group and others have shown that FOXP3 recruits EZH2 to repress gene expression [16,17]. Transgenic mouse studies in which the EZH2 SET domain (which carries the methyltransferase activity of EZH2) was deleted in FOXP3+ cells demonstrated that Ezh2ΔSET/ΔSET-Tregs did not show a regulator phenotype and secreted proinflammatory cytokines, unlike Tregs from Ezh2+/+ mice. These studies showed that FOXP3 and the Polycomb repressor complex 2 (PRC2) form a repressive complex and that IL6 disrupts this complex through a JAK-STAT signaling pathway. In vivo, mice with Ezh2 SET domain knockout in FOXP3+ cells developed lethal autoimmunity, including spontaneous IBD. These studies suggest that EZH2 plays a role in epigenetic regulation of Tregs in IBD. RNA-seq of CD4+ lymphocytes from ilia of patients with Crohn’s disease vs. normal controls demonstrated differential gene expression between the two groups. Among the differentially expressed genes, gene network analysis identified an enrichment of gene targets shared by FOXP3 and EZH2 [18]. Together, these investigations support a possible dysfunction of EZH2 in Tregs in IBD.
The above-mentioned studies illustrate the involvement of the repressive EZH2/H3K27me3 epigenetic mechanism in the differentiation of different cell types (ICCs, cholangiocytes, and Tregs) involved in non-neoplastic diseases. These observations demonstrate the complexity of trying to target EZH2 to treat disease, as inhibition of EZH2 will promote ICC and cholangiocyte differentiation, but inhibit the function of normal Tregs. On the horizon, these presentations also indicate that much more needs to be learned about epigenetic processes in these diseases. For example, are the demethylases that oppose EZH2 and remove the H3K27me3 mark also regulated in these model systems? Further, regulation of these GI diseases by other histone modifying enzymes has been poorly studied.
Epigenetic mechanisms in neoplastic diseases
The presence of gene mutations in tumor cells has been well-characterized as a primary driver of cancer initiation and progression. However, mutations in epigenetic modifying genes, and the identification of epigenetic aberrations in cancer suggest that epigenetic factors play as important a role in cancer [19,20]. Dr. Daniel Billadeau discussed his work on the epigenetic regulation of autophagy in cancer cells. His group demonstrated that depletion of the histone methyltransferase G9a by short hairpin (sh) RNA or using chemical inhibitors of G9a in pancreatic cancer cells resulted in increased expression of genes promoting the cell cycle (e.g., p21/Cdkn1a and p27/Cdkn1b), enlargement of lysosomes, and increased levels of LC3B-II, a protein marker of autophagy. They further found that G9a is normally associated with the promoters of autophagy related genes LC3B, WIPI1, and DOR, where G9a mediates repression of these genes [21]. Upon induction of autophagy by glucose starvation, G9a, H3K9me3, and H3K9me2 were decreased at these promoters, contributing to increased expression of LC3B, WIPI1 and DOR. p62 aggregation, another marker for autophagy, was increased by inhibition of G9a, but p62 degradation was only increased when mTOR was also inhibited, indicating that G9a inhibition by itself was insufficient to increase autophagic flux. These studies indicate that the derepression of gene expression caused by the dissociation of G9a from gene promoters plays a key role in autophagy.
The involvement of the nucleosome remodeling protein BRG1 (Brahma-related gene-1; SMARCA4) in pancreatic cancer was discussed by Dr. Nilotpal Roy. His group used a genetic mouse model in which mutant Kras is expressed in pancreatic exocrine cells to demonstrate that loss of Brg1 in these cells inhibited the formation of intraductal papillary mucinous neoplasms (IPMNs) arising from pancreatic duct cells, but accelerated the development of pancreatic intraepithelial neoplasia (PanIN) and pancreatic adenocarcinoma (PDAC) from pancreatic acinar cells [22,23]. They further demonstrated that BRG1 expression in duct cells supported the expression of the transcription factor SOX9, a protein that maintains the differentiated state of adult pancreatic duct cells. Thus, in the absence of BRG1, Sox9 was decreased, allowing for the de-differentiation of duct cells and increased incidence of IPMN.
The role of epigenetic reprogramming in pancreatitis and the link to pancreatic ductal adenocarcinoma (PDAC) are poorly understood. Dr. Christopher Pin has developed several genetically modified mouse models that allow for inducible loss of epigenetic regulators, such as EZH2 or ATRX, in adult acinar cells ± the inducible activation of oncogenic KRAS. These mouse lines show that altering epigenetic regulating factors increases the susceptibility and progression of pancreatitis and KRAS-induced PDAC. These studies have relevance to the concept that environmental factors (e.g., alcohol and tobacco use) may alter the pancreatic epigenome towards an increased susceptibility to pancreatitis and cancer. In addition, Dr. Pin pointed out that targeting factors that alter the epigenome for therapeutic purposes in pancreatic disease could have negative consequences and such therapies need to be developed with caution.
Epigenetic heterogeneity was a major theme of work presented by Dr. Keith Robertson on liver disease and hepatocellular cancer (HCC). Hepatic cirrhosis is often an outcome of chronic infection by hepatitis viruses B or C, with alcoholism also playing a major role, and most HCC appear to develop by progression from premalignant cirrhotic nodules in the liver [24]. Dr. Robertson’s laboratory studied global DNA methylation patterns in multiple cirrhotic nodules and HCC from individual human liver samples. DNA methylation patterns in nodules were quite heterogeneous but, overall, nodules exhibited a pattern of progression towards the aberrant DNA methylation seen in HCC. Classification of epigenetic landscapes from individual cirrhotic nodules, which were summarized as ‘epigenetic progression’ (EP) scores, demonstrated that the EP score is significantly linked to cellular proliferation. Further examination of loci that drive EP scores shows that many are correlated with altered expression, are deregulated in HCC, and stratify patient survival. Taken together, this approach allows the formulation of a model of epigenetic deregulation in liver disease progression, where CpGs with highly variable methylation are enriched in key pathways early in disease, are further deregulated in HCC, and may ultimately point to novel epigenetic drivers of HCC.
Dr. George Calin reviewed progress in the discovery and characterization of noncoding RNAs and their relevance to human diseases. By using pattern discovery methods, Dr. Calin’s group identified multiple types of long noncoding RNAs (lncRNAs), including pyknon-transcripts [25]. Pyknons are nonrandom repeated elements found most often in the 3′ untranslated regions of genes. Pyknons are species-specific and are believed to buffer the effects of short noncoding micro RNAs (miRNAs), possibly by acting as ‘sponges’. Differential pyknon expression is observed in normal and disease tissues, and studies support a role in fundamental cellular processes, such as epithelial-to-mesenchymal transition, cell proliferation, and cell death. For example, increased expression of pyknon pyk90 correlated to decreased expression of E-cadherin, likely through a miRNA-dependent process. In addition to these lncRNAs, miRNAs have already been shown to be important in the initiation, progression, and metastases of human cancer. Expression correlating these ‘new and old’ ncRNAs with cancer metastatic potential and overall survival rates suggests that ncRNAs could potentially be used as biomarkers or novel therapeutic targets in human disease.
Dr. Muller Fabbri presented work describing the exosomal transfer of miRNAs between cancer cells and immune cells. His work showed that non-small-cell lung carcinoma (NSCLC) cells release exosomes containing miR-16, miR-21, and miR-29a, which are taken up by immune cells [26]. However, rather than acting by binding specific mRNAs and inhibiting translation, miR-21 and miR-29a bind Toll-like receptor 8 (TLR8; and mouse Tlr7), causing receptor activation and initiating a pro-oncogenic inflammatory response mediated by NFκB. This group also showed that neuroblastoma cells released exosomes containing miR-21, which are taken up by monocytes, thus activating TLR8 and leading to their release of miR-155 [27]. miR155 is in turn taken up by the neuroblastoma cells, where it suppresses expression of the telomerase inhibitor, Telomeric repeat-binding factor (TERF1), thus contributing to tumor growth. Importantly, miR155 expression is correlated with therapeutic resistance, suggesting it may be a target for suppression in combinatorial therapies.
Dr. Marina Koutsioumpa described work on Inhibitor of growth family member 3 (ING3), a protein that is reduced in pancreatic cancer as well as in mouse models of PDAC. ING3 is a component of the NuA4 histone acetyltransferase (HAT) complex, which acetylates histones H4 and H2A, promoting gene activation [28]. ING3 binds H3K4Me3 and its overexpression promotes apoptosis. Depletion of ING3 increases miR-539 expression, resulting in increased proliferation, invasiveness, migration and enhanced gemcitabine resistance in pancreatic cancer cells. Her studies suggest epigenetic mechanisms can play roles in regulating miRNA expression that are relevant in pancreatic cancer pathogenesis and the response to chemotherapeutic agents. Together, these presentations emphasize that multiple epigenetic mechanisms (histone methyltransferases, DNA methylation, chromatin remodeling complexes, and mi- and lnc-RNAs) affect the pathogenesis of cancer by affecting susceptibility, initiation, and progression.
Translational value of epigenetics for GI disease diagnosis and treatment
The study of epigenetic mechanisms in GI diseases is leading to advances in the areas of therapeutics, precision medicine, and cancer biomarkers. Several studies were presented concerning efforts to use epigenetically-targeted pharmaceuticals to treat GI cancers.
Dr. Gwen Lomberk described work from her laboratory on Aurora A kinase (AURKA), a Ser/Thr kinase that induces G2/M transition and phosphorylates heterochromatin protein (HP) 1γ [29]. AURKA inhibitor (MLN8237) was used synergistically with a pan-H3K9 methyltransferase (HMT) inhibitor (chaetocin) to inhibit growth and kill pancreatic cancer cells both in vitro and in mouse xenografts. Mechanistically, combined inhibition of AURKA mitotic kinase and H3K9 HMTs triggers uncoordinated checkpoint responses in G2/M, disrupting the cell cycle and leading to mitotic catastrophe. Dr. Jens Siveke presented recent work highlighting the importance of patient stratification in treating pancreatic cancer, as 30% of PDAC patients have alterations in epigenomic regulators [30]. His group showed synergism between the histone deacetylase (HDAC) inhibitor suberoylanilide hydroxamic acid (SAHA) and JQ, an inhibitor of bromodomain and extraterminal (BET) proteins, in suppressing PDAC, but only in a subset of tumor xenografts. These results suggest that a drug screening program should be developed to identify candidates for dual BET/HDAC inhibition. Dr. Nita Ahuja reviewed the status of DNA methyltransferase (DMNT) inhibitors and HDAC inhibitors for use in solid tumors [31]. These drugs have been FDA approved and are used with success for hematological malignancies, but have thus far not been successfully utilized in solid tumors due to factors such as toxicity and rapid degradation of the compounds. Current trials are now evaluating epigenetic compounds as priming agents to reprogram the epigenetic state of tumor cells with subsequent use of chemotherapy or immunotherapy for cancer treatment.
Attempts have been made to identify subtypes of PDAC in hopes of developing more specific and effective therapies for use within subtypes [32–34]. Thus far, genome-wide studies of gene mutations in PDAC have not led to selective treatments of PDAC subtypes based on mutation type. However, transcriptome studies of PDAC samples have suggested the division of this disease into classical, quasimesenchymal, and exocrine-like subtypes on the basis of differential gene expression and histological differences within subtypes [34]. As mentioned above, Dr. Jens Siveke identified different PDAC subtypes based on differential sensitivity to drugs such as HDAC and BET inhibitors [30]. Higher levels of the lncRNA CCAT1 correlated with increased sensitivity to HDAC inhibitors, which is already used to predict sensitivity to BET inhibitors in colorectal cancers. Using a patient-derived xenograft (PDX) approach, this group identified a transcriptomic signature that identifies PDAC cells dependent upon c-MYC. PDX with a c-MYC signature were more likely to respond to JQ, which inhibits BET proteins required for c-MYC function.
Drs. Raul Urrutia and Juan Iovanna both talked about the significance of stratifying patient populations to patient-specific or ‘precision’ medicine. Dr. Urrutia focused on the bioinformatics analysis of epigenetic signatures as they related to histone modifications and RNA expression. Dr. Iovanna noted that most genome-wide studies of mutations and gene expression in PDAC have been performed on the small percentage of patients with resectable tumors. Thus, samples from unresectable PDAC patients have been underrepresented in genome-wide investigations due to lack of tissue. To circumvent this limitation, PDAC biopsies have been used to develop patient derived xenografts (PDX) that can be used for multi-omics characterization. By combining global DNA methylation and histone modification patterns with RNA expression, two general subtypes of PDAC were described: classical and basal [35]. These two PDAC subtypes are mainly defined by differential DNA methylation patterns. Dr. Urrutia suggested that further stratification within these groups could help predict PDAC aggressiveness and response to therapy. In support of this work, Dr. Iovanna presented data on PDAC patients with varying responses to the existing treatments, gemcitabine and Folfirinox, although the mechanisms underlying the differential responses are unknown.
Dr. Kisiel reviewed the use of DNA methylation patterns in stool to detect cancer. The use of these samples allows for a non-invasive approach to assess targeted aberrancies of DNA methylation. Multi-target stool DNA assays detect 94% of Stage I-II colorectal cancer (CRC) and 70% of advanced adenomas with high-grade dysplasia; the assay technology used in the COLOGUARD system developed in collaboration with Exact Sciences (Madison, WI) is expected to allow for detection of DNA methylation patterns in other GI cancers as well [36]. Dr. Kisiel’s group has shown that aberrant DNA methylation patterns in stool is more broadly informative and sensitive for detection of neoplasia than individual DNA mutations. Importantly, DNA methylation patterns in stool and plasma, especially at CpG islands, appear to be unique for specific tumor types. Through a comprehensive comparison of the range of particular DNA methylation patterns observed for different tumor types, it may be possible to screen for multiple GI tumors with a single test. Indeed, a feasibility study showed that two methylation markers, BMP3 and QKI, were able to discriminate CRC from pancreatic cancer from normal controls at 74% accuracy, using plasma samples from advanced-stage patients.
Conclusions
Despite the recent progress made, it is clear that a better understanding of epigenomic changes and how they relate to gene expression and drug sensitivity/resistance is necessary to realize the potential of precision medicine-based approaches in therapy for GI diseases. Specialized approaches to target epigenetic modifying complexes in a cell specific fashion need to be developed to limit adverse side effects. Finally, much of the work presented in this workshop, and most of the studies in the literature, point to epigenetic repression as a key target for therapy. Further studies are needed to examine aberrant activity of epigenetic activating complexes in the context of GI disease. This is particularly true for PDAC, in which changes in global demethylation can correlate with tumor progression.
In conclusion, these talks highlighted numerous mechanisms by which epigenetic alterations contribute to GI disease initiation and progression. All three classes of epigenetic regulation (DNA methylation, chromatin remodeling, and ncRNAs) show promise as both diagnostic and therapeutic targets. Epigenetic changes can be equally informative as genetic mutations regarding cancer severity and treatment sensitivity, and epigenomic profiles can aid in stratification of cancer-subtypes. In addition, epigenetic modifying proteins/complexes may be excellent targets for combinatorial therapies, and many drugs inhibiting epigenetic complexes are already in clinical trials or approved for therapy.
Funding Statement
This work was supported by the National Institutes of Health [CA015083];National Institutes of Health [CA102701];National Institutes of Health [DK090728];National Institutes of Health [DK084567].
Acknowledgments
We would like to thank Charyl M. Dutton Gibbs and Lori R. Anderson for their expert secretarial assistance in the organization of the workshop. This meeting was supported by Mayo Clinic Center for Cell Signaling in Gastroenterology (C-SiG), GI Program of Mayo Clinic Comprehensive Cancer Center (MCCC), Basic Sciences Program of MCCC, Mayo Clinic SPORE in Pancreatic Cancer, Mayo Translational PKD Center (MTPC), Mayo Clinic Center for Biomedical Discovery (CBD) and Epigenomics Program of the Mayo Clinic Center for Individualized Medicine (CIM).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Czaja AJ. Epigenetic changes and their implications in autoimmune hepatitis. Eur J Clin Invest. 2018. January 31;48:e12899 PubMed PMID: 29383703. [DOI] [PubMed] [Google Scholar]
- 2.HSP de Souza, Fiocchi C, Iliopoulos D.. The IBD interactome: an integrated view of aetiology, pathogenesis and therapy. Nat Rev Gastroenterol Hepatol. 2017. December;14(12):739–749. PubMed PMID: 28831186. [DOI] [PubMed] [Google Scholar]
- 3.Duell EJ. Epidemiology and potential mechanisms of tobacco smoking and heavy alcohol consumption in pancreatic cancer. Mol Carcinog. 2012. January;51(1):40–52. PubMed PMID: 22162230. [DOI] [PubMed] [Google Scholar]
- 4.Kleeff J, Whitcomb DC, Shimosegawa T, et al. Chronic pancreatitis. Nat Rev Dis Primers. 2017. September 7;3:17060 PubMed PMID: 28880010. [DOI] [PubMed] [Google Scholar]
- 5.Ley D, Desseyn JL, Mischke M, et al. Early-life origin of intestinal inflammatory disorders. Nutr Rev. 2017. March 1;75(3):175–187. PubMed PMID: 28340001. [DOI] [PubMed] [Google Scholar]
- 6.Moran-Salvador E, Mann J. Epigenetics and liver fibrosis. Cell Mol Gastroenterol Hepatol. 2017. July;4(1):125–134. PubMed PMID: 28593184; PubMed Central PMCID: PMCPMC5453904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reichert MC, Hall RA, Krawczyk M, et al. Genetic determinants of cholangiopathies: molecular and systems genetics. Biochim Biophys Acta. 2017. July 27 PubMed PMID: 28757171 DOI: 10.1016/j.bbadis.2017.07.029 [DOI] [PubMed] [Google Scholar]
- 8.Robertson CC, Rich SS. Genetics of type 1 diabetes. Curr Opin Genet Dev. 2018. February 13;50:7–16. PubMed PMID: 29453110. [DOI] [PubMed] [Google Scholar]
- 9.Webb GJ, Siminovitch KA, Hirschfield GM. The immunogenetics of primary biliary cirrhosis: A comprehensive review. J Autoimmun. 2015. November;64:42–52. PubMed PMID: 26250073; PubMed Central PMCID: PMCPMC5014907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018. February;14(2):88–98. PubMed PMID: 29219149. [DOI] [PubMed] [Google Scholar]
- 11.Grover M, Bernard CE, Pasricha PJ, et al. Diabetic and idiopathic gastroparesis is associated with loss of CD206-positive macrophages in the gastric antrum. Neurogastroenterol Motil. 2017. June;29(6). PubMed PMID: 28066953; PubMed Central PMCID: PMCPMC5423829 DOI: 10.1111/nmo.13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi Y, Toyomasu Y, Saravanaperumal SA, et al. Hyperglycemia increases interstitial cells of cajal via MAPK1 and MAPK3 signaling to ETV1 and KIT, leading to rapid gastric emptying. Gastroenterology. 2017. August;153(2):521–535 e20. PubMed PMID: 28438610; PubMed Central PMCID: PMCPMC5526732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ordog T, Takayama I, Cheung WK, et al. Remodeling of networks of interstitial cells of Cajal in a murine model of diabetic gastroparesis. Diabetes. 2000. October;49(10):1731–1739. PubMed PMID: 11016458. [DOI] [PubMed] [Google Scholar]
- 14.Jalan-Sakrikar N, De Assuncao TM, Lu J, et al. Hedgehog signaling overcomes an EZH2-dependent epigenetic barrier to promote cholangiocyte expansion. PLoS One. 2016;11(12):e0168266 PubMed PMID: 27936185; PubMed Central PMCID: PMCPMC5148157. This does not alter our adherence to PLOS ONE policies on sharing data and materials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo CT, Liao W, Dadi S, et al. Graded Foxo1 activity in Treg cells differentiates tumour immunity from spontaneous autoimmunity. Nature. 2016. January 28;529(7587):532–536. PubMed PMID: 26789248; PubMed Central PMCID: PMCPMC4978705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arvey A, van der Veeken J, Samstein RM, et al. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol. 2014. June;15(6):580–587. PubMed PMID: 24728351; PubMed Central PMCID: PMCPMC4112080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiong Y, Khanna S, Grzenda AL, et al. Polycomb antagonizes p300/CREB-binding protein-associated factor to silence FOXP3 in a Kruppel-like factor-dependent manner. J Biol Chem. 2012. October 5;287(41):34372–34385. PubMed PMID: 22896699; PubMed Central PMCID: PMCPMC3464543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarmento OF, Svingen PA, Xiong Y, et al. The role of the histone methyltransferase enhancer of Zeste Homolog 2 (EZH2) in the pathobiological mechanisms underlying Inflammatory Bowel Disease (IBD). J Biol Chem. 2017. January 13;292(2):706–722. PubMed PMID: 27909059; PubMed Central PMCID: PMCPMC5241744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iguchi E, Safgren SL, Marks DL, et al. Pancreatic cancer, a mis-interpreter of the epigenetic language. Yale J Biol Med. 2016. December;89(4):575–590. PubMed PMID: 28018146; PubMed Central PMCID: PMCPMC5168833. [PMC free article] [PubMed] [Google Scholar]
- 20.Ribich S, Harvey D, Copeland RA. Drug discovery and chemical biology of cancer epigenetics. Cell Chem Biol. 2017. September 21;24(9):1120–1147. PubMed PMID: 28938089. [DOI] [PubMed] [Google Scholar]
- 21.Artal-Martinez de Narvajas A, Gomez TS, Zhang JS, et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol. 2013. October;33(20):3983–3993. PubMed PMID: 23918802; PubMed Central PMCID: PMCPMC3811684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy N, Malik S, Villanueva KE, et al. Brg1 promotes both tumor-suppressive and oncogenic activities at distinct stages of pancreatic cancer formation. Genes Dev. 2015. March 15;29(6):658–671. PubMed PMID: 25792600; PubMed Central PMCID: PMCPMC4378197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.von Figura G, Fukuda A, Roy N, et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat Cell Biol. 2014. March;16(3):255–267. PubMed PMID: 24561622; PubMed Central PMCID: PMCPMC4684081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghouri YA, Mian I, Rowe JH. Review of hepatocellular carcinoma: epidemiology, etiology, and carcinogenesis. J Carcinog. 2017;16:1 PubMed PMID: 28694740; PubMed Central PMCID: PMCPMC5490340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rigoutsos I, Lee SK, Nam SY, et al. N-BLR, a primate-specific non-coding transcript leads to colorectal cancer invasion and migration. Genome Biol. 2017. May 24;18(1):98 PubMed PMID: 28535802; PubMed Central PMCID: PMCPMC5442648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fabbri M, Paone A, Calore F, et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc Natl Acad Sci USA. 2012. July 31;109(31):E2110–6. PubMed PMID: 22753494; PubMed Central PMCID: PMCPMC3412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Challagundla KB, Wise PM, Neviani P, et al. Exosome-mediated transfer of microRNAs within the tumor microenvironment and neuroblastoma resistance to chemotherapy. J Natl Cancer Inst. 2015. July;107(7). PubMed PMID: 25972604; PubMed Central PMCID: PMCPMC4651042 DOI: 10.1093/jnci/djv135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tallen G, Riabowol K. Keep-ING balance: tumor suppression by epigenetic regulation. FEBS Lett. 2014. August 19;588(16):2728–2742. PubMed PMID: 24632289. [DOI] [PubMed] [Google Scholar]
- 29.Mathison A, Salmonson A, Missfeldt M, et al. Combined AURKA and H3K9 methyltransferase targeting inhibits cell growth by inducing mitotic catastrophe. Mol Cancer Res. 2017. August;15(8):984–997. PubMed PMID: 28442587; PubMed Central PMCID: PMCPMC5705946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazur PK, Herner A, Mello SS, et al. Combined inhibition of BET family proteins and histone deacetylases as a potential epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat Med. 2015. October;21(10):1163–1171. PubMed PMID: 26390243; PubMed Central PMCID: PMCPMC4959788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdelfatah E, Kerner Z, Nanda N, et al. Epigenetic therapy in gastrointestinal cancer: the right combination. Therap Adv Gastroenterol. 2016. July;9(4):560–579. PubMed PMID: 27366224; PubMed Central PMCID: PMCPMC4913338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016. March 3;531(7592):47–52. PubMed PMID: 26909576. [DOI] [PubMed] [Google Scholar]
- 33.Birnbaum DJ, Finetti P, Birnbaum D, et al. Validation and comparison of the molecular classifications of pancreatic carcinomas. Mol Cancer. 2017. November 6;16(1):168 PubMed PMID: 29110659; PubMed Central PMCID: PMCPMC5674743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011. April;17(4):500–503. PubMed PMID: 21460848; PubMed Central PMCID: PMCPMC3755490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicolle R, Blum Y, Marisa L, et al. Pancreatic adenocarcinoma therapeutic targets revealed by tumor-stroma cross-talk analyses in patient-derived xenografts. Cell Rep. 2017. November 28;21(9):2458–2470. PubMed PMID: 29186684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kisiel JB, Raimondo M, Taylor WR, et al. New DNA methylation markers for pancreatic cancer: discovery, tissue validation, and pilot testing in pancreatic juice. Clin Cancer Res. 2015. October 1;21(19):4473–4481. PubMed PMID: 26023084; PubMed Central PMCID: PMCPMC4592385. [DOI] [PMC free article] [PubMed] [Google Scholar]