
Figure 3.
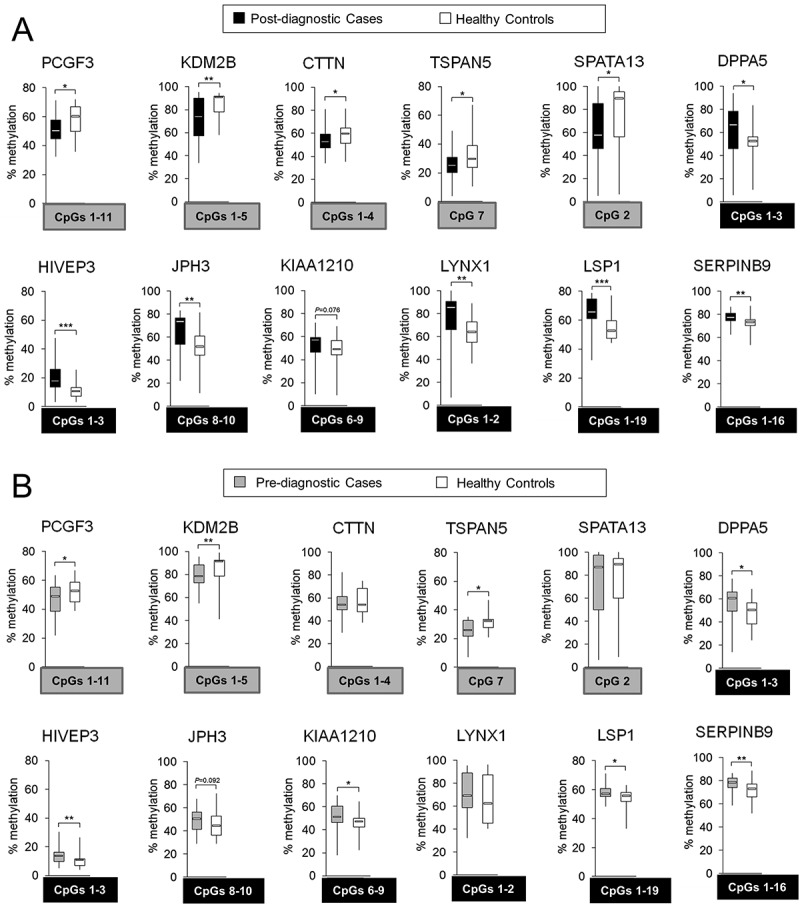
Differentially methylated regions within 5 hypomethylated probes: PCGF3, KDM2B, CTTN, TSPAN5, and SPATA13; and 7 hypermethylated probes: DPPA5, HIVEP3, JPH3, KIAA1210, LYNX1, LSP1, and SERPINB9, in hepatocellular carcinoma (HCC) cases vs. matched healthy controls. Using Illumina 450K microarray, DNA methylation was determined in blood DNA collected from individuals after conventional diagnosis with HCC. Based on the microarray data, 5 hypomethylated and 9 hypermethylated CpG sites corresponding to 5 and 7 genes (probes), respectively, were chosen for validation by pyrosequencing. The difference in DNA methylation, statistical significance, consistency of the difference, location of the CpG site, and the function of a corresponding gene were taken into account in the selection. (A,B) Average methylation level across selected CpG sites within a given probe in (A) post-diagnostic HCC cases (n = 24) and matched healthy controls (n = 24), and (B) pre-diagnostic HCC cases (n = 21) and matched healthy controls (n = 21), as determined by pyrosequencing in PCGF3, KDM2B, CTTN, TSPAN5, SPATA13, DPPA5, HIVEP3, JPH3, KIAA1210, LYNX1, LSP1, and SERPINB9. Blood samples of pre-diagnostic HCC cases (n = 21) were collected at the time when the patients were clinically considered cancer free and developed HCC within 4 years of follow-up. CpG sites included in calculating the average methylation levels of the probe are shown in grey or black square box where grey refers to hypomethylation and black to hypermethylation. The CpG sites were selected based on the consistency of DNA methylation differences in pre- and post-diagnostic samples. ***P <0.001, **P <0.01, *P <0.05.