

Abstract
Although the cause of hypertension among individuals with obesity and insulin resistance is unknown, increased plasma insulin, acting in the kidney to increase sodium reabsorption, has been proposed as a potential mechanism. Insulin may also stimulate glucose uptake, but the contributions of tubular insulin signaling to sodium or glucose transport in the setting of insulin resistance is unknown. To directly study the role of insulin signaling in the kidney, we generated inducible renal tubule–specific insulin receptor–KO mice and used high-fat feeding and mineralocorticoids to model obesity and insulin resistance. Insulin receptor deletion did not alter blood pressure or sodium excretion in mice on a high-fat diet alone, but it mildly attenuated the increase in blood pressure with mineralocorticoid supplementation. Under these conditions, KO mice developed profound glucosuria. Insulin receptor deletion significantly reduced SGLT2 expression and increased urinary glucose excretion and urine flow. These data demonstrate a direct role for insulin receptor–stimulated sodium and glucose transport and a functional interaction of insulin signaling with mineralocorticoids in vivo. These studies uncover a potential mechanistic link between preserved insulin sensitivity and renal glucose handling in obesity and insulin resistance.
Keywords: Endocrinology, Nephrology
Keywords: Diabetes, Hypertension, Insulin signaling

With peripheral insulin resistance and hyperinsulinemia, renal tubular insulin signaling minimally regulates blood pressure but profoundly regulates glucose reabsorption.
Introduction
Obesity and insulin resistance are associated with significantly higher rates of sodium-sensitive hypertension (1, 2). Approximately 50% of individuals with essential hypertension are also insulin resistant (3), and 80% of obese individuals have hypertension (1). In this form of hypertension, impaired sodium excretion has been attributed to several mechanisms, including increased activity of the sympathetic nervous system (4) and the renin-angiotensin-aldosterone system (5), increased renal interstitial pressure (6), and hyperinsulinemia (7).
Reaven (7) hypothesized that, in states of peripheral insulin resistance, the kidney remains insulin sensitive. As an “unwilling accomplice,” compensatory hyperinsulinemia would enhance sodium reabsorption, which, in turn, increases plasma volume and raises blood pressure. Several investigators have demonstrated insulin-dependent renal sodium reabsorption (8–10). Indeed, insulin infusion increases expression and/or activity of several sodium transporters (11) and channels (9, 12) in the kidney. Additionally, mammalian models of obesity associated with hyperinsulinemia and hypertension exhibit increased expression and activity of renal sodium transporters and channels (13–15).
The long-term effects of insulin on blood pressure are species specific. In rats, long-term insulin infusion increases blood pressure (16). Brands and colleagues (17) recently reported that infusion of insulin into the renal artery of rats raises blood pressure independently of sodium reabsorption. However, in mice (10), dogs (18), and humans (19), long-term insulin infusion does not increase blood pressure (20). Findings from Brands and colleagues support an indirect effect of insulin on blood pressure, with the stimulation of sodium reabsorption counteracting hyperglycemia-induced sodium and water loss (21). A direct role of insulin on sodium and glucose reabsorption has not been established in vivo.
To study the direct role of insulin in the kidney to regulate blood pressure, sodium, and glucose handling, we generated an inducible tubule and collecting duct–specific epithelial insulin receptor–KO (InsrPax8/TetOCre, hereafter referred to as iTIRKO) mouse. We employed a diet-induced model of obesity and peripheral insulin resistance with attendant physiologic hyperinsulinemia in order to induce hypertension. In mice, a high-fat diet alone induces obesity and insulin resistance, impairs sodium excretion, and mildly increases blood pressure (22). Unlike humans (23, 24) and other mammalian models (6) of obesity or the metabolic syndrome, high-fat–fed mice do not develop hyperaldosteronism. Mineralocorticoids are a potent regulator of blood pressure, and mineralocorticoid receptor antagonism lowers blood pressure in this disorder (25, 26). Thus, to model the human condition in mice, we utilized high-fat feeding with mineralocorticoid supplementation.
Results
Characterization of inducible iTIRKO mice.
As expected from the breeding scheme, approximately 25% of pups were iTIRKO mice. Depicted in Figure 1, iTIRKO mice were homozygous for the floxed insulin receptor allele and hemizygous for both the Pax8-rtTA and TetOCre transgenes. After 2 weeks of doxycycline in chow, there was a marked reduction in β insulin receptor abundance in whole kidney lysates of iTIRKO compared with control mice (iTIRKO 0.17 ± 0.03 vs. control 1.00 ± 0.06 arbitrary units, P < 0.001) that persisted 8 weeks after discontinuation of treatment. In contrast, β insulin receptor abundance was similar in skeletal muscle (iTIRKO 0.66 ± 0.22 vs. control 1.00 ± 0.33 arbitrary units, P = 0.62). Growth curves with high-fat feeding, to generate obesity and hyperinsulinemia (22), were not significantly different between control and iTIRKO mice. Food and water intake and plasma electrolytes were similar and are provided in Supplemental Tables 1 and 2 (supplemental material available online with this article; https://doi.org/10.1172/jci.insight.95107DS1).
Figure 1. Generation of iTIRKO mice.
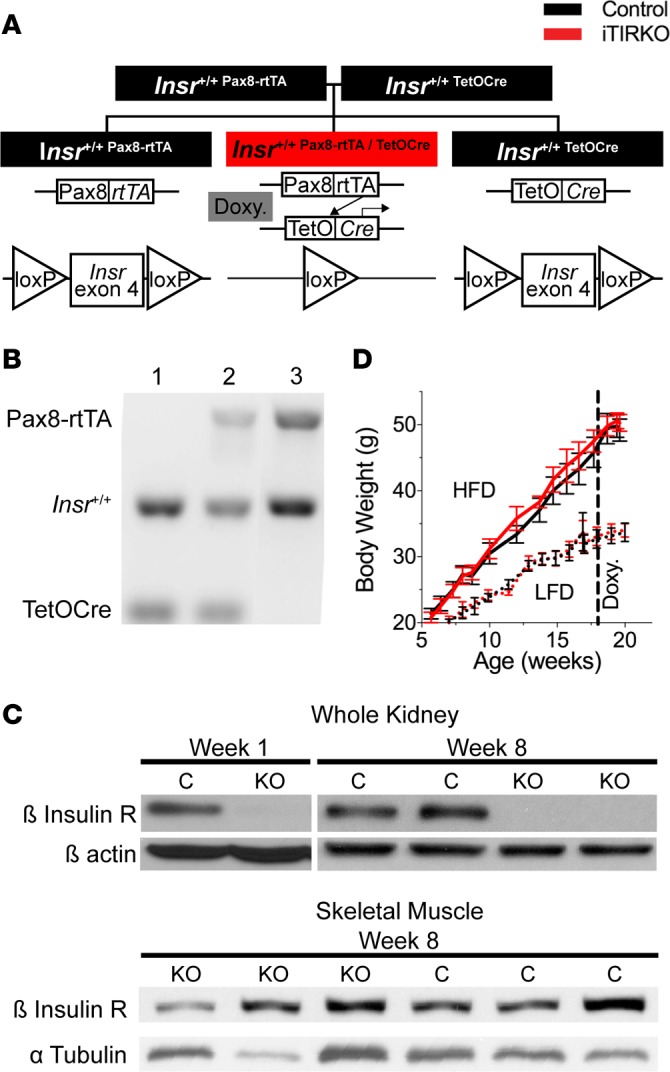
(A) Breeding strategy and schematic of a tetracycline-inducible Cre-loxP system to generate inducible renal tubular insulin receptor–KO (Insr-KO) mice. (B) Genotyping DNA gel of Insr+/+;TetOCre (control, lane 1), Insr+/+;Pax8-rtTA/TetOCre (iTIRKO, lane 2), and Insr+/+;Pax8-rtTA (control, lane 3) mice. (C) Representative immunoblots of whole kidney and skeletal muscle lysates were probed for the β-subunit of the insulin receptor (β Insulin R) at weeks 1 and 8 after completing 2 weeks of doxycycline administration and were then stripped and reprobed for β-actin or α tubulin (n = 19, control; 15, iTIRKO). (D) Growth curve of control (black) and iTIRKO (red) mice on a low-fat diet (LFD, dotted line) or high-fat diet (HFD, solid line) (n = 9, LFD control; 7, LFD iTIRKO; 12, HFD control; 10, HFD iTIRKO). Control indicates age-matched littermates of iTIRKO mice. iTIRKO, inducible renal tubular insulin receptor knockout mice; Doxy., doxycycline.
To experimentally model increased plasma mineralocorticoid concentrations in obesity and insulin resistance, which is not recapitulated in high-fat–fed mice (Supplemental Figure 1), we supplemented high-sodium– and high-fat–fed mice with oral fludrocortisone.
Attenuated hypertension in obese and insulin-resistant iTIRKO mice with mineralocorticoid supplementation.
Depicted in Figure 2, we measured blood pressure on a normal- and high-sodium diet, during 24 hours of vehicle or fludrocortisone administration. On a normal- and high-sodium diet, mean arterial pressure was not significantly different between groups with a similar magnitude of sodium-dependent increase in mean arterial pressure (iTIRKO 1.8 ± 0.5 vs. control 4.2 ± 1.7 mmHg, P = 0.98). In response to fludrocortisone administration, iTIRKO mice had a lower mean arterial pressure than controls when averaged over 24 hours (iTIRKO 118.9 ± 0.5 vs. control 120.8 ± 0.5 mmHg, P < 0.001). The increase in mean arterial pressure with fludrocortisone was less in iTIRKO compared with control mice (iTIRKO 5.4 ± 0.4 vs. control 7.7 ± 0.9 mmHg, P = 0.04). Sodium intake was not different between groups for the duration of the experiment (Supplemental Figure 2).
Figure 2. Blood pressure in high-fat–fed iTIRKO mice.

(A) Experimental scheme. After high-fat feeding and doxycycline administration to induce Cre expression, we implanted radiotelemetric blood pressure catheters in control (black, n = 7) and iTIRKO (red, n = 5) mice. We measured mean arterial pressure (MAP) on high-fat–fed normal or high-sodium diets and, subsequently, on high-sodium diet supplemented with fludrocortisone or vehicle for the intervals as indicated. Blue line denotes duration of the blood pressure recording. (B) Twenty-four–hour MAP on a normal- and high-sodium diet in control and iTIRKO mice. (C and D) Time-averaged MAP tracing over 24 hours on vehicle (C) and (D) fludrocortisone in the same mice. (E) Change in 24-hour MAP between vehicle and fludrocortisone. Control, age-matched littermates of iTIRKO mice; iTIRKO, inducible renal tubular insulin receptor–KO mice; w, week; d, day. Black *P < 0.05, control > iTIRKO mice by 2-tailed t test; red *P < 0.05, iTIRKO > control mice by 2-tailed t test. #P < 0.05 by 2-way ANOVA with repeated measures and Tukey correction for multiple comparisons. In box and whisker plots, the whiskers represent minimum and maximum values, and box borders represent the 25th percentile, median, and 75th percentile.
We found no significant difference in water intake or urine flow between high-fat–fed, hyperinsulinemic control and iTIRKO mice. Similarly, sodium excretion rates after selective inhibition of sodium transporters and channels, sodium transporter transcript, and protein abundance and phosphorylation — a regulator of activation — were unchanged in iTIRKO compared with control mice (Supplemental Figure 3).
Fludrocortisone treatment increased Na-K-2Cl cotransporter (NKCC2) and Na-Cl cotransporter (NCC) transcript abundance in control mice but was blunted in iTIRKO mice (NKCC2, iTIRKO 0.85 ± 0.5 vs. control 1.54 ± 0.14 arbitrary units compared with untreated control mice, P < 0.001; NCC, iTIRKO 1.33 ± 0.07 vs. control 1.75 ± 0.10 arbitrary units compared with untreated control mice, P = 0.004). Unlike mRNA, fludrocortisone affected other sodium transporters and channel protein expression similarly between control and iTIRKO mice (Supplemental Figure 3).
Polyuria and glucosuria in obese and insulin-resistant iTIRKO mice.
In response to fludrocortisone, we noted that iTIRKO mice demonstrated strikingly high water intake and urine flow rates (fourth day of fludrocortisone, 14.7 ± 1.1 ml/day vs. 8.9 ± 0.4 ml/day, iTIRKO vs. control mice, respectively; P < 0.001, Figure 3). Coincident with significantly higher urine flow rate, fludrocortisone-treated iTIRKO mice developed glucosuria that was absent in all control and vehicle-treated iTIRKO mice (treated iTIRKO 0.99 ± 0.27 μmol vs. all other groups <0.01 ± <0.01 μmol glucose/min, P < 0.001, Figure 3). Fludrocortisone lowered fasting plasma glucose concentrations and enhanced glucose tolerance (3,690 ± 1,078 and 2,695 ± 1,581 arbitrary units, iTIRKO and control mice, respectively; P < 0.001 compared with pretreatment area under curve in the same mice) and significantly increased creatinine clearance in both control and iTIRKO mice (iTIRKO, treated 1,228 ± 589 μmol/min, untreated 379 ± 72 μmol/min; control, treated 1,113 ± 49 μmol/min, untreated 203 ± 27 μmol/min; P = 0.009 treated vs. untreated).
Figure 3. Effect of Fludrocortisone on high-fat–fed control and iTIRKO mice.

(A) Experimental scheme. After high-fat feeding and doxycycline administration to induce Cre expression, we collected urine from control (black, n = 11) and iTIRKO (red, n = 9) mice fed vehicle followed by fludrocortisone. Blue line denotes interval with daily urine collection. (B and C) Daily water intake (B) and urine output (C) of high-sodium– and high-fat–fed control and iTIRKO mice. (D) Urinary glucose excretion on vehicle and on the fourth day of fludrocortisone. (E) Glucose tolerance test tracing before (circles, n = 11, control; 5, iTIRKO) and after (squares, n = 11, control; 5, iTIRKO) treatment with fludrocortisone (squares), and (F) calculated AUC. (G) Creatinine clearance on fourth day of fludrocortisone (control, n = 6; iTIRKO, n = 6) and separate mice on the same diet without fludrocortisone (control, n = 6; iTIRKO, n = 6). Control represents age-matched littermates of iTIRKO mice. iTIRKO, inducible renal tubular insulin receptor–KO mice; w, week; d, day; IPGTT, i.p. glucose tolerance test. *P < 0.05 by 2-tailed t test compared with control mice (C and D). *P < 0.05 by 2-way ANOVA with Tukey correction for multiple comparisons (D and F). #P < 0.05 for effect of fludrocortisone independent of genotype by 2-way ANOVA. In box and whisker plots, the whiskers represent minimum and maximum values, and box borders represent the 25th percentile, median, and 75th percentile.
Compared with control mice, iTIRKO mice have a mild liver insulin receptor deficiency and higher plasma insulin levels (Figure 4). To address whether either feature influences the observed glucosuria in iTIRKO mice, we utilized a single additional reagent, the hyperinsulinemic constitutive liver insulin receptor–KO mice (27) (Insrfl/fl; AlbCre, LIRKO). Liver homogenates from low-fat–fed LIRKO mice had a significant reduction of the β insulin receptor and elevated fasting insulin levels. However, in contrast to iTIRKO mice, LIRKO mice did not develop glucosuria with (0.01 ± 0.02 μmol glucose/min) or without (0.01 ± 0.01 μmol glucose/min) fludrocortisone.
Figure 4. Liver insulin receptor–KO (LIRKO) increases insulin resistance but does not cause glucosuria.
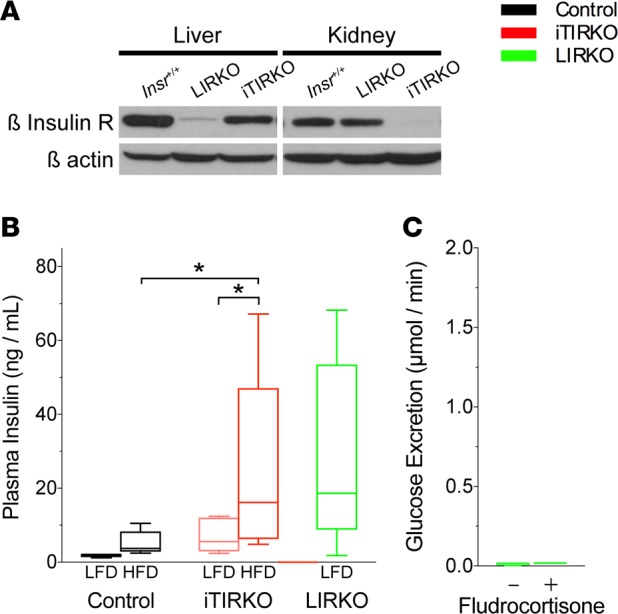
(A) Representative immunoblot of whole kidney from control (Insr+/+, n = 3), LIRKO (n = 3), and iTIRKO (n = 3) mice probed for β insulin receptor and then stripped and reprobed for β-actin. (B) Fasting plasma insulin concentration from high -sodium–, low-fat–fed control (n = 5) and high-fat–fed control (n = 8); low fat-fed (n = 5) and high-fat–fed (n = 9) iTIRKO; and low-fat–fed LIRKO (n = 10) mice. (C) Urinary glucose excretion rate in fludrocortisone-treated (n = 4) and untreated (n = 4) LIRKO mice. Control represents age-matched littermates of iTIRKO mice. iTIRKO, inducible renal tubular insulin receptor–KO mice. *P < 0.05 by 2-way ANOVA with Bonferroni correction for multiple comparisons. In box and whisker plots, the whiskers represent minimum and maximum values, and box borders represent the 25th percentile, median, and 75th percentile.
Lower SGLT2 mRNA expression and protein abundance in iTIRKO mice.
To determine the cause of glucosuria in iTIRKO mice, we measured expression of sodium-glucose cotransporters 1 and 2 (SGLT1 and SGLT2). Transcript abundance of Slc5a1, which encodes SGLT1, was not different between control and iTIRKO mice with or without fludrocortisone treatment (iTIRKO, treated 1.36 ± 0.06, untreated 1.10 ± 0.27 arbitrary units; control, treated 0.35 ± 0.09, untreated 1.00 ± 0.15 arbitrary units; P = 0.1 between all groups). While there was no significant difference in SGLT1 protein abundance between genotypes (iTIRKO, treated 0.88 ± 0.11, untreated 0.79 ± 0.18 arbitrary units; control, treated 0.92 ± 0.08, untreated 1.00 ± 0.16 arbitrary units; P = 0.69 between all groups), the immunoreactive band migrated slower in fludrocortisone-treated iTIRKO compared with control mice (Figure 5, A–C).
Figure 5. Fludrocortisone decreases SGLT1 migration and SGLT2 abundance in iTIRKO mice.

(A) Representative SGLT1 immunoblots of non–fludrocortisone-treated and fludrocortisone-treated high-sodium– and high-fat–fed control (black, n = 10 nontreated, n = 9 treated) and iTIRKO (red, n = 8 nontreated, n = 7 treated) mice. Arrows indicate different migration locations. (B) SGLT1 mRNA abundance normalized to GAPDH and relative to nontreated control mice. (C) Densitometry of SGLT1 immunoblots of nontreated and treated high-sodium– and high-fat–fed iTIRKO mice relative to control mice. (D) Representative SGLT2 immunoblots of the same nontreated and treated control and iTIRKO mice. (E) SGLT2 mRNA abundance normalized to GAPDH relative to nontreated high-sodium– and high-fat–fed control mice. (F) Densitometry of SGLT2 immunoblots of nontreated and treated high-fat–fed iTIRKO mice relative to control mice. (G) Representative images of fluorescent-conjugated LTL and SGLT2 immunoreactivity in fludrocortisone-treated and untreated control and iTIRKO mice. Scale bar: 50 μm. Control represents age-matched littermates of iTIRKO mice. iTIRKO, inducible renal tubular insulin receptor–KO mice. *P < 0.05 by 2-way ANOVA with Tukey correction for multiple comparisons. For B, C, E, and F, each dot represents data from 1 mouse with mean ± SEM.
In contrast, transcript abundance of Slc5a2, which encodes SGLT2, was increased by fludrocortisone in control mice but not in iTIRKO mice. Deletion of the insulin receptor did not change SGLT2 abundance on a high-sodium and high-fat diet, but administration of fludrocortisone resulted in an approximately 50% reduction in protein abundance in iTIRKO (treated 0.44 ± 0.16 vs. untreated 1.04 ± 0.13 arbitrary units, P = 0.002) but not control (treated 0.76 ± 0.1 vs. untreated 1.00 ± 0.11 arbitrary units, P = 0.14) mice (Figure 5, D–F). There was no difference in SGLT2 localization between fludrocortisone-treated or untreated controls and iTIRKO mice, with expression at or nearby the apical surface (Figure 5G).
Discussion
Insulin resistance and hyperinsulinemia are significantly associated with hypertension (3) and have been hypothesized to increase tubular sodium reabsorption and, therefore, directly contribute to hypertension (7). While several investigators have shown that insulin can stimulate sodium reabsorption in the kidney (9, 21, 28), no prior study has tested the ability of insulin signaling in the renal tubule to raise blood pressure in the setting of peripheral (i.e., extrarenal) insulin resistance. Skøtt and colleagues (29) showed that individuals with type 2 diabetes mellitus have preserved antinatriuretic sensitivity to insulin infusion, but these investigators did not assess for changes in blood pressure. In diabetes mellitus, Brands and colleagues (30) have shown that coincident insulin and hyperglycemia stimulate sodium reabsorption in rats and dogs, and they hypothesize that insulin in the kidney stimulates sodium reabsorption to prevent glucosuria-induced volume loss and, perhaps, hypotension in the setting of hyperglycemia. Acute or chronic insulin infusion in the absence of peripheral insulin resistance increases blood pressure in rats (16) but not mice or dogs (30). Ecelbarger and colleagues (9, 10, 12) have described several phenotypes of renal tubular insulin receptor deficiency. However, these studies were not performed in the setting of insulin resistance (8–10, 31) and did not employ insulin receptor deletion in the adult nephron, allowing for a possible confounding role of insulin receptor activation during tubular morphogenesis (32). To extend previously published work, and to elucidate mechanisms of blood pressure elevation with obesity and peripheral insulin resistance, we addressed the question of whether renal tubular epithelial insulin receptor signaling is necessary to effectuate sodium and glucose reabsorption and systemic hypertension.
We previously characterized high-fat–fed mice as a model of obesity and insulin resistance, as these mice exhibit sodium retention and elevated blood pressure (22). However, this model lacks the increase in circulating aldosterone found in dogs (25) and humans (23) with obesity and insulin resistance. Plasma aldosterone concentrations correlate directly with the severity of obesity and hyperinsulinemia (33), and mineralocorticoid receptor blockade is an effective treatment for hypertension in individuals with this disorder (34). Moreover, as we have previously demonstrated, the elevated blood pressure in obese, hyperinsulinemic high-fat–fed mice is mild (22). Therefore, we utilized a mineralocorticoid analog to more closely mimic the disease in humans. We chose fludrocortisone in lieu of aldosterone due to its longer half-life and ability to administer the compound via food without causing undue stress. Moreover, this method also allowed for us to measure acute changes in renal solute handling and blood pressure, which is not possible with minipump implantation. While our data show that absence of the renal insulin receptor does not reduce sodium sensitivity of blood pressure with diet-induced obesity and insulin resistance alone, hypertension is attenuated in KO mice with mineralocorticoid supplementation.
Attenuated hypertension in mineralocorticoid-treated iTIRKO mice.
The modest decline in blood pressure in iTIRKO mice may translate to a clinically significant difference in humans. C57BL/6 mice are notoriously resistant to changes in blood pressure. For example, patients with hypertension due to Liddle’s syndrome (i.e., gain of function mutations in epithelial sodium channel (ENaC) subunits) have an average mean arterial pressure increase of 43 mmHg (35). Transgenic mice carrying similar mutations (36) have an increase in mean arterial pressure of only about 10 mmHg on a high-sodium diet.
Mineralocorticoids increase blood pressure via increased sodium reabsorption, vasoconstriction, and stimulation of the CNS. The mild decline in blood pressure in iTIRKO mice does not easily fit into any of these pathways of mineralocorticoid-mediated blood pressure regulation. Comparison of renal sodium transporter and channel expression in fludrocortisone-treated control and iTIRKO mice demonstrated an attenuation of NKCC2 mRNA in iTIRKO mice compared with controls. However, there was no difference in total or phosphorylated NKCC2 abundance. We cannot exclude a role for inhibition of NHE3 or ENaC in attenuation of hypertension in iTIRKO mice, but KO mice did not exhibit the biochemical features associated with inhibition of these sodium transporters/channels (i.e. lower plasma bicarbonate or higher plasma potassium, respectively). The most striking phenotype of these mice with attenuated hypertension was significant polyuria and glucosuria, revealing a unifying defect in SGLT2 expression and glucose reabsorption.
Mineralocorticoid-induced glucosuria in iTIRKO mice.
Mineralocorticoids have a number of effects beyond sodium and fluid retention. Hall and colleagues (37, 38) elegantly demonstrated that aldosterone acts at the macula densa to disrupt tubuloglomerular feedback and increase glomerular filtration rate, promoting sodium excretion to return to sodium balance during hyperaldosteronism (i.e., aldosterone escape). We demonstrated that fludrocortisone similarly increases creatinine clearance, a proxy for glomerular filtration rate, consistent with prior reports. A higher glomerular filtration rate increases (39) the filtered load of glucose, which would mandate a higher rate of tubular reabsorption to prevent urinary loss. Thus, our findings would indicate that tubular insulin receptors are required for maximal SGLT2 expression and SGLT2-mediated glucose reabsorption. Indeed, some individuals with SGLT2 gene defects (OMIM #233100; familial renal glucosuria) only develop glucosuria in the setting of an increased filtered load of glucose (e.g., hyperfiltration in pregnancy; ref. 40). As the increase in glomerular filtration rate was proportionately larger than the decrease in fasting and postprandial serum glucose with fludrocortisone, it is likely that the rate of glucose filtration was higher on mineralocorticoids than on a high-sodium diet alone.
Off-target Cre expression can confound phenotypes in Cre-loxP systems. Consistent with previous reports of rtTA expression in periportal hepatocytes (41), immunoblotting of liver homogenates showed a mild reduction of insulin receptor abundance in iTIRKO vs. control mice. We therefore utilized LIRKO mice with complete deletion of liver insulin receptors and preserved renal tubular insulin receptor expression to provide evidence that fludrocortisone-induced glucosuria was due to deletion of renal tubular insulin receptors.
To further explore the mechanism of glucosuria, we observed that insulin has been shown to modulate SGLT2 expression (42). High-fat–fed iTIRKO mice were hyperinsulinemic relative to controls, and fludrocortisone increased insulin levels in our iTIRKO mice. Therefore, we sought to determine whether hyperinsulinemia itself led to glucosuria. We utilized LIRKO mice that are hyperinsulinemic, similar to high-fat–fed iTIRKO mice, but that lack obesity and high-fat feeding. The absence of glucosuria in hyperinsulinemic low-fat–fed LIRKO mice treated with or without fludrocortisone suggests that hyperinsulinemia alone is insufficient to induce the glucosuria observed in iTIRKO mice. Taken together, these data demonstrate that — in a diet-induced mouse model of obesity, hyperinsulinemia, and mineralocorticoid supplementation — tubular insulin receptor deficiency causes renal glucosuria.
Insulin and sodium-glucose cotransport.
SGLT1 and SGLT2 are expressed in the proximal tubule and are responsible, under normal conditions, for the reabsorption of 97% (43) and 3% (44) of filtered glucose, respectively. SGLT1 activity can increase in the setting of SGLT2 deletion and reduce glucose (and water) wasting (43). Rieg and colleagues (have shown that SGLT2 expression is increased in hyperinsulinemic db/db mice (45) and decreased in hypoinsulinemic Akita mice. Thus, in in vivo models of diabetes mellitus, hyperglycemia is a driver of SGLT2 expression. In contrast, in vitro studies show that supraphysiologic insulin concentrations increase SGLT2 expression via oxidative stress (46) and that insulin can increase SGLT2-mediated transport via a known site of phosphorylation (47, 48). Surprisingly, iTIRKO mice had lower SGLT2 expression despite higher glucose levels. To our knowledge, our studies represent the first evidence of the critical role of tubular insulin receptor signaling to stimulate SGLT2 expression in vivo. Apical abundance of SGLT2 was similar between control and KO mice, and we cannot exclude effects of insulin on SGLT2 activity. SGLT1 protein abundance was similar between control and iTIRKO; however, there is a molecular weight shift in fludrocortisone-treated iTIRKO mice. In the intestine, a similar migration of anti-SGLT1 immunoreactivity is caused by increased glycosylation, which increases activity (49). We speculate that a similar mechanism occurs in proximal tubular cells of iTIRKO mice in an attempt to compensate for diminished SGLT2 abundance or diminished renal tubular insulin signaling. Why this would occur remains elusive, particularly as direct loss or inhibition of SGLT2 induces increased SGLT1 protein expression (50).
SGLT2 inhibitors are US Food and Drug Administration (FDA) approved for glucose control (51) and reduction of cardiovascular risk (52). These drugs have also been shown to diminish the likelihood of decline in kidney function (53). Despite the beneficial effects of insulin on glucose disposal, individuals with diabetes and the highest levels of plasma insulin have the highest rates of kidney disease (54). Our data show that insulin receptor signaling stimulates SGLT2 function in the setting of hyperinsulinemia. Thus, future studies are needed to explore whether insulin signaling in the setting of obesity and/or type 2 diabetes mellitus promotes progression of kidney and/or cardiovascular disease.
Interaction of insulin receptor signaling and mineralocorticoids.
Our data are consistent with an indirect (via hypertension and/or hyperfiltration) or a direct interaction of insulin and mineralocorticoid signaling. The presence of a robust phenotype in high-fat–fed iTIRKO mice only, with mineralocorticoid supplementation, may signify that insulin receptor signaling is relevant to human hypertension only in the subset of patients with elevated aldosterone levels. Interestingly, these individuals are known to have the highest rates of obesity, insulin resistance, and hypertension (24). Regarding a possible direct interaction, convergence of insulin and mineralocorticoids signaling pathways are well described in the distal nephron, but no examples exist in the proximal tubule. For example, serum and glucocorticoid kinase 1 (SGK1) is a serine-threonine kinase that is transcriptionally regulated by mineralocorticoids in the kidney (55), and its activity is induced by insulin (56). These 2 extracellular signaling systems converge in principal cells to increase ENaC-mediated sodium transport (57). SGK1 (58) and mineralocorticoid receptors (59) are also expressed in segments of the proximal tubule that overlap with SGLT2, and diabetic SGK1-KO mice with elevated aldosterone have diminished glucose reabsorption (60). While fludrocortisone is a potent mineralocorticoid, we cannot exclude binding and activation of the glucocorticoid receptor; however, glucose tolerance improved with fludrocortisone treatment, which is inconsistent with a predominant glucocorticoid effect (61).
Renal insulin sensitivity in obesity and peripheral insulin resistance.
Reaven’s “unwilling accomplice” hypothesis (7) links insulin resistance and hyperinsulinemia with the impaired sodium excretion and sodium-sensitive blood pressure observed in human and mouse models. This hypothesis predicts no renal tubular resistance to insulin receptor signaling and amplified signal transduction due to hyperinsulinemia. Nakamura and colleagues (62) showed preserved insulin reception substrate 2 (IRS2) signal transduction despite attenuated IRS1 signaling in the kidneys of insulin-resistant mice and in human proximal tubule cells, demonstrating selective insulin resistance in the kidney. IRS2 contributes to insulin-mediated bicarbonate reabsorption and, thus, may be upregulated in the metabolic syndrome. In contrast, an increase in ENaC-mediated sodium transport is absent in hyperinsulinemic high-fat–fed mice (22), despite the contribution of insulin receptor signaling to ENaC activity in insulin-sensitive mice (9). These data suggest that principal cells, which express ENaC, may be insulin resistant. Here, we demonstrate for the first time to our knowledge that high-fat–fed hyperinsulinemic control mice modestly increase blood pressure and prevent glucosuria directly through renal tubular epithelial insulin receptors, supporting a hypothesis of preserved renal insulin sensitivity in specific pathways in the setting of peripheral, extrarenal insulin resistance.
Conclusion.
These findings support a role for renal insulin receptor signaling for sodium and glucose cotransport in the setting of obesity and insulin resistance. Although our studies indicate a limited role for renal tubular insulin signaling as a mechanism for hypertension, these data suggest that insulin, rather than glucose, may primarily regulate SGLT2 abundance and glucose transport. These data contribute to our understanding of insulin sensitivity in the kidney and have implications for management of hypertension, hyperglycemia, and kidney disease.
Methods
Generation of iTIRKO mice.
Ronald Kahn (Joslin Diabetes Center, Boston, Massachusetts, USA) provided Insrfl/fl mice, and we obtained Pax8-rtTA and TetOCre mice from the Jackson Laboratory. Each strain was on C57BL/6 background. Pax8-rtTA is expressed along the entire renal tubule and collecting system, and it is absent in vessels and glomeruli (41). We bred Insrfl/fl mice with either Pax8-rtTA or TetOCre mice, and we then intercrossed the progeny with Insrfl/fl mice to generate either InsrPax8 or InsrTetOCre mice (Figure 1). We then interbred these lines to generate iTIRKO mice. We used InsrPax8 or InsrTetOCre littermates of iTIRKO mice as controls because there were no differences in experimental observations between these 2 strains (Supplemental Figure 4). We performed genotyping using previously published primers (41, 63). We confirmed insulin receptor–KO by immunoblotting of whole kidney lysate, as described below.
Pax8 is also expressed in periportal hepatocytes, and thus — for additional control experiments — we generated and characterized LIRKO mice (Supplemental Figure 5).
High-fat model of obesity and insulin resistance.
We fed male mice either a low-fat diet (Research Diets Inc., D12450B) or high-fat diet (D12492) for 12 weeks and then included 1 mg doxycycline hyclate (MilliporeSigma) per gram of dry food as gel food for an additional 2 weeks to induce Cre expression. We generated custom gel diets to vary dietary water and sodium content of low- and high-fat diets using established protocols (22).
Blood pressure measurements.
Depicted in Figure 2, we implanted radiotelemetric blood pressure transmitters (PA-C10, Data Sciences International) into the left carotid artery of high-fat–fed control and iTIRKO mice. After 14 days of recovery, we recorded blood pressure on a normal-sodium and high-fat diet (0.13% w/w in high-fat diet) for 7 days, using established protocols (64). To test for sodium-sensitive hypertension, we then increased dietary sodium content (high-sodium diet, 2.1% w/w in high-fat diet) for 14 days and recorded blood pressure for 7 days. For mineralocorticoid supplementation, we added weight-based dosing of vehicle (methanol) or 3.6 mg fludrocortisone/kg body weight (MilliporeSigma) to high-sodium and high-fat diets for 1 day, according to established protocols (64). After 4 days of washout, we repeated blood pressure recording on both vehicle- and fludrocortisone-treated mice, and we calculated the average mean arterial pressure.
Metabolic cage experiments.
To assess the effect of mineralocorticoids on sodium balance, we fed a cohort of high-fat–fed control and iTIRKO mice a high-sodium and high-fat diet and acclimated them to metabolic cages. We then added vehicle to gel food and collected urine for 1 day. We added 3.6 mg/kg fludrocortisone to gel food and collected urine daily for 4 days. To study glucose handling, we performed i.p. glucose tolerance testing at the end of metabolic cage acclimation and on day 4 of fludrocortisone. The following day, after we fasted the mice for 4 hours, we anesthetized the mice with isoflurane (VetOne), obtained blood by cardiac puncture, perfused the mice with PBS (Thermo Fisher Scientific), and collected plasma and tissue.
Real-time semi-quantitative PCR.
We lysed whole kidney and extracted RNA using RNeasy mini kits (Qiagen) and prepared cDNA using Improm2 reverse transcriptase. We designed primers (Supplemental Table 3) for real-time PCR using Primer3, tested them using PrimerBLAST, and confirmed precision and amplification efficiency using a relative standard curve from kidney cDNA from control mice. We amplified cDNA using Fast SYBR Green Master Mix (Thermo Fisher Scientific) and the appropriate primers, and we performed relative quantification by the ΔΔCt method using control mice as the reference. We excluded genomic contamination, as reactions lacking reverse transcriptase demonstrated CT values >33.
Antibodies.
We used rabbit polyclonal antibodies to probe for NKCC2 and phospho-T96,101 NKCC2 (gifts from Pablo Ortiz, Henry Ford Hospital, Detroit, Michigan, USA), NCC, phospho-T53 NCC (gifts from David Ellison and James McCormick, Oregon Health Sciences University, Portland, Oregon, USA), SGLT1, SGLT2, and the β-subunit of the insulin receptor (SC-711, polyclonal, Santa Cruz Biotechnology). We used a mouse monoclonal antibody to probe for β-actin (MAB1501, clone C4, MilliporeSigma).
Western blotting.
We homogenized whole kidney in buffer (250 mM sucrose and 10 mM triethanolamine, pH 7.6) containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], 1 mM benzamidine, and 1× Complete Protease Inhibitor Mixture; Roche Applied Science) and phosphatase inhibitors (2 μM microcystin-LR, and 1× phosphatase-Inhibitor Sets I and II; Calbiochem). We centrifuged homogenates at 4,000 g for 15 minutes to remove debris, then at 16,500 g for 60 minutes to obtain a membrane-enriched fraction. We determined protein concentration via the Lowry method (Bio-Rad DC protein assay) and generated immunoblots by SDS-PAGE. With the exception of blots to probe for SGLT1, we reduced lysate in the sample buffer containing dithiothreitol (DTT; MilliporeSigma) and β-mercaptoethanol (MilliporeSigma). We quantified protein abundance by densitometry using ImageJ (NIH). Separate blots were used for total and phosphorylated NKCC2 and NCC, and they were stripped and reprobed for proteins at significantly different molecular weights.
Immunofluorescence.
At the conclusion of the study, we obtained kidneys from mice, drop fixed in 10% buffered formalin (Thermo Fisher Scientific), and embedded them in OCT. We stained 8-μm cryosections for SGLT2, as described previously. Briefly, we subjected sections to antigen retrieval in a citrate buffer, pH 6.0, followed by Triton X-100 (Thermo Fisher Scientific) permeabilization (0.5% for 15 minutes and 2% for 30 minutes). After blocking in 1% BSA, we incubated sections with the polyclonal SGLT2 (1:500) antibodies overnight at 4°C. We washed sections with PBS and detected with Alexa Fluor 594 secondary antibodies. We then labeled proximal tubules with LTL (Vector Laboratories) and post-fixed with 4% paraformaldehyde. No primary controls are depicted in Supplemental Figure 6. We captured confocal with a Leica SP8 confocal microscope at 63× and compressed Z stacks with Leica LAS software.
Plasma and urine chemistries.
We measured plasma electrolytes using an iSTAT portable analyzer (Abaxis). We measured plasma insulin by ELISA (MilliporeSigma). The Mouse Metabolic Phenotyping Core at Vanderbilt University measured plasma aldosterone by ELISA. We measured urine sodium concentration by flame photometry (BWB). We measured the urine glucose concentration using an Xpand chemistry analyzer (Siemens) and blood glucose using a commercial glucometer (Bayer Contour).
Statistics.
In experiments using 4 groups in a 2-by-2 scheme, we analyzed data by 2-way ANOVA followed by the Tukey method to correct for multiple comparisons (Prism 6). To compare mean arterial pressure between groups during vehicle and fludrocortisone administration, we used 1-way ANOVA with repeated measures and Tukey correction for multiple comparisons. We compared mean arterial pressure at specific time points using a 2-tailed t test. We compared data from glucose tolerance tests by AUC analysis. In experiments comparing more than 4 groups, we analyzed by 2-way ANOVA followed by the Bonferroni correction for multiple comparisons. We defined statistical significance as P < 0.05 for all methods. Data represent mean ± SEM.
Study approval.
We performed all animal experiments in accordance with the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) and obtained approval from the Stanford University IACUC.
Author contributions
JMN and VB conceived of experiments. JMN, VTV, and BDS performed experiments. JMN, VTV, BDS, and VB analyzed data. JMN and VB drafted the manuscript. JMN and VB edited the manuscript. All authors approved of the final manuscript.
Supplementary Material
Acknowledgments
The authors thank Herman Koepsell, David Ellison, Jim McCormick, and Pablo Ortiz for providing antibodies. The authors also thank Wuxing Dong and Nona Velarde for their technical assistance, Mingming Zhao and Dan Bernstein for their surgical assistance, and Justin Annes, Timo Rieg, and the late Gerald Reaven for helpful discussions. JMN is supported by the NIH/NIDDK (K08 DK114567-01) and was previously supported by an American Heart Association Postdoctoral Fellowship on behalf of the Grossman Family and by the Tashia and John Morgridge Endowed Postdoctoral Fellowship, Child Health Research Institute at Stanford University, and Stanford CTSA (UL1 TR001085). BDS is supported by the NIH/NIDDK (K01 DK106400). VTV was supported by the Stanford Institutes of Medicine Research Internship. VB received support from the NIH/NIDDK (1R01 DK091565).
Version 1. 08/23/2018
Electronic publication
Funding Statement
to Jonathan Nizar
to Jonathan Nizar
to Blythe Shepard
to Vivek Bhalla
to Jonathan Nizar
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(16):e95107. https://doi.org/10.1172/jci.insight.95107.
Contributor Information
Blythe D. Shepard, Email: Blythe.Shepard@georgetown.edu.
Vivek Bhalla, Email: vbhalla@stanford.edu.
References
- 1.Landsberg L, et al. Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment: a position paper of The Obesity Society and the American Society of Hypertension. J Clin Hypertens (Greenwich) 2013;15(1):14–33. doi: 10.1111/jch.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen F, Cham JL, Badoer E. High-fat feeding alters the cardiovascular role of the hypothalamic paraventricular nucleus. Am J Physiol Regul Integr Comp Physiol. 2010;298(3):R799–R807. doi: 10.1152/ajpregu.00558.2009. [DOI] [PubMed] [Google Scholar]
- 3.Lima NK, Abbasi F, Lamendola C, Reaven GM. Prevalence of insulin resistance and related risk factors for cardiovascular disease in patients with essential hypertension. Am J Hypertens. 2009;22(1):106–111. doi: 10.1038/ajh.2008.263. [DOI] [PubMed] [Google Scholar]
- 4.Armitage JA, et al. Rapid onset of renal sympathetic nerve activation in rabbits fed a high-fat diet. Hypertension. 2012;60(1):163–171. doi: 10.1161/HYPERTENSIONAHA.111.190413. [DOI] [PubMed] [Google Scholar]
- 5.Byrd JB, Brook RD. A critical review of the evidence supporting aldosterone in the etiology and its blockade in the treatment of obesity-associated hypertension. J Hum Hypertens. 2014;28(1):3–9. doi: 10.1038/jhh.2013.42. [DOI] [PubMed] [Google Scholar]
- 6.Hall JE, do Carmo JM, da Silva AA, Wang Z, Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116(6):991–1006. doi: 10.1161/CIRCRESAHA.116.305697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reaven GM. The kidney: an unwilling accomplice in syndrome X. Am J Kidney Dis. 1997;30(6):928–931. doi: 10.1016/S0272-6386(97)90106-2. [DOI] [PubMed] [Google Scholar]
- 8.Pandey G, et al. Reduced Insulin Receptor Expression Enhances Proximal Tubule Gluconeogenesis. J Cell Biochem. 2017;118(2):276–285. doi: 10.1002/jcb.25632. [DOI] [PubMed] [Google Scholar]
- 9.Pavlov TS, Ilatovskaya DV, Levchenko V, Li L, Ecelbarger CM, Staruschenko A. Regulation of ENaC in mice lacking renal insulin receptors in the collecting duct. FASEB J. 2013;27(7):2723–2732. doi: 10.1096/fj.12-223792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tiwari S, et al. Impaired sodium excretion and increased blood pressure in mice with targeted deletion of renal epithelial insulin receptor. Proc Natl Acad Sci USA. 2008;105(17):6469–6474. doi: 10.1073/pnas.0711283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chávez-Canales M, et al. Insulin increases the functional activity of the renal NaCl cotransporter. J Hypertens. 2013;31(2):303–311. doi: 10.1097/HJH.0b013e32835bbb83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiwari S, Nordquist L, Halagappa VK, Ecelbarger CA. Trafficking of ENaC subunits in response to acute insulin in mouse kidney. Am J Physiol Renal Physiol. 2007;293(1):F178–F185. doi: 10.1152/ajprenal.00447.2006. [DOI] [PubMed] [Google Scholar]
- 13.Nishida H, et al. Phosphatidylinositol 3-kinase/Akt signaling pathway activates the WNK-OSR1/SPAK-NCC phosphorylation cascade in hyperinsulinemic db/db mice. Hypertension. 2012;60(4):981–990. doi: 10.1161/HYPERTENSIONAHA.112.201509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riazi S, Tiwari S, Sharma N, Rash A, Ecelbarger CM. Abundance of the Na-K-2Cl cotransporter NKCC2 is increased by high-fat feeding in Fischer 344 X Brown Norway (F1) rats. Am J Physiol Renal Physiol. 2009;296(4):F762–F770. doi: 10.1152/ajprenal.90484.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bickel CA, Verbalis JG, Knepper MA, Ecelbarger CA. Increased renal Na-K-ATPase, NCC, and beta-ENaC abundance in obese Zucker rats. Am J Physiol Renal Physiol. 2001;281(4):F639–F648. doi: 10.1152/ajprenal.2001.281.4.F639. [DOI] [PubMed] [Google Scholar]
- 16.Song J, Hu X, Riazi S, Tiwari S, Wade JB, Ecelbarger CA. Regulation of blood pressure, the epithelial sodium channel (ENaC), and other key renal sodium transporters by chronic insulin infusion in rats. Am J Physiol Renal Physiol. 2006;290(5):F1055–F1064. doi: 10.1152/ajprenal.00108.2005. [DOI] [PubMed] [Google Scholar]
- 17.Irsik DL, Chen JK, Brands MW. Chronic renal artery insulin infusion increases mean arterial pressure in male Sprague-Dawley rats. Am J Physiol Renal Physiol. 2018;314(1):F81–F88. doi: 10.1152/ajprenal.00374.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hall JE, Coleman TG, Mizelle HL, Smith MJ. Chronic hyperinsulinemia and blood pressure regulation. Am J Physiol. 1990;258(3 Pt 2):F722–F731. doi: 10.1152/ajprenal.1990.258.3.F722. [DOI] [PubMed] [Google Scholar]
- 19.Rocchini AP, et al. Insulin and renal sodium retention in obese adolescents. Hypertension. 1989;14(4):367–374. doi: 10.1161/01.HYP.14.4.367. [DOI] [PubMed] [Google Scholar]
- 20.Hall JE, Brands MW, Mizelle HL, Gaillard CA, Hildebrandt DA. Chronic intrarenal hyperinsulinemia does not cause hypertension. Am J Physiol. 1991;260(5 Pt 2):F663–F669. doi: 10.1152/ajprenal.1991.260.5.F663. [DOI] [PubMed] [Google Scholar]
- 21.Manhiani MM, Cormican MT, Brands MW. Chronic sodium-retaining action of insulin in diabetic dogs. Am J Physiol Renal Physiol. 2011;300(4):F957–F965. doi: 10.1152/ajprenal.00395.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nizar JM, et al. Na+-sensitive elevation in blood pressure is ENaC independent in diet-induced obesity and insulin resistance. Am J Physiol Renal Physiol. 2016;310(9):F812–F820. doi: 10.1152/ajprenal.00265.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bochud M, et al. Plasma ald sterone is independently associated with the metabolic syndrome. Hypertension. 2006;48(2):239–245. doi: 10.1161/01.HYP.0000231338.41548.fc. [DOI] [PubMed] [Google Scholar]
- 24.Goodfriend TL, Kelley DE, Goodpaster BH, Winters SJ. Visceral obesity and insulin resistance are associated with plasma aldosterone levels in women. Obes Res. 1999;7(4):355–362. doi: 10.1002/j.1550-8528.1999.tb00418.x. [DOI] [PubMed] [Google Scholar]
- 25.de Paula RB, da Silva AA, Hall JE. Aldosterone antagonism attenuates obesity-induced hypertension and glomerular hyperfiltration. Hypertension. 2004;43(1):41–47. doi: 10.1161/01.HYP.0000105624.68174.00. [DOI] [PubMed] [Google Scholar]
- 26.Chapman N, et al. Effect of spironolactone on blood pressure in subjects with resistant hypertension. Hypertension. 2007;49(4):839–845. doi: 10.1161/01.HYP.0000259805.18468.8c. [DOI] [PubMed] [Google Scholar]
- 27.Michael MD, et al. Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol Cell. 2000;6(1):87–97. doi: 10.1016/S1097-2765(05)00015-8. [DOI] [PubMed] [Google Scholar]
- 28.DeFronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. The effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55(4):845–855. doi: 10.1172/JCI107996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skøtt P, et al. Effect of insulin on renal sodium handling in hyperinsulinaemic type 2 (non-insulin-dependent) diabetic patients with peripheral insulin resistance. Diabetologia. 1991;34(4):275–281. doi: 10.1007/BF00405088. [DOI] [PubMed] [Google Scholar]
- 30.Brands MW, Manhiani MM. Sodium-retaining effect of insulin in diabetes. Am J Physiol Regul Integr Comp Physiol. 2012;303(11):R1101–R1109. doi: 10.1152/ajpregu.00390.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, et al. Reduced ENaC activity and blood pressure in mice with genetic knockout of the insulin receptor in the renal collecting duct. Am J Physiol Renal Physiol. 2013;304(3):F279–F288. doi: 10.1152/ajprenal.00161.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu ZZ, Kumar A, Ota K, Wallner EI, Kanwar YS. Developmental regulation and the role of insulin and insulin receptor in metanephrogenesis. Proc Natl Acad Sci USA. 1997;94(13):6758–6763. doi: 10.1073/pnas.94.13.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kidambi S, et al. Association of adrenal steroids with hypertension and the metabolic syndrome in blacks. Hypertension. 2007;49(3):704–711. doi: 10.1161/01.HYP.0000253258.36141.c7. [DOI] [PubMed] [Google Scholar]
- 34.Sowers JR, Whaley-Connell A, Epstein M. Narrative review: the emerging clinical implications of the role of aldosterone in the metabolic syndrome and resistant hypertension. Ann Intern Med. 2009;150(11):776–783. doi: 10.7326/0003-4819-150-11-200906020-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsushita T, et al. Liddle’s syndrome in an elderly woman. Intern Med. 1998;37(4):391–395. doi: 10.2169/internalmedicine.37.391. [DOI] [PubMed] [Google Scholar]
- 36.Pradervand S, et al. A mouse model for Liddle’s syndrome. J Am Soc Nephrol. 1999;10(12):2527–2533. doi: 10.1681/ASN.V10122527. [DOI] [PubMed] [Google Scholar]
- 37.Hall JE, Granger JP, Smith MJ, Premen AJ. Role of renal hemodynamics and arterial pressure in aldosterone “escape”. Hypertension. 1984;6(2 Pt 2):I183–I192. doi: 10.1161/01.hyp.6.2_pt_2.i183. [DOI] [PubMed] [Google Scholar]
- 38.Fu Y, et al. Aldosterone blunts tubuloglomerular feedback by activating macula densa mineralocorticoid receptors. Hypertension. 2012;59(3):599–606. doi: 10.1161/HYPERTENSIONAHA.111.173195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Campen TJ, Vaughn DA, Fanestil DD. Mineralo- and glucocorticoid effects on renal excretion of electrolytes. Pflugers Arch. 1983;399(2):93–101. doi: 10.1007/BF00663903. [DOI] [PubMed] [Google Scholar]
- 40.Toka HR, Yang J, Zera CA, Duffield JS, Pollak MR, Mount DB. Pregnancy-associated polyuria in familial renal glycosuria. Am J Kidney Dis. 2013;62(6):1160–1164. doi: 10.1053/j.ajkd.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 41.Traykova-Brauch M, et al. An efficient and versatile system for acute and chronic modulation of renal tubular function in transgenic mice. Nat Med. 2008;14(9):979–984. doi: 10.1038/nm.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freitas HS, et al. Na(+) -glucose transporter-2 messenger ribonucleic acid expression in kidney of diabetic rats correlates with glycemic levels: involvement of hepatocyte nuclear factor-1alpha expression and activity. Endocrinology. 2008;149(2):717–724. doi: 10.1210/en.2007-1088. [DOI] [PubMed] [Google Scholar]
- 43.Vallon V, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22(1):104–112. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dominguez Rieg JA, Chirasani VR, Koepsell H, Senapati S, Mahata SK, Rieg T. Regulation of intestinal SGLT1 by catestatin in hyperleptinemic type 2 diabetic mice. Lab Invest. 2016;96(1):98–111. doi: 10.1038/labinvest.2015.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vallon V, et al. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol. 2013;304(2):F156–F167. doi: 10.1152/ajprenal.00409.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakamura N, Matsui T, Ishibashi Y, Yamagishi S. Insulin stimulates SGLT2-mediated tubular glucose absorption via oxidative stress generation. Diabetol Metab Syndr. 2015;7:48. doi: 10.1186/s13098-015-0044-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghezzi C, Wright EM. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am J Physiol, Cell Physiol. 2012;303(3):C348–C354. doi: 10.1152/ajpcell.00115.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Feric M, Zhao B, Hoffert JD, Pisitkun T, Knepper MA. Large-scale phosphoproteomic analysis of membrane proteins in renal proximal and distal tubule. Am J Physiol, Cell Physiol. 2011;300(4):C755–C770. doi: 10.1152/ajpcell.00360.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arthur S, Coon S, Kekuda R, Sundaram U. Regulation of sodium glucose co-transporter SGLT1 through altered glycosylation in the intestinal epithelial cells. Biochim Biophys Acta. 2014;1838(5):1208–1214. doi: 10.1016/j.bbamem.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Rieg T, et al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol. 2014;306(2):F188–F193. doi: 10.1152/ajprenal.00518.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey CJ, Gross JL, Hennicken D, Iqbal N, Mansfield TA, List JF. Dapagliflozin add-on to metformin in type 2 diabetes inadequately controlled with metformin: a randomized, double-blind, placebo-controlled 102-week trial. BMC Med. 2013;11:43. doi: 10.1186/1741-7015-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wanner Ch, Inzucchi SE, Zinman B. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med. 2016;375(18):1801–1802. doi: 10.1056/NEJMc1611290. [DOI] [PubMed] [Google Scholar]
- 53.Zinman B, et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373(22):2117–2128. doi: 10.1056/NEJMoa1504720. [DOI] [PubMed] [Google Scholar]
- 54.Ahlqvist E, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6(5):361–369. doi: 10.1016/S2213-8587(18)30051-2. [DOI] [PubMed] [Google Scholar]
- 55.Loffing J, et al. Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol. 2001;280(4):F675–F682. doi: 10.1152/ajprenal.2001.280.4.F675. [DOI] [PubMed] [Google Scholar]
- 56.Wang J, et al. SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am J Physiol Renal Physiol. 2001;280(2):F303–F313. doi: 10.1152/ajprenal.2001.280.2.F303. [DOI] [PubMed] [Google Scholar]
- 57.Pearce D. The role of SGK1 in hormone-regulated sodium transport. Trends Endocrinol Metab. 2001;12(8):341–347. doi: 10.1016/S1043-2760(01)00439-8. [DOI] [PubMed] [Google Scholar]
- 58.Satoh N, Nakamura M, Suzuki M, Suzuki A, Seki G, Horita S. Roles of Akt and SGK1 in the Regulation of Renal Tubular Transport. Biomed Res Int. 2015;2015:971697. doi: 10.1155/2015/971697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Todd-Turla KM, et al. Distribution of mineralocorticoid and glucocorticoid receptor mRNA along the nephron. Am J Physiol. 1993;264(5 Pt 2):F781–F791. doi: 10.1152/ajprenal.1993.264.5.F781. [DOI] [PubMed] [Google Scholar]
- 60.Ackermann TF, et al. SGK1-sensitive renal tubular glucose reabsorption in diabetes. Am J Physiol Renal Physiol. 2009;296(4):F859–F866. doi: 10.1152/ajprenal.90238.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Andrews RC, Walker BR. Glucocorticoids and insulin resistance: old hormones, new targets. Clin Sci. 1999;96(5):513–523. doi: 10.1042/cs0960513. [DOI] [PubMed] [Google Scholar]
- 62.Nakamura M, et al. Stimulatory effect of insulin on renal proximal tubule sodium transport is preserved in type 2 diabetes with nephropathy. Biochem Biophys Res Commun. 2015;461(1):154–158. doi: 10.1016/j.bbrc.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Brüning JC, et al. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell. 1998;2(5):559–569. doi: 10.1016/S1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 64.Veeneman JM, de Jong PE, Huisman RM, Reijngoud DJ. Re: Adey et al. Reduced synthesis of muscle proteins in chronic renal failure. Am J Physiol Endocrinol Metab 278: E219-E225, 2000. Am J Physiol Endocrinol Metab. 2001;280(1):E197–E198. doi: 10.1152/ajpendo.2001.280.1.E197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.