

Abstract
N-methylation of amino acids is an effective way to create protease resistance in both natural and synthetic peptides. However, alkyl substituents other than N-methyl have not been extensively studied. Here, we prepare and examine a series of N-substituted peptides in which the size and length of the alkyl group is modulated. These design insights provide a unique and modular handle for tuning proteolysis in oligopeptides.
In this work we demonstrate a strategy for tuning proteolysis of oligopeptides by expanding the N-alkylation of peptides beyond the common methyl group.

Synthetic peptides have been shown to have excellent in-vitro activity in biological systems owing to their high affinity and specificity to various targets. However, despite their potential, the clinical utility of peptides is limited due to their low proteolytic stability and reduced in-vivo life-times.1–3 Several approaches have been examined to overcome this challenge, including synthetic modifications such as the insertion of unnatural β-amino acids, D-amino acids or N-methylated units.3–9 N-methylation is of particular interest as it most closely resembles nature’s basic amino acid units with respect to backbone bond lengths and chirality. For example, it has been shown that upon multiple N-methylations of somatostatin cyclopeptidic analogue, the enzymatic stability is enhanced fivefold, from half-life time of 15.5±2 to 74±6 min, with a significant improved bioavailability. Notably, this performance was achieved without modifying biological activity and selectivity.10 This unique performance may also explain the sub-set of natural systems that exploit N-methylation to modulate biological functions.6 Despite these promising examples of N-methylation for tuning reactivity, the introduction of other N-alkyl substituents have not been explored.11–13 For example, the ability to further expand the regulation of proteolysis rates via simple chemical modifications would be beneficial when a clinical treatment may be too acute and decreased circulation times preferred.14
One challenge to the synthesis of modified peptides with larger N-alkyl groups is increased steric hindrance leading to significantly lower coupling reactions.15,16 Recently, we have successfully developed synthetic strategies for sterically demanding N/Cα-disubstituted peptides (DS-peptides) with a high degree of control over preferred conformation of the biomimetic oligomers being observed.17 Based on this synthetic pathway, we sought to study how variation in N-alkylation affects proteolysis of oligopeptides.
For this purpose, a simple model oligopeptide platform was designed to elucidate the effects of N-alkylation of a single modified amino acid. short peptides were preferred to eliminate any influence from secondary structures. In addition, oligopeptides have found wide use in applications ranging from sensing, enzyme inhibition to therapeutics.18–23 A model pentapeptide was targeted based on an N-terminal glutamic acid unit for water solubility followed by four alanine residues.
The central amino acid unit could then be systematically modified with various N-alkylated amino acid derivatives spanning a range of sizes of substituents from methyl (Me) and ethyl (Et), to benzyl (Bn), ethylphenyl (EtPh), and propylphenyl (PrPh) groups, as shown in Scheme 1. These N-alkyl units were selected as they systematically increase the length of the alkyl chain while also introducing steric bulk through the phenyl ring. In addition, the phenyl unit opens up a variety of substituted derivatives for secondary functionalization. With the design of the oligopeptide series identified, elastase was selected as a model protease enzyme based on its specific targeting of alanine and glycine units. Furthermore, uncontrolled proteolytic degradation by elastase has been implicated in a number of pathological conditions such as pancreatitis and other inflammatory disorders.24 The identification of design principles for regulating elastase protease activity is therefore important from both an application viewpoint and for future synthetic peptide modifications.
Scheme 1.

The pentapeptide used in this study and the elastase protease resistance trend upon N-substations. Orange arrow: the site that is cleaved by elastase upon N-alkylation (R1≠H).
All of the N-alkylated amino acid building blocks were synthesized in-house using a reductive amination protocol, followed by Fmoc protection (Scheme 2A). A longstanding challenge in peptide synthesis is the coupling of sterically hindered amino acid residues. Since the oligopeptide targets identified in this study all have a central N/Cα-disubstituted repeat unit, COMU, 1-[(1-(cyano-2-ethoxy-2-oxoethylideneaminooxy)-dimethylamino-morpholinomethylene)]methanaminium hexafluorophosphate coupling agent (Scheme 2B, Table S1) was examined and found to be particularly effective. For all cases, except for the most hindered example, R1=Bn, high yield (>90%) of the coupled product were obtained.25 In this case of R1=Bn, triphosgene was found to be the only coupling agent to give the desired N/Cα-disubstituted peptides. Detailed procedures for the monomer and peptide syntheses can be found in the ESI.
Scheme 2.

A) Monomer synthesis according to the reductive amination protocol. B) Solid phase synthesis of oligopeptides.
In order to understand the effect of N-substitution on the proteolysis rate, the synthetic peptides were incubated at 37°C and pH 7.8 in the presence of elastase. Reverse phase high performance liquid chromatography (RP-HPLC) was then used to analyze aliquots taken at different time points with the cleavage products identified by mass spectrometry (Figures S1-S3). The resulting half-lives (τ1/2) were determined for each peptide sequence and are shown in Figure 1. As expected, the N-methylated (R1= Me) derivative showed significantly slower proteolysis with the half-life increasing threefold when compared to the natural amino acid sequence (R1=H) (Figure 1). Unexpectedly, increasing the steric bulk and hydrophobicity of the N-alkyl group led to a decrease in half-life with a systematic decrease being observed on going from ethyl (R1=Et) to phenyl (R1=Bn) and ethylphenyl (R1=EtPh). For longer alkyl groups, (R1=PrPh), the half-life time was further decreased to values approaching the natural sequence (R1=H) (Figure 1) with multiple experimental repeats being performed to allow a general trend to be distinguished.
Figure 1.
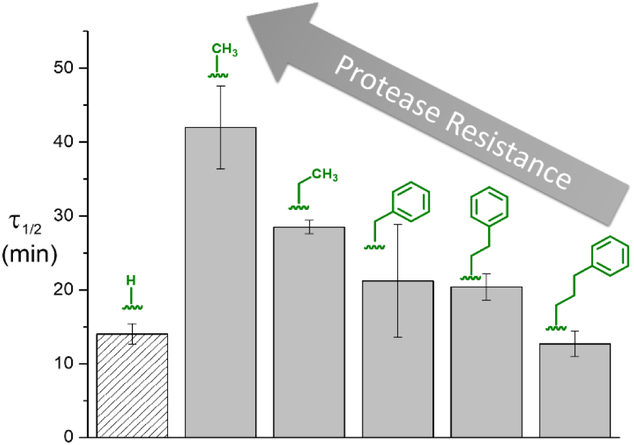
Change in half-life values for alanine-based pentapeptides with different R1 N-substituents.
It is worth noting that when a peptide consisting of a single alkylated unit is exposed to the corresponded protease, no degradation is observed. For example, peptides having a single N-substituted phenylalanine (R1=Me or Bn) units shows no reactivity with chymotrypsin (Figure S2). In the case of multiple alanine units, the site of proteolysis was observed to change with the introduction of the unnatural amino acid units. RP-HPLC data revealed that the natural peptide sequence (R1=H) undergoes cleavage at multiple locations along the backbone leading to multiple smaller fragments after proteolysis. In direct contrast, quantitative transformation to a single fragment was observed for all N-alkylated pentapeptides (Figure 2, Figure S1). This is attributed to proteolysis of the unnatural series only occurring at the lone alanine residue at the C-terminus due to the N-alkyl group blocking cleavage at other sites along the backbone. In agreement with this observation, ESI-MS analysis of each reaction mixture demonstrated that the N-alkylated pentapeptides were cleaved at the last peptide bond resulting in a tetrapeptide, while avoiding two potential sites before and after the N-alkylated units (Figure 2). These results confirm that the N-modified unit inhibits the protease activity at those sites, presumably due to the hydrogen bond disruption of the amidic nitrogen substituent within the catalytic triad of the serine protease pocket, as was observed in prior studies for peptoids (N-substituted polyglycine).4 The ability to control peptide hydrolysis through N-alkylation of a site distant from the active center is of great interest especially when there is a need to maintain bioactivity. Interestingly, while all substitution patterns result in a change in site of cleavage, the rate of proteolysis is tunable, possibly reflecting steric differences in binding to elastase leading to variable substrate inhibition. In addition, since our N-modified sequence resembles the motif in proline-containing elastase inhibitor, Ale-Ala-Pro-Ala, it may exhibit similar (but tunable) conformational effects on elastase activity.26–30
Figure 2.

Schematic presentation of proteolysis upon N-alkylation and the observed fragments as measured by HPLC and ESI-MS. Orange arrows are the cleavage positions.
To further expand the scope of this system beyond N-modified building blocks, N-modified glycine and phenylalanine derivatives were also examined for tenability of proteolysis rates across different amino acid families (Figure 3). In the case of glycine (R2=H), a sixfold increase in half-life from the natural amino acid to the N-methylated derivative (Sarcosine) was observed, again reinforcing the increased protease resistance of N-methylation. Significantly, upon increasing substituent size and steric constraints through N-benzylation, the half-life of proteolysis in the presence of elastase decreased to less than 50% of the natural system. In contrast, a more hydrophilic N-substituent increased resistance dramatically with a half-life value similar to the N-methylated derivative (210 ± 40 min) (Figure S1-S3). This ability to tune reactivity was also observed for larger amino acid units (Figure 3).26 For phenylalanine in particular, R2=Bn, the effect of N-methylation is enhanced with 45 fold increase in stability and further N-benzylation did not decrease the half-life substantially. In this case, the sterically hindered N/Cα-disubstituted repeat unit (R1=Bn, R2=Bn) may significantly decrease enzyme-peptide interactions leading to a decrease in the rate of proteolysis. In conclusion, the effect of N-alkylation on tuning protease reactivity for short peptide sequences is reported. Through optimization of coupling agents and reaction conditions, a library of pentapeptides containing an N/Cα-disubstituted central unit were prepared and the effect of varying substituent size examined. Interestingly, all N-Me derivatives showed the highest stability to cleavage in the presence of elastase with a systematic decrease in stability with increasing steric size. The principles found in this study have potential for aiding the design of synthetic peptides and functional inhibitors with improved performance and/or clinical utility.
Figure 3.

Variation in half-life for the proteolysis of pentapeptides based on glycine, alanine, or phenylalanine derivatives with the N-substituent (R1) being modified from hydrogen (natural amino acid) to methyl and benzyl.
Supplementary Material
Acknowledgements
We acknowledge use of Materials Research Laboratory (MRL) Central Facilities supported by the National Science Foundation (NSF) through the Materials Research Science and Engineering Centers under Grant DMR 1720256. The authors thank the Defense Advanced Research Projects Agency (DARPA) #N66001–14-2–4055 Encode-Sort-Decode (ESD): Integrated System for Discovery of Non-Natural Affinity Reagents program for funding of this work. W.R.G. thanks the NIH for a postdoctoral fellowship (F32GM108323). A.A. is grateful to the European Union (EU) for a Marie Curie Postdoctoral Fellowship. A.A and R.K thank the California NanoSystems Institute for an Elings Prize Fellowship in Experimental Science R.K. is also an awardee of the Weizmann Institute of Science- National Postdoctoral Award for Advancing Women in Science.
Footnotes
Conflicts of interest
The authors have no conflicts to declare.
Notes and references
- 1.Fosgerau K and Hoffmann T, Drug Discov. Today, 2015, 20, 122–128. [DOI] [PubMed] [Google Scholar]
- 2.Otvos LJ and Wade JD, Chem. Biol, 2014, 2, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henninot A, Collins JC and Nuss JM, J. Med. Chem, 2018, 61, 1382–1414. [DOI] [PubMed] [Google Scholar]
- 4.Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM and Moos WH, Drug Dev. Res, 1995, 35, 20–32. [Google Scholar]
- 5.Kirshenbaum K, Barron AE, Goldsmith RA, Armand P, Bradley EK, Truong KTV, Dill KA, Cohen FE and Zuckermann RN, Proc. Natl. Acad. Sci, 1998, 95, 4303–4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee J, Rechenmacher F and Kessler H, Angew. Chem. Int. Ed, 2013, 52, 254–269. [DOI] [PubMed] [Google Scholar]
- 7.Garton M, Nim S, Stone TA, Wang KE, Deber CM and Kim PM, Proc. Natl. Acad. Sci, 2018, 201711837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laursen JS, Engel-Andreasen J and Olsen CA, Acc. Chem. Res, 2015, 48, 2696–2704. [DOI] [PubMed] [Google Scholar]
- 9.Werner HM, Cabalteja CC and Horne WS, ChemBioChem, 2016, 17, 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A and Kessler H, Angew. Chem. Int. Ed, 2008, 47, 2595–2599. [DOI] [PubMed] [Google Scholar]
- 11.Pels K and Kodadek T, ACS Comb. Sci, 2015, 17, 152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernández-Llamazares AI, García J, Soto-Cerrato V, Pérez-Tomás R, Spengler J and Albericio F, Chem. Commun, 2013, 49, 6430–6432. [DOI] [PubMed] [Google Scholar]
- 13.Gao Y and Kodadek T, Chem. Biol, 2013, 20, 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiśniewski K, Galyean R, Tariga H, Alagarsamy S, Croston G, Heitzmann J, Kohan A, Wiśniewska H, Laporte R, Rivière PJ-M and Schteingart CD, J. Med. Chem, 2011, 54, 4388–4398. [DOI] [PubMed] [Google Scholar]
- 15.Paradís-Bas M, Tulla-Puche J and Albericio F, Chem. Soc. Rev, 2016, 45, 631–654. [DOI] [PubMed] [Google Scholar]
- 16.Fernández-Llamazares AI, Spengler J and Albericio F, Pept. Sci, 2015, 104, 435–452. [DOI] [PubMed] [Google Scholar]
- 17.Kaminker R, Kaminker I, Gutekunst WR, Luo Y, Lee S-H, Niu J, Hawker CJ and Han S, Chem. Commun, DOI: 10.1039/C8CC01356J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura C, Inuyama Y, Goto H, Obataya I, Kaneko N, Nakamura N, Santo N and Miyake J, Anal. Chem, 2005, 77, 7750–7757. [DOI] [PubMed] [Google Scholar]
- 19.Fu J, Larini L, Cooper AJ, Whittaker JW, Ahmed A, Dong J, Lee M and Zhang T, PLOS ONE, 2017, 12, e0182847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lampel A, McPhee SA, Park H-A, Scott GG, Humagain S, Hekstra DR, Yoo B, Frederix PWJM, Li T-D, Abzalimov RR, Greenbaum SG, Tuttle T, Hu C, Bettinger CJ and Ulijn RV, Science, 2017, 356, 1064–1068. [DOI] [PubMed] [Google Scholar]
- 21.Gross S, Lennerz V, Gallerani E, Mach N, Böhm S, Hess D, von Boehmer L, Knuth A, Ochsenbein A, Gnad-Vogt U, Forssmann U, Woelfel T and Kaempgen E, Cancer Immunol. Res, 2016, 4, 18–25. [DOI] [PubMed] [Google Scholar]
- 22.Rufo CM, Moroz YS, Moroz OV, Stöhr J, Smith TA, Hu X, DeGrado WF and Korendovych IV, Nat. Chem, 2014, 6, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tenidis K, Waldner M, Bernhagen J, Fischle W, Bergmann M, Weber M, Merkle M-L, Voelter W, Brunner H and Kapurniotu A, J. Mol. Biol, 2000, 295, 1055–1071. [DOI] [PubMed] [Google Scholar]
- 24.Tamada T, Kinoshita T, Kurihara K, Adachi M, Ohhara T, Imai K, Kuroki R and Tada T, J. Am. Chem. Soc, 2009, 131, 11033–11040. [DOI] [PubMed] [Google Scholar]
- 25.El-Faham A, Funosas RS, Prohens R and Albericio F, Chem. – Eur. J, 2009, 15, 9404–9416. [DOI] [PubMed] [Google Scholar]
- 26.Theillet F-X, Kalmar L, Tompa P, Han K-H, Selenko P, Dunker AK, Daughdrill GW and Uversky VN, Intrinsically Disord. Proteins, 2013, 1, e24360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butterfoss GL, Yoo B, Jaworski JN, Chorny I, Dill KA, Zuckermann RN, Bonneau R, Kirshenbaum K and Voelz VA, Proc. Natl. Acad. Sci, 2012, 109, 14320–14325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sui Q, Borchardt D and Rabenstein DL, J. Am. Chem. Soc, 2007, 129, 12042–12048. [DOI] [PubMed] [Google Scholar]
- 29.Bode W, Meyer E and Powers JC, Biochemistry (Mosc.), 1989, 28, 1951–1963. [DOI] [PubMed] [Google Scholar]
- 30.Thompson RC and Blout ER, Biochemistry (Mosc.), 1973, 12, 44–47. [DOI] [PubMed] [Google Scholar]
- 31.Avbelj F, Grdadolnik SG, Grdadolnik J and Baldwin RL, Proc. Natl. Acad. Sci, 2006, 103, 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.