

Abstract
Purpose
Irinotecan (CPT-11) and SN-38 – its active metabolite – are alkaloid-derived topoisomerase I interactive compounds widely used in various cancer therapy protocols. To solve the problems associated with the instability of their lactone ring at physiological pH and with the extreme insolubility of SN-38, the development of delivery carriers (eg, liposomes) has been considered a subject of unquestionable medical interest. This article focuses on the development of an alternative protocol to the classical lipid-film hydration procedures to obtain a pharmaceutical formulation for SN-38.
Methods
SN-38-loaded liposomes (SN-38lip) were produced by microemulsification, without a prior lipid-film preparation step, and characterized by different methods. Formulation parameters were determined by photon correlation spectroscopy, and the SN-38 entrapment efficiency was evaluated by absorbance spectroscopy. SN-38lip was obtained as a dry, white powder by lyophilization. MTT and LDH assays were conducted to assess the cytotoxic effect of SN-38, both in liposomal (SN-38lip) and solubilized form (SN-38sol); flow cytometry was used to quantify SN-38 uptake and to analyze cell-cycle phase distribution after drug exposure.
Results
Microfluidic, stable, and controlled sized, negatively charged liposomes, with high SN-38 incorporation efficiency into egg yolk phosphatidylcholine (EPC)/L-α-dioleoyl-phospathidylserine (DOPS) (9:1) vesicles (SN-38lip), were prepared. A lyophilized powder of SN-38lip, easily reconstitutable while retaining physicochemical parameters, was finally obtained. The efficacy of SN-38lip was assessed by in vitro studies with two tumor cell lines (HeLa and Caco-2) and compared with that of SN-38sol. It demonstrated the highest uptake of SN-38lip, in accordance with its highest cytotoxicity effect, in comparison with that of SN-38sol. In addition, different cell-cycle alterations were induced in both cell lines by the liposomal formulation.
Conclusion
The results highlight the potential usefulness of the procured SN-38 liposomal formulation and provide the basis for conducting in vivo studies that allow the development of alternative strategies for colorectal cancer treatment.
Keywords: microfluidic liposomes, drug delivery, SN-38, cytotoxicity, drug uptake, cell-cycle analysis
Introduction
Camptothecins are efficient antineoplastic alkaloid-derived compounds that belong to the family of the so-called topoisomerase I (Topo I) interactive compounds.1,2 They are natural molecules or semisynthetic analogs, and their solubility properties and antitumor activity are determined by different substituted five-ring backbone structure.3 Camptothecins cause cell death because of their ability to bind to DNA and Topo I as well as to stabilize the complex they both form during replication.4,5 Topotecan and irinotecan (CPT-11) are two camptothecins that have already been approved by the US Food and Drug Administration (FDA). Topotecan was approved in 1996 for the treatment of recurrent ovarian cancer, in 1998 as a second-line therapeutic agent in small cell lung cancer, and in 2006 for the treatment of advanced, recurrent, and metastatic cervical cancer.6 CPT-11, in turn, is a first-line drug approved for the treatment of a variety of human tumors, including colorectal, lung, and gynecological cancers.7 It has been administered in combination with 5-fluorouracil (5-FU) and as a rescue therapy in 5-FU-refractory disease. CPT-11 is a water-soluble molecule that can be converted by carboxylesterase-catalyzed hydrolysis to its metabolite SN-38 and has been reported to have at least 100-fold higher activity.8,9 There are, however, certain clinical limitations for the use of all of these drugs. These include: 1) spontaneous inactivation to a carboxylate form in blood, 2) rapid reversal of the trapped cleavable complex after drug removal, requiring prolonged infusions, 3) resistance of cancer cells overexpressing membrane transporters, and 4) dose-limiting side effects of diarrhea, myelosuppression, neutropenia, and an acute cholinergic-like syndrome.10 In the case of SN-38, another important drawback is its great insolubility in almost all solvents that could be used to properly formulate this drug for clinical purposes.
To solve these problems and to optimize the therapeutic effectiveness of these drugs, several strategies have been examined. Among these, the development of controlled-delivery carriers, such as liposomes, polymeric nanoparticles, or microspheres, provides promising alternatives in the field of cancer therapy.11–14
The liposome platform has been extensively studied as a tool to encapsulate drugs, and it is considered a subject of unquestionable medical interest.15–21 Liposomal devices, which have emerged as one of the most studied and useful drug delivery systems in the last two decades, provide suitable strategies to improve the efficacy of chemotherapeutics in cancer treatment. Liposomes can modify the pharmacokinetics of the encapsulated drugs, promote their intracellular uptake, and allow selective delivery to tumor cells, resulting in a decrease in some of the undesirable side effects associated with chemotherapy and an increase in the maximum tolerated dose.11 The US FDA has already approved various liposome formulations for clinical use, and many others have been tested in clinical trials.22–24
Liposomes offer a wide variety of possibilities to formulate chemically different molecules because of the two well-separated environments of their structure. Moreover, they are biodegradable, biocompatible, and provide a galenic form endowed with drug-sustained-release abilities. The pharmacological efficacy of any liposomal formulation is dependent on several factors such as physical structure, physical stability, and bilayer dynamics. All of these factors can be controlled by parameters related to liposome formulation, including lipid composition, preparation method, and specific labeling with recognizing molecules. In addition, the partition coefficients of the drug molecules in a biphasic organic/aqueous system and the interactions that they can establish with the lipid components of the liposomal bilayer influence the delivery of the encapsulated drugs and the stability of the liposomal formulation.25
Recent innovations and developments in nanotechnologies have encouraged new research programs to engineer nanotechnology products for therapeutic applications, have enabled the development and marketing of new bioactive macromolecules that require a precise intracellular location for biological activity and have revolutionized the form of administering medicines.23,26,27 Moreover, smart chemistry has recently emerged in developing stimuli-responsive nanocarriers that change their structures, composition, or conformations in response to external or internal physical or biochemical stimuli, which leads to the release of the encapsulated drug.28
The development of sustained-release nanodevices, with encapsulated products, is especially useful in the case of camptothecins.29,30 There are two main advantages of using liposomes for the delivery of these drugs: the protection of the liposomal form against hydrolysis of the lactone to the inactive carboxylate form and the possibility of solubilizing molecules that are as insoluble as SN-38, whose great hydrophobicity limits its application. Until recently, CPT-11 – as acidic solution (Camptosar®, Pfizer, USA) – was the only form approved for indirect administration of SN-38. In October 2015, the US FDA approved an encapsulated form of CPT-11 (previously known as MM-398) – the prodrug of SN-38 – in liposomes (Onivyde, Merrimack Pharmaceuticals, Inc).31 Although it was demonstrated by in vitro experiments that CPT-11 could be converted into SN-38 in intact liposomes after their incubation with carboxylesterase,32 it was also established that only a small fraction of the prodrug generates the active metabolite.33 At present, both CPT-11 and SN-38 liposomal formulations are being tested in clinical trials.19
Sadzuka et al34 reported a comparative study of the application of different procedures to incorporate SN-38 into liposomes. The classic protocol – the Bangham method35 – showed an encapsulation efficiency of only 5.6%, which could be increased up to 20.3% by the application of a modified remote-loading method, and up to 53.0% by using a novel film-loading method. The poor solubility of SN-38 would explain the very low encapsulation efficiency achieved when conventional methods, involving a hydration step of a lipid film, were used. Bala et al33 published a review in which some interesting considerations with regard to the preparation of nanomedicines for the delivery of SN-38 were made. Nevertheless, all approaches to incorporate this drug in liposomes are variations of the classic procedure based on the hydration of a lipid film.
Improving the formulation of SN-38 is, therefore, an interesting approach from a biomedical point of view, given its great antitumor activity. Microfluidic techniques constitute an efficacious alternative strategy for liposome production. They provide a simple, automatized, continuous process that makes possible the scaling of the process for the manufacture of large volumes of liposomal suspensions. In addition, microfluidic devices allow an accurate control of the physical characteristics of liposome vesicles by establishing the most appropriate flow conditions in the microfluidic channels, without further processing steps as in film hydration methods.36–38 Thus, the pressure, flow rate, and number of cycles of circulation of the lipid and drug dispersion through the system are key parameters for the standardization and optimization of the preparation protocol. Furthermore, it has been reported that microfluidic procedures enhance the loading efficiency of both water-soluble and hydrophobic drugs.36
This article describes the development and characterization of a liposomal-based medicine for SN-38 that constitutes a simpler – as well as effective drug-loading – alternative than that provided by other published methods. The uptake of the liposomal drug by cells and their cytotoxic activity has been analyzed and compared with those corresponding to the drug solubilized in dimethyl sulfoxide (DMSO) with promising results.
Materials and methods
Materials
L-α-distearoyl-phosphatidylcholine (DSPC), egg yolk phosphatidylcholine (EPC), soy bean lipid extract (SLE), L-α-dioleoyl-phosphatidylserine (DOPS), and cholesterol (CHOL) were purchased from Avanti Polar Lipids (Birmingham, AL, USA). SN-38 was from Tocris Bioscience (Bristol, UK). All the organic solvents (Panreac, Montcada y Reixac, Barcelona, Spain) were distilled before use. Milli-Q water (resistivity 18 MΩ⋅cm; Millipore, Bedford, MA, USA) was used. All other chemicals and solvents were of analytical grade. Dulbecco’s Modified Eagle’s Medium (DMEM) with 4.5 g glucose/L, fetal calf serum, L-glutamine, and penicillin–streptomycin solutions were provided by Biological Industries (Crowell, CT, USA). Sterile Dulbecco’s phosphate buffered saline (PBS), DMSO, 3-[4,5-dimethylthiazol-2-yl] 2,5-diphenyltetrazolium bromide (MTT), Trypan blue, toluidine blue, and Hoechst-33258 (H-33258) were from Sigma-Aldrich Chemical Co. (St Louis, MO, USA). The CytoScan-Fluoro Assay Kit was provided by G-Biosciences (St Louis, MO, USA). Sterilized plastic material was purchased from Corning Incorporated (Corning, NY, USA).
Preparation and characterization of SN-38-loaded liposomes
Liposomes were prepared by an initial dispersion of the lipids/SN-38 mixture in the preparation buffer with a high-performance IKA Ultra-Turrax® homogenizer (T25 Digital, IKA-Werke GmbH & Co. KG). DSPC, DPPC, DOPS, CHOL, SLE, and EPC were used and mixed in binary and ternary combinations, at different molar ratios, without preparation of the standard lipid films. Dry lipids and SN-38 were weighted in the same glass recipient, and the proper volume of 10 mM, pH 4.4, lactate buffer was added to obtain a lipid concentration of 10 mg/mL. After a brief bath sonication, intermediate unilamellar liposomes (IUVs) were obtained by processing lipids/SN-38 aqueous buffered dispersions with a high-pressure Microfluidic device (Avestin M-110EH-30 Microfluidizer® Processor or Emulsiflex C3). The lipids/SN-38 suspension was injected into the central channel of the Microfluidizer and processed by circulating the suspension ten times through the circuit at a pressure of 15,000 psi and a flow rate of 50 mL/min. The protocol was standardized, and the reproducibility of the different batches obtained was analyzed. A white liposomal dry powder was finally obtained by lyophilization using trehalose as the cryoprotectant. Two milliliters of liposomal suspension were placed in 4-mL glass vials and frozen at −80°C (liquid nitrogen) over 3–5 h. Vials were subsequently dried for 24 h at −55°C and 0.04 mbar (Freeze Dryer Alpha 1–2/LD, Martin Christ GmbH, Osterode, Germany).
Liposomes – both in suspension and lyophilized forms – were visualized by transmission electron microscopy (TEM) and scanning electron microscopy (SEM) and systematically characterized by measuring particle size and surface charge (photon correlation spectroscopy [PCS]) and by quantification of the drug loaded into vesicles (absorbance spectroscopy). Drug encapsulation efficiency (DEE, %), drug bulk concentration, and drug-loading efficiency (mg SN-38/mmol lipid) were obtained from the difference between total and non-entrapped SN-38. Lipid quantity was measured according to Stewart’s method.39
Cell cultures and treatments
Experiments were conducted on the tumor epithelial cell line HeLa (from cervix adenocarcinoma, ATCC CCL-2) and on human Caco-2 colon adenocarcinoma cells (ATCC HTB-37) – two well-established models for in vitro testing of the efficacy of anticancer drugs.40,41 Cells were grown in DMEM supplemented with penicillin, streptomycin, nonessential amino acids, and fetal bovine serum (FBS). Cell cultures were carried out in a humidified sterile atmosphere (95% air, 5% CO2), at 37°C, in a Nuaire NU-4750 incubator (Plymouth, MN, USA). Cells were incubated with different concentrations of SN-38 – trapped in liposomes (SN-38lip) or solubilized in DMSO (SN-38sol) – for different durations before measuring the parameters corresponding to each assay.
Cell survival assessment
Thiazolyl blue (MTT, Sigma) reduction42 was used for the assessment of cell survival. Production of MTT formazan precipitates, dissolved in 100 µL DMSO per well, was measured as absorbance at 540 nm in a Synergy H1 microplate spectrofluorimeter (BioTek Instruments, Inc., Winooski, VT, USA). Cell survival was expressed as the percentage of absorption of treated cells in comparison with that of control cells (100% survival). The results presented were the mean value and SD from at least six independent experiments.
SN-38 uptake by flow cytometry
Flow cytometry was used to quantify the uptake of SN-38 by HeLa and Caco-2 cells. Cells were treated with 10 µM SN-38sol or SN-38lip for up to 48 h. SN-38 fluorescence was quickly measured within the cell population (after debris exclusion) with a Gallios multicolor flow cytometer instrument (Beckman Coulter, Inc, Fullerton, CA, USA). Permeabilized (death) cells incorporating propidium iodide (PI) were excluded and the uptake was measured according to the blue fluorescence (λexc=405 nm, λem=450 nm).
Cell-cycle analysis
HeLa and Caco-2 cell-cycle distribution was analyzed by flow cytometry after staining the cells to label cellular DNA content with PI.43 Flasks of controls and 10 µM SN-38lip-treated cells were trypsinized (harvesting also detached cells) and centrifuged at 1,200 rpm for 5 min for pelleting after prior fixing with cold 70% ethanol solution (15 min). After centrifugation, the pellet was resuspended in 1 mL fresh PBS with 50 µL of a 100 µg/mL solution of RNase and incubated for 30 min at 37°C. Immediately prior to measurement, DNA was stained by adding 25 µL of 1 mg/mL PI solution. All reagents were from Sigma. Measurements were undertaken with an Epics XL flow cytometer (Beckman Coulter, Brea, CA, USA) with an argon laser line at 488 nm and complemented with the appropriate filters. Cell fractions in Sub-G1, G0/G1, S, G2/M, and >4C phases were quantified in histograms with Summit software. Identification of apoptotic cells (Sub-G1 region) was achieved by determination of hypoploid cell populations. Moreover, polyploid cells were identified in the >4C region. For each experiment, a minimum of 20,000 events were analyzed.
Necrosis analysis
The possible contribution of necrosis to general cell death was analyzed by measuring the activity of LDH, released into the culture medium of necrotic cells after rupture of the plasma membrane44 by using a fluorometric kit (CytoScan-Fluoro Assay Kit).45 Fluorescence was measured (λexc 560 nm, λem 590 nm) after shaking the plates for 15 s in a Synergy H1 microplate spectrofluorimeter (BioTek Instruments, Inc., Winooski, VT, USA). The percentage of cytotoxicity was calculated by using the average fluorescence values from experimental, maximum LDH release, and culture medium background.
Statistical analysis
All data are presented as the mean ± SD from separate samples that were analyzed. The statistical significance of differences was evaluated by using a Student’s paired t-test, where necessary. Data with P<0.05 was considered significant (significance levels are indicated in the figure legends).
A more detailed version of the other Methods employed in this study is given in Supplementary materials.
Results and discussion
Formulation and characterization of SN-38-loaded liposomes
Liposome formulations were prepared from DSPC/DOPS/CHOL (DOC) and EPC/DOPS mixtures, at the appropriate molar ratios, and from the natural extracts SLE and EPC. The great hydrophobicity of SN-38 imposes substantial restrictions on using standard lipid film hydration-based procedures: the solubility characteristics of this molecule, as inferred by its partition coefficient,25 must be considered when designing a liposomal carrier for its delivery as they influence the mechanical properties of lipid bilayers. On the other hand, the dynamic properties of the bilayer could greatly hinder or facilitate the incorporation of hydrophobic molecules such as SN-38. Although carriers with rigid bilayers have evident advantages such as low permeability and stability in vivo,46 they suffer from considerable restrictions in terms of achieving useful pharmaceutical formulations with drug concentrations according to therapeutic doses. In contrast, fluid bilayers more easily accommodate hydrophobic molecules inside their hydrophobic core. The design of the liposomal formulation for SN-38 has taken the above considerations into account.
There are some references in the literature about the procurement of suitable liposomal devices to encapsulate CPT-11 – the prodrug of SN-38. However, despite belonging to the same family of drugs, they have different structural features that will determine the overall characteristics of the carrier synthesis.47,48 Thus, each drug has specific requirements to be efficiently and stably encapsulated into liposomes. Among these, the lipid composition and the manufacturing process – which determine the size, surface charge, and bilayer fluidity – are decisive factors for the success of the final formulation. Moreover, the efficiency of drug loading will depend on its encapsulation in the aqueous core, its incorporation into the liposomal bilayer, or its partition between these two phases. In this way, SN-38 can be inserted within the hydrophobic core of a mixed lipid bilayer, without disturbing its structure or affecting the stability of the formulated liposomes.25
Taking into account the decisive influence of lipid composition in the characteristics of liposome populations, we have considered different options for the design of liposomal formulations. SN-38-loaded liposomes were first prepared from a natural SLE. The lipid concentration of the liposomal suspensions was fixed at 10 mg/mL. In addition, the influence of the lipid/SN-38 molar ratio on the physical properties and encapsulation efficiency of liposomes was analyzed. Further, it was considered whether the DSPC/DOPS/CHOL lipid mixture used to encapsulate CPT-11 was capable of efficiently incorporating its metabolite, SN-38. Alternatively, other compositions were assessed: the criterion to select them was to obtain liposomes with more fluid bilayers than those given by the DSPC/DOPS/CHOL solid mixture and in the results of the studies on molecular interactions between this camptothecin and the abovementioned phospholipids. Thus, an EPC extract and the binary composition EPC/DOPS were also assayed. The results are given in Table 1.
Table 1.
Physicochemical characteristics of SN-38-loaded liposomes and SN-38 entrapment inside vesicles
| Sample
|
Physical parameters
|
Liposomal SN-38
|
||||
|---|---|---|---|---|---|---|
| Liposome composition | Lipid/SN-38 (molar ratio) | Particle sizea (nm) | PDIb | Zeta-potentialc (mV) | [SN-38]d (μM) | mg SN-38/mmol lipide |
| SLE | (7.5:1) | 355.3±13.2 | 0.472±0.011 | −47.7±8.8 | 1,770.0±10.2 | 52.2±0.3 |
| (10:1) | 332.8±19.6 | 0.525±0.058 | −54.3±10.3 | 1,329.0±11.3 | 39.2±0.3 | |
| (20:1) | 196.2±9.4 | 0.415±0.033 | −45.3±16.3 | 652.6±7.6 | 19.3±2.2 | |
| (40:1) | 119.1±1.9 | 0.398±0.081 | −36.1±8.7 | 331.3±8.2 | 9.8±0.2 | |
| DSPC/DOPS/CHOL (65:35:30) | (20:1) | 248.3±9.5 | 0.368±0.016 | −32.1±17.5 | 713.6±6.2 | 19.7±1.7 |
| EPC | (20:1) | 146.3±14.2 | 0.401±0.120 | −12.6±1.5 | 637.5±8.9 | 19.1±2.6 |
| EPC/DOPS (9:1) | (20:1) | 147.2±7.9 | 0.345±0.072 | −31.4±7.4 | 670.2±5.7 | 20.3±1.6 |
Notes:
Particle size measured as Z-average mean.
Polydispersity index.
Zeta potential.
Bulk SN-38 concentration in the liposome suspension.
Drug-loading efficiency, expressed as the amount of SN-38 relative to lipid (w/w). Lipid concentration in all the samples was 10 mg/mL. Three independent batches were processed and analyzed for physicochemical characterization. The data are the mean ± SD values corresponding to three independent preparations.
Abbreviations: SN-38, irinotecan metabolite; SLE, soy bean lipid extract; DSPC, L-α-distearoyl-phosphatidylcholine; DOPS, L-α-dioleoyl-phosphatidylserine; CHOL, cholesterol; EPC, egg yolk phosphatidylcholine.
Otherwise, a profound amendment of the methods described to date has been made to solve the problems associated with the insolubility of SN-38 and in thinking about the design of an alternative protocol to obtain an improved liposomal formulation for this drug. After discarding methods based on the preparation and hydration of lipid films, assayed with modest results, we have considered alternative procedures such as microfluidic techniques. This method has proved useful to procure a controlled-size liposome population.36,37 In recent reviews, Garg et al49 and Carugo et al50 have emphasized the advantages of applying micro-fluidic procedures to liposome production as they provide improved drug-loading efficiencies, have better binding of targeting agents to desired carriers, and ensure reproducible manufacturing and uniform scaling.
Liposomes were thus prepared by processing lipid/SN-38 aqueous buffered dispersions with a high-pressure Microfluidic device (Avestin M-110EH-30 Microfluidizer® Processor). First, the appropriate amounts of dry powder lipids and SN-38 were weighed and dispersed in 10 mM lactate (pH 4.4) buffer with a T 25 digital ULTRA-TURRAX® dispersing device (IKA, Staufen, Germany). Liposome suspensions were stored at 4°C, in the dark, and systematically characterized by different methods.
It was observed that the lipid/drug molar ratio strongly influenced the size and the amount of SN-38 inside vesicles, whereas the entrapment efficiency was not affected. The results showed that the size of liposomes increased as the molar ratio of SN-38 in the lipid/drug mixture increased. By considering the PCS data (size analysis) obtained for SLE liposomes, the molar ratio selected as the most suitable for liposome formulation was 20:1. With this ratio, the mean hydrodynamic diameter of the EPC and EPC/DOPS liposomes was approximately 150 nm, in the suitable range for the biomedical applications for which they are designed, whereas the SLE and DOC vesicles were too large. Moreover, the results indicate that the efficacy of SN-38 incorporation into liposomes was always greater than 95% for fresh preparations, irrespective of the lipid mixture and the lipid/drug molar ratio used in the formulation. Obviously, the drug bulk concentration in the liposomal/buffer suspension was higher for the highest SN-38 molar ratio in the lipid/drug mixture, ranging from 3.31×10−4 to 17.7×10−4 M. This represents an improvement over other liposome formulations reported for SN-38 that were based on film hydration procedures.34 However, these results – together with the consideration of the stability of the liposomes suspension for all the assayed lipid combinations (see below) and the results of previously published biophysical data on interfacial interactions of CPT-11 and SN-38 with membrane mimetic models using monolayer techniques and differential scanning calorimetry25 – argue in favor of the choice of the binary EPC/DOPS phospholipid mixture to obtain the most suitable formulation for SN-38. The most remarkable aspect of the procedure we developed is its simplicity, without the need of further processing steps as in film hydration methods, and the possibility of formulating within liposomes a molecule as insoluble and unstable at physiological pH (due to its lactone ring) as SN-38. In addition, the established protocol provides vesicles with reproducible physical characteristics in terms of particle size, zeta potential values, ability to incorporate SN-38 camptothecin, and cytotoxic activity on the cancer cell lines analyzed, in the different batches obtained.
If this protocol is compared with others published in the literature, the advantages are obvious. Water-soluble camptothecins are usually encapsulated into liposomes by active loading methods using citrate to create pH gradients, with trapping agents to form intraliposomal drug–polyanion complexes, ammonium sulfate gradients, or divalent cation/ionophores. It is more difficult to work with hydrophobic molecules and, in many cases, the incorporation processes in liposomes are less effective. The restrictions imposed by their low solubility in aqueous media make it difficult to use methods that are based on the preparation of dry lipid films. Zhang et al51 published an alternative procedure to derive a liposomal SN-38 formulation that takes advantage of the fact that the solubility of this drug increases significantly at alkaline pH. However, the method involves undertaking the preparation at basic pH (≈10.0) and returning the lyophilized sample to an acidic pH (<3.0) during the resuspension procedure. All of these alternatives are multistage, highly time-consuming procedures that need additional products whose effect should be taken into account when evaluating the activity of the drug.
Physical stability of SN-38lip suspensions
During storage, SN-38lip vesicles could aggregate and precipitate, with consequent changes in their mean size and optical density (OD). These changes can be used to monitor their physical stability. The stability of the IUV suspensions and lyophilized powders was monitored by periodic determination of the drug and lipid content in liposomes, the average size and polydispersity of the vesicles, and by turbidimetric analysis (absorbance spectroscopy). Samples were kept in the dark at 4°C during successive measurements. Firstly, the analysis was carried out during 168 h by measuring the OD of the different liposome suspensions at 400 nm. Data (Figure 1A) show that their turbidity did not undergo significant changes during storage. Accordingly, it was ascertained that the increases in vesicle size were never greater than 10%.
Figure 1.

Physical stability of SN-38 liposome preparations.
Notes: (A) Optical density measurements of SN-38 liposome suspensions. The measurements were conducted for each formulation indicated in the x-axis. The grup of bars for each formulation corresponds to samples immediately after preparation, and also 24, 72, and 168 h after preparation, from dark blue to light blue. Samples were stored at 4°C in the dark. (B) Size and (C) SN-38 retention rate during storage of lyophilized SN-38 loaded liposomes. The measurements were undertaken for each formulation indicated in the x-axis. The grup of bars for each formulation corresponds to samples after rehydration of freshly lyophilized samples and at 24 and 168 h after storage, from dark pink to light pink. The measurements were performed, from left to right, after rehydration of freshly lyophilized samples and 24, and 168 h after storage. Data correspond to mean values ± SD of, at least, three different experiments.
Abbreviations: SLE, soy bean lipid extract; SN-38, irinotecan metabolite; DOC, DSPC/DOPS/CHOL; DSPC, L-α-distearoyl-phospathidylcholine; DOPS, L-α-dioleoyl-phospathidylserine; CHOL, cholesterol; EPC, egg yolk phosphatidylcholine; Zave, Z-average mean.
To enhance the stability during storage, liposome suspensions were lyophilized using 5% trehalose as the cryoprotectant. Samples were rehydrated immediately and at 24 h and 168 h after lyophilization; then, the particle size and polydispersity index, zeta potential, and turbidity were measured. As shown in Figure 1B, and in comparison with the data in Table 1, there were no significant differences in the particle size from before to after lyophilization. Likewise, the values of zeta potential and turbidity were almost identical before and after the lyophilization process. These results show that this methodology is suitable to achieve sufficiently stable liposomal formulations.
As a measure of stability, the ability of the different liposomal formulations to retain the drug inside the vesicles was also assessed, in terms of the evolution of this parameter during storage. The results show that EPC/SN-38 and EPC/DOPS/SN-38lip exhibit a retention rate of ~95% over the time period analyzed (Figure 1C).
The morphology of lyophilized liposomes was characterized by TEM, using the technique of negative contrast with 2% uranyl acetate, and by SEM. Both SEM (Figure 2A) and TEM (Figure 2B) micrograph images showed that liposomes appeared as rounded and intact vesicles with a size close to that determined by PCS. A small population of smaller vesicles was also observed by TEM.
Figure 2.
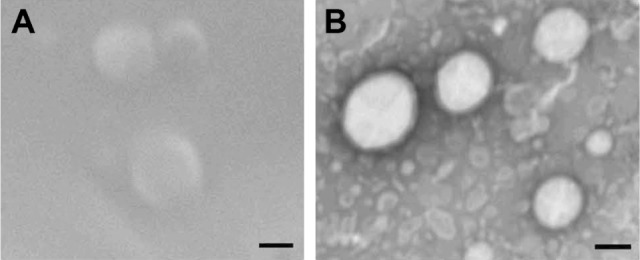
SEM and TEM images.
Notes: SEM (A) and TEM (B) images of a lyophilized preparation of SN-38-loaded EPC/DOPS liposomes. The bar is equivalent to 100 nm.
Abbreviations: TEM, transmission electron microscopy; SEM, scanning electron microscopy; SN-38, irinotecan metabolite; EPC, egg yolk phosphatidylcholine; DOPS, L-α-dioleoyl-phosphatidylserine.
Taken together, all of the physicochemical data have allowed selection of the best lipid/drug ratio (20:1) and bilayer composition (EPC/DOPS, 9:1) to develop the final formulation for SN-38. This option was supported by previous biophysical studies on lipid/camptothecin interactions.25
In vitro assessment of liposomal SN-38 cytotoxic activity
The quantification of cell cytotoxicity is used to assess the efficacy of new drugs and new pharmaceutical formulations. The MTT assay was conducted to determine the effect of SN-38lip, in comparison with that of SN-38sol, on the survival and growth of the cell lines HeLa and Caco-2.
A first series of experiments were carried out with HeLa cells to establish the importance of the liposome bilayer composition on the cytotoxic effect of the drug. The results are shown in Figure 3A and B. Before the study of cell survival in the presence of SN-38 solubilized in DMSO or incorporated into liposomes (Figure 3), we confirmed that blank liposomes and DMSO did not have any effect on cell viability. It can be observed that the cytotoxic effectiveness of the SN-38lip formulations was, in general, higher than that of the drug solubilized in DMSO. The possible toxicity of DMSO on HeLa and Caco-2 cells was taken into consideration before comparatively analyzing the results of the MTT assay. DMSO is widely used to solubilize poorly soluble drugs in permeation assays because of its well-known effect as a chemical penetration enhancer to deliver active molecules through the skin and into the cells.52 However, the maximum usable concentration of DMSO in cell cultures has not been perfectly established, although the toxicity of this amphiphilic solvent has already been evaluated in model membranes53 and with different cell culture lines.54,55 The general consensus is that concentrations of DMSO below or equal to 1% are safe for cells in culture, and that 1% DMSO does not cause any toxicity to cells.
Figure 3.

Cytotoxic effect of SN-38 solubilized in dimethyl sulfoxide (DMSO) and encapsulated in liposomes.
Notes: (A) Survival (%) of HeLa cells after treatment with SN-38 encapsulated in SLE liposomes at different molar ratios. (B) Survival (%) of HeLa cells after treatment with SN-38 encapsulated in liposomes with the compositions indicated, at the same lipid/drug molar ratio (20:1) and in DMSO solution. For (A) and (B), cells were incubated for 24 h with the drug and survival was evaluated 3, 24, and 48 h after drug removal by the MTT assay. Cell survival in the presence of the amount of blank liposomes or DMSO in the incubation medium, which would contain the amount of drug to provide all tested SN-38 concentrations, was always between 96% and 98%. Data correspond to mean values ± SD of at least three different experiments.
Abbreviations: SLE, soy bean lipid extract; SN-38, irinotecan metabolite; DOC, DSPC/DOPS/CHOL; DSPC, L-α-distearoyl-phospathidylcholine; DOPS, L-α-dioleoyl-phosphatidylserine; CHOL, cholesterol; EPC, egg yolk phosphatidylcholine.
At this stage, it is important to underline that, despite the effect of DMSO as a permeation enhancer, the SN-38lip formulation was more effective both in increasing the uptake of SN-38 by cells (see below) and to improve its cytotoxic activity. All of these considerations further highlight the importance of using delivery systems for drug administration.
Taking into account the results of the characterization of all the liposomal formulations (data in Table 1), together with those of MTT assays, to evaluate its potential as SN-38 carriers (data in Figure 3A and B), we conducted a second series of MTT experiments, both with HeLa and Caco-2 cells, with the SN-38lip formulation chosen as the more appropriate option in terms of stability and cytotoxic activity. EPC/DOPS (9:1 molar ratio) liposomes were loaded with SN-38 at a 20:1 molar ratio and used in these series. Cells were incubated with a 10 µM SN-38 concentration, both in its liposomal form and solubilized in DMSO, for 24 and 48 h; quantification measures were carried out immediately (0 h) and at 24 and 48 h after drug removal. In a first series of experiments, it was checked that the survival of HeLa and Caco-2 cells was not affected after its incubation with the amount of blank EPC/DOPS liposomes and DMSO necessary to provide a 10 µM SN-38 concentration in the incubation medium. The results are given in Figure 4.
Figure 4.

Assessment of survival of HeLa and Caco-2 cells.
Notes: Cells were incubated with 10 µM SN-38, both in liposomal form and in dimethyl sulfoxide (DMSO) solution, for 24 (left panels) and 48 h (right panels). The surviving fraction was evaluated 0, 24, and 48 h after drug removal by the MTT assay. Cell survival in the presence of the amount of blank liposomes and DMSO, which would contain the amount of drug to provide 10 µM SN-38 in the incubation medium, was always between 96% and 98%. Data correspond to mean values ± SD of at least six different experiments. P-values <0.03 (*), 0.008 (**), and 0.0001 (***) were statistically significant.
Abbreviations: SN-38sol, irinotecan metabolite solution in DMSO; SN-38lip, irinotecan metabolite in liposomal form.
To select the 10 µM SN-38 concentration, we considered the IC50 value that could be approximately estimated from the data shown in Figure 2B for the chosen liposome formulation (1 µM for incubation times of ~20 h) and the criterion of choosing a drug concentration high enough to induce a large cytotoxic effect.
The results show the acute cytotoxicity induced by SN-38 on both HeLa and Caco-2 cell lines: a dramatic decrease in cell survival for SN-38-treated cells in relation to control cells was observed, which was that the cytotoxicity of the drug increased with the incubation time and was dependent on the time elapsed after the end of treatment in both cell lines. Moreover, the liposomal SN-38 action was always stronger than that observed when the drug was administered in DMSO, at all of the assayed conditions, despite the effect of this solvent as permeation enhancer. The cytotoxic effect was particularly high after 48-h treatment with the liposomal formulation (never reached 20% survival), and the most pronounced differences between the action of the liposomal formulation and that of the molecule solubilized in DMSO were observed in the case of Caco-2 cells. The results are validated by a statistical significance analysis of the differences observed between cell survival in HeLa and Caco-2 cells incubated with the same amount of SN-38 solubilized in DMSO (SN-38sol) or incorporated into liposomes (SN-38lip) for all the experimental conditions assayed. These results are not surprising when taking into account the many bibliographic references showing that liposome formulations are one of the best-known alternatives for improving the therapeutic index of many drugs. Previously, we had already demonstrated that liposomes increase the cytotoxic effect of CPT-11 (the prodrug of SN-38) on HeLa and Caco-2 cells and that HeLa cells are more sensitive to the effect of different photosensitizers when loaded into liposomes (uncoated or coated with folate), with regard to the effect of these drugs solubilized in the appropriate media (buffer or DMSO).48,56,57
Evaluation of the uptake of liposomal SN-38 by HeLa and Caco-2 cells
The study of the ability of a cell to internalize a drug provides useful information in relation with its possible therapeutic effect and has been widely investigated in biomedical research when designing products for pharmaceutical applications. The quantitative analysis of the uptake of SN-38 – both free and in the liposomal form – by HeLa and Caco-2 cells, was carried out by flow cytometry measuring the intrinsic fluorescence of this camptothecin derivative under ultraviolet excitation. Cells were incubated with 10 µM SN-38 incorporated into liposomes (SN-38lip) or solubilized in DMSO (SN-38sol) for different times, and their fluorescence was quantified with the flow cytometer. The results are plotted in Figure 5.
Figure 5.

Uptake of SN-38 by HeLa and Caco-2 cells.
Notes: The internalization of the drug was quantified by flow cytometry. Emission fluorescence at 450 nm was determined after excitation at 405 nm. Data are the mean ± SD from three independent experiments. Significance was assessed using a Student’s paired t-test; P-values <0.02 (*), <0.06 (**), and <0.003 (***). Fluorescence profiles are representative of the three experiments analyzed. The color of the curves indicate the different incubation times: 0 h (black); 3 h (turquoise blue); 6 h (blue); 24 h (red) and 48 h (lilac).
Abbreviations: SN-38sol, irinotecan metabolite solution in DMSO; SN-38lip, irinotecan metabolite in liposomal form.
Data showed that the amount of SN-38 within cells was dependent on the incubation time and that, in both cell lines, the uptake was higher for SN-38lip than for SN-38sol. After 48-h incubation, HeLa cells internalized 28.4% more SN-38 in their liposomal form than when solubilized in DMSO, whereas for Caco-2 this difference increased slightly to 32.9%. However, the greatest difference was observed after 24-h incubation (54.8% and 89.4%, respectively). The uptake kinetics are somewhat different for HeLa and Caco-2 cells and it was observed that, at a given time, HeLa cells internal-ized a greater amount of drug than Caco-2 cells, irrespective of the SN-38 formulation used (liposomal or solubilized). These results agree with those of the MTT analysis and account for the highest cytotoxicity of the liposomal form.
In a similar way to what was observed for the cytotoxic activity, we had already demonstrated that liposomes increase the uptake of CPT-11 (the prodrug of SN-38) by HeLa and Caco-2 cells. Furthermore, our studies showed different uptake kinetics and different amounts of CPT-11 in cells depending on the cellular type. However, we do not have additional data to justify the reasons as to why HeLa cells internalize, to a greater extent, both the SN-38lip and the SN-38sol formulations, as it happens with CPT-11. It seems that the different characteristics of these two cell types, HeLa and Caco-2, are responsible for the different degrees of internalization. Otherwise, the result is in total accordance with the cytotoxicity assessment of SN-38 on both tumor cell lines.
Cell-cycle distribution
The effect of liposomal SN-38 on cell-cycle phase distribution, after drug exposure, was evaluated by flow cytometry. HeLa and Caco-2 cells were treated with the EPC/DOPS/SN-38 (18:2:1 molar ratio) liposomal formulation for 24 and 48 h and, in each case, cell-cycle profiles were acquired immediately (0 h) and 24 h after the drug treatment and removal of detached cells. Those profiles were compared with the ones obtained for untreated controls. In all cases, the SN-38lip concentration was 10 µM. The images given in Figure 6A show that, under the same experimental conditions, SN-38lip induces different alterations in the cell-cycle profile of HeLa and Caco-2 when compared with untreated cells. Similar results were obtained by Ramasamy et al58 when SCC-7 and MDA-MB-231 cells were treated with different formulations of doxorubicin and quercetin.
Figure 6.

Flow cytometry analysis of cell-cycle phase distribution.
Notes: Cell-cycle phase distribution was analyzed after incubation with SN-38lip (10 µM) for 24 and 48 h. (A) HeLa (upper panels) and Caco-2 (lower panels) cells. Cell-cycle profiles were acquired for non-treated cells (control) and immediately (24+0 h, 48+0 h) after treatment and 24 h after drug removal (24+24 h, 48+24 h). For comparative purposes, typical profiles that were acquired while maintaining the same scale for total counts (y-axis, from 0 to 1,100) and DNA content (x-axis, from 0 to 256), are shown. (B) Percentage of cells in each cell-cycle phase calculated from the histograms. (C) Percentages of detached cells in each phase. For each experiment, a minimum of 20,000 events were analyzed.
Figure 6B shows the different response of HeLa and Caco-2 cells to SN-38 action – the marked influence of introducing post-incubation periods after drug treatment and before recording the cycle profile and the dramatic changes that occur in HeLa cells after SN-38lip treatment. In this case, the most significant changes were the huge increase in the number of cells in the Sub-G1 phase and the large decrease in the percentage of cells at the G0/G1 sector in the two assayed conditions (without and with post-incubation times; Figure 6B, left panel).
The effect of the post-incubation time on the cell-cycle profile showed notable differences with regard to those observed when it was acquired immediately after drug incubation. In this case, the number of cells in the G0/G1 phase decreased dramatically again, but the increase of Sub-G1 percentage was much more moderate. The changes in the S, G2/M, and >4C sectors were also rather different: cells in the S phase showed sustained levels, whereas the G2/M and polyploid cells (>4C) showed progressive increases. It should be considered that the percentage of cells in all cycle phases at 24 h and 48 h after treatment corresponds to the evolution of cells remaining after removing the detached ones immediately after incubation (0 h). The visualization of the histograms shows the profound changes in HeLa cells induced by SN-38lip. Previous studies about the effect of CPT-11 – the SN-38 prodrug – on different cell lines have shown some contradictory results, from cell-cycle arrest at S and G2/M phases to apoptotic responses.59,60
The response of Caco-2 cells to SN-38lip treatment was quite different (Figure 6B, right panel). The following features were observed: 1) a decrease in the proportion of cells at the G0/G1 sector, although more moderate than what was detected for HeLa cells (from 52.7% for control cells, down to ~32.1% for treated cells for the longest incubation time, without a post-incubation period); 2) a cell-cycle arrest at the S phase, for which the peak increased from 19.5% (control cells) up to 35.2% (48-h-treated cells); 3) an almost sustained percentage of G2/M cells, irrespective of the incubation time, but which slightly decreased when post-incubation time was applied; and 4) a lower percentage of cells that evolve both toward the Sub-G1 sector and the polyploid section, which was more perceptible at 24 h post-incubation. In this case, after removing SN-38 and the detached cells, the remaining cells, with the internalized drug, evolved as they did before. There are significant differences such as the important decrease in the G0/G1 population (drops to 15%) and the great increase in the number of cells in the S phase (up to 49%).
An additional table in Figure 6C shows the analysis of the percentage of cells in all cycle phases calculated from the cycle profiles corresponding to HeLa and Caco-2 detached cells. As it can be observed, almost all detached HeLa cells were in the Sub-G1 phase (apoptotic population), whereas the highest percentage of Caco-2 cells were in the S region. These results are in accordance with the evolution of both, HeLa and Caco-2, attached cells when their cycle profiles were analyzed at different times, without or with post-incubation times.
The cytotoxic activity of a CPT-11 liposomal formulation on Caco-2 cells, as well as a deep study about the mechanism of the death induced by this camptothecin on HeLa and Hs68 cells, has been reported in the literature.48,56 However, SN-38 is considered the active metabolite of CPT-11, and diverse references point out the convenience of using it instead of its prodrug, because of the limited metabolic transformation of the latter in vivo.61 The CPT-11lip formulation produced severe alterations in the characteristic diploid DNA histogram of untreated Caco-2 cells: as suggested, the changes were consistent with the progress of an increasing apoptotic process induced by this drug in its liposomal form. In contrast, the response of Caco-2 cells to SN-38lip is quite different in terms of the cell death mechanism, as commented earlier. Without analyzing in depth the features associated with apoptotic or necrotic processes, it is clear that the cell-cycle profile undergoes different changes than those observed after incubation with CPT-11lip. From this result, it could be interpreted that the cell cycle is arrested at the S phase when incubated with SN-38lip, without significant changes in the Sub-G1 region (apoptotic population).
The response of colon cancer cells to CPT-11 in solubi-lized form has also been published.59,62 In general, the most common effects observed are arrests of cell cycle at the G2/M phase (and at S, sometimes) together with decreases of the G1 population. However, it should be noted that there are discrepancies in some aspects of the entire cell-cycle profiles presented in the most relevant bibliographic references consulted.
Considering previous published results and analyzing the corresponding data, some similarities and differences with those presented here can be observed.56 Thus, the effect of SN-38lip on HeLa cells is somewhat similar to that induced by CPT-11lip: essentially, there were substantial increases of the Sub-G1 peak, as the change induced by SN-38lip was much greater. HeLa cells respond to treatment with CPT-11lip mainly with increases of the Sub-G1, G2/M, and >4C peaks, suggesting an arrest of the cell cycle at G2/M, besides the existence of apoptotic cells after incubation with this formulation. The response of HeLa cells to SN-38lip treatment was a strong increase of the peak in the Sub-G1 sector together with decreases, from dramatic to more moderate, of the peaks at the G0/G1, S, and G2/M regions.
With regard to the results of the analysis of the Caco-2 cycle profile, the comparison under the same treatment conditions with CPT-11lip and SN-38lip of cell percentages in the different phases of the cell cycle shows not only noticeable similarities but also significant differences. As it happens after CPT-11lip treatment, it could be interpreted that the cell cycle is arrested at the S phase when incubated with SN-38lip; however, unlike those results, changes in the Sub-G1 region are considerably lower than those observed after treatment with CPT-11lip.
Lactate dehydrogenase leakage assay
Cell death induced by the cytotoxic action of a drug can be the result of chemical or physical cytoplasmic membrane damage associated with a necrotic membrane disruption or an apoptotic-mediated membrane blebbing. There are several assays to study these processes. The MTT test and the LDH assay are two experimental methods used together for measuring the levels and the possible causes of cellular cytotoxicity as well as for obtaining reliable information about the cell death mechanism.63 The measure of the release of intracellular enzymes, such as LDH, through permeabilized plasma membranes has proven to be useful to evaluate cellular membrane damage and to verify the results of the viability assay (MTT).
The results, plotted in Figure 7, showed that the effect of SN-38 – either in liposomal suspension or in DMSO solution – depended on both the drug concentration and the incubation time, with the one caused by the liposomal formulation being slightly more pronounced. The differences between HeLa and Caco-2 cell survival after treatment were very small. Cell survival decreased bŷ40% for the highest SN-38lip concentration and the longest incubation time and ~30% when the SN-38sol formulation was used, under the same conditions. The consideration of the statistical significance of the differences observed shows its validity for the highest SN-38 concentrations.
Figure 7.

LDH assay after treatment of HeLa and Caco-2 cells with SN-38.
Notes: HeLa and Caco-2 cells were treated with SN-38lip and SN-38sol formulations. The numbers along the x-axis indicate the SN-38 concentration; 1, 10, and 50 µM. LDH release was measured at 24 (left panels) and 48 h (right panels). The columns represent the mean percentage of cell survival calculated from LDH release and the error bars (±SD) from three independent experiments. Significance was assessed using a Student’s paired t-test; P-values <0.05 (*), <0.03 (**) and <0.02 (***). The data of LDH activity in the culture medium was normalized with the total protein content.
Abbreviations: SN-38sol, irinotecan metabolite solution in DMSO; SN-38lip, irinotecan metabolite in liposomal form.
Both MTT and LDH assays showed the cytotoxic effect of SN-38lip and SN-38sol, although the degree of survival after treatment is extremely different depending on the method used to evaluate it. Only 17.9% of Caco-2 and 20.6% of HeLa cells survived after 48-h incubation with 10 µM SN-38lip, as measured by the MTT test, whereas survival values of 77.7% and 64.6% were determined under the same conditions for Caco-2 and HeLa cells, respectively, by the LDH assay. This disagreement could indicate that membrane damage does not seem to be the main factor involved in the decrease of cell viability.
The results described herein are not sufficient to propose a mechanism to justify the pathway by which SN-38 exerts its cytotoxic action. It should be noted that the release of LDH is considered an early event in necrosis, but a late event in apoptosis and in long-term assays of both processes could be a confounding factor. Our results were observed after an incubation time of 24 h with SN-38lip, when the survival of Caco-2 and HeLa cells increased up to values of 91% and 96.2%, respectively, and when the SN-38sol was almost non-cytotoxic, are in accordance with this comment. Therefore, more research is needed to draw conclusions about the mechanism of the cytotoxic effect of SN-38, and studies are currently underway to more precisely establish the action mode of SN-38 in its liposomal form.
Conclusion
The development of controlled-delivery, sustained-release nanodevices, such as liposomes, is providing promising alternative tools in the field of cancer therapy and it is especially useful in the case of camptothecins because of their chemical characteristics. The present article reports the preparation of a new liposomal formulation for SN-38 designed with a careful selection of the liposomal lipid components, the optimal drug lipid ratio, and the proper methodology, taking into account our previous drug–lipid interaction studies. Its potential for cancer chemotherapy is determined in vitro as a first and necessary step before in vivo assays.64 The results obtained have allowed establishment of the following considerations: 1) SN-38-loaded liposomes have been successfully formulated and obtained by a microfluidic protocol which gives stable and well-characterized vesicle suspensions. The SN-38lip formulation has been lyophilized to provide a readily reconstitutable white liposomal powder which maintains its physicochemical properties and drug retention ability practically unchanged after resuspension; 2) SN-38 was better internalized by HeLa and Caco-2 cells when formulated in liposomal devices than when solubilized in DMSO, despite the effect of this solvent as a permeation enhancer; 3) the cytotoxicity of SN-38, both in liposomal and solubilized form, has been demonstrated by means of two complementary experimental methods – the MTT reduction assay and the LDH release test. The improved cytotoxic effect of SN-38 when liposomes were used as delivery carriers could be directly related with the greater internalization of the liposomal form of the drug; 4) the cellular response to SN-38 of HeLa and Caco-2 cells showed noticeable differences, being the more significant the huge increase in the number of HeLa cells in the Sub-G1 phase, that could be related to an apoptotic cell death process, and an arrest of the Caco-2 cell cycle in the S phase, which may lead to the suppression of cell proliferation. In any case, it has been proposed that both actions could constitute a barrier for cancer progression.
The results obtained highlight the potential of the developed formulation and prove the benefits of using liposomes in cancer therapy. Furthermore, they underline the need for more research to draw conclusions about the exact mechanism by which SN-38 exerts its cytotoxic action, and studies to elucidate this are currently underway.
Supplementary materials
Preparation of SN-38-loaded liposomes
A suspension of multilamellar liposomes (MLVs) was first prepared by direct hydration of lipids and SN-38. L-α-distearoyl-phosphatidylcholine (DSPC), DPPC, L-α-dioleoyl-phosphatidylserine (DOPS), cholesterol (CHOL), soy bean lipid extract (SLE), and egg yolk phosphatidyl-choline (EPC) were used and mixed in binary and ternary combinations, at different molar ratios, without preparation of the standard lipid films. The appropriate amounts of lipids and SN-38 were weighed together, and MLVs were formed by hydrating the dry powder of lipid/SN-38 mixtures with 10 mM lactate (pH 4.4) buffer to a final lipid concentration of 10–20 mg lipid/mL. The hydration was done with a T 25 digital ULTRA-TURRAX® dispersing device (IKA, Staufen, Germany). MLV dispersions were sonicated for 10 min in an ultrasonic bath (Branson, Danbury, CT, USA) above the phase-transition temperature (Tm) and processed by a high-pressure Microfluidic device (Avestin M-110EH-30 Microfluidizer® Processor or Emulsiflex C3). The Emulsiflex C3 processor is a high-pressure homogenizer designed for laboratory-scale production by continuous, high-shear, fluid processing, easily scalable up to pilot and/or production volumes with guarantee of repeatability. Liposomes size can be controlled by adjusting the fluid flow rates in the microfluidic network. Liposomes were stored in the dark at 4°C.
Lyophilization process
To enhance stability during storage, liposomes were lyo-philized using 5% trehalose as the cryoprotectant agent. The protocol was established as follows: 2 mL liposomal suspension was placed in 4 mL glass vials and frozen at −80°C (liquid nitrogen) for 3–5 h. Vials were subsequently dried for 24 h at −55°C and 0.04 mbar (Freeze Dryer Alpha 1–2/LD, Martin Christ GmbH, Germany). Lyophilized liposomes were rehydrated immediately before the experiments by adding 2 mL of 10 mM lactate (pH 4.4) buffer. The resulted suspension was prewarmed at 45°C for 15 min and vortexed for 30 min (alternating 30 s periods of heating/vortexing).
Characterization of liposomes
Determination of the lipid content
The lipid amount in liposome suspensions was measured following Stewart’s method.1 Calibration curves for each lipid composition (0.01–0.2 mg lipid) were obtained. Chloroform and ammonium ferrothiocyanate (1:1, v/v) were added to each dried sample. The chloroformic phase, extracted after vortexing and centrifugation (2,680×g, 10 min), was used to select the absorbance maximum. Measures were carried out in a Specord 205 UV/VIS spectrophotometer (Analytical Jena AG, Jena, Germany).
Vesicle size and zeta potential
The average particle size, particle size distribution, and zeta potential of unilamellar liposomes were determined by dynamic light scattering (DLS) and laser Doppler micro-electrophoresis in a Malvern Zetasizer NANO-ZS device (Malvern Instruments Ltd, Malvern, UK) equipped with an optic unit containing a 5 mW He-Ne laser (Spectra Physics) and an electrophoresis cell. The device was calibrated with standard carboxy-modified polystyrene latex samples. Measurements were carried out at 25°C and λexc=633 nm, using liposomal suspensions containing 0.2 mg lipid/mL of 10 mM, pH 4.4, lactate buffer. Data were collected with a Malvern data channel correlator and the mean hydrodynamic diameter was calculated from a cumulative analysis of the intensity autocorrelation function.
Transmission electron microscopy
A direct observation of liposomes was carried out by transmission electron microscopy (TEM). Ammonium molybdate (2.0%) was used for negative staining to visualize vesicles. Liposomal dispersion was placed on a carbon-Formvar-coated copper grid for 1 min, after which 10 µL negative stain was used for 30 s and the excess sample and stain was blotted with filter paper. Afterward, the copper grid was air-dried for imaging using an electron microscope JEOL JEM 1010 100 kv, with CCD Megaview 1 k×1 k (JEOL Ltd., Tokyo, Japan).
Entrapment efficiency and SN-38 liposomal content
Total and entrapped SN-38 was systematically quantified by UV-vis absorption spectroscopy. A calibration curve for SN-38 (1–30 µM) was previously obtained in dimethyl sulfoxide (DMSO) at λmax (390 nm). Liposome-entrapped SN-38 was quantified as the difference between the total SN-38 used to prepare the film and the amount of non-encapsulated drug. An aliquot of the liposomal-SN-38 suspension was centrifuged using Centricon YM-10 Filter Devices (Millipore Corporation, Billerica, MA, USA) to obtain a filtrate containing the non-entrapped drug, without liposomes. The concentration of non-entrapped SN-38 was determined by comparison with the calibration curve obtained in the same conditions. Drug encapsulation efficiency (DEE%) and drug-loading efficiency (mg SN-38/mmol lipid) were calculated as the amount of drug inside liposomes with respect to the total amount of drug or lipids added in preparing formulation, respectively.
Liposomes stability
To control the stability of the formulations, both before and after the lyophilization process, the drug and lipid content in liposomes as well as the average size and polydispersity of the vesicles were determined. The turbidity of all samples was determined by diluting the suspension in 10 mM, pH 4.4 lactate buffer and measuring the absorbance of the sample at 400 nm in a Specord 205 UV/VIS spectrophotometer (Analytical Jena AG, Jena, Germany). The size and the drug content were determined as described previously.
Cell cultures
Experiments were conducted on the tumor epithelial cell line HeLa (originated from a cervix adenocarcinoma, ATCC CCL-2) and the human Caco-2 colon adenocarcinoma cells (ATCC HTB-37). Both of these are well-established models for in vitro testing of the efficacy of anticancer drugs.2,3 Both cell lines were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 50 U/mL penicillin, 50 µg/mL streptomycin, 1% nonessential amino acids, and FBS at a final concentration of 10%. Cell cultures were carried out in a humidified sterile atmosphere of 95% air and 5% CO2, at 37°C, in a Nuaire NU-4750 incubator (Plymouth, MN, USA). Cells were grown for 72 h and treated when cultures were in exponential growth.
In vitro studies of liposomal SN-38 cytotoxic activity
Cells were incubated with different concentrations of SN-38 – either entrapped in liposomes (SN-38lip) or solu-bilized in DMSO (SN-38sol) – at different times between 0 and 48 h to assess the cellular uptake of the drugs using flow cytometry. For cell survival studies and cell-cycle analysis, 24 h and 48 h of incubation with post-incubation times of up to 48 h were used.
Cell survival
HeLa and Caco-2 cells were incubated with concentrations of SN-38lip or SN-38sol ranging from 1 to 50 µM for 3, 24, and 48 h. Thiazolyl blue (MTT, Sigma) reduction4 was used for the assessment of cell survival. Briefly, a MTT stock solution in PBS (1 mg/mL) was prepared immediately prior to use and diluted in complete medium to achieve a final concentration of 50 µg/mL (100 µL MTT stock solution to each 2 mL of medium). Cells were incubated for 2 h to allow formazan crystals to precipitate. Formazan precipitates were dissolved in 100 µL DMSO per well and the absorbance was measured at 540 nm in a Synergy H1 microplate spectrofluorimeter (BioTek Instruments, Inc., Winooski, VT, USA). Cell survival was expressed as the percentage of absorption of treated cells in comparison with that of control cells (100% survival). The results presented were the mean value and SD from at least six independent experiments. Another set of experiments with the 10 µM concentration of SN-38 was conducted with incubation times of 24 h or 48 h and post-incubations times up to 48 h.
SN-38 uptake by flow cytometry
Flow cytometry was used to quantify the uptake of SN-38 by HeLa and Caco-2 cells. Cells cultured on 25 cm2 flasks were treated with 10 µM SN-38sol or SN-38lip for up to 48 h. After incubation, cells were washed twice with PBS, in order to remove the drug outside the cells, and cells were trypsinized and centrifuged at 1,200 rpm before final resuspension in 1 mL PBS. SN-38 fluorescence was quickly measured within the cell population (after debris exclusion). The analysis was carried out using a Gallios multi-color flow cytometer (Beckman Coulter, Inc, Fullerton, CA, USA) set up with the three-lasers, ten-colors standard configuration. Excitation was done using a blue (488 nm) laser for forward scatter (FS) and side scatter (SS). A gate on cells was settled on this dot plot, in order to exclude small debris and aggregates. Red fluorescence from propidium iodide (PI; 2–4 µg/mL) was collected in a log scale (λexc=488 nm, λem=620 nm). Permeabilized (death) cells incorporating PI were excluded. Uptake was measured according to the blue fluorescence (λexc=405 nm, λem=450 nm). Results were projected on a 1024-channel histogram, where the percentage of positive cells was measured.
Cell-cycle analysis
HeLa and Caco-2 cell-cycle distribution was analyzed by flow cytometry after staining the cells, to label cellular DNA content, with PI.5 Flasks of controls and 10 µM SN-38lip-treated cells were trypsinized (harvesting also detached cells) and centrifuged at 1,200 rpm for 5 min for pelleting before fixing with cold 70% ethanol solution (15 min). After centrifugation, the pellet was resuspended in 1 mL fresh PBS with 50 µL of a 100 µg/mL solution of RNase and incubated for 30 min at 37°C. Immediately prior to measurement, DNA was stained by adding 25 µL of 1 mg/mL PI solution. All reagents were from Sigma. Measurements were carried out with an Epics XL flow cytometer (Beckman Coulter, Brea, CA, USA) with an argon laser line at 488 nm, and complemented with the appropriate filters. Cell fractions in Sub-G1, G0/G1, S, G2/M, and >4C phases were quantified in histograms with Summit software. Identification of apoptotic cells (Sub-G1 region) was achieved by determination of hypoploid cell populations. Polyploid cells were also identified in the >4C region. For each experiment, a minimum of 20,000 events were analyzed.
Necrosis analysis
To study the possible contribution of necrosis to the general death, we conducted a test to measure the activity of the enzyme LDH, which is released into the culture medium of necrotic cells following rupture of the plasma membrane.6 For this purpose, a fluorometric kit (CytoScan-Fluoro Assay Kit) for measuring LDH release was employed according to the manufacturer’s instructions (G-Biosciences, St Louis, MO, USA).7 Cells were seeded in black 96-well plates and treated for 24 h or 48 h with 100 µL culture medium containing concentrations ranging from 1 to 50 µM of SN-38 either as SN-38lip or SN-38sol. Plates were removed from the incubator and equilibrated to room temperature for 20–30 min. In total, 100 µL of the reaction buffer, containing the LDH substrates, were added to each well, mixed with the medium, and incubated for 10 min at room temperature prior to the addition of 50 µL of the stop solution. Fluorescence was measured with an excitation at 560 nm and emission at 590 nm after shaking the plates for 15 s in a Synergy H1 microplate spectrofluorimeter (BioTek Instruments, Inc., Winooski, VT, USA). The percentage of cytotoxicity was calculated by using the average fluorescence values from experimental, maximum LDH release, and culture medium background.
References
- 1.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104(1):10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 2.Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 2001;46(1–3):27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 3.Ferreira D, Adega F, Chaves R. The importance of cancer cell lines as in vitro models in cancer methylome analysis and anticancer drugs testing. In: López-Camarillo C, Aréchaga-Ocampo E, editors. Oncogenomics and Cancer Proteomics-Novel Approaches in Biomarkers Discovery and Therapeutic Targets in Cancer. InTechOpen; 2013. pp. 139–166. Available from: https://cdn.intechopen.com/pdfs-wm/43632.pdf. [Google Scholar]
- 4.Mosmann T. Rapid calorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 5.Fried J, Perez AG, Clarkson BD. Flow cytofluorometric analysis of cell cycle distributions using propidium iodide. Properties of the method and mathematical analysis of the data. J Cell Biol. 1976;71(1):172–181. doi: 10.1083/jcb.71.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan FK, Moriwaki K, De Rosa MJ. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol Biol. 2013;979:65–70. doi: 10.1007/978-1-62703-290-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wolterbeek HT, van der Meer AJ. Optimization, application, and interpretation of lactate dehydrogenase measurements in microwell determination of cell number and toxicity. Assay Drug Dev Technol. 2005;3(6):675–682. doi: 10.1089/adt.2005.3.675. [DOI] [PubMed] [Google Scholar]
Acknowledgments
This work was supported by the Spanish Ministry of Economy and Competitiveness (grant no CTQ2013-48767-C3-1-R) and by the Govern de la Generalitat de Catalunya (grant no 2009SGR-367). A Casadó thanks the University of Barcelona for a predoctoral fellowship. The authors are grateful to Prof ML Garcia (Faculty of Pharmacy, University of Barcelona) for the use of the Malvern Zetasizer NANO-ZS device, and thank the helpful technical support and valuable contribution of Dr Jaume Coma (Centres Científics i Tecnològics, University of Barcelona).
Footnotes
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Chen AY, Liu LF. DNA topoisomerases: essential enzymes and lethal targets. Annu Rev Pharmacol Toxicol. 1994;34:191–218. doi: 10.1146/annurev.pa.34.040194.001203. [DOI] [PubMed] [Google Scholar]
- 2.Li TK, Liu LF. Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol. 2001;41:53–77. doi: 10.1146/annurev.pharmtox.41.1.53. [DOI] [PubMed] [Google Scholar]
- 3.Teicher BA. Next generation topoisomerase I inhibitors: rationale and biomarker strategies. Biochem Pharmacol. 2008;75(6):1262–1271. doi: 10.1016/j.bcp.2007.10.016. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer. 2006;6(10):789–802. doi: 10.1038/nrc1977. [DOI] [PubMed] [Google Scholar]
- 5.Lacombe OK, Zuma AA, da Silva CC, de Souza W, Motta MC. Effects of camptothecin derivatives and topoisomerase dual inhibitors on Trypanosoma cruzi growth and ultrastructure. J Negat Results Biomed. 2014;13(1):11. doi: 10.1186/1477-5751-13-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Randall-Whitis LM, Monk BJ. Topotecan in the management of cervical cancer. Expert Opin Pharmacother. 2007;8(2):227–236. doi: 10.1517/14656566.8.2.227. [DOI] [PubMed] [Google Scholar]
- 7.Liew ST, Yang LX. Design, synthesis and development of novel camptothecin drugs. Curr Pharm Des. 2008;14(11):1078–1097. doi: 10.2174/138161208784246180. [DOI] [PubMed] [Google Scholar]
- 8.Slatter JG, Su P, Sams JP, Schaaf LJ, Wienkers LC. Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions. Drug Metab Dispos. 1997;25(10):1157–1164. [PubMed] [Google Scholar]
- 9.Wu MH, Yan B, Humerickhouse R, Dolan ME. Irinotecan activation by human carboxylesterases in colorectal adenocarcinoma cells. Clin Cancer Res. 2002;8(8):2696–2700. [PubMed] [Google Scholar]
- 10.Garcia-Carbonero R, Supko JG. Current perspectives on the clinical, experience pharmacology, and continued development of the camptothecins. Clin Cancer Res. 2002;8(3):641–661. [PubMed] [Google Scholar]
- 11.Dawidczyk CM, Kim C, Park JH, et al. State-of-the-art in design rules for drug delivery platforms: lessons learned from FDA-approved nanomedicines. J Control Release. 2014;187:133–144. doi: 10.1016/j.jconrel.2014.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Estanqueiro M, Amaral MH, Conceição J, Sousa Lobo JM. Nanotechnological carriers for cancer chemotherapy: the state of the art. Colloids Surf B Biointerfaces. 2015;126:631–648. doi: 10.1016/j.colsurfb.2014.12.041. [DOI] [PubMed] [Google Scholar]
- 13.Pérez-Herrero E, Fernández-Medarde A. Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm. 2015;93:52–79. doi: 10.1016/j.ejpb.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 14.Zylberberg C, Matosevic S. Pharmaceutical liposomal drug delivery: a review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016;23(9):3319–3329. doi: 10.1080/10717544.2016.1177136. [DOI] [PubMed] [Google Scholar]
- 15.Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65(1):36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 16.Perche F, Torchilin VP. Recent trends in multifunctional liposomal nanocarriers for enhanced tumor targeting. J Drug Deliv. 2013;2013:705265. doi: 10.1155/2013/705265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen K, Mandal M. Second generation liposomal cancer therapeutics: transition from laboratory to clinic. Int J Pharm. 2013;448(1):28–43. doi: 10.1016/j.ijpharm.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Kraft JC, Freeling JP, Wang Z, Ho RJ. Emerging research and clinical development trends of liposome and lipid nanoparticle drug delivery systems. J Pharm Sci. 2014;103(1):29–52. doi: 10.1002/jps.23773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torchilin VP. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat Rev Drug Discov. 2014;13(11):813–827. doi: 10.1038/nrd4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bozzuto G, Molinari A. Liposomes as nanomedical devices. Int J Nanomedicine. 2015;10:975–999. doi: 10.2147/IJN.S68861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi JY, Ramasamy T, Tran TH, et al. Systemic delivery of axitinib with nanohybrid liposomal nanoparticles inhibits hypoxic tumor growth. J Mater Chem B. 2015;3:408–416. doi: 10.1039/c4tb01442a. [DOI] [PubMed] [Google Scholar]
- 22.Madni A, Sarfraz M, Rehman M, et al. Liposomal drug delivery: a versatile platform for challenging clinical applications. J Pharm Pharm Sci. 2014;17(3):401–426. doi: 10.18433/j3cp55. [DOI] [PubMed] [Google Scholar]
- 23.Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. doi: 10.1016/j.jconrel.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 24.Panahi Y, Farshbaf M, Mohammadhosseini M, et al. Recent advances on liposomal nanoparticles: synthesis, characterization and biomedical applications. Artif Cells Nanomed Biotechnol. 2017;45(4):788–799. doi: 10.1080/21691401.2017.1282496. [DOI] [PubMed] [Google Scholar]
- 25.Casadó A, Giuffrida MC, Sagristá ML, et al. Langmuir monolayers and differential scanning calorimetry for the study of the interactions between camptothecin drugs and biomembrane models. Biochim Biophys Acta. 2016;1858(2):422–433. doi: 10.1016/j.bbamem.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Farokhzad OC, Langer R. Impact of nanotechnology on drug delivery. ACS Nano. 2009;3(1):16–20. doi: 10.1021/nn900002m. [DOI] [PubMed] [Google Scholar]
- 27.Ramasamy T, Haidar ZS, Tran TH, et al. Layer-by-layer assembly of liposomal nanoparticles with PEGylated polyelectrolytes enhances systemic delivery of multiple anticancer drugs. Acta Biomater. 2014;10(12):5116–5127. doi: 10.1016/j.actbio.2014.08.021. [DOI] [PubMed] [Google Scholar]
- 28.Ramasamy T, Ruttala HB, Gupta B, et al. Smart chemistry-based nanosized drug delivery systems for systemic applications: a comprehensive review. J Control Release. 2017;258:226–253. doi: 10.1016/j.jconrel.2017.04.043. [DOI] [PubMed] [Google Scholar]
- 29.Emerson DL. Liposomal delivery of camptothecins. Pharm Sci Technolo Today. 2000;3(6):205–209. doi: 10.1016/s1461-5347(00)00268-6. [DOI] [PubMed] [Google Scholar]
- 30.Hatefi A, Amsden B. Camptothecin delivery methods. Pharm Res. 2002;19(10):1389–1399. doi: 10.1023/a:1020427227285. [DOI] [PubMed] [Google Scholar]
- 31.Chustecka Z. Liposomal Irinotecan (MM-398) Approved in Pancreatic Cancer. Medscape News. FDA approvals: [2p.] [Accessed October 22, 2015]. Available from: https://www.medscape.com/viewarticle/853072.
- 32.Sadzuka Y, Hirotsu S, Hirota S. Effective irinotecan (CPT-11)- containing liposomes: intraliposomal conversion to the active metabolite SN-38. Jpn J Cancer Res. 1999;90(2):226–232. doi: 10.1111/j.1349-7006.1999.tb00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bala V, Rao S, Boyd BJ, Prestidge CA. Prodrug and nanomedicine approaches for the delivery of the camptothecin analogue SN38. J Control Release. 2013;172(1):48–61. doi: 10.1016/j.jconrel.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 34.Sadzuka Y, Takabe H, Sonobe T. Liposomalization of SN-38 as active metabolite of CPT-11. J Control Release. 2005;108(2–3):453–459. doi: 10.1016/j.jconrel.2005.08.023. [DOI] [PubMed] [Google Scholar]
- 35.Bangham AD, Standish MM, Watkins JC. Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol. 1965;13(1):238–252. doi: 10.1016/s0022-2836(65)80093-6. [DOI] [PubMed] [Google Scholar]
- 36.Jahn A, Vreeland WN, DeVoe DL, Locascio LE, Gaitan M. Microfluidic directed formation of liposomes of controlled size. Langmuir. 2007;23(11):6289–6293. doi: 10.1021/la070051a. [DOI] [PubMed] [Google Scholar]
- 37.Yu B, Lee RJ, Lee LJ. Microfluidic methods for production of liposomes. Methods Enzymol. 2009;465:129–141. doi: 10.1016/S0076-6879(09)65007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guimarães Sá Correia M, Briuglia ML, Niosi F, Lamprou DA. Microfluidic manufacturing of phospholipid nanoparticles: stability, encapsulation efficacy, and drug release. Int J Pharm. 2017;516(1–2):91–99. doi: 10.1016/j.ijpharm.2016.11.025. [DOI] [PubMed] [Google Scholar]
- 39.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104(1):10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 40.Artursson P, Palm K, Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv Drug Deliv Rev. 2001;46(1–3):27–43. doi: 10.1016/s0169-409x(00)00128-9. [DOI] [PubMed] [Google Scholar]
- 41.Ferreira D, Adega F, Chaves R. The importance of cancer cell lines as in vitro models in cancer methylome analysis and anticancer drugs testing. In: López-Camarillo C, Aréchaga-Ocampo E, editors. Oncogenomics and Cancer Proteomics-Novel Approaches in Biomarkers Discovery and Therapeutic Targets in Cancer. InTechOpen; 2013. pp. 139–166. Available from: https://cdn.intechopen.com/pdfs-wm/43632.pdf. [Google Scholar]
- 42.Mosmann T. Rapid calorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 43.Fried J, Perez AG, Clarkson BD. Flow cytofluorometric analysis of cell cycle distributions using propidium iodide. Properties of the method and mathematical analysis of the data. J Cell Biol. 1976;71(1):172–181. doi: 10.1083/jcb.71.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chan FK, Moriwaki K, De Rosa MJ. Detection of necrosis by release of lactate dehydrogenase activity. Methods Mol Biol. 2013;979:65–70. doi: 10.1007/978-1-62703-290-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wolterbeek HT, van der Meer AJ. Optimization, application, and interpretation of lactate dehydrogenase measurements in microwell determination of cell number and toxicity. Assay Drug Dev Technol. 2005;3(6):675–682. doi: 10.1089/adt.2005.3.675. [DOI] [PubMed] [Google Scholar]
- 46.Kong G, Dewhirst MW. Hyperthermia and liposomes. Int J Hyperthermia. 1999;15(5):345–370. doi: 10.1080/026567399285558. [DOI] [PubMed] [Google Scholar]
- 47.Hattori Y, Shi L, Ding W, et al. Novel irinotecan-loaded liposome using phytic acid with high therapeutic efficacy for colon tumors. J Control Release. 2009;136(1):30–37. doi: 10.1016/j.jconrel.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 48.Casadó A, Sagristá ML, Mora M. Formulation and in vitro characterization of thermosensitive liposomes for the delivery of irinotecan. J Pharm Sci. 2014;103(10):3127–3138. doi: 10.1002/jps.24097. [DOI] [PubMed] [Google Scholar]
- 49.Garg S, Heuck S, Ip S, Ramsay E. Microfluidics: a transformational tool for nanomedicine development and production. J Drug Target. 2016;24(9):821–835. doi: 10.1080/1061186X.2016.1198354. [DOI] [PubMed] [Google Scholar]
- 50.Carugo D, Bottaro E, Owen J, Stride E, Nastruzzi C. Liposome production by microfluidics: potential and limiting factors. Sci Rep. 2016;6:25876. doi: 10.1038/srep25876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang JA, Xuan T, Parmar M, et al. Development and characterization of a novel liposome-based formulation of SN-38. Int J Pharm. 2004;270(1–2):93–107. doi: 10.1016/j.ijpharm.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 52.Kligman AM. Topical pharmacology and toxicology of dimethyl sulfoxide. 1. JAMA. 1965;193:796–804. doi: 10.1001/jama.1965.03090100042010. [DOI] [PubMed] [Google Scholar]
- 53.de Ménorval MA, Mir LM, Fernández ML, Reigada R. Effects of dimethyl sulfoxide in cholesterol-containing lipid membranes: a comparative study of experiments in silico and with cells. PLoS One. 2012;7(7):e41733. doi: 10.1371/journal.pone.0041733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Malinin TI, Perry VP. Toxicity of dimethyl sulfoxide on HeLa cells. Cryobiology. 1967;4(2):90–96. doi: 10.1016/s0011-2240(67)80216-5. [DOI] [PubMed] [Google Scholar]
- 55.Da Violante G, Zerrouk N, Richard I, Provot G, Chaumeil JC, Arnaud P. Evaluation of the cytotoxicity effect of dimethyl sulfoxide (DMSO) on Caco2/TC7 colon tumor cell cultures. Biol Pharm Bull. 2002;25(12):1600–1603. doi: 10.1248/bpb.25.1600. [DOI] [PubMed] [Google Scholar]
- 56.Casadó A, Mora M, Sagristá ML, et al. Improved selectivity and cytotoxic effects of irinotecan via liposomal delivery: a comparative study on Hs68 and HeLa cells. Eur J Pharm Sci. 2017;109:65–77. doi: 10.1016/j.ejps.2017.07.024. [DOI] [PubMed] [Google Scholar]
- 57.García-Díaz M, Nonell S, Villanueva A, et al. Do folate-receptor targeted liposomal photosensitizers enhance photodynamic therapy selectivity? Biochim Biophys Acta. 2011;1808(4):1063–1071. doi: 10.1016/j.bbamem.2010.12.014. [DOI] [PubMed] [Google Scholar]
- 58.Ramasamy T, Ruttala HB, Chitrapriya N, et al. Engineering of cell microenvironment-responsive polypeptide nanovehicle co-encapsulating a synergistic combination of small molecules for effective chemotherapy in solid tumors. Acta Biomater. 2017;48:131–143. doi: 10.1016/j.actbio.2016.10.034. [DOI] [PubMed] [Google Scholar]
- 59.Kaku Y, Tsuchiya A, Kanno T, Nishizaki T. Irinotecan induces cell cycle arrest, but not apoptosis or necrosis, in Caco-2 and CW2 colorectal cancer cell lines. Pharmacology. 2015;95(3–4):154–159. doi: 10.1159/000381029. [DOI] [PubMed] [Google Scholar]
- 60.Morandi E, Severini C, Quercioli D, et al. Gene expression time-series analysis of camptothecin effects in U87-MG and DBTRG-05 glioblastoma cell lines. Mol Cancer. 2008;7:66. doi: 10.1186/1476-4598-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rivory LP, Bowles MR, Robert J, Pond SM. Conversion of irinotecan (CPT-11) to its active metabolite, 7-ethyl-10-hydroxycamptothecin (SN-38), by human liver carboxylesterase. Biochem Pharmacol. 1996;52(7):1103–1111. doi: 10.1016/0006-2952(96)00457-1. [DOI] [PubMed] [Google Scholar]
- 62.Haug K, Kravik KL, De Angelis PM. Cellular response to irinotecan in colon cancer cell lines showing differential response to 5-fluorouracil. Anticancer Res. 2008;28(2A):583–592. [PubMed] [Google Scholar]
- 63.Kim H, Yoon SC, Lee TY, Jeong D. Discriminative cytotoxicity assessment based on various cellular damages. Toxicol Lett. 2009;184(1):13–17. doi: 10.1016/j.toxlet.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 64.He R, Du Y, Ling L, et al. Nanoformulation of dual bexarotene-tailed phospholipid conjugate with high drug loading. Eur J Pharm Sci. 2017;100:197–204. doi: 10.1016/j.ejps.2017.01.012. [DOI] [PubMed] [Google Scholar]