

Abstract
ER stress in type II alveolar epithelial cells (AECs) is common in idiopathic pulmonary fibrosis (IPF), but the contribution of ER stress to lung fibrosis is poorly understood. We found that mice deficient in C/EBP homologous protein (CHOP), an ER stress–regulated transcription factor, were protected from lung fibrosis and AEC apoptosis in 3 separate models where substantial ER stress was identified. In mice treated with repetitive intratracheal bleomycin, we identified localized hypoxia in type II AECs as a potential mechanism explaining ER stress. To test the role of hypoxia in lung fibrosis, we treated mice with bleomycin, followed by exposure to 14% O2, which exacerbated ER stress and lung fibrosis. Under these experimental conditions, CHOP–/– mice, but not mice with epithelial HIF (HIF1/HIF2) deletion, were protected from AEC apoptosis and fibrosis. In vitro studies revealed that CHOP regulates hypoxia-induced apoptosis in AECs via the inositol-requiring enzyme 1α (IRE1α) and the PKR-like ER kinase (PERK) pathways. In human IPF lungs, CHOP and hypoxia markers were both upregulated in type II AECs, supporting a conclusion that localized hypoxia results in ER stress–induced CHOP expression, thereby augmenting type II AEC apoptosis and potentiating lung fibrosis.
Keywords: Pulmonology
Keywords: Cell stress, Fibrosis, hypoxia
In the alveolar epithelium, localized hypoxia induces endoplasmic reticulum stress and promotes lung fibrosis through induction of the transcription factor C/EBP homologous protein (CHOP).
Introduction
Idiopathic pulmonary fibrosis (IPF) is characterized by progressive dyspnea, exercise intolerance, and hypoxemia, which eventually lead to respiratory failure (1). The pathology of IPF, termed usual interstitial pneumonia, consists of patchy regions of collagen and matrix deposition in the lung parenchyma, accompanied by fibroblastic foci and hyperplastic type II alveolar epithelial cells (AECs) lining areas of active remodeling (1, 2). Although the cause of IPF remains unknown, current evidence indicates that both genetic and environmental factors contribute to disease pathogenesis (1, 3). The first genetic cause of IPF was identified in 2002, with the discovery that a rare missense variant (L188Q) in the gene encoding surfactant protein C (SFTPC) was shared by adults diagnosed with familial IPF in one large kindred (4). This rare variant in SFTPC is adjacent to a cysteine residue in the proprotein required for proper folding, and subsequent studies showed that misfolded prosurfactant protein C (pro-SPC) aggregates in the ER and activates the unfolded protein response (UPR) (5, 6). Identification of ER stress in type II AECs expressing mutant forms of SP-C prompted additional studies that showed that ER stress and UPR activation are common features of IPF and are primarily localized to hyperplastic type II AECs in areas of fibrotic remodeling (7, 8). While available studies provide compelling evidence for ER stress in IPF, the etiology of ER stress in the vast majority of IPF lungs is unknown.
In addition to uncertainty regarding the cause of ER stress in IPF, the mechanisms by which ER stress contributes to fibrosis in the lungs have been challenging to unravel. Transgenic mice inducibly expressing L188Q SFTPC in type II AECs show evidence of ER stress but normal lung histology in the absence of a fibrogenic stimulus (5). However, these mice develop increased type II AEC apoptosis and exaggerated lung fibrosis in response to bleomycin treatment (5). Reduced AEC survival can inhibit reepithelialization after injury and impair barrier functions of the epithelium, thus facilitating fibrotic remodeling in the lungs (1). Proapoptotic signaling can occur through each of the 3 arms of the UPR cascade through induction or activation of several effector molecules, including C/EBP homologous protein (CHOP). CHOP is a transcription factor that regulates target gene expression through interactions with C/EBP family members or independent binding to a different set of enhancer sites (9–11). In these studies, we examined the role of CHOP in lung fibrosis and found that CHOP regulates fibrosis and AEC apoptosis following repetitive bleomycin treatment and in other models where substantial ER stress is induced in AECs. In addition, we identified localized hypoxia in areas of injured lung parenchyma as an important etiological factor in ER stress–induced CHOP expression. Together, these findings represent an important advance in understanding the mechanisms linking hypoxia, ER stress, epithelial dysfunction, and progressive lung fibrosis.
Results
CHOP regulates epithelial cell survival and mediates lung fibrosis in the presence of ER stress.
We investigated the mechanisms by which ER stress contributes to pulmonary fibrosis using a repetitive intratracheal (i.t.) bleomycin injury model; we have previously demonstrated that this model recapitulates important features of human IPF, including hyperplastic AECs and persistent fibrosis (12). In this model, we treated 8- to 10-week-old male and female WT mice (C57BL/6J background) with 6 doses of bleomycin (0.04 units) delivered by direct i.t. instillation at 2-week intervals. Mice were euthanized and lungs were harvested 2 weeks after the final bleomycin instillation. Consistent with previous findings (12), we observed distinct areas of parenchymal fibrosis with type II AEC hyperplasia (Figure 1A). Markers of ER stress were markedly upregulated, as shown by Western blotting of lung lysates, including CHOP, protein disulphide isomerase (PDI), activating transcription factor 4 (ATF4), and spliced X-box binding protein 1 (XBP1s) (Figure 1B). By IHC, we found that CHOP expression was localized primarily to type II AECs in areas of fibrosis (Figure 1C).
Figure 1. Repetitive bleomycin treatment results in ER stress and lung fibrosis.

(A) Representative Masson’s trichrome–stained lung sections and immunostaining for prosurfactant protein C (pro-SPC) showing areas of fibrosis and hyperplastic type II alveolar epithelial cells (AECs) following intratracheal (i.t.) injection of bleomycin (0.04 units) every 2 weeks for 6 doses (Rep Bleo) compared with untreated controls. Scale bars: 800 μm (low magnification): 60 μm (high magnification). (B) Western blots and densitometry for CHOP, PDI, ATF4, and XBP1s from lung tissue lysates. β-Actin was used as a loading control. *P < 0.05 compared with untreated WT by unpaired, 2-tailed Student’s t test. (C) Representative immunostaining for CHOP (arrows indicate CHOP staining) and quantification of CHOP+ AECs per high-power field on lung sections. Scale bars: 60μM. *P < 0.05 compared with other groups by 1-way ANOVA with Tukey’s post hoc test.
Next, we performed experiments using repetitive i.t. bleomycin in CHOP-deficient (CHOP–/–) mice to determine whether CHOP is an important profibrotic mediator in this model (Figure 2, A–E). In these studies, we observed significant protection of CHOP–/– mice from development of lung fibrosis, with a marked reduction in fibrotic area, reduced soluble collagen content, and decreased fibronectin level by ELISA (Figure 2, A–D). Since AEC apoptosis is an important antecedent of bleomycin-induced fibrosis, we quantified apoptotic AECs by colocalization of pro-SPC and TUNEL staining. We found that the number of TUNEL+ AECs was significantly lower in repetitive bleomycin-treated CHOP–/– mice compared with WT controls (Figure 2E). Together, these studies suggest that CHOP deficiency protects from lung fibrosis following repetitive bleomycin by reducing AEC apoptosis.
Figure 2. CHOP mediates the effects of ER stress on epithelial cell apoptosis and lung fibrosis.

(A–E) WT and CHOP–/– mice were studied using the repetitive bleomycin model (Rep Bleo), and lungs were harvested 2 weeks after the last dose. (A) Representative Masson’s trichrome–stained lung sections. Scale bars: 800 μm. (B) Evaluation of fibrosis by morphometry. (C) Quantification of total soluble collagen in right lower lobe (RLL) by Sircol assay. (D) Fibronectin measurement by ELISA. (E) Quantification of dual immunofluorescence for pro-SPC+ and TUNEL+ cells per high-power field (HPF) on lung sections. Comparisons between groups were made using unpaired, 2-tailed Student’s t test (B and D) or 1-way ANOVA with Tukey’s post hoc test (C and E). *P < 0.05 compared with WT + rep bleo, **P < 0.05 compared with untreated WT. (F–H) Inducible transgenic mice expressing L188Q mutant surfactant protein C (L188Q SFTPC), CHOP–/– mice, L188Q SFTPC/CHOP–/– mice, and WT mice were treated with doxycycline (2 g/l) for 1 week followed by intratracheal injection of bleomycin (0.08 units), and lungs were harvested 3 weeks after bleomycin. (F) Morphometric evaluation of fibrosis. (G) Total soluble collagen in RLL by Sircol assay. (H) Densitometry on Western blot for CHOP from lung tissue lysates. β-Actin was used for normalization. *P < 0.05 compared with other groups by 1-way ANOVA with Tukey’s post hoc test (F–H).
To more directly test the role of CHOP in AECs, we investigated whether selective induction of ER stress in type II AECs affects lung fibrosis via CHOP induction. For these studies, we utilized a doxycycline-inducible transgenic model in which expression of a mutant form of human SFTPC (L188Q SFTPC) is limited to type II AECs (5), resulting in ER stress and enhanced fibrotic susceptibility (5). After crossing CHOP–/– mice into this model to generate L188Q SFTPC/CHOP–/– mice, we treated these mice with doxycycline in drinking water (2 g/l) for 1 week and then administered a single i.t. injection of bleomycin (0.08 units). At 3 weeks after bleomycin instillation, L188Q SFTPC mice had increased lung fibrosis compared with WT controls, but CHOP deficiency (L188Q SFTPC/CHOP–/– mice) reduced fibrosis to WT levels (Figure 2, F and G). Notably, no difference in fibrosis was observed between WT and CHOP–/– mice used as controls in these experiments. Single-dose i.t. bleomycin did not induce robust CHOP upregulation in WT mice, potentially explaining why CHOP deletion was not protective in this model (Figure 2H and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.99543DS1). Collectively, these studies in two different fibrosis models indicate that CHOP is a key mediator of lung fibrosis when expressed in AECs.
CHOP regulates epithelial cell responses and inflammation following repetitive bleomycin treatment.
To evaluate the effect of CHOP on potential profibrotic pathways, we analyzed gene expression by RNA sequencing of EpCAM+ (EpCAM is also known as CD326) cells isolated from lungs of WT and CHOP–/– mice following repetitive bleomycin treatment (Supplemental Figure 2). Compared with that in lung epithelial cells from untreated WT mice, 2,156 genes were differentially expressed (fold change > 2, P < 0.05, FDR < 0.05), including some ER stress genes, in repetitive bleomycin-treated WT mice (Supplemental Figure 2, A and B). From among this set of differentially expressed genes, 127 genes were differentially expressed in epithelial cells from WT and CHOP–/– mice following repetitive bleomycin (Supplemental Figure 2A). By gene ontology analysis, 46 of these gene products were grouped into the following 4 categories: (a) cellular response to growth factor stimulus, (b) epithelial cell differentiation, (c) epithelium development, and (d) cell migration (Supplemental Figure 2C), thereby suggesting several profibrotic pathways in the epithelial compartment that could be regulated by CHOP.
We also investigated whether CHOP regulates inflammatory cell recruitment or function. We compared the inflammatory response in CHOP–/– mice with that of WT controls following repetitive bleomycin treatment. As shown in Supplemental Figure 3, relevant differences were not detected in numbers of immune/inflammatory cells in the lungs of WT and CHOP–/– mice (alveolar macrophages, interstitial macrophages, dendritic cells, B or T lymphocytes), with the exception of neutrophils, which were reduced in the lungs of CHOP–/– mice.
Repetitive bleomycin injury results in localized tissue hypoxia.
Although a variety of factors can effect protein processing in the ER, the mechanism by which repetitive lung injury causes ER stress is unknown. Since hypoxia has been shown to cause ER stress in vitro (13, 14), we postulated that localized areas of hypoxia in lung parenchyma could induce ER stress during repetitive cycles of lung injury and repair. To evaluate this possibility, we utilized pimonidazole as a chemical marker of cellular hypoxia (15) delivered by i.p. injection (60 μg/g) 3 hours prior to euthanasia in the repetitive bleomycin model. We then harvested lungs and performed IHC for pimonidazole adducts. Strong immunostaining was observed in areas of fibrosis, while minimal pimonidazole staining was identified in lungs of control (nonbleomycin-treated) mice and in nonfibrotic areas of lungs of mice treated with repetitive bleomycin (Figure 3A). Using dual immunofluorescence for pimonidazole adducts and the type II AEC marker pro-SPC, we found that pimonidazole staining colocalized in this cell population. Quantification of dual immunofluorescence revealed that 65.3% ± 1.2% of pro-SPC+ cells were also positive for pimonidazole staining in areas of fibrosis, whereas almost none of the pro-SPC+ cells in control (nonbleomycin-treated) mice stained for pimonidazole (Figure 3, B and C). We also performed dual immunofluorescence for pimonidazole adducts and markers of type I AECs, fibroblasts, endothelial cells, and macrophages and found minimal colocalization in all of these cell types (Supplemental Figure 4, A–D). Based on these data, we concluded that repetitive bleomycin injury results in localized hypoxia in lung parenchyma that is primarily detectable in type II AECs.
Figure 3. Repetitive bleomycin treatment results in cellular hypoxia localized to type II AECs.

(A) Pimonidazole injection (i.p., 60 μg/g) was performed 3 hours before euthanasia in mice treated with repetitive intratracheal bleomycin, followed by immunostaining for pimonidazole adducts on lung sections. Scale bars: 800 μm (low magnification): 80 μm (high magnification). (B) Dual immunofluorescence for pimonidazole adducts (red) and pro-SPC for type II AECs (green). Nuclei were counterstained with DAPI (blue). White arrows point to cells with colocalization (yellow). Scale bar: 80 μm. (C) Quantification of the percentage of pro-SPC+ cells that were also positive for pimonidazole per high-power field (HPF).
Exposure to hypoxia following bleomycin injury increases ER stress and worsens lung fibrosis.
Since our studies suggested that cellular hypoxia could cause ER stress and promote CHOP expression in type II AECs, we sought to develop a model to test this idea. We reasoned that exposure of bleomycin-treated mice to normobaric hypoxia could exacerbate local hypoxia in the lungs and worsen fibrosis. Therefore, we treated mice with a single dose of i.t. bleomycin (0.04 units) and then exposed mice to hypoxia from day 7 to day 21 after bleomycin (Figure 4A). As shown in Figure 4B, 10% O2 was uniformly lethal and 12% O2 resulted in 50% mortality by day 21; however, 14% O2 was relatively well tolerated (10% mortality). To determine whether exposure to 14% O2 was sufficient to exacerbate cellular hypoxia in the lungs, we injected pimonidazole and performed IHC on lung sections obtained at day 21 after bleomycin treatment (14 days of hypoxia). While no pimonidazole staining was observed in unchallenged mice exposed to 14% O2 and minimal staining was detected in bleomycin-challenged mice maintained in normoxia, there was robust pimonidazole staining in areas of bleomycin-induced fibrosis in mice exposed to 14% O2 (Figure 4C). As in the repetitive bleomycin model, pimonidazole staining strongly colocalized with pro-SPC+ type II AECs (Supplemental Figure 5, A and B), while S100A4+ fibroblasts, T1α+ type I AECs, CD34+ endothelial cells, and F4/80+ macrophages did not have detectable colocalization with pimonidazole adducts (Supplemental Figure 5, C–F).
Figure 4. Exposure to hypoxia following bleomycin treatment increases ER stress and worsens lung fibrosis.

(A) Schematic representation of experimental design in which a single dose of bleomycin (0.04 units) was injected intratracheally, followed 7 days later by exposure to normobaric hypoxia or normoxia (21% O2) for an additional 14 days. (B) Kaplan-Meier survival plot for mice treated with bleomycin and exposed to 21%, 14%, 12%, or 10% O2. n = 48 (in 21% O2), 35 (in 14% O2), 16 (in 12% O2), and 12 (in 10% O2). (C) Representative immunostaining for CHOP expression and pimonidazole adducts in mice injected with pimonidazole (60 μg/g) at 3 hours before lung harvest. Mice were treated with bleomycin, followed 7 days later by exposure to 14% O2 for an additional 14 days or maintenance in 21% O2. Additional controls were only exposed to 14% O2 for 2 weeks or were maintained in 21% O2. Scale bars: 800 μm (low magnification): 60 μm (high magnification). (D) Western blots for ER stress markers CHOP, PDI, ATF4, and XBP1s in lung tissue lysates. β-Actin was used as a loading control. (E) Quantification of CHOP+ AECs per high-power field (HPF) on lung sections. (F) Masson’s trichrome staining and (G) morphometric evaluation of lung fibrosis. (H) Total soluble collagen in right lower lobe (RLL) by Sircol assay. Comparisons between groups were made using unpaired, 2-tailed Student’s t test (G) or 1-way ANOVA with Tukey’s post hoc test (E and H). *P < 0.05 compared with single bleo + 21% O2, **P < 0.05 compared with untreated + 21% O2.
With evidence that exposure to 14% O2 following bleomycin injury was associated with cellular hypoxia in AECs, we evaluated ER stress markers and found that CHOP, PDI, ATF4, and XBP1s were upregulated in lung tissue from mice treated with bleomycin followed by 14% O2 (Figure 4D). By quantification of IHC, we found a marked induction of CHOP in AECs in lungs of mice treated with bleomycin followed by 14% O2 (Figure 4, C and E). Consistent with our findings in other fibrosis models characterized by robust induction of ER stress, mice treated with bleomycin followed by 14% O2 had a marked exacerbation of fibrosis compared with bleomycin-treated mice maintained in normoxia (Figure 4, F–H).
CHOP–/– mice are protected from exaggerated lung fibrosis induced by exposure to hypoxia.
To test whether CHOP mediates the excess lung fibrosis resulting from exposure to hypoxia following bleomycin treatment, we treated WT and CHOP–/– mice with a single i.t. injection of bleomycin (0.04 units) followed by randomization to normoxia (21% O2) or hypoxia (14% O2) between day 7 and day 21 after bleomycin. As shown in Figure 5, A–C, the hypoxia-induced exacerbation of lung fibrosis was completely abrogated in CHOP–/– mice. Similar to our findings above (see Figure 2), CHOP deficiency had no detectable effects in mice treated with single-dose bleomycin and maintained in normoxia.
Figure 5. CHOP–/– mice are protected from exaggerated lung fibrosis induced by exposure to hypoxia.

(A–D) WT and CHOP–/– mice received an intratracheal injection of bleomycin (0.04 units), followed 7 days later by exposure to 14% O2 or 21% O2. (A) Representative Masson’s trichrome staining (scale bars: 800 μm) and (B) morphometric evaluation of lung fibrosis at 21 days after bleomycin injection. (C) Total soluble collagen in right lower lobe (RLL) by Sircol assay. (D) Quantification of dual immunofluorescence for pro-SPC+ and TUNEL+ cells per high-power field (HPF) on sections from lungs harvested 3 days after randomization to 21% O2 or 14% O2 (10 days after bleomycin injection). (E–G) Mice with targeted deletion of HIF1/2 in lung epithelium (HIF1/2Δ/Δ) and controls (HIF1/2fl/fl ) were studied. (E) Morphometric evaluation of fibrosis at 21 days after bleomycin injection. (F) Total soluble collagen in RLL by Sircol assay. (G) Quantification of TUNEL+ AECs per HPF on sections from lungs harvested 3 days after randomization to 21% O2 or 14% O2. Comparisons between groups were made using 1-way ANOVA with Tukey’s post hoc test. *P < 0.05 compared with WT + single bleo + 14% O2, **P < 0.05 compared with WT + single bleo + 21% O2.
Since AEC apoptosis following single-dose bleomycin is most prominent within the first 2 weeks after treatment (16), we tested whether exposure of bleomycin-treated mice to 14% O2 for 3 days (between day 7–10 after bleomycin) exacerbates AEC apoptosis and if this effect is mediated by CHOP. In lungs harvested at 10 days after bleomycin, quantification of pro-SPC and TUNEL dual-positive cells showed that AEC apoptosis was increased in WT mice exposed to hypoxia compared with WT mice maintained in normoxia and that this increase was mitigated in lungs of CHOP–/– mice (Figure 5D). We also examined whether CHOP–/– mice had differences in lung immune/inflammatory cell populations in the bleomycin followed by hypoxia model. At 21 days after bleomycin treatment, we found no differences in myeloid cell subsets (Supplemental Figure 6A) or lymphocytes (Supplemental Figure 6B) between CHOP–/– and WT mice exposed to 14% O2 or maintained in normoxia. Since M2 macrophage polarization has been associated with lung fibrosis (17–19), we also isolated lung macrophages and examined whether CHOP deficiency alters macrophage polarization; however, no M1/M2 phenotype shift was observed in CHOP–/– mice following bleomycin treatment in either the normoxia or hypoxia groups (Supplemental Figure 6C). Since bleomycin results in inflammation that peaks between 1 and 2 weeks after exposure, we also evaluated inflammation during this time period at 10 days after bleomycin injection (exposure to 21% O2 or 14% O2 for 3 days between 7 and 10 days after bleomycin). Similar to our findings at day 21 after bleomycin, numbers of lymphocytes were similar in the lungs of WT and CHOP–/– mice in normoxia and hypoxia. In contrast, neutrophils and interstitial macrophages were reduced in bleomycin-treated CHOP–/– mice compared with WT mice in hypoxia, while numbers of alveolar macrophages and dendritic cells were similar (Supplemental Figure 7, A and B). Regarding macrophage polarization, no difference in the pattern of M1/M2 markers was observed between WT and CHOP–/– mice following bleomycin treatment in normoxia or hypoxia (Supplemental Figure 7C). Consistent with similar macrophage polarization among treatment groups, no evidence of CHOP induction was observed in macrophages isolated from bleomycin-treated WT mice in either hypoxia or normoxia (Supplemental Figure 8).
Because many of the effects of hypoxia are mediated through HIF signaling (20–24), we investigated whether HIF signaling could affect CHOP expression and lung fibrosis by generating mice with lung epithelial cell–specific deletion of HIF1α and HIF2α. Epithelial HIF1/2-deficient (HIF1/2Δ/Δ) mice and littermate controls (HIF1/2fl/fl) were treated with bleomycin (0.04 units), followed by exposure to 14% O2 or 21% O2 between day 7 and 21 (for evaluation of fibrosis) or between day 7 and 10 (for quantification of AEC apoptosis). Despite substantial HIF activation (Supplemental Figure 9A), epithelial HIF1/2 deletion did not alter fibrosis or reduce AEC apoptosis in mice treated with bleomycin followed by hypoxia exposure (Figure 5, E–G, and Supplemental Figure 9B). In addition, CHOP expression was similar in HIF1/2Δ/Δ mice and controls treated with bleomycin followed by hypoxia (Supplemental Figure 10). Taken together, these studies demonstrate that HIF is not the primary pathway mediating the effects of hypoxia on lung fibrosis in this model. Rather, non-HIF–dependent induction of CHOP appears to be critical for determining fibrosis under these conditions.
Hypoxia induces CHOP expression in AECs through the IRE1α/XBP1 and PERK/ATF4 pathways.
To investigate the regulation of CHOP expression by hypoxia, we exposed a type II AEC line (MLE12 cells) to hypoxia (1.5% O2) for 24–48 hours. By Western blotting and qPCR, we found upregulation of CHOP in hypoxic cells compared with cells cultured in room air (21% O2) (Figure 6A). In addition, we found increased expression of ATF4 (a measure of activation of PKR-like ER kinase [PERK]) and spliced XBP1 (a measure of activation of inositol-requiring enzyme 1 [IRE1]) following hypoxia exposure. In contrast, cleaved activating transcription factor 6 (ATF6) and IRE1α levels remained unchanged after exposure to hypoxia (Figure 6, B and C). To determine the regulation of CHOP expression in hypoxia, we used small-molecule inhibitors to disrupt IRE1α or PERK activity in MLE12 cells. We treated MLE12 cells with a small-molecule inhibitor of IRE1α (PCP-101, Mannkind Corporation), which interacts directly with the RNase active site of IRE1α and blocks XBP1 mRNA splicing (25–27), or DMSO as vehicle control. Following treatment, cells were exposed to 1.5% O2 or room air for 48 hours. Treatment of MLE12 cells with PCP-101 efficiently blocked XBP1 splicing and markedly reduced CHOP induction in hypoxia, without affecting ATF4 induction (Figure 6, D and E). These data show an important role of the IRE1α/XBP1 pathway in hypoxia-induced CHOP expression. In confirmation of this finding, treatment of MLE12 cells with an IRE1 siRNA also prevented CHOP induction in hypoxia (Supplemental Figure 11). Next, to evaluate the role of the PERK/ATF4 pathway, we treated MLE12 cells with a small-molecule inhibitor of PERK (GSK2606414) or DMSO as vehicle control, followed by exposure to hypoxia for 48 hours. PERK inhibition led to a reduction in ATF4 expression and a concomitant decrease in CHOP expression, without affecting XBP1 splicing (Figure 6F). siRNA-mediated inhibition of ATF4 signaling led to a slight reduction in CHOP levels in hypoxia (Supplemental Figure 11). Taken together, these data suggest that both the IRE1/XBP1s and the PERK/ATF4 arms of the UPR pathways contribute to CHOP induction in hypoxia.
Figure 6. Hypoxia induces CHOP expression in AECs through the IRE1α/XBP1 pathway.

MLE12 cells were exposed to normoxia (21% O2) or hypoxia (1.5% O2) for 24 or 48 hours. (A) qPCR for CHOP normalized to RPL19 and Western blot for CHOP at 48 hours. (B) Western blots for ATF4, ATF6, IRE1, and β-actin at 48 hours. (C) qPCR for ATF4 normalized to RPL19 and spliced XBP1 normalized to unspliced XBP1 at 24 hours. Comparisons between groups were made using unpaired, 2-tailed Student’s t test (A and C) *P < 0.05 compared with MLE12 + 21% O2. (D) MLE12 cells were treated with a small-molecule inhibitor of IRE1 (PCP101) or DMSO (vehicle control) and exposed to hypoxia for 48 hours. Western blots for CHOP, IRE1, and ATF4 and RT-PCR gel showing XBP1 splicing. XBP1u, XBP1 unspliced; XBP1s, XBP1 spliced. (E) Densitometry of CHOP normalized to β-actin. (F) MLE12 cells were treated with a small-molecule inhibitor of PERK (GSK2606414) or DMSO (vehicle control) and exposed to hypoxia for 48 hours. qPCR for ATF4 and CHOP normalized to RPL19 and spliced XBP1 normalized to unspliced XBP1. (G) MLE12 cells were transfected with HIF1α siRNA or control nontarget (NT) siRNA and exposed to hypoxia for 48 hours. Comparisons between groups were made using unpaired, 2-tailed Student’s t test (F) or 1-way ANOVA with Tukey’s post hoc test (E and G). *P < 0.05 compared with vehicle + 1.5% O2, **P < 0.05 compared with vehicle + 21% O2.
To evaluate the role of HIF signaling in regulating CHOP expression in AECs, we treated MLE12 cells with siRNA directed toward HIF1α, followed by exposure to hypoxia (1.5% O2). While we observed efficient HIF1α knockdown (Supplemental Figure 12), hypoxia-induced CHOP expression was unchanged (Figure 6G), thus supporting findings from in vivo studies that HIF and CHOP are independently regulated.
CHOP regulates hypoxia-induced apoptosis of AECs.
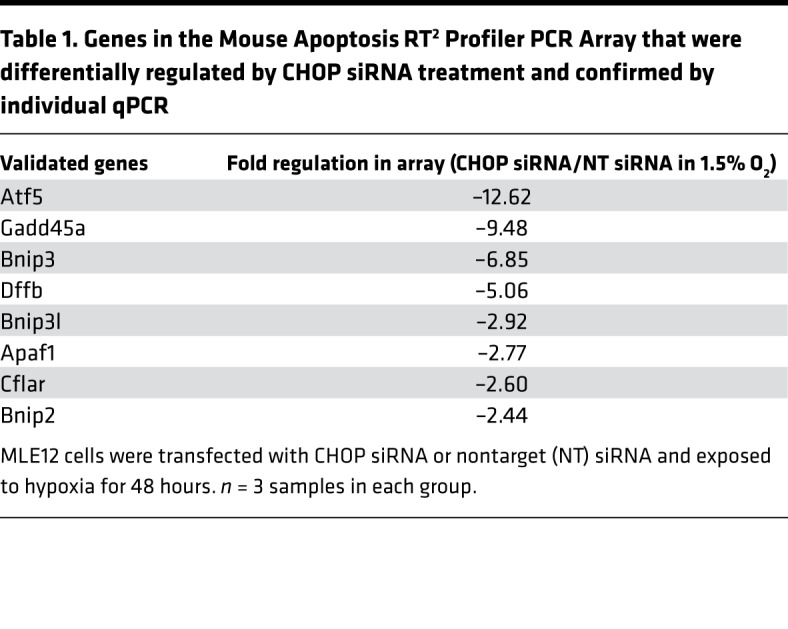
To determine the mechanism through which CHOP regulates hypoxia-induced apoptosis of AECs, we transfected MLE12 cells with an siRNA targeting CHOP (or nontargeted control siRNA), followed by exposure to 1.5% O2 for 48 hours. We confirmed efficient CHOP knockdown and found that CHOP–/– cells were protected from hypoxia-induced cell death (Figure 7, A and B). To identify the mediators regulated by CHOP in AECs, we utilized the Mouse Apoptosis RT2 Profiler PCR Array (Qiagen) to assess mRNA expression of proapoptotic and antiapoptotic genes in MLE12 cells treated with CHOP siRNA or nontargeted siRNA and exposed to 1.5% O2. Of the 59 genes expressed in MLE12 cells (cycle threshold < 30), 13 were significantly downregulated and 1 was upregulated in CHOP–/– cells (Figure 7C and Supplemental Figure 13). Following PCR validation of this set of apoptosis-related genes, 8 genes remained that were differentially expressed (all downregulated) in the setting of CHOP deficiency (Table 1). Subsequently, we evaluated these targets in vivo and found that expression of activating transcription factor 5 (ATF5), growth arrest and DNA damage inducible α (GADD45A), and BCL2-interacting protein 3 like (BNIP3L) were reduced in lungs of CHOP–/– mice compared with WT mice after repetitive bleomycin injury (Supplemental Figure 14), thus validating these targets as potential downstream CHOP mediators affecting AEC survival in this model.
Figure 7. CHOP mediates hypoxia-induced apoptosis of AECs through expression of apoptosis-regulating proteins.

MLE12 cells were transfected with CHOP siRNA or nontarget (NT) siRNA and exposed to hypoxia for 48 hours. (A) Western blot for CHOP. β-Actin was used as a loading control. (B) Cell death as measured by LDH assay. Comparisons between groups were made using 1-way ANOVA with Tukey’s post hoc test. *P < 0.05 compared with NT siRNA + 1.5% O2, **P < 0.05 compared with NT siRNA + 21% O2. (C) Volcano plot showing differentially regulated genes (cycle threshold < 30) using the Mouse Apoptosis RT2 Profiler PCR Array comparing MLE12 cells treated with CHOP siRNA or NT siRNA, followed by hypoxia exposure for 48 hours. n = 3 samples in each group.
Table 1. Genes in the Mouse Apoptosis RT2 Profiler PCR Array that were differentially regulated by CHOP siRNA treatment and confirmed by individual qPCR.
CHOP and markers of hypoxia are prominently expressed in lungs of IPF patients.
We evaluated the expression of CHOP and hypoxia markers in lungs of IPF patients and non-IPF controls. We observed induction of CHOP in IPF lungs by IHC on lung sections and by Western blotting of lung tissue homogenates. CHOP was predominantly expressed in hyperplastic type II AECs in areas of fibrosis. Additionally, we observed prominent expression of hypoxia markers HIF1α, pyruvate kinase, and carbonic anhydrase IX in hyperplastic type II AECs in IPF lungs, with a pattern similar to CHOP staining (Figure 8). Together, these studies in lung specimens from IPF patients provide further evidence for a connection between localized hypoxia and ER stress in this disease.
Figure 8. CHOP and markers of hypoxia are prominently expressed in lungs of IPF patients.

(A) Representative immunostaining for CHOP, HIF1α, pyruvate kinase, and carbonic anhydrase IX (CA-IX) on lung sections of IPF patients and non-IPF controls. Arrows indicate positive staining in type II AECs. Scale bars: 60 μm. (B) Western blot and densitometry for CHOP on protein from lung tissue lysates of IPF patients and non-IPF controls. β-Actin was used as a loading control. Comparisons between groups were made using unpaired, 2-tailed Student’s t test. *P < 0.05.
Discussion
Our studies indicate that localized hypoxia augments ER stress and induces CHOP expression in AECs, thus favoring fibrotic remodeling over effective repair. In hypoxia, CHOP expression in AECs was unrelated to HIF signaling but was induced by ER stress through the IRE1α/XBP1 and PERK/ATF4 pathways. Induction of apoptosis in AECs, likely via expression of CHOP target genes, appears to be an important mechanism by which CHOP promotes fibrotic remodeling. These studies point to a distinction between the standard injury-repair model (single-dose i.t. bleomycin) and models involving sequential or repetitive injuries that worsen or prolong fibrotic remodeling. CHOP was not required for fibrosis in the single-dose bleomycin model, where CHOP induction was minimal. In contrast, CHOP was shown to be a critical mediator of fibrosis in models with substantial ER stress, including repetitive-dose bleomycin, bleomycin treatment in mice with L188Q SFTPC-induced ER stress in AECs, and bleomycin followed by hypoxia, thereby suggesting that CHOP (and ER stress) may be more important in progression, rather than initiation, of fibrosis. Since repetitive injuries in the lung parenchyma are thought to underlie progressive fibrosis in IPF, factors identified as important in multiple-injury models may have added relevance when designing new therapeutic strategies for humans. This idea is further supported by our findings of increased expression of hypoxia markers and CHOP in hyperplastic type II AECs in IPF lungs.
CHOP is a multifunctional transcription factor that regulates ER stress–induced apoptosis (28). In our studies, the effect of CHOP on fibrosis was mirrored by its effects on AEC apoptosis, thus implicating CHOP-induced AEC apoptosis as an important mechanism influencing fibrosis severity. Although the molecular mechanisms underlying CHOP-induced apoptosis may be context dependent, our studies showed that CHOP upregulates expression of numerous apoptosis-related genes in AECs following exposure to hypoxia, including GADD45A, ATF5, and BNIP3L, which we verified in lungs of CHOP–/– mice after recurrent bleomycin injury. GADD45A can regulate cell cycle checkpoints, apoptosis, and DNA repair by contributing to activation of p53 (29), a protein that is upregulated in AECs in IPF (30). ATF5 has been identified as a direct downstream target of CHOP in mouse embryonic fibroblasts and has been reported to cooperate with CHOP for full induction of a specific subset of downstream genes (31). BNIP3L functions in B cell lymphoma 2–mediated (Bcl-2–mediated) cell death, production of reactive oxygen species, and mitophagy (32). While the effect of CHOP may be multifactorial, further work is needed to precisely define the pathways through which CHOP regulates AEC apoptosis during lung fibrosis. Although we have focused on the role of CHOP in mediating ER stress–induced apoptosis, JNK and caspase-4/12 are additional ER stress–induced mediators that could effect AEC apoptosis and fibrosis (33–35).
In addition to apoptosis, CHOP can regulate a variety of cellular processes, such as differentiation, polarization, and inflammatory signaling, through direct transcriptional regulation and interactions with other C/EBP family members (28, 36–40). In this regard, CHOP was reported to regulate M2 macrophage polarization in lung fibrosis induced by single-dose bleomycin (41); however, in our studies, we did not observe induction of CHOP in macrophages and CHOP deficiency did not effect macrophage polarization in any of our models. CHOP deficiency did reduce the number of neutrophils in the lungs following repetitive-bleomycin treatment and at day 10 in the bleomycin followed by hypoxia model (interstitial macrophages were reduced at day 10 in this model as well). It is unclear whether these changes in inflammatory cells effect fibrosis in these models; we speculate that the observed decrease in these inflammatory cell subtypes may be secondary to reduced epithelial injury and death, rather than a direct effect of CHOP in inflammatory cells. In addition to epithelial cell survival and regulation of inflammation, our epithelial cell expression profiling studies suggested that CHOP may affect a number of other cellular phenotypes that could effect fibrotic remodeling, including epithelial differentiation, growth factor responses, and cell migration.
CHOP expression has been linked to a number of ER stress–mediated pathologies in the kidneys, pancreas, heart, and liver, including diseases with progressive remodeling and organ dysfunction (3, 42–44). In lung fibrosis, prior studies regarding the role of CHOP have yielded conflicting results, with two reports showing that CHOP–/– mice have reduced fibrosis after single-dose i.t. bleomycin treatment (41, 45) and another report showing markedly worse survival and increased fibrosis in CHOP–/– mice after single-dose i.t. bleomycin treatment (46). The latter study also found that mice with heterozygous deletion of binding immunoglobulin protein (BiP), which should exacerbate ER stress through reduced chaperone function, were protected from lung fibrosis through increased CHOP-dependent macrophage apoptosis. In contrast to these studies, we found no effects of CHOP deficiency in the standard single-dose bleomycin model using two different doses of bleomycin (0.04 units and 0.08 units). Although the explanation for the discrepancy between these studies is not obvious, our findings suggest that CHOP regulation of fibrosis differs depending on the ER stress–dependent contribution to the fibrotic phenotype. Specifically, CHOP deficiency in bleomycin-treated L188Q SFTPC/CHOP–/– mice reduced fibrosis only to the level observed in bleomycin-treated WT mice, and similarly, CHOP–/– mice exposed to hypoxia following single-dose bleomycin developed lung fibrosis equivalent to that of WT mice treated with single-dose bleomycin and maintained in normoxia.
Although the PERK/ATF4 pathway is generally considered to be predominant in CHOP activation (47), our studies showed that the IRE1α/XBP1 arm of the UPR also plays an important role in hypoxia-induced CHOP upregulation in AECs. In disease models, the PERK pathway has been implicated in liver fibrosis (48), and the IRE1α pathway has been reported to regulate both liver and skin fibrosis (49–51). Together with our studies, these data indicate that these UPR pathways may be the most relevant for regulating fibrotic remodeling.
The finding of localized hypoxia in the repetitive bleomycin model is an important advance in understanding how ER stress could be generated in IPF. Our findings support the idea that hypoxia can develop in areas of alveolar collapse and consolidation through reduced ventilation, hypoxic vasoconstriction, and increased oxygen demand. Increased expression of a repertoire of hypoxia-inducible genes has been reported in IPF lungs and in single-cell analysis of AECs in IPF (52–54), consistent with our findings of increased expression of markers of hypoxia in lung tissue of IPF patients. In experimental models, regions of hypoxia have been observed in lungs of rats following radiation-induced injury (55) and in injured lungs of mice following H1N1 influenza infection (53). In our studies, we identified pimonidazole adducts, which require a very low pO2 (≤10 mmHg) for generation, as a marker of cellular hypoxia in type II AECs in the repetitive bleomycin treatment model and the single-dose bleomycin followed by hypoxia model. Depletion of oxygen to the level required for pimonidazole adduct formation may occur preferentially in AECs because of the high metabolic activity of these cells (16). Our model of exposure to 14% O2, which is equivalent to ascension to an altitude of approximately 10,500 feet above sea level, after bleomycin injury makes it possible for future studies to further investigate the specific contribution of hypoxia to development of lung fibrosis.
Although hypoxia can induce both HIF and ER stress (40), the relationship between these two pathways is incompletely understood. We found that HIF did not regulate CHOP expression in vivo or in vitro and that epithelial deletion of both HIF1 and HIF2 did not affect bleomycin-induced fibrosis or AEC apoptosis (in either hypoxic or normoxic conditions). These findings are consistent with our previous report that endothelial HIF deletion reduced pulmonary hypertension but not lung fibrosis following IP bleomycin treatment (56). However, HIF signaling augments fibrosis in several organs (20, 23, 57–59), and Xi et al. (53) recently demonstrated in an H1N1 influenza injury model that hypoxia drives persistent Notch activity in lineage– alveolar progenitors cells in a HIF1α-dependent manner, leading to impaired epithelial regeneration in lung parenchyma (53). Therefore, additional investigations regarding the role of HIF signaling in lung remodeling may be warranted.
In summary, we have furthered knowledge in this area by (a) discovering that ER stress is mechanistically linked to lung fibrosis through CHOP in 3 distinct multiple injury-induced lung fibrosis mouse models, (b) demonstrating that CHOP is a critical molecular mediator of ER stress–induced apoptosis and may also regulate other important profibrotic functions of AECs in lung fibrosis, and (c) showing that localized hypoxia in areas of parenchymal damage could explain the etiology of ER stress in lung fibrosis. Based on these findings, CHOP and its downstream targets may be attractive targets for novel therapeutic strategies in IPF.
Methods
Human samples.
Lung samples were obtained from surgical lung biopsies (n = 4) or explanted lung tissue obtained at the time of organ transplantation (n = 6). Explanted donor lungs that were rejected for transplantation were used as controls (n = 10).
Mice.
We used 8- to 10-week-old male and female mice for these experiments (C57BL/6J background). WT and CHOP–/– mice were purchased from The Jackson Laboratory. Doxycycline-inducible transgenic mice that express a mutant form of human SFTPC (L188Q SFTPC) (5) driven by a murine SFTPC promoter construct were crossed with CHOP–/– mice to generate L188Q SFTPC/CHOP–/– mice. L188Q SFTPC mice were used as controls. HIF1α-floxed (58) and HIF2α-floxed (60) mice were crossed to generate HIF1α/HIF2α-floxed mice. Transgenic mice in which both HIF1α and HIF2α are deleted in the lung epithelium were generated by crossing the HIF1α/HIF2α-floxed mice with SPC.Cre.Rosa.STOP.LacZ mice, in which Cre recombinase expression is under the control of a 3.7-kb human SPC promoter as described previously (12, 58, 61, 62). Mice with HIF1α/HIF2α deleted in lung epithelium were designated as HIFΔ/Δ. Littermate controls used were negative for Cre expression and designated as HIFfl/fl. Mice were housed in the animal care facility at the Vanderbilt University Medical Center and given food and water ad libitum.
Lung fibrosis models.
Bleomycin (Hospira Inc.) was purchased from Vanderbilt University Medical Center pharmacy. Bleomycin (0.04 units or 0.08 units) in 100 μl saline was delivered by direct i.t. instillation under anesthesia as described previously (12, 62, 63). Mice were administered a single-dose or repetitive doses (6 doses at intervals of 2 weeks) of bleomycin, depending on the experimental strategy. Doxycycline-inducible transgenic mice were treated with doxycycline in drinking water (2 g/l) for 1 week prior to bleomycin instillation, and doxycycline was continued through the course of experiments. Lungs were harvested following euthanasia by exposure to carbon dioxide at designated time points. Right lungs were tied off and snap frozen for estimation of collagen and extraction of RNA and protein, and left lungs were inflated with 10% formalin for histology under constant pressure using a 25-cm pressure column as previously described (12).
Exposure of mice to hypoxia.
Mice were exposed to hypoxia in a normobaric hypoxia chamber (Biospherix), in which oxygen concentration is controlled through flow of nitrogen to achieve the desired FiO2. Ventilation ensures that carbon dioxide concentration remains below 1,000 parts per million.
Detection of cellular hypoxia.
Mice were treated with pimonidazole (60 μg/g body weight; HypoxyProbe Inc.) in 75 μl sterile normal saline by i.p. injection at 3 hours prior to lung harvest. Subsequently, IHC for pimonidazole adducts was performed on lung sections.
Morphometry.
Left lungs were formalin-fixed, paraffin-embedded, sectioned, and stained with Masson’s trichrome as previously described (12, 62). Lung morphometry was performed as previously described (12). For each lung sample, images were captured at ×4 using an Olympus BX81 research microscope, and areas of fibrosis were measured using Cell Sens Dimension (Olympus).
IHC and immunofluorescence.
IHC was performed on paraffin-embedded lung tissue sections as previously published (12, 62) using the following primary antibodies: pro-SPC (1:1,000 dilution, AB3786, MilliporeSigma), CHOP (1:300 dilution, SC-575, Santa Cruz Biotechnology), HIF1α (1:300 dilution, SC-10790, Santa Cruz), pyruvate kinase (1:300 dilution, GTX107977, Genetex), CA-IX (1:300 dilution, NB100-417, Novus Biologicals), pimonidazole (biotin-conjugated antibody, Mouse-Biotin-Mab, HypoxyProbe Inc.). Primary antibodies were developed using Vectastain Elite anti-rabbit ABC peroxidase kits and Vectastain Elite anti-mouse ABC peroxidase kits (both from Vector Laboratories). Peroxidase substrate was developed with VECTOR NovaRED Peroxidase (HRP) Substrate Kit (Vector Laboratories). For quantification of CHOP, CHOP+ AECs (based on morphology and location) were counted in 10 high-power fields (HPFs) on each lung section.
To detect cellular hypoxia in different cell types, dual immunofluorescence staining for pimonidazole adducts was performed with pro-SPC (1:1,000 dilution, AB3786, MilliporeSigma), S100A4 (1:400 dilution) (64), T1α (1:500 dilution, 8.1.1, Developmental Studies Hybridoma Bank, Iowa City, Iowa, USA), CD34 (1:300 dilution, 119302, BioLegend), and F4/80 (1:300 dilution, MCA497G, AbD Serotec). Following primary antibody incubation, appropriate fluorescent secondary antibodies (Jackson Immunoresearch) were used and nuclear counterstaining was performed using Vectashield mounting media with DAPI (Vector Laboratories). Dual fluorescent images were captured using an Olympus IX81 Inverted Research Microscope configured with an Olympus IX2 Biological Disk Scanning Unit (Olympus). Quantification of pro-SPC, pimonidazole, and dual-positive cells was performed in 10 HPFs on each lung section.
Evaluation of cell death.
For evaluation of apoptosis in lung tissue, TUNEL staining was performed using the Fluorescent In Situ Cell Death Detection Kit (MilliporeSigma). The total number of TUNEL+ type II AECs (based on morphology and location) was quantified in 10 HPFs on each lung section as previously published (65). In addition, dual immunofluorescence was performed for TUNEL and pro-SPC, and double-positive cells were quantified in 10 HPFs on each lung section. To detect cytotoxicity in vitro, the lactate dehydrogenase (LDH) assay (Promega) was performed as per the manufacturer’s instructions.
Measurement of collagen and fibronectin.
Total soluble collagen in right lower lobes of the lung was measured using the Sircol assay (Biocolor; Accurate Chemical And Scientific Corporation) as per the manufacturer’s instructions and as previously published (66). Fibronectin in lung tissue was measured using the Mouse Fibronectin ELISA Kit (Abcam) as per the manufacturer’s instructions.
Quantification of immune/inflammatory cell types by flow cytometry.
Lungs were enzymatically digested using collagenase XI (MilliporeSigma) and DNase IV (Macs Miltenyi Biotec) for 45 minutes at 37°C to obtain single-cell suspensions, followed by incubation with viability dye (Thermo Fisher Scientific) for 20 minutes according to the manufacturer’s instructions. Cells were then incubated with anti-CD16/32 antibodies and further labeled with the following fluorescent dye–conjugated mAbs: CD45-efluor 650 (95-0451-42, eBioscience/Thermo Fisher Scientific), Ly6C-APC-Cy7 (128025, BioLegend), Ly6G-Alexa Fluor 700 (127621, BioLegend), CD11b-APC (101212, BioLegend), CD11c-PE-Cy7 (25-01140-81, eBioscience/Thermo Fisher Scientific), F4/80-PE-Cy5 (123111, BioLegend), CD103-PE (12-1031-81, eBioscience/Thermo Fisher Scientific), CD19-PE (12-0193-81, eBioscience/Thermo Fisher Scientific), and CD3-Alexa Fluor 488 (100321, BioLegend). Samples were acquired on an LSRII machine (BD Biosciences), and data were analyzed using FlowJo software.
M1/M2 macrophage polarization.
The M1 and M2 macrophage phenotype was determined as previously published (67). In brief, freshly isolated lungs were digested with collagenase XI (0.7 mg/ml; MilliporeSigma) and type IV bovine pancreatic DNase (30 mg/ml; MilliporeSigma) in RPMI-1640 media to obtain single-cell suspensions. Red blood cells (RBCs) were lysed in RBC Lysis Buffer (BioLegend), followed by adherence on 6-well culture plates for 2 hours. Cells were supplemented with RPMI-1640 media with 10% FBS. Supernatant was discarded, mRNA was isolated from adherent macrophages, and qPCR was performed. The following primer sets were used: TNF-α — forward, 5′-AAGCCTGTAGCCCACGTCGTA-3′, reverse, 5′-GGCACCACTAGTTGGTTGTCTTTG-3′; IL-6 — forward, 5′-TCCTCTGGTCTTCTGGAGTA-3′, reverse, 5′-CTTAGCCACTCCTTCTGTGA-3′; IL-12p35 — forward, 5′-TGGACCTGCCAGGTGTCTTAG-3′, reverse, 5′-CAATGTGCTGGTTTGGTCCC-3′; iNOS — forward, 5′-CACCTTGGAGTTCACCCAGT-3′, reverse, 5′-ACCACTCGTACTTGGGATGC-3′; IL-10 — forward, 5′-ATAACTGCACCCACTTCCCA-3′, reverse, 5′-GGGCATCACTTCTACCAGGT-3′; arginase-1 — 5′-ATGGAAGAGACCTTCAGCTAC-3′, reverse, 5′-GCTGTCTTCCCAAGAGTTGGG-3′; Ym1 — forward, 5′-GGGCATACCTTTATCCTGAG-3′, reverse, 5′-CCACTGAAGTCATCCATGTC-3′; mannose receptor — forward, 5′-CAAGGAAGGTTGGCATTTGT-3′, reverse, 5′-CCTTTCAGTCCTTTGCAAGC-3′; and GAPDH — forward, 5′-TGAGGACCAGGTTGTCTCCT-3′, reverse: 5′-CCCTGTTGCTGTAGCCGTAT-3′. mRNA expression in each sample was normalized to GAPDH.
Cell culture and hypoxia exposure.
Mouse lung epithelial cells (MLE12) were purchased from ATCC. The cells were maintained in DMEM (Invitrogen, Life Technologies) with 2 mM L-glutamine, supplemented with FBS (10%) and antibiotics (100 units/ml penicillin and 100 μg/ml streptomycin). MLE12 cells were plated at 70%–80% confluence in 6-well plates. Twenty-four hours later, media were changed to fresh DMEM with FBS and antibiotics, and cells were exposed to hypoxia (1.5% O2) using an in vitro hypoxia chamber (Coy Laboratory) or normoxia (21% O2) for an additional 24–48 hours.
siRNA studies.
On-Target Smartpool siGenome siRNAs were used to target ER stress pathways and HIF1α in MLE12 cells as previously described (68). The following mouse constructs were used for siRNA targeting: IRE1α (L-041030-00), ATF4 (L-042737-01), HIF1α (L-040368-00), CHOP (L-062068-00), and nontarget control (D-001210-04-05) (all from Dharmacon). In brief, MLE12 cells were plated at 40%–50% confluence in 6-well plates in DMEM with FBS but no antibiotics and cultured overnight in a standard cell culture incubator. Cells were transfected with 25 nM Smartpool siRNA using Lipofectamine RNAi Max (Life Technologies) as per the manufacturer’s instructions. Twenty-four hours later, the media were replaced with fresh DMEM (with FBS and antibiotics), and cells were exposed to hypoxia (1.5% O2) or normoxia (21% O2) for an additional 48 hours.
Small-molecule–mediated inhibition of UPR pathways.
For chemical inhibition of IRE1α and PERK, MLE12 cells were plated at 70% confluence in 6-well plates in DMEM. Twenty-four hours later, media were replaced with 2 ml fresh DMEM in each well. 10 μM PCP-101 (Mannkind Corporation) (39–41) or 0.3 μM GSK2606414 (MilliporeSigma) in DMSO was added to the treatment groups, and 2 μl DMSO (MilliporeSigma) was added to the vehicle control groups.
Western blotting.
Total protein extracts were prepared from lungs using RIPA lysis buffer with protease and phosphatase inhibitors (Life Technologies Corporation) as previously described (8, 68). The following primary antibodies were used: CHOP (1:300 dilution, SC-7351, Santa Cruz Biotechnology), PDI (1:1,000 dilution, C81H6, Cell Signaling), ATF4 (1:1,000 dilution, 10835-1-AD, Proteintech Group Inc.), XBP1s (1:1,000 dilution, 658802, BioLegend), β-actin (1:25,000 dilution, A5316, MilliporeSigma), IRE1α (1:1,000 dilution, 14C10, Cell Signaling), ATF6 (1:500 dilution, NB1-40256SS, Novus Biologicals), and HIF1α (1:500 dilution, NB-100-479, Novus Biologicals). Western blots were developed using corresponding fluorescent-conjugated IRDye secondary antibodies and the Odyssey Infrared Imaging System (LI-COR Biosciences). Densitometry was performed using ImageJ software (NIH), and band intensities were normalized to β-actin.
RNA isolation and quantitative PCR.
Total mRNA was isolated from lung tissue or MLE12 cells using a RNeasy Mini kit (Qiagen) as per the manufacturer’s instructions. Following RNA isolation, DNA digestion was performed using the DNA-free DNA Removal kit (Ambion Thermo Scientific), and cDNA was synthesized using Superscript III Reverse Transcriptase (Invitrogen) or Superscript VILO Master Mix (Invitrogen) as per the manufacturer’s instructions. qPCR was performed using SYBR green PCR master mix (Applied Biosystems) as per the manufacturer’s instructions. The following primer sets were used: XBP1 spliced forward 5′-GAGTCCGCAGCAGGTG-3′ and XBP1 spliced reverse 5′-GTGTCAGAGTCCATGGGA-3′, XBP1 unspliced forward 5′-GACTATGTGCACCTCTGCAG-3′ and XBP1 unspliced reverse 5′-CTGGGAGTTCCTCCAGACTA-3′, ATF4 forward 5′-GGGTTCTGTCTTCCACTCCA-3′ and ATF4 reverse 5′-AAGCAGCAGAGTCAGGCTTTC-3′, CHOP forward 5′- CCACCACACCTGAAAGCAGAA-3′ and CHOP reverse 5′-AGGTGAAAGGCAGGGACTCA-3′, HIF1α forward 5′-TGGAGATGCTGGCTCCCTAT-3′ and HIF1α reverse 5′-TGGAGGGCTTGGAGAATTGC-3′, GADD45A forward 5′-TGCGAGAACGACATCAACAT-3′ and GADD45A reverse 5′-TCCCGGCAAAAACAAATAAG-3′, ATF5 forward 5′-GGCTGGCTCGTAGACTATGG-3′ and ATF5 reverse 5′CCAGAGGAAGGAGAGCTGTG-3′, BNIP3L forward 5′-CCTCGTCTTCCATCCACAAT-3′ and BNIP3L reverse 5′-GTCCCTGCTGGTATGCATCT-3′, and RPL19 forward 5′-ATGCCAACTCCCGTCAGCAG-3′ and RPL19reverse 5′-TCATCCTTCTCATCCAGGTCACC-3. Expression values of XBP1s were normalized to that of XBP1us using the 2ΔCt method. Expression values of all other genes were normalized to RPL19 also using the 2ΔCt method. Evaluation of XBP1 splicing by separation of spliced and unspliced cDNA products on an agarose gel was performed as previously described (69). For cDNA synthesis for this evaluation, the following primers were used: XBP1 forward 5′-CTGGAAAGCAAGTGGTAGA-3′ and XBP1 reverse 5′-CTGGGTCCTTCTGGGTAGAC-3′.
Mouse apoptosis PCR array.
The Mouse Apoptosis RT2 Profiler PCR Array (Qiagen) was used. MLE12 cells were plated at 40%–50% confluence in 6-well plates in antibiotic-free DMEM media. Twenty-four hours later, MLE12 cells were transfected with Smartpool siRNA for CHOP or control nontargeted siRNA. Following transfection, cells were exposed to hypoxia (1.5% O2) or normoxia (21% O2) for an additional 48 hours. After mRNA isolation, cDNA was synthesized using the RT2 First Strand Kit (Qiagen) and qPCR was performed using RT2 qPCR master mixes (Qiagen).
Isolation of EpCAM+ cells.
Lungs were enzymatically digested using Dispase II (MilliporeSigma) for 45 minutes at 37°C and passed through 100-μM and 40-μM SwiftStrain Cell Strainers (Denville), followed by the 20-μM Steriflip vacuum filter (EMD Millipore) to obtain single-cell suspensions. The single-cell suspension was then coated with mouse CD45 microbeads as per the manufacturer’s instructions (Macs Miltenyi Biotech). CD45+ cells were depleted by passing the CD45-coated single-cell suspension through LD columns on a QuadroMACS Separator (Macs Miltenyi Biotech) as per the manufacturer’s instructions. The flow through containing CD45– cells was then coated with mouse EpCAM microbeads as per the manufacturer’s instructions (Macs Miltenyi Biotech). EpCAM+ cells were collected by passing EpCAM-coated single-cell suspension through LS columns on a QuadroMACS Separator as per the manufacturer’s instructions.
RNA sequencing.
Total RNA was isolated from EpCAM+ cells using Direct-Zol RNA MiniPrep (Zymo Research), followed by DNA Digestion (Ambion, Invitrogen). The quality check of the RNA, mRNA enrichment, and cDNA library preparation utilizing stranded mRNA (polyA selected) were conducted at Vanderbilt Technologies for Advanced Genomics. RNA sequencing was performed on an Illumina NovaSeq6000 system with a paired-end mRNA library prep, PE-150, with 30 million reads. Initial alignment and quantification were performed using the Partek Flow package. STAR 2.5.3a was used to align RNA-sequencing reads, with quantification performed by Ensembl Transcripts Release 83 using Partek E/M. Reads were normalized to total count. Differentially expressed genes were determined based on >2-fold change (log2 ratio > 1) between groups, with P < 0.05 and FDR < 0.05. Annotation of overrepresented pathways was done using WebGestalt (WEB-based Gene SeT AnaLysis Toolkit).
Statistics.
Statistical analyses were performed using GraphPad Prism Software. Data are presented as mean ± SEM. Survival data were assessed using a Kaplan-Meier survival analysis. Pairwise comparisons were made using 2-tailed, unpaired Student’s t test. Comparisons between groups were made by 1-way ANOVA, followed by Tukey’s post hoc test. P values of less than 0.05 were considered significant.
Study approval.
Tissue collection and studies were approved by the Vanderbilt University Institutional Review Board (060165). All experimental protocols were approved by the Animal Care and Utilization Institutional Committee at Vanderbilt University Medical Center.
Author contributions
AB designed and performed research, conducted experiments, analyzed data, and wrote the paper. JAK designed research and wrote paper. CLC performed research and analyzed data. APS performed research, conducted experiments, and analyzed data. BDP performed research, conducted experiments, and analyzed data. WH performed research, conducted experiments, and analyzed data. TS performed research and analyzed data. LG performed research and analyzed data. WEL performed research and analyzed data. LRY designed research and wrote paper. TSB designed and performed research, analyzed data, and wrote the paper. HT designed and performed research, conducted experiments, analyzed data, and wrote the paper.
Supplementary Material
Acknowledgments
Funding was provided by the NIH/National Heart, Lung, and Blood Institute (R01 HL-119503 to LRY; R01 HL 085317 and P01 HL 092870 to TSB; R01 HL 105479 to WEL; K08 HL130595 to JAK); the Francis Family Foundation (to JAK); the Pulmonary Fibrosis Foundation (to JAK); the Vanderbilt International Scholar Program (to AB); an American Heart Association Predoctoral Fellowship (to AB); the American Thoracic Society/Hermansky-Pudlak Syndrome Network (to HT); and the Department of Veterans Affairs (to TSB and WEL).
Version 1. 08/23/2018
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2018;3(16):e99543. https://doi.org/10.1172/jci.insight.99543.
Contributor Information
Ankita Burman, Email: ankita.burman@vanderbilt.edu.
Wei Han, Email: wei.han@vanderbilt.edu.
Taylor Sherrill, Email: taylor.sherrill@vanderbilt.edu.
Linda Gleaves, Email: linda.gleaves@vanderbilt.edu.
Lisa R. Young, Email: lisa.young@vanderbilt.edu.
Harikrishna Tanjore, Email: harikrishna.tanjore@vanderbilt.edu.
References
- 1.Mora AL, Rojas M, Pardo A, Selman M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat Rev Drug Discov. 2017;16(11):755–772. doi: 10.1038/nrd.2017.170. [DOI] [PubMed] [Google Scholar]
- 2.Raghu G, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanjore H, Blackwell TS, Lawson WE. Emerging evidence for endoplasmic reticulum stress in the pathogenesis of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2012;302(8):L721–L729. doi: 10.1152/ajplung.00410.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas AQ, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002;165(9):1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 5.Lawson WE, et al. Endoplasmic reticulum stress enhances fibrotic remodeling in the lungs. Proc Natl Acad Sci USA. 2011;108(26):10562–10567. doi: 10.1073/pnas.1107559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kabore AF, Wang WJ, Russo SJ, Beers MF. Biosynthesis of surfactant protein C: characterization of aggresome formation by EGFP chimeras containing propeptide mutants lacking conserved cysteine residues. J Cell Sci. 2001;114(Pt 2):293–302. doi: 10.1242/jcs.114.2.293. [DOI] [PubMed] [Google Scholar]
- 7.Korfei M, et al. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2008;178(8):838–846. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lawson WE, et al. Endoplasmic reticulum stress in alveolar epithelial cells is prominent in IPF: association with altered surfactant protein processing and herpesvirus infection. Am J Physiol Lung Cell Mol Physiol. 2008;294(6):L1119–L1126. doi: 10.1152/ajplung.00382.2007. [DOI] [PubMed] [Google Scholar]
- 9.Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13(8):477–491. doi: 10.1038/nrneurol.2017.99. [DOI] [PubMed] [Google Scholar]
- 10.Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2008;7(12):1013–1030. doi: 10.1038/nrd2755. [DOI] [PubMed] [Google Scholar]
- 11.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 12.Degryse AL, et al. Repetitive intratracheal bleomycin models several features of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2010;299(4):L442–L452. doi: 10.1152/ajplung.00026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koumenis C, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22(21):7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008;3:399–425. doi: 10.1146/annurev.pathmechdis.3.121806.151434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varia MA, et al. Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol Oncol. 1998;71(2):270–277. doi: 10.1006/gyno.1998.5163. [DOI] [PubMed] [Google Scholar]
- 16.Polosukhin VV, et al. Intratracheal bleomycin causes airway remodeling and airflow obstruction in mice. Exp Lung Res. 2012;38(3):135–146. doi: 10.3109/01902148.2012.658595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wojtan P, Mierzejewski M, Osińska I, Domagała-Kulawik J. Macrophage polarization in interstitial lung diseases. Cent Eur J Immunol. 2016;41(2):159–164. doi: 10.5114/ceji.2016.60990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32(5):593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Nair MG, et al. Alternatively activated macrophage-derived RELM-{alpha} is a negative regulator of type 2 inflammation in the lung. J Exp Med. 2009;206(4):937–952. doi: 10.1084/jem.20082048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Higgins DF, Kimura K, Iwano M, Haase VH. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle. 2008;7(9):1128–1132. doi: 10.4161/cc.7.9.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nauta TD, van Hinsbergh VW, Koolwijk P. Hypoxic signaling during tissue repair and regenerative medicine. Int J Mol Sci. 2014;15(11):19791–19815. doi: 10.3390/ijms151119791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med. 2011;183(2):152–156. doi: 10.1164/rccm.201009-1393PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haase VH. Hypoxia-inducible factor signaling in the development of kidney fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S16. doi: 10.1186/1755-1536-5-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mimura N, et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood. 2012;119(24):5772–5781. doi: 10.1182/blood-2011-07-366633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanches M, et al. Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat Commun. 2014;5:4202. doi: 10.1038/ncomms5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Volkmann K, et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J Biol Chem. 2011;286(14):12743–12755. doi: 10.1074/jbc.M110.199737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang Y, Liu L, Naik I, Braunstein Z, Zhong J, Ren B. Transcription factor C/EBP homologous protein in health and diseases. Front Immunol. 2017;8:1612. doi: 10.3389/fimmu.2017.01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Salvador JsM, Brown-Clay JD, Fornace AJ. Gadd45 in Stress Signaling, Cell Cycle Control, and Apoptosis. In: Liebermann DA, Hoffman B, eds. Gadd45 Stress Sensor Genes. New York, NY: Springer New York; 2013:1–19. [DOI] [PubMed] [Google Scholar]
- 30.Steele MP, et al. Relationship between gene expression and lung function in Idiopathic Interstitial Pneumonias. BMC Genomics. 2015;16:869. doi: 10.1186/s12864-015-2102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teske BF, et al. CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol Biol Cell. 2013;24(15):2477–2490. doi: 10.1091/mbc.e13-01-0067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ney PA. Mitochondrial autophagy: Origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta. 2015;1853(10 Pt B):2775–2783. doi: 10.1016/j.bbamcr.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 33.Casson CN, et al. Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc Natl Acad Sci USA. 2015;112(21):6688–6693. doi: 10.1073/pnas.1421699112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277(37):34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 35.Nakagawa T, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 36.Endo M, Mori M, Akira S, Gotoh T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J Immunol. 2006;176(10):6245–6253. doi: 10.4049/jimmunol.176.10.6245. [DOI] [PubMed] [Google Scholar]
- 37.Frakes AE, Dillin A. The UPRER: Sensor and coordinator of organismal homeostasis. Mol Cell. 2017;66(6):761–771. doi: 10.1016/j.molcel.2017.05.031. [DOI] [PubMed] [Google Scholar]
- 38.Han J, Kaufman RJ. Physiological/pathological ramifications of transcription factors in the unfolded protein response. Genes Dev. 2017;31(14):1417–1438. doi: 10.1101/gad.297374.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Willy JA, Young SK, Stevens JL, Masuoka HC, Wek RC. CHOP links endoplasmic reticulum stress to NF-κB activation in the pathogenesis of nonalcoholic steatohepatitis. Mol Biol Cell. 2015;26(12):2190–2204. doi: 10.1091/mbc.e15-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–864. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 41.Yao Y, et al. Chop deficiency protects mice against bleomycin-induced pulmonary fibrosis by attenuating M2 macrophage production. Mol Ther. 2016;24(5):915–925. doi: 10.1038/mt.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oyadomari S, Araki E, Mori M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic beta-cells. Apoptosis. 2002;7(4):335–345. doi: 10.1023/A:1016175429877. [DOI] [PubMed] [Google Scholar]
- 43.Oyadomari S, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109(4):525–532. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanjore H, Lawson WE, Blackwell TS. Endoplasmic reticulum stress as a pro-fibrotic stimulus. Biochim Biophys Acta. 2013;1832(7):940–947. doi: 10.1016/j.bbadis.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka Y, et al. The exacerbating roles of CCAAT/enhancer-binding protein homologous protein (CHOP) in the development of bleomycin-induced pulmonary fibrosis and the preventive effects of tauroursodeoxycholic acid (TUDCA) against pulmonary fibrosis in mice. Pharmacol Res. 2015;99:52–62. doi: 10.1016/j.phrs.2015.05.004. [DOI] [PubMed] [Google Scholar]
- 46.Ayaub EA, et al. GRP78 and CHOP modulate macrophage apoptosis and the development of bleomycin-induced pulmonary fibrosis. J Pathol. 2016;239(4):411–425. doi: 10.1002/path.4738. [DOI] [PubMed] [Google Scholar]
- 47.Harding HP, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 48.Koo JH, Lee HJ, Kim W, Kim SG. Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK-mediated degradation of HNRNPA1 and up-regulation of SMAD2. Gastroenterology. 2016;150(1):181–193.e8. doi: 10.1053/j.gastro.2015.09.039. [DOI] [PubMed] [Google Scholar]
- 49.Hassler J, Cao SS, Kaufman RJ. IRE1, a double-edged sword in pre-miRNA slicing and cell death. Dev Cell. 2012;23(5):921–923. doi: 10.1016/j.devcel.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heindryckx F, et al. Endoplasmic reticulum stress enhances fibrosis through IRE1α-mediated degradation of miR-150 and XBP-1 splicing. EMBO Mol Med. 2016;8(7):729–744. doi: 10.15252/emmm.201505925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maurel M, Chevet E, Tavernier J, Gerlo S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem Sci. 2014;39(5):245–254. doi: 10.1016/j.tibs.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 52.Tzouvelekis A, et al. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med. 2007;176(11):1108–1119. doi: 10.1164/rccm.200705-683OC. [DOI] [PubMed] [Google Scholar]
- 53.Xi Y, et al. Local lung hypoxia determines epithelial fate decisions during alveolar regeneration. Nat Cell Biol. 2017;19(8):904–914. doi: 10.1038/ncb3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kusko RL, et al. Integrated genomics reveals convergent transcriptomic networks underlying chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194(8):948–960. doi: 10.1164/rccm.201510-2026OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vujaskovic Z, et al. Radiation-induced hypoxia may perpetuate late normal tissue injury. Int J Radiat Oncol Biol Phys. 2001;50(4):851–855. doi: 10.1016/S0360-3016(01)01593-0. [DOI] [PubMed] [Google Scholar]
- 56.Bryant AJ, et al. Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2016;310(3):L249–L262. doi: 10.1152/ajplung.00258.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haase VH. Hypoxia-inducible factor signaling in the development of kidney fibrosis. Fibrogenesis Tissue Repair. 2012;5(Suppl 1):S16. doi: 10.1186/1755-1536-5-S1-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Higgins DF, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117(12):3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. 2013;93(1):1–21. doi: 10.1152/physrev.00017.2012. [DOI] [PubMed] [Google Scholar]
- 60.Kapitsinou PP, et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. 2010;116(16):3039–3048. doi: 10.1182/blood-2010-02-270322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Degryse AL, et al. TGFβ signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am J Physiol Lung Cell Mol Physiol. 2011;300(6):L887–L897. doi: 10.1152/ajplung.00397.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanjore H, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am J Respir Crit Care Med. 2009;180(7):657–665. doi: 10.1164/rccm.200903-0322OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lawson WE, et al. Increased and prolonged pulmonary fibrosis in surfactant protein C-deficient mice following intratracheal bleomycin. Am J Pathol. 2005;167(5):1267–1277. doi: 10.1016/S0002-9440(10)61214-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lawson WE, et al. Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am J Respir Crit Care Med. 2005;171(8):899–907. doi: 10.1164/rccm.200311-1535OC. [DOI] [PubMed] [Google Scholar]
- 65.Tanjore H, et al. β-catenin in the alveolar epithelium protects from lung fibrosis after intratracheal bleomycin. Am J Respir Crit Care Med. 2013;187(6):630–639. doi: 10.1164/rccm.201205-0972OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Young LR, et al. The alveolar epithelium determines susceptibility to lung fibrosis in Hermansky-Pudlak syndrome. Am J Respir Crit Care Med. 2012;186(10):1014–1024. doi: 10.1164/rccm.201207-1206OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Han W, et al. Molecular imaging of folate receptor β-positive macrophages during acute lung inflammation. Am J Respir Cell Mol Biol. 2015;53(1):50–59. doi: 10.1165/rcmb.2014-0289OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tanjore H, et al. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2011;286(35):30972–30980. doi: 10.1074/jbc.M110.181164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cheng DS, et al. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J Immunol. 2007;178(10):6504–6513. doi: 10.4049/jimmunol.178.10.6504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.