

Abstract
A central goal in biochemistry is to explain the causes of protein sequence, structure, and function. Mainstream approaches seek to rationalize sequence and structure in terms of their effects on function and to identify function’s underlying determinants by comparing related proteins to each other. Although productive, both strategies suffer from intrinsic limitations that have left important aspects of many proteins unexplained. These limits can be overcome by reconstructing ancient proteins, experimentally characterizing their properties, and retracing their evolution through time. This approach has proven to be a powerful means for discovering how historical changes in sequence produced the functions, structures, and other physical/chemical characteristics of modern proteins. It has also illuminated whether protein features evolved because of functional optimization, historical constraint, or blind chance. Here we review recent studies employing ancestral protein reconstruction and show how they have produced new knowledge not only of molecular evolutionary processes but also of the underlying determinants of modern proteins’ physical, chemical, and biological properties.
Keywords: ancestral reconstruction, evolutionary biochemistry, vertical analysis, epistasis, historical contingency
1. STRATEGIES TO REVEAL SEQUENCE–STRUCTURE–FUNCTION RELATIONS
1.1. Functionalist Biochemistry
The quest to understand why proteins have their particular sequences, structures, and functions lies at the heart of both protein biochemistry and molecular evolution. Some early structural biologists were interested in how evolution produced modern proteins (60, 64, 83), but evolutionary analysis never entered the mainstream of the field. Instead, the major program in protein biochemistry has been to rationalize and explain protein sequence and structure by how they enable biological function. Francis Crick (17, p. 150) famously asserted, “If you want to understand function, study structure,” and Zuckerkandl & Pauling (83, p. 97) wrote that the search for molecular explanations of biological phenomena were “what counts most in the life sciences today.” In this framework, protein structure is the proximate cause of biological function, and function, in turn, explains why any protein structure exists in its particular form.
This functionalist mindset has been enormously productive. First, it has advanced the reductionist program of explaining biological phenomena in terms of the properties and laws of the physical objects that underlie them. Second, it distills the extraordinary complexity of a protein— thousands of atomic interactions in a dynamic three-dimensional topology—down to the much smaller set of features that “matter” for its specific function. For example, the atomic structure of the potassium channel explains how it can be permeable to only potassium ions and not the smaller sodium ions: backbone atoms in the so-called selectivity filter are positioned to exactly replace the hydration shell of potassium but not sodium (22). The channel’s complex structure is thus conceptually reduced to the useful abstraction of a precisely sized electrostatic pore that acts as a surrogate hydration shell across an apolar barrier.
But functionalist biochemistry has some limitations, which can be addressed in large part by incorporating evolutionary analysis into protein biochemistry. First, many aspects of protein structure cannot be explained by their contribution to biochemical function. For example, certain functionally defined groups of proteins—the carbonic anhydrases (51), alcohol dehydrogenases(25), and serine proteases (24)—contain members that have the same biochemical activity but very dissimilar overall structures because they evolved independently from different ancestral proteins. The functionalist paradigm provides no way to explain these structural differences among proteins that have the same function, because it excludes as inexplicable and irrelevant any properties of a protein that cannot be causally linked to its functions.
A second limitation is that functionalism implicitly assumes that all aspects of proteins have been optimized to perform their functions. This assumption may often be false and excludes many questions and phenomena that may be interesting in their own right, including how the physical architecture of a protein may constrain, drive, or allow nonoptimal forms. Decades of work in evolutionary biology and biochemistry have shown that a protein’s sequence, structure, stability, affinity for ligands, and other physical properties can and do drift dramatically along many degrees of freedom, as long as they remain compatible with the action of purifying selection (10, 32, 39, 47). Further, they often reflect the constraints imposed by their evolutionary history and the limits of “tinkering” to produce the optimal form (42). In organismal biology, a similar functionalist assumption was replaced long ago by a search for explanations that supplement functional optimization with historical analysis and the importance of stochastic factors (34). In biochemistry and biophysics, the assumption of functional optimality has lingered, largely because the evolutionary history of proteins cannot be directly examined in the same way that changes in organismal features can be traced in the fossil record.
A third limitation is that the functionalist approach often struggles to answer its own ultimate question: How does a protein’s sequence encode its structure and, in turn, its biochemical function? In its unbounded form, this question is unanswerable. Our knowledge of biophysics is inadequate for us to explain or predict from first principles how a protein’s structure and biochemical activities will emerge from its particular primary sequence. We might try to approach this problem empirically, mapping the association of possible sequences with possible functions, but the space of protein sequences and functions is far too vast. There are simply too many degrees of freedom in sequence, structure, and function to identify the causal links among these phenomena in abstract terms. But it is tractable to ask instead how evolutionary divergence in sequence from a common ancestral protein caused structure and function to diverge, thus producing specific distinct properties of modern proteins.
1.2. Comparative Biochemistry: Horizontal Analysis of Protein Diversity
Comparative analysis of proteins partially addresses this third limitation by asking how differences in the biochemical functions of two related proteins are caused by differences in their sequences and structures. A protein of interest is compared with at least one homologous protein that has identifiable similarity in sequence and structure but a distinct function. To identify the causal sequence differences for the functional variation, amino acid states in one protein are replaced with the corresponding states from the other. If residue swaps can be identified that switch the function to that of the homolog, they can be mapped onto the structure to identify structural elements that confer the difference in function (18, 48).
In practice, horizontal swap experiments frequently fail to identify sequence differences that are necessary and sufficient for functional differences (31, 50). There are two major reasons. First, horizontal comparisons are inefficient: They must address all sequence differences between the homologs, which reflect all the changes that occurred along the lineages from the last common ancestor to the present-day proteins. Many or most of these changes had nothing to do with the acquisition of the functional difference of interest and occurred during different temporal intervals (Figure 1). Even a moderate increase in the number of sequence differences results in an astronomical increase in the number of experiments necessary to characterize their functional effects and interactions (28). This makes horizontal comparisons particularly ill-suited for functions or phenomena—such as differences in conformational dynamics or protein stability—for which a small set of obvious candidate substitutions is not readily identifiable a priori.
Figure 1.
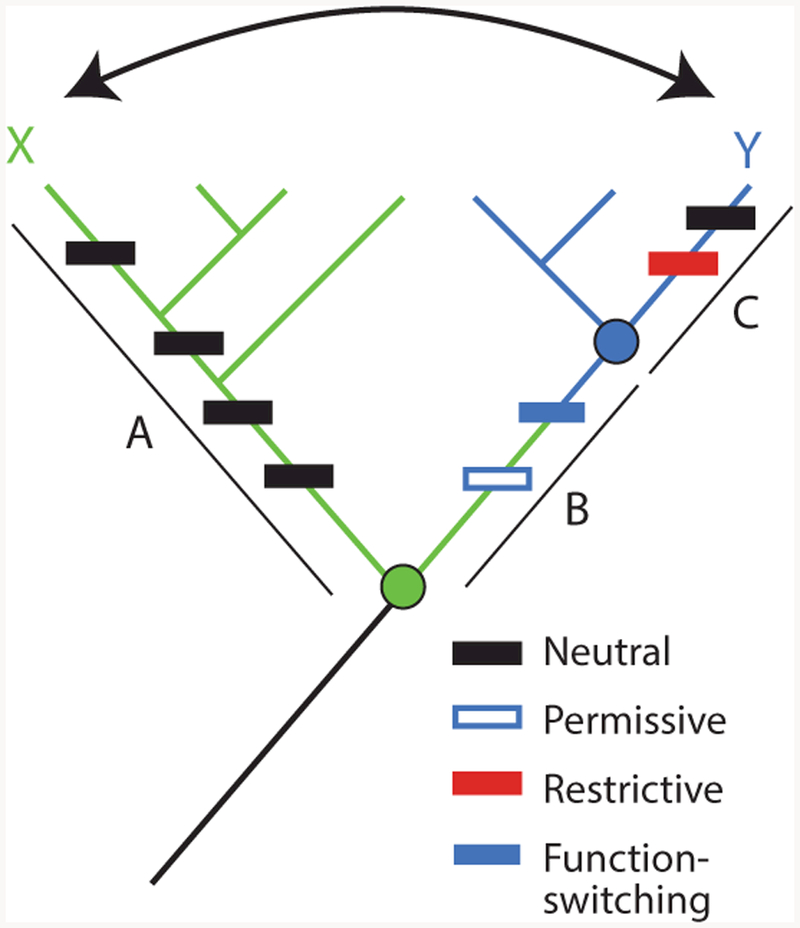
Horizontal and vertical analysis of sequence–function relations. To identify the sequence differences that confer different functions ( green or blue) between paralogous proteins X and Y, a horizontal comparison (arrow) would include all sequence changes that occurred on branches A, B, and C (rectangles, colored by their functional and epistatic effects). Permissive substitutions in isolation do not affect function but allow the protein to tolerate function-switching changes; horizontally swapping function-switching residues from Y into X would yield a nonfunctional protein, because it lacks the permissive substitution. Restrictive substitutions make the ancestral state at the function-switching sites deleterious; swapping these residues from X into Y would also yield a nonfunctional protein. Vertical analysis would determine the function of ancestral nodes (circles, colored by their functions) and isolate the change in function to branch B, reducing the number of changes to consider and minimizing the confounding effect of epistasis.
The second problem is that horizontal comparisons often produce nonfunctional proteins (52) because of epistasis—a genetic term that refers to context-dependence, such that the phenotypic effects of a mutation depend on the genetic state at other sites (69). A horizontal swap will yield a nonfunctional protein if an amino acid state (or set of states) from one homolog is incompatible with the sequence background of the homolog into which it is introduced. This may occur either because permissive residues required for the state to be tolerated are absent from the receiving homolog or because restrictive residues that prevent it from being tolerated are present (Figure 1) (9, 13, 59). In either case, sequence differences that are in fact causal factors for a functional difference cannot be identified because their effect is masked by the presence or absence of other residues that modify their functional effects.
1.3. Historical Biochemistry: Vertical Evolutionary Analysis Using Ancestral Reconstruction
These limitations of functionalism and horizontal comparison can be addressed by explicitly analyzing evolution vertically by reconstructing how a protein family’s sequence, structures, and functions changed over time. The idea that ancestral proteins could be reconstructed and characterized was first proposed in the 1960s (60). With the development of statistical phylogenetic methods, it became possible to infer the sequences of extinct proteins from the sequences of their extant descendants (82). Then, as gene synthesis methods improved, it became possible to express reconstructed ancestral proteins and experimentally characterize their biological and biochemical properties (30, 68, 72).
An experimental strategy for dissecting the mechanisms underlying historical protein evolution has been built on this foundation. By reconstructing successive ancestors on a protein family phylogeny, a shift in function and/or structure can be isolated to a specific branch, which represents a discrete interval of evolutionary time (Figure 2). Candidate causal sequence changes that may have conferred these shifts are the ones that occurred on this branch; their effects can be tested experimentally by introducing them into the reconstructed protein. This approach dramatically reduces the number of sequence differences between proteins with divergent functions, making it much easier to identify the function-switching set of residues and the structural elements they affect. It also reduces the effect of epistasis, because historical substitutions are reintroduced into ancestral sequence backgrounds identical or very similar to those in which they actually occurred. And vertical analysis also allows all features of sequence and structure to be investigated, irrespective of whether they were caused by functional optimization, historical contingency, or tinkering with ancestral forms.
Figure 2.
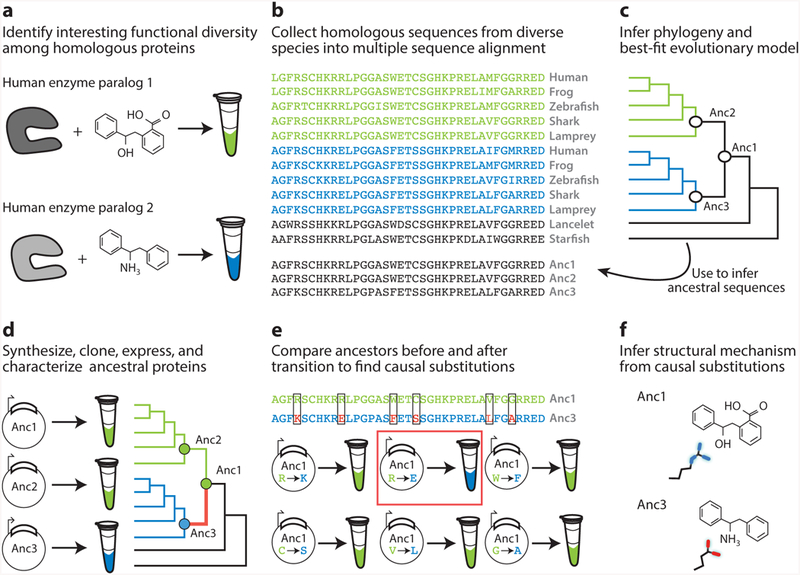
Workflow for vertical analysis of the genetic and structural causes of functional differences between related proteins, shown for a hypothetical family of enzymes. (a) Two paralogous enzymes catalyze similar reactions on different substrates, yielding different products (colors). (b) Sequences of both paralogs ( green and blue) are collected and aligned from many species, including outgroups (black). (c) The alignment is used to computationally infer the best-fit evolutionary model and a phylogeny. Ancestral sequences are inferred by maximum likelihood at nodes representing the last common ancestor of each paralog group (Anc2, Anc3) and at the gene duplication ancestral to both groups. (d ) DNA sequences coding for ancestral proteins are synthesized and cloned; ancestral proteins are expressed and their functions experimentally characterized. This allows the branch on which a new function evolved (red ) to be identified. (e) The substitutions that conferred the derived (blue) function must be among the differences between Anc1 and Anc3 (boxed sites). To identify causal substitutions, amino acid states from Anc3 (red states in blue sequence) are introduced into Anc1 and the resulting proteins tested experimentally (bottom). In the example, an arginine to glutamate substitution (red box) recapitulates the switch in specificity. ( f ) Structures or homology models of ancestral proteins are determined to infer the mechanism by which causal substitutions conferred the new function. In this case, the derived glutamate of Anc3 satisfied the hydrogen bonding potential of the amine group unique to the derived ligand.
Ancestral sequence reconstruction (ASR) begins with some form of biologically or structurally interesting diversity, the evolution and biochemical determinants of which we would like to understand (Figure 2a). Modern ASR requires a sequence alignment, a probabilistic model of evolution, and a phylogenetic tree that describes the relationships among the sequences (71) (Figure 2b,c). Probabilistic models of evolution specify the equilibrium frequencies of all amino acid states and the relative rates of all pairwise exchanges among them (26); more complex versions are mixture models that specify distributions of variable substitution rates among sites (80), or even different exchange rates or state frequencies (70). A large number of such models are available, so the typical strategy is to assess the fit of a set of candidate models to the aligned sequence data using statistical criteria and select the best-fitting option (1). The phylogenetic relationships among the sequences can then be inferred from the sequence data and model using maximum likelihood or Bayesian methods; alternatively, a well-corroborated phylogeny can be used if it is known a priori—as is often the case for the species from which the proteins have been extracted.
With the aligned sequences, model, and phylogeny in hand, ancestral sequences at every internal node of the tree can be statistically inferred using maximum likelihood or Bayesian approaches. Sites in the sequence are reconstructed individually; at any site, the likelihood of some ancestral amino acid state at a node of interest is defined as the conditional probability that all the observed data—the states in all extant proteins at that site in the alignment—would have evolved, given the ancestral state, the model, and the phylogeny. The posterior probability of each ancestral state is defined as the ratio of its likelihood (weighted by the prior probability of that state) to the sum of the prior-weighted likelihoods of all 20 possible amino acids. For each node on the tree, the output of the procedure is a list of the posterior probabilities of every possible sequence state at each site (35, 81, 82). The best estimate of the ancestral state is the maximum a posteriori (MAP) state, and the MAP ancestor—colloquially referred to as the maximum likelihood (ML) ancestor—is the string of MAP states. This ML sequence represents the single ancestral protein sequence with the highest probability, given all the observed sequence data, the model, and the phylogeny. DNA molecules that code for the reconstructed ancestral protein sequence can then be synthesized and cloned, and the proteins can be expressed in the appropriate systems for biological and/or biochemical analysis (Figure 2d ).
There are usually some sites that are inferred ambiguously, with two or more amino acid states having nonnegligible posterior probabilities. Rather than providing a single reconstruction of the ancestral protein, ASR should therefore be thought of as providing an ensemble of plausible ancestral sequences, with the MAP sequence as the single best estimate. Exploring the robustness of functional inferences to uncertainty about the ancestral protein’s precise sequence is therefore a particularly important part of the process. This is typically accomplished by experimentally characterizing alternate reconstructions that are less likely than the maximum likelihood sequence but still statistically plausible (7, 57, 74). A study of several protein families showed that the qualitative functions of reconstructed proteins—but not always the precise values of quantitative parameters—appear to be quite robust to uncertainty about the precise ancestral sequence, apparently because the sites and states that are ambiguously reconstructed are weakly constrained by function, whereas strongly constrained states are reconstructed with confidence (23).
In the following sections, we discuss recent studies that use ASR, with particular attention to the ways that vertical analysis can address some of the limitations of other modes of evolutionary and biochemical analysis. We highlight new insights that these studies provide into sequence– structure–function relations and their evolution.
2. EVOLUTIONARY ANALYSIS NARROWS SEQUENCE AND STRUCTURAL SEARCH SPACE
One advantage of vertical evolutionary analysis is its power to identify the specific sequence determinants that confer particular differences in function between related proteins. This approach has allowed structural mechanisms for function to be revealed that would otherwise have remained cryptic.
2.1. Structural Mechanisms of Action for a Cancer Drug
A study of the sensitivity of protein targets to Gleevec, an anticancer drug, provides a compelling example (78). Gleevec inhibits the kinase Abl by occupying Abl’s active site (58); it does not efficiently inhibit the structurally similar kinase Src. Gleevec’s selectivity for Abl is clinically important, but its structural basis remained a mystery for decades despite considerable effort. Abl binds Gleevec more tightly than Src does, primarily because an induced-fit step after the initial binding event reverses rapidly in Src but is much slower to reverse in Abl, leading to a slower off-rate and higher affinity (2). The structures of Abl and Src are very similar, except that a loop in Abl folds over Gleevec but occupies a different conformation in Src (Figure 3a, subpanel i ). Swapping residues within the loop between the two proteins did not affect Gleevec binding (66), so the loop conformation was deemed unimportant for affinity. The large number of sequence differences throughout the two proteins—and their structural similarity—frustrated further attempts to identify the physical basis for the difference in Gleevec binding (Figure 3a, subpanel ii ).
Figure 3.
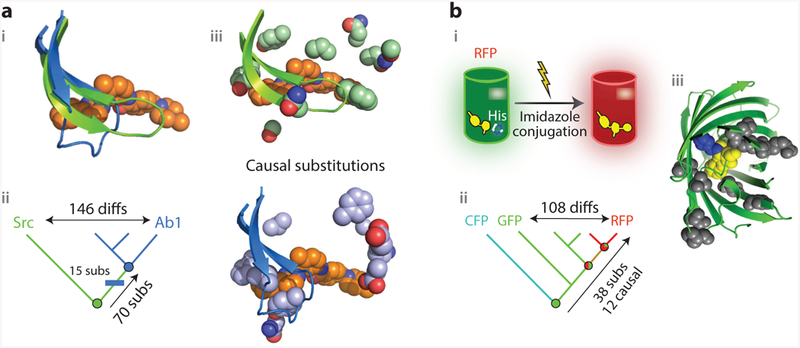
Vertical analysis has revealed genetic and structural mechanisms for the evolution of new functions. (a) Evolution of sensitivity to the inhibitor Gleevec in two related kinases (78). (i ) Superposition of the active sites of Abl (blue), a kinase sensitive to the inhibitor Gleevec (orange spheres), and of Gleevec-insensitive kinase Src ( green). In Abl, a loop folds over Gleevec. (ii ) Vertical analysis isolated the origin of Gleevec-sensitivity to the branch between two reconstructed ancestors (circles, colored by sensitivity). Fifteen substitutions on this branch (blue rectangle) were sufficient to confer sensitivity when introduced into the deepest ancestor. (iii ) Position of causal residues in Src (top; PDB 2OIQ) and Ab1 (bottom; PDB 1OPJ). (b) Evolution of emission wavelength in red, green, and cyan fluorescent proteins of Faviina corals (46, 74). (i ) In RFP (cylinder), the imidazole group from a histidine residue unique to RFP is covalently incorporated into the chromophore ( yellow) during maturation of the protein, causing it to emit red light. (ii ) Vertical analysis showed that RFPs evolved from a green-emitting ancestor and pointed to 38 potential causal substitutions along that lineage; 12 of these, including the derived histidine, were sufficient to recapitulate the evolution of red fluorescence when introduced into the common ancestor ( green circle).(iii ) Structural location of the causal substitutions, plotted on the structure of the reconstructed common ancestor ( green cartoon; PDB 4DXN). Yellow, chromophore; blue, incorporated imidazole ring from histidine residue; gray, other causal residues, most of which are far from the chromophore and are thought to allow a conformational rearrangement necessary for imidazole incorporation (46). Abbreviations: CFP, cyan fluorescent protein; diffs, amino acid differences; GFP, green fluorescent protein; His, histidine side chain; RFP, red fluorescent protein; subs, substitutions.
A recent study solved this problem using vertical analysis. The authors reconstructed and characterized ancient proteins along the historical trajectories that Abl and Src took from their common ancestor. By focusing on the phylogenetic interval in which Gleevec sensitivity emerged, the authors were able to identify 15 sequence substitutions in the Abl lineage that confer Gleevec sensitivity when introduced into the ancestral protein (Figure 3a, subpanel ii ). Kinetic analysis showed that these sequence changes slow reversal of the induced-fit step and thereby increase affinity for the drug. X-ray crystallography of the reconstructed protein showed that these residues were distributed in the body of the protein near the base of the loop. The ancestral states at these sites, before Gleevec sensitivity evolved, participated in a network of contacts that apparently stabilized the entire binding pocket and thereby kept the loop in a straight conformation (Figure 3a, subpanel iii ), whereas the derived states disrupted these contacts, allowing the folded-over iii ). Thus ASR provided a well-supported account that linked evolutionary changes in protein sequence to changes in structure and onward to changes in kinetics and ultimately to changes in biological function that, together, explain a biomedically important aspect of functional diversity among present-day proteins.
2.2. Evolution of Fluorescence in Coral Proteins
A series of studies on the evolution of red fluorescence in coral proteins further demonstrates how isolating functional change to a specific phylogenetic interval can reveal the genetic and structural basis of complex functions. Corals in the order Faviina contain members with considerable diversity in the color of their fluorescent proteins (FPs). In all FPs, the chromophore is generated in an autocatalytic reaction in the core of the molecule. The basic chromophore emits green light; in red FPs, this chromophore undergoes an additional step in which it incorporates the imidazole of a nearby histidine side chain, causing it to emit red (Figure 3b, subpanel i ). Red FPs uniquely possess the histidine, but introducing that residue into a green FP is not sufficient to make it emit red (27).
Matz and colleagues (74) conducted vertical evolutionary analysis to understand the mechanistic basis for fluorescence color variation. Ancestral protein reconstruction showed that the last common ancestor of all the Faviina FPs was green; red fluorescence evolved gradually through a series of intermediate ancestral proteins that became progressively redder (Figure 3b, subpanel ii ). To capture the full transformation to red fluorescence, Field & Matz (28) focused on the combined set of branches leading from the last completely green ancestor to the red FP of the great star coral Monastrea cavernosa. Along this lineage, 37 substitutions occurred, compared to 108 differences between M. cavernosa and its closest green relative (Figure 3b, subpanel ii ). To identify the causal residues, Field & Matz created a library of shuffled proteins containing various combinations of ancestral or derived states at the 37 sites and statistically analyzed the association of each state with the emitted color. Twelve substitutions turned out to be necessary and sufficient to completely shift the wavelength to red when introduced into the ancestral background.
This genetic dissection allowed the structural mechanism to be revealed. Most of the causal sites were far from the chromophore (Figure 3b, subpanel iii ), and the only one with an obvious physical explanation was the histidine that is incorporated into the chromophore. Crystal structures of the ancestral protein with and without the red-shifting substitutions showed that these residues have virtually no effect on the overall geometry of the structure (46). But molecular dynamics simulations of these proteins suggested that the key substitutions allow a transient intermediate conformational step to be occupied that makes incorporation of the imidazole into the red chromophore possible; specifically, the chromophore must undergo a light-activated internal twist that organizes the surrounding functional groups properly relative to the histidine (45, 46). The derived residues at the 11 other key sites make the backbone around the chromophore flexible and enable this conformation to be accommodated (46). In the ancestral state, the backbone is more rigid and would clash with the twisted intermediate chromophore, so the imidazole cannot be incorporated, even if the histidine alone were present (45, 46).
The capacity to occupy multiple conformations is thought to be important for some present-day proteins’ functional mechanisms (8), but how that flexibility is encoded in protein sequence has often been unclear. Both the Gleevec and FP examples solve this problem and establish that a handful of substitutions far from a protein’s active site can cause dramatic differences in functional specificity through their effects on the protein’s conformational ensemble. Several other vertical analyses of protein evolution have suggested similar mechanisms for the evolution of functional variation (21, 36, 62).
3. EVOLUTIONARY ANALYSIS OVERCOMES AND REVEALS EPISTASIS
A second advantage of vertical evolutionary analysis is that it can overcome the obscuring effect that epistasis often has on sequence–structure–function studies that focus on present-day proteins. By introducing sequence changes into the historical background in which they occurred, ancestral protein reconstruction avoids epistatic interactions between key function-changing substitutions and other subsequently acquired sequence differences that may make the former incompatible with the derived protein or ineffective at changing its function. Vertical evolutionary analysis has therefore identified the structural and genetic basis for functional diversity in cases where horizontal comparisons have failed and revealed the ways that epistatic substitutions during history can dramatically change a protein’s capacity to evolve new functions.
3.1. Permissive and Restrictive Epistasis: Mechanisms of Hormone Specificity
Permissive mutations allow a protein to tolerate substitutions that would otherwise make it nonfunctional. Restrictive mutations make a protein unable to tolerate substitutions that otherwise did not make it nonfunctional (Figure 1). Either of these can undermine the capacity of horizontal comparative analysis to identify key function-switching mutations. Vertical analysis of glucocorticoid and mineralocorticoid receptor evolution (GR and MR, respectively) established that permissive and restrictive mutations played important roles in these proteins’ evolution and revealed the structural basis for these paralogs’ ligand specificity.
GR and MR are ligand-activated transcription factors related to each other by a gene duplication that occurred early in vertebrate evolution. MR’s major physiological ligands are aldosterone and deoxycorticosterone, but GR is activated by cortisol, which differs from mineralocorticoids by having a hydroxyl at the C17 position. Crystallographic studies of extant mammalian GR and MR proteins revealed differences in active-site geometry (50). At the end of a helix lining the active site, MR contains a serine, but GR contains a proline, a difference thought to change the position of the helix and result in a narrower active site in MR than in GR (Figure 4a). In GR, a conserved glutamine on that helix makes a specific contact with cortisol’s unique C17 hydroxyl, whereas MR’s leucine makes an apolar contact with its ligands at the same position (50). Horizontal swaps at those two positions produced nonfunctional receptors (50). At the time, this was taken to mean that these two sites are important to the receptors’ functions but not sufficient to change ligand specificity. As a result, hypothesized structural effects of the GR- and MR-specific states at those sites could not be directly tested.
Figure 4.
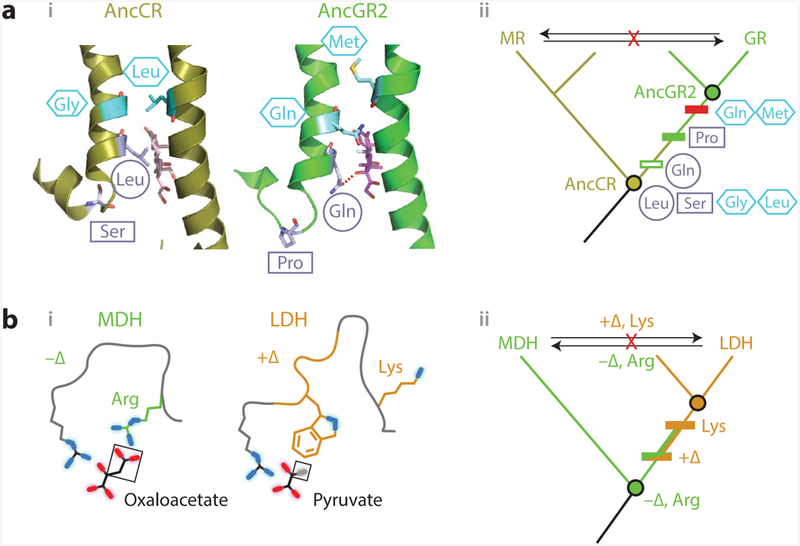
Vertical analysis has illuminated mechanisms of epistasis and functional change in protein evolution.(a) Evolution of hormone specificity in glucocorticoid and mineralocorticoid receptors. (i, left) Position of key helices in the crystal structure of AncCR, the ancestor of all MRs and GRs (olive; 2Q1H) with aldosterone bound (sticks); (right) position of helices in AncGR2, the ancestor of cortisol-specific GRs ( green; 3GN8), with cortisol bound (59). Sites that change specificity are shown as light blue sticks. The Ser–Pro substitution (rectangle) repositions one helix, allowing the Leu–Gln substitution (circle) to form a cortisol-specific hydrogen bond (dashed red line). Restrictive substitutions (hexagons) introduce residues into AncGR2 that would clash in the ancestral helix conformation (13). (ii ) Phylogeny showing the inferred historical order of the substitutions between AncCR and AncGR. Horizontally swapping key residues between paralogs (arrows) yields nonfunctional proteins. (b) Evolution of substrate specificity in apicomplexan malate and lactate dehydrogenases (11). (i ) Active-site geometry of extant DHs, with the key side chain and loop insertion (+Δ) highlighted in green and orange. Substrates (black lines, with oxygen atoms in red and methyl group in gray) are labeled; functional groups unique to each substrate are boxed. (ii ) Phylogeny and ancestral reconstruction showed that LDH function (orange) evolved from an MDH-like ancestor (green). Introducing the derived loop and Lysine residue into the deepest ancestor confers pyruvate specificity. Horizontal swaps of these features (arrows) failed to confer on either protein its paralog’s functional specificity. Abbreviations: AncCR, ancestral corticoid receptor; AncGR, ancestral glucocorticoid receptor; Arg, arginine; DH, dehydrogenase; Gln, glutamine; Gly, glycine; GR, glucocorticoid receptors; LDH, lactate dehydrogenase; Leu, leucine; Lys, lysine; MDH, malate dehydrogenase; Met, methionine; MR, mineralocorticoid; Pro, proline; Ser, serine.
When the last common ancestor of GRs and MRs was resurrected and characterized, it was found to have MR-like specificity and amino acid states at the two candidate sites (12) (Figure 4a). This indicated that GR’s specificity was derived, and the task became to understand how the MR-like ligand recognition of the ancestor was transformed into the GR’s specificity for cortisol. By reconstructing and characterizing successive ancestral proteins along the GR lineage, this shift was isolated to a specific interval of phylogenetic time—the same interval in which the two focal substitutions occurred. When the derived residues from the descendant GR were introduced into the ancestral mineralocorticoid-preferring protein, they not only could be tolerated but also conferred a new preference for cortisol (Figure 4a, subpanel ii ) (59). Crystal structures of the ancestral proteins showed how (59): The serine–proline substitution relocated the helix from its ancestral, MR-like state, reducing affinity for all ligands and also bringing the other site close to the ligand’s C17, where the leucine–glutamine substitution established a cortisolspecific hydrogen bond, restoring affinity for that ligand. Further analysis showed that five more substitutions from the same interval were sufficient to completely recapitulate the shift to cortisol specificity.
This analysis also revealed why the horizontal swap failed. After the functional transition, GRs acquired additional, restrictive substitutions that sterically clash with the ancestral helix conformation (Figure 4a) (13). This explained why extant GRs cannot tolerate the MR’s residues at the key sites. In addition, permissive substitutions, which occurred earlier in the GR lineage, were required to stabilize elements of the structure destabilized by the function-switching substitutions(38); this explained why extant MRs, lacking these states, could not tolerate the function-switching residues from the GR. Despite their profound effects on the evolvability of the proteins, none of these epistatically acting mutations had detectable effects on ligand preference when introduced on their own.
3.2. Obscuring Epistatic Mutations: Mechanisms of Enzyme Substrate Specificity
In the GR/MR case, epistatic substitutions obscured the effect of key historical mutations and made the mechanism for ligand specificity seem more complex than it actually was. A study of metabolic enzymes in apicomplexan parasites illustrates how vertical analysis can reveal structural mechanisms for function that because of epistasis are different from those that might be suggested by analysis of extant proteins (11).
Apicomplexan parasites possess two homologous 2-ketoacid oxidoreductases that have very similar structures and a common catalytic mechanism. One paralog, malate dehydrogenase (MDH), catalyzes the interconversion of oxaloacetate and malate, whereas lactate dehydrogenase (LDH) catalyzes the interconversion of pyruvate and lactate. Structural analysis and alanine scans suggested that the different activities might be due to two key differences—a single residue and one insertion/deletion—that result in complementary interfaces between each protein and its preferred ligand (11). Specifically, MDH binds its ligand using an arginine that makes electrostatic contacts to oxaloacetate’s charged carboxylate group (Figure 4b), whereas LDH has a lysine at this residue. LDH binds its ligand using a loop, absent from MDH, that contains a hydrophobic side chain that packs against the methyl group of pyruvate (Figure 4b, subpanel i ). Horizontal swaps of these two features did not switch the function: Inserting the loop and the lysine into an extant MDH failed to yield pyruvate activity, and removing the loop and substituting arginine into an extant LDH yielded only very weak oxaloacetate activity. These observations seemed to suggest that more than these two sequence features determine MDH and LDH substrate specificity (11).
Reconstructing ancestral dehydrogenases revealed that epistasis in the extant enzymes was in fact obscuring the function-switching effect of these features (Figure 4b, subpanel ii ). The last common ancestor of LDH and MDH was oxaloacetate-specific, just like extant MDH, and a crystal structure showed the ancient protein had the same active-site geometry. Inserting the loop and the derived lysine into this protein completely recapitulated the evolution of LDH function, conferring pyruvate activity and abolishing oxaloacetate activity. This result indicates that subsequent epistatic substitutions along the MDH lineage made the mechanism by which pyruvate binding was acquired in LDH ineffective at conferring pyruvate activity. Conversely, substitutions along the LDH lineage came to interfere with the ancestral mechanism of oxaloacetate activity, making it more difficult to restore that function.
By dissecting the effects of switching the loop and the arginine–lysine residue individually, Boucher et al. (11) found that the structural mechanisms of these features in the ancestral background were very different from what might be supposed from studying the extant proteins. First, the work revised the interpretation of the basis for LDH’s pyruvate activity. Inserting LDH’s loop into the extant MDH confers no pyruvate activity, irrespective of whether arginine or lysine is present at the other site. In the ancestral protein, however, inserting the loop yields strong pyruvate activity. This shows that the packing of the hydrophobic surface of the loop against pyruvate is in fact the key determinant of activity on pyruvate.
Second, the structural basis for MDH’s oxaloacetate activity turned out to be more subtle and interesting than it might have appeared. In the extant MDH, adding the loop alone dramatically reduced oxaloacetate activity, suggesting that its hydrophobic surface might be incompatible with oxaloacetate’s charged carboxylate group. In the ancestral protein, however, adding just the loop had no effect on oxaloacetate activity, indicating that the loop was perfectly compatible with that function, probably because the ancestral protein is flexible enough to rotate the loop away from the substrate. Further, replacing just the arginine with lysine in the extant MDH abolished oxaloacetate activity altogether, which might suggest an absolute requirement for the interaction between that side chain and the substrate’s carboxylate; in the ancestral protein, however, changing this residue only weakly affected oxaloacetate activity, indicating that the arginine interaction was in fact unnecessary. Because both the loop and the arginine must be changed to eliminate the ancestral function, oxaloacetate activity depends not on the absence of the loop or on the arginine– carboxylate interaction per se but rather on the protein’s capacity to occupy a conformation in which some positively charged residue can access the substrate, whether or not the loop insertion is present.
In both the GR/MR and MDH/LDH cases, the mechanism of functional specificity turned out to be simpler than horizontal analysis would have suggested, because subsequent epistatic substitutions were incompatible with those ancient features. Mechanisms that were once sufficient were no longer so; additional features had to be set back to their ancestral state for the determinants of function to be revealed. Several other studies using ancestral protein reconstruction have identified major functional transitions that can be recapitulated by just a few historical substitutions (5, 36), suggesting that discrete shifts in function may not be as difficult to evolve as has been proposed based on horizontal comparisons (3, 44, 73).
4. KNOWING ANCESTRAL STATES IMPROVES STRUCTURAL INTERPRETATION
When extant proteins are compared to each other horizontally, differences in sequence, structure, and function are not polarized in time: Which characteristics are ancestral and which are derived remains unresolved. Not knowing the directionality of functional change limits our ability to interpret sequence–structure–function relations. Sequence substitutions can change function in several possible ways: by conferring a new function, modifying an existing one, eliminating an ancestral function, or even restoring an ancestral function that had been lost. Only if the ancestral states are known can we tell these scenarios apart. Vertical evolutionary analysis polarizes these states and leads to clearer inferences concerning how sequence determines structure and how structure, in turn, determines function (Figure 5a).
Figure 5.
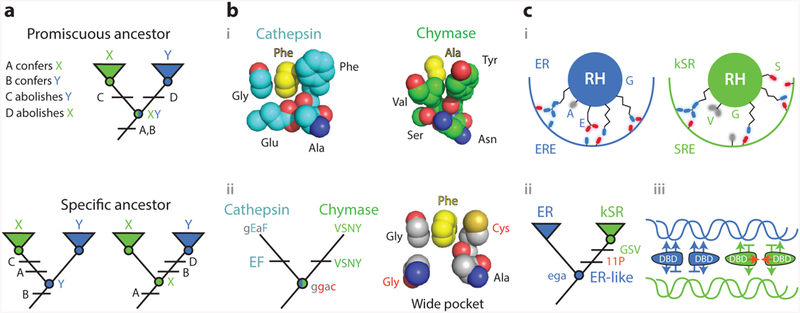
Knowledge of ancestral states clarifies structure-function mechanisms. (a) Simplified example of the implications of vertical analysis. Homologs with distinct functions X and Y can be generated by partitioning functions from a multifunctional ancestral protein (top) or by a discrete change in function from a specific ancestor (bottom). Different trajectories imply different effects of key sequence differences (A–D). (b) Mechanism of evolution of serine protease specificity. (i ) Specialized tight binding pockets of the extant serine proteases cathepsin (1CGH) and chymase (2RDL). (ii ) Their reconstructed last common ancestor had both activities and a wide binding pocket (79). Lower- and upper-case letters show ancestral and derived amino acid states for key residues, using the single-letter code. Ancestral states that confer the promiscuous wide pocket are highlighted in red. (c) Evolution of DNA specificity in steroid hormone receptors. Estrogen and ketosteroid receptors bind different DNA sequences (ERE and SRE). (i ) Schematic of the receptors’ recognition helices bound to the DNA major groove. Residues at variable sites are labeled. kSRs ( green) make fewer specific interactions than ERs (blue). (ii ) Vertical analysis showed that ERs and kSRs evolved from an ER-like ancestor (57). Specificity-switching substitutions (ancestral ega to derived GSV in single-letter code) and permissive substitutions (11P) are labeled.(iii ) Interactions that characterize ER/ERE (blue) and kSR/SRE ( green) complexes are shown, with favorable interactions as arrows and exclusionary interactions as horizontal lines. Permissive substitutions enhanced dimer formation and cooperativity of binding (red arrows) in kSRs. Abbreviations: Ala, alanine; Asn, asparagine; Cys, cysteine; DBD, DNA-binding domain; ER, estrogen receptors; ERE, estrogen response element; Glu, glutamic acid; Gly, glycine; kSR, ketosteroid receptors; Phe, phenylalanine; RH, recognition helice; Ser, seranine; SRE, steroid response element; Tyr, tyrosine; Val, valine.
4.1. Promiscuous Ancestors: Serine Protease Activity and Specificity
Serine proteases show how knowing ancestral states can clarify the functional significance of sequence and structural differences between homologs. These proteases specifically cleave peptides with a particular side chain immediately N-terminal of the scissile peptide bond; different proteases recognize different side chains at this position. Crystallographic evidence and horizontal mutagenesis showed that the scissile bond is precisely positioned relative to the catalytic triad because of specific contacts between the substrate’s side chain and a highly complementary binding pocket (Figure 5b, subpanel i ) (41, 75). This was taken to mean that the ability to cleave depends on a tight fit to the substrate, which stabilizes the transition state (40, 63).
It was therefore surprising when the last common ancestor of four clades of serine proteases, each with a different specificity, was reconstructed and shown to have broad substrate recognition. The ancestral protein, in fact, could cleave all the various substrates of its descendants, indicating that there was no gain of activity with any substrates during their divergence; rather, the evolutionary change in function along each lineage was the loss of activity against all but one ancestral substrate. Further, structural modeling of the ancestral enzyme showed that it had a wide binding pocket that could not achieve the kind of tight packing with any substrate that the extant proteins have with theirs (79) (Figure 5b, subpanel ii ). Precise positioning of the N-terminal side chain in the pocket is therefore unnecessary for protease activity. Instead, the effect of the tight geometrical complementarity between substrate and extant proteases is to exclude nonpreferred substrates, contributing not to the enzyme’s activity but to its specificity. Manipulation of the ancestral protein confirmed that a single historical substitution conferred substrate specificity on the ancestral protein by destabilizing the transition states of what became nonpreferred substrates, rather than stabilizing that of the preferred substrate.
A general insight from this work is that the primary effect of structural differences between related proteins may often be to exclude potential substrates, including those that were bound by multifunctional ancestors, rather than to enhance the protein’s primary function. Several other historical trajectories leading from multifunctional ancestors to specialized proteins have now been documented using vertical analysis (16, 21, 76). Negative interactions that exclude certain ancestral substrates feature prominently in these examples, although novel positive interactions with the preferred substrate sometimes contribute to narrower derived specificities (16, 76).
4.2. Specific Ancestors: Mechanisms for Transcription Factor/DNA Recognition
The above example shows how vertical evolutionary analysis is uniquely able to identify the underlying determinants of function when the ancestral protein was multifunctional. The same is true when paralogs with different functions descend from a specific ancestor (Figure 5a). In such cases, the key sequence and structural differences confer the derived function and abolish the ancestral one. Without knowing the ancestral state, however, there is no way to know which function is affected in which way. Because the sequence states that transform or abolish an ancestral function are usually different from those that confer it in the first place, horizontal analysis is therefore prone to uncertainty in its causal inferences.
A recent study of the evolution of steroid hormone receptors’ DNA-binding domains (DBDs) illustrates this point. Steroid receptors bind as dimers to DNA response elements that are palindromic repeats of specific half sites. The two major clades of receptors differ in their half-site specificity: estrogen receptors (ERs) bind to estrogen response elements (EREs), which differ at two nucleotides from the half site bound by their sister clade, the ketosteroid receptors (kSRs), which bind to steroid response elements (SREs). Horizontal comparison of crystal structures of extant DBDs from both clades had shown that ERs make base-specific hydrogen bonds to EREs that are absent from the kSR–SRE interaction (65), and these differences are mediated by sequence differences at three sites in a recognition helix (RH) that lies in the DNA’s major groove (Figure 5c, subpanel i ). Substituting the ER states at those sites into an extant kSR conferred ERE specificity (19), which was taken to mean that these states and the contacts they make are the primary causes of ERE recognition.
Reconstruction of the ancestral DBD, however, showed that the primordial function was to specifically bind EREs. The determinants of ERE binding must therefore be more ancient than could be revealed in a comparison of ER and kSR. Instead, the evolutionary difference in function between the two groups must be caused by states in the kSR, which actively abolished ERE binding and allowed SRE recognition (Figure 5c, subpanel ii ) (57).
Biochemical, crystallographic, and molecular dynamics analyses of the ancestral proteins with and without the key substitutions revealed two major features of the mechanisms that drive specific DNA recognition in these proteins (6, 57). First, the primary means by which the key substitutions reduced the ancestral ERE affinity was not by abolishing positive contacts but by establishing new negative interactions against ERE, including a steric clash and unpaired hydrogen bond donors in the DBD–DNA interface. Second, they improved affinity for SRE without adding any new positive interactions; rather, they relieved negative interactions—particularly a major steric clash—between the ancestral side chains and the SRE DNA. Thus, specificity was achieved primarily through the gain and loss of negative, exclusionary interactions rather than through the gain and loss of favorable interactions.
The vertical analysis revealed one more important structural feature of SRE recognition. When the three recognition helix substitutions were introduced into the ancestral background, they fully recapitulated the dramatic shift in the DBD’s relative preference for SRE over ERE, but they yielded a protein with affinity too low to activate transcription effectively from either binding site. Further experiments revealed that 11 other permissive substitutions occurred during the same historical interval that increased affinity for DNA in a nonspecific fashion; they therefore allowed the three key preference-switching substitutions to be tolerated. These permissive changes, some of which primarily affected the interface for dimer formation in the DBD, increased the cooperativity of dimeric binding to the palindromic repeat, while others apparently affected nonspecific binding to the DNA backbone (Figure 5c, subpanel iii ).
Taken together, these experiments indicated that the energetics of binding were redistributed through the protein-DNA complex during the evolution of SRE specificity. Changes in cooperativity, conferred by substitutions near the dimeric interface, permitted changes in specificity to be triggered by substitutions at the interface to DNA. This rather nonparsimonious evolutionary pathway was required because the shift in specificity was conferred by the gain and loss of negative, exclusionary interactions at the DNA interface, without acquiring any sequence-specific positive contacts. McKeown et al. suggested that this complex mechanism may reflect the fact that there are likely to be more mutational opportunities to gain and lose unfavorable contacts than to create favorable ones (57). None of these inferences would have been possible if the functional, structural, and sequence states had been polarized incorrectly or not at all.
4.3. PyrR Transcription Factors: Mechanisms for Forming Dimers and Tetramers
A final instructive example of the importance of determining ancestral states for structural interpretation comes from a study of homologs in the PyrR family of transcription factors. Some of these proteins form dimers; others make tetramers. It might seem reasonable to assume that the tetramer is the derived state in this comparison, because dimers are topologically simpler than tetramers and complexity is often assumed to be derived from simpler progenitors (49). By that assumption, structural differences between dimers and tetramers would provide information about the structural determinants required to form the tetrameric interface.
But when the last common ancestor of dimeric and tetrameric PyrRs was reconstructed, it was a tetramer, indicating that dimers are the derived state (61). Crystal structures of the ancestral proteins showed that the substitutions that converted a tetramer into a dimer did not affect the sites that compose the tetrameric interface. Instead, they changed the geometry among the subunits so that they could no longer be arranged into a tetramer.
This work revealed that interfaces can be broken during evolution through long-range effects within a protein, experimentally confirming an earlier theoretical prediction about the geometric requirements for oligomerization (61). The underlying causes of tetramer formation cannot be revealed by a horizontal comparison of PyrR proteins, because the determinants that made the intersubunit angles tetramer-compatible in the first place did not necessarily involve the same sites and structural features that made them tetramer-incompatible later. Further, a key aspect of evolving the ancient tetramer must have been the acquisition of multiple complementary surfaces for high-affinity binding among subunits, and these were not directly affected by the evolutionary transition from tetramer to dimer. Thus, the vertical analysis was crucial to identify the structural mechanism that confers the differences between dimer- and tetramer-forming family members and to determine whether those features are sufficient to confer higher-order complex formation or whether they are sufficient to prevent it.
5. EVOLUTIONARY ANALYSIS EXPLAINS STRUCTURAL FEATURES THAT ARE NOT FUNCTIONALLY OPTIMAL
Natural selection has great power to optimize proteins, just as it does for biological entities at higher levels of organization (20), but this does not mean that all features of such systems are adaptations that have been driven to their optimal forms by selection for their present-day functions (34). Some are the result of chance events and historical constraint. Knowledge of historical trajectories can help identify and explain features of proteins that are not the result of functional optimization.
5.1. Complexity of a Molecular Machine: Vacuolar ATPases
Molecular systems often change in complexity over time. For example, protein complexes often gain new subunits through gene duplication (56), and the new subunits become obligate members of the complex. Complexity is often thought to increase because it enhances function (56), but this contention is difficult to test decisively because a required part of a complex may become necessary for it to work later even if it did nothing to improve the function when it first evolved.
Vertical evolutionary analysis makes the problem tractable because it reveals the mechanism by which new components are incorporated into a system and then become essential. One study addressed this question by using ASR to dissect the causes of changes in the subunit composition of a six-membered ring in an ATPase that pumps protons across vacuolar membranes (29). In most animals, the ring is made of five subunits of one type and one of another (Figure 6a, bottom); in Fungi, the protein contributing five subunits duplicated and diverged, resulting in a ring with three obligate protein types, each of which occupies specific positions relative to the others (Figure 6a, top).
Figure 6.
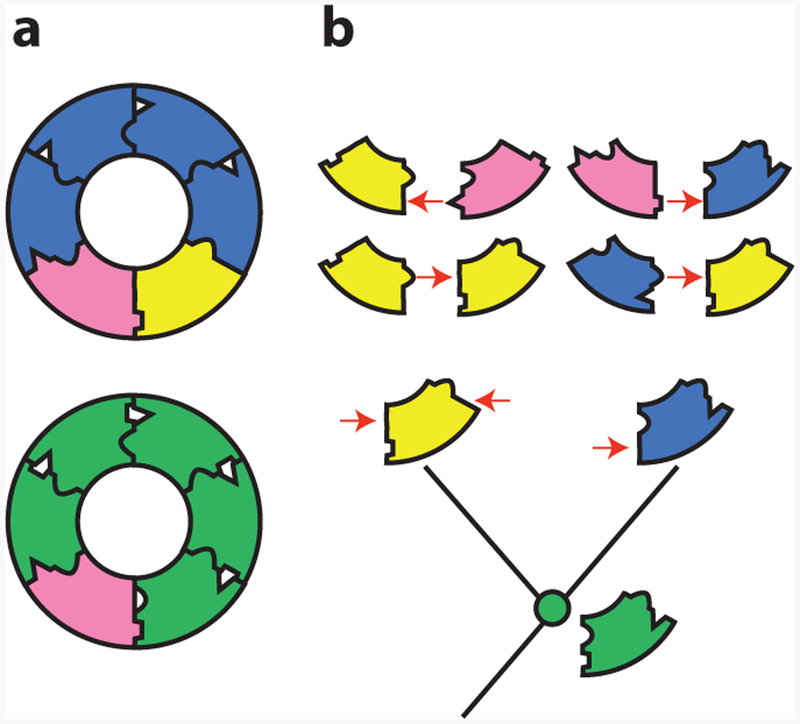
A neutral increase in complexity in a molecular machine (29). (a) Structure of the transmembrane ring of the vacuolar ATPase of fungi (top) and animals (bottom). The fungal ring contains three unique obligate subunits, which occupy specific relative positions. The animal ring contains only two subunits. (b) Evolution of paralogous subunits in yeast. (Bottom) Two subunits in the fungal ring (blue and yellow) are paralogs duplicated from one ancestral subunit ( green). The reconstructed ancestral subunit can form all required interfaces and reconstitute a functional ring in extant Fungi (when expressed with the pink subunit). The duplicated subunits became required because they lost specific interfaces to other subunits in a complementary fashion (red arrows). They thus could occupy only a subset of the ancestral positions relative to other subunits.
Vertical analysis showed that this increase in complexity evolved through degenerative processes that did not confer any new functions on the ring (Figure 6b). The authors reconstructed the ancestral ring proteins before and after the duplication and studied their capacity to form a functional ATPase. They also used fusion constructs to discern which positions in the ring each protein could occupy. The ancestor from before the duplication could replace both of its descendants, and it could occupy any of the ring positions, forming interfaces with itself or with the other component; even in present-day yeast, the ancestral two-member ring could carry out all the functions of the more complex extant version.
Retracing the subunits’ evolution showed that soon after the duplication, each of its daughters lost the capacity to form some of the ancestral interfaces. As a result, they could occupy only specific positions in the ring, and both became necessary to form a complete ATPase and carry out the ancestral functions (Figure 6b, top). The increase in complexity was therefore not driven by the optimization of the ring’s function but by a kind of molecular bureaucratization: after duplication, proteins “forgot” how to carry out some of the functions of their ancestors and became increasingly specialized. Remarkably, a single historical substitution from the lineages leading to each daughter protein was sufficient to cause the complementary loss of ancestral interfaces and confer the requirement for the more complex ring.
Neutral subfunctionalization of protein–protein interfaces also caused an irreversible increase in the complexity of a transcriptional network in yeasts (7). Similar complexity ratchets may have been important in the evolution and retention of eukaryotic protein complexes that contain more genetically different subunits than their bacterial homologs do (55). Naively, this observation might be interpreted in light of their higher cellular and regulatory complexity, but it may in fact reflect just how easy it is to compromise protein–protein interfaces.
6. THE FUTURE OF RETRACING THE PAST FOR STRUCTURAL BIOLOGY
The studies we have discussed here illustrate how vertical analysis of evolution through time can deepen our understanding of sequence–structure–function relationships in present-day proteins. We anticipate that the advantages of ASR will secure it an important place in the tool kit of structural biologists and biochemists. To date, the strategy has been employed primarily to answer questions about molecular evolution (36, 77), rather than to address questions about protein biochemistry per se (but see 39 and 78). Future work can employ ASR to reveal the mechanisms that cause functional diversity in specific protein families, as many of the studies reviewed here did. In addition, many more general phenomena in protein biophysics—such as protein conformational plasticity and fractionally populated excited states (8)—seem ripe to benefit from vertical analysis.
ASR can help to reveal how these phenomena have been conferred by sequence differences and to test the hypothesis that they provide a starting point for the evolution of new functions (73). Identifying the determinants of protein quaternary structure is another potential focus for historical analysis. Higher-order complexes are ubiquitous in biology (33), and the mechanisms by which variation in quaternary structure evolves have attracted intense theoretical interest (4, 49, 53, 54). ASR will make it possible to explicitly test these predictions.
Increasing computer power and falling costs for gene sequencing and synthesis have overcome what were once practical barriers to ASR, but the strategy still cannot be applied to all proteins. Many proteins lack sufficient phylogenetic signal—because they are too divergent or difficult to align in parts of their sequence—for ancestral sequences to be inferred with confidence, even with optimal sampling. For example, intrinsically disordered proteins usually evolve so rapidly that ASR cannot be used (14, 15). Even when alignment is possible, there may be insufficient information to reconstruct ancestral proteins accurately or with confidence. For example, entire lineages might have gone extinct, and if information about their sequences and functions were included in a phylogenetic analysis, different ancestral states might be reconstructed, or the same states reconstructed with different estimates of statistical confidence.
Some limitations of ASR may be overcome with future technical improvements, such as the development of better models. In membrane-associated proteins, for example, the soluble and membrane-spanning portions can undergo very different substitution processes (43), but most current ASR models assume identical models across sites within a protein. Further advances in the application of structurally informed and other kinds of heterogeneous models—especially family-specific models—will be necessary before these kinds of ancestral proteins can be reconstructed with confidence.
Some systematic limitations of ASR are unlikely to be overcome in the near future. Ancestral proteins are usually characterized in vitro or in cell culture. These assays almost certainly do not fully represent the range or subtlety of functions that proteins had in their ancestral host organisms, which might have required particular interaction partners or posttranslational modifications. Testing the function of ancestral proteins in vivo in model organisms should become a more important part of ASR in the future (67). But even this approach may be inadequate to understand the effects that some ancestral proteins had in their ancestral biological context. ASR should therefore be used to ask questions about intrinsic functions and activities of proteins that can be assessed using assays that are relatively insensitive to differences between the extant assay system and ancestral cells or organisms. For example, studies of X-ray crystal structures, or of an ancestral protein’s affinity for or catalysis of conserved molecules should be largely robust to this concern, whereas studies of a protein’s effect on whole-organism physiology or development are much harder to assess using reductionist experiments.
A final issue is the accessibility of expertise to carry out ASR in a rigorous fashion. Automated web servers to reconstruct ancestral sequences are now available (35), but a solid understanding of the capacities and pitfalls of statistical phylogenetics remains important to accurately reconstruct ancestral proteins. In particular, users of ASR should resist the dual temptations to uncritically accept the maximum likelihood reconstruction as true per se or to dismiss any surprising inference as the result of statistical uncertainty or phylogenetic bias; rather, the extent to which ancestral proteins’ observed properties are sensitive to various sources of uncertainty and bias should be examined critically and experimentally, whenever possible. Not many biochemists have this expertise, so for now collaborations or disciplined self-study are required. We hope that education and training in molecular evolution and phylogenetics become more accessible to students in biochemistry and that protein biochemistry becomes a routine part of training in molecular evolution.
In our view, the potential of ASR is considerably greater than the strategy’s limitations. Technical barriers to obtaining protein structures are shrinking, creating countless opportunities to dissect the connection between sequence, structure, and function. But for the reasons we have detailed here, the accessibility of protein structures—even complemented by rich functional information— will not be sufficient to explain how and why those structures exist in their particular forms. Ancient proteins contain rich information about the sequence–structure–function relationship, and the capacity to genetically manipulate those proteins provides a focused means to test hypotheses about the causal links between levels of biochemical phenomena. Extending Crick’s directive, we therefore suggest that if you want to understand proteins, you should study their evolution.
SUMMARY POINTS.
By identifying the historical sequence changes that changed structure and function during evolution, ancestral protein reconstruction can provide strong causal explanations for the functional diversity of present-day proteins.
Reconstructing a protein family’s evolutionary history is an efficient way to identify the genetic and structural causes of functional diversity, because it enables a focused identification of causal sequence and structural features and minimizes the confounding effects of epistasis.
Characterizing the ancestral state from which related proteins with different functions evolved clarifies whether the causal sequence and structural features confer new functions or abolish ancestral activities, thereby leading to the discovery of novel, unexpected aspects of structural mechanisms.
Ancestral protein reconstruction has shown that new functions are often conferred by small numbers of sequence substitutions and relatively simple biochemical mechanisms, but this simplicity often remains obscure when extant proteins are compared to each other.
When related proteins have distinct functions, their specificities are often conferred not by the gain of new favorable interactions but by the gain and loss of negative, exclusionary interactions.
Dissecting the historical trajectory of functional or structural change can explain features of structure and mechanism that were caused by historical constraint and chance rather than functional optimization. Complex aspects of proteins, normally thought to be functionally important, are often the result of evolutionary tinkering with and subtle degradation of ancestral forms.
ACKNOWLEDGMENTS
We thank the members of the Thornton group for helpful discussions and critical reading of the manuscript. G.K.A.H. was supported by a Chicago Fellowship. J.W.T. was supported by NIH grant R01GM104397.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–5 [DOI] [PubMed] [Google Scholar]
- 2.Agafonov RV, Wilson C, Otten R, Buosi V, Kern D. 2014. Energetic dissection of Gleevec’s selectivity toward human tyrosine kinases. Nat. Struct. Mol. Biol 21:848–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aharoni A, Gaidukov L, Khersonsky O, McQ Gould S, Roodveldt C, Tawfik DS. 2005. The ‘evolvability’ of promiscuous protein functions. Nat. Genet 37:73–76 [DOI] [PubMed] [Google Scholar]
- 4.Ahnert SE, Marsh JA, Hernandez H, Robinson CV, Teichmann SA. 2015. Principles of assembly reveal a periodic table of protein complexes. Science 350:aaa2245. [DOI] [PubMed] [Google Scholar]
- 5.Anderson DP, Whitney DS, Hanson-Smith V, Woznica A, Campodonico-Burnett W, et al. 2016. Evolution of an ancient protein function involved in organized multicellularity in animals. eLife 5:e10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson DW, McKeown AN, Thornton JW. 2015. Intermolecular epistasis shaped the function and evolution of an ancient transcription factor and its DNA binding sites. eLife 4:e07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker CR, Hanson-Smith V, Johnson AD. 2013. Following gene duplication, paralog interference constrains transcriptional circuit evolution. Science 342:104–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baldwin AJ, Kay LE. 2009. NMR spectroscopy brings invisible protein states into focus. Nat. Chem. Biol 5:8–14 [DOI] [PubMed] [Google Scholar]
- 9.Bloom JD, Gong LI, Baltimore D. 2010. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 328:1272–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bloom JD, Romero PA, Lu Z, Arnold FH. 2007. Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biol. Direct 2:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boucher JI, Jacobowitz JR, Beckett BC, Classen S, Theobald DL. 2014. An atomic-resolution view of neofunctionalization in the evolution of apicomplexan lactate dehydrogenases. eLife 3:e02304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bridgham JT, Carroll SM, Thornton JW. 2006. Evolution of hormone-receptor complexity by molecular exploitation. Science 312:97–101 [DOI] [PubMed] [Google Scholar]
- 13.Bridgham JT, Ortlund EA, Thornton JW. 2009. An epistatic ratchet constrains the direction of glucocorticoid receptor evolution. Nature 461:515–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown CJ, Johnson AK, Daughdril GW. 2010. Comparing models of evolution for ordered and disordered proteins. Mol. Biol. Evol 27:609–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown CJ, Takayama S, Campen AM, Vise P, Marshall TW et al. 2002. Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol 55:104–10 [DOI] [PubMed] [Google Scholar]
- 16.Clifton BE, Jackson CJ. 2016. Ancestral protein reconstruction yields insights into adaptive evolution of binding specificity in solute-binding proteins. Cell Chem. Biol 2:236–45An ancestral amino acid binding protein was promiscuous because of large-scale active-site conformational plasticity.
- 17.Crick F 1988. What Mad Pursuit: A Personal View of Scientific Discovery. New York: Basic Books [Google Scholar]
- 18.Cunningham BC, Jhurani P, Ng P, Wells JA. 1989. Receptor and antibody epitopes in human growth hormone identified by homolog-scanning mutagenesis. Science 243:1330–36 [DOI] [PubMed] [Google Scholar]
- 19.Danielsen M, Hinck L, Ringold GM. 1989. Two amino acids within the knuckle of the first zinc finger specify DNA response element activation by the glucocorticoid receptor. Cell 57:1131–38 [DOI] [PubMed] [Google Scholar]
- 20.Dawkins R 1986. The Blind Watchmaker. New York: Norton [Google Scholar]
- 21.Devamani T, Rauwerdink AM, Lunzer M, Jones BJ. 2016. Catalytic promiscuity of ancestral esterases and hydroxynitrile lyases. J. Am. Chem. Soc 138:1046–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, et al. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280:69–77 [DOI] [PubMed] [Google Scholar]
- 23.Eick GN, Bridgham JT, Anderson DP, Harms MJ, Thornton JW. 2017. Robustness of reconstructed ancestral protein functions to statistical uncertainty. Mol. Biol. Evol 34(2):247–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ekici OD, Paetzel M, Dalbey RE. 2008. Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration. Protein Sci 17:2023–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elleuche S, Fodor K, von der Heyde A, Klippel B, Wilmanns M, Antranikian G. 2014. Group III alcohol dehydrogenase from Pectobacterium atrosepticum: insights into enzymatic activity and organization of the metal ion-containing region. Appl. Microbiol. Biotechnol 98:4041–51 [DOI] [PubMed] [Google Scholar]
- 26.Felsenstein J 2004. Inferring Phylogenies. Sunderland, Mass.: Sinauer Assoc. [Google Scholar]
- 27.Field SF, Bulina MY, Kelmanson IV, Bielawski JP, Matz MV. 2006. Adaptive evolution of multicolored fluorescent proteins in reef-building corals. J. Mol. Evol 62:332–39 [DOI] [PubMed] [Google Scholar]
- 28.Field SF, Matz MV. 2010. Retracing evolution of red fluorescence in GFP-like proteins from Faviina corals. Mol. Biol. Evol 27:225–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Finnigan GC, Hanson-Smith V, Stevens TH, Thornton JW. 2012. Evolution of increased complexity in a molecular machine. Nature 481:360–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaucher EA, Thomson JM, Burgan MF, Benner SA. 2003. Inferring the palaeoenvironment of ancient bacteria on the basis of resurrected proteins. Nature 425:285–88This study and Reference 72 were the first to use modern phylogenetic methods to reconstruct and characterize very ancient proteins.
- 31.Gerlt JA, Babbitt PC. 2009. Enzyme (re)design: lessons from natural evolution and computation. Curr. Opin. Chem. Biol 13:10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goldstein RA. 2011. The evolution and evolutionary consequences of marginal thermostability in proteins. Proteins 79:1396–407 [DOI] [PubMed] [Google Scholar]
- 33.Goodsell DS, Olson AJ. 2000. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct 29:105–53 [DOI] [PubMed] [Google Scholar]
- 34.Gould SJ, Lewontin RC. 1979. The spandrels of San Marco and the Panglossian paradigm: a critique of the adaptationist programme. Proc. R. Soc. Lond. B 205:581–98 [DOI] [PubMed] [Google Scholar]
- 35.Hanson-Smith V, Johnson A. 2016. PhyloBot: a web portal for automated phylogenetics, ancestral sequence reconstruction, and exploration of mutational trajectories. PLOS Comput. Biol 12:e1004976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harms MJ, Eick GN, Goswami D, Colucci JK, Griffin PR, et al. 2013. Biophysical mechanisms for large-effect mutations in the evolution of steroid hormone receptors. PNAS 110:11475–80Used ancestral sequence reconstruction to trace apparently neutral fluctuations in the biophysical causes of protein stability during ancient evolution of RNaseHs.
- 37.Harms MJ, Thornton JW. 2013. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat. Rev. Genet 14:559–71First study using ancestral sequence reconstruction to show how evolution of a protein’s conformational ensemble conferred a new function.
- 38.Harms MJ, Thornton JW. 2014. Historical contingency and its biophysical basis in glucocorticoid receptor evolution. Nature 512:203–7First mutational scan of a reconstructed protein found strong historical contingency in functional evolution.
- 39.Hart KM, Harms MJ, Schmidt BH, Elya C, Thornton JW, Marqusee S. 2014. Thermodynamic system drift in protein evolution. PLOS Biol 12:e1001994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hedstrom L, Farr-Jones S, Kettner CA, Rutter WJ. 1994. Converting trypsin to chymotrypsin: ground-state binding does not determine substrate specificity. Biochemistry 33:8764–69 [DOI] [PubMed] [Google Scholar]
- 41.Hung S- H, Hedstrom L. 1998. Converting trypsin to elastase: Substitution of the S1 site and adjacent loops reconstitutes esterase specificity but not amidase activity. Protein Eng 11:669–73 [DOI] [PubMed] [Google Scholar]
- 42.Jacob F 1977. Evolution and tinkering. Science 196:1161–66 [DOI] [PubMed] [Google Scholar]
- 43.Jones DT, Taylor WR, Thornton JM. 1994. A mutation data matrix for transmembrane proteins. FEBS Lett 339:269–75 [DOI] [PubMed] [Google Scholar]
- 44.Khersonsky O, Tawfik DS. 2010. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem 79:471–505 [DOI] [PubMed] [Google Scholar]
- 45.Kim H, Grunkemeyer TJ, Modi C, Chen L, Fromme R, et al. 2013. Acid-base catalysis and crystal structures of a least evolved ancestral GFP-like protein undergoing green-to-red photoconversion. Biochemistry 52:8048–59 [DOI] [PubMed] [Google Scholar]
- 46.Kim H, Zou T, Modi C, Dorner¨ K, Grunkemeyer TJ, et al. 2015. A hinge migration mechanism unlocks the evolution of green-to-red photoconversion in GFP-like proteins. Structure 23:34–43Last of an impressive series of studies that dissect the evolution of red fluorescent proteins.
- 47.Kimura M 1983. The Neutral Theory of Molecular Evolution. New York: Cambridge Univ. Press [Google Scholar]
- 48.Kobilka BK, Kobilka TS, Daniel K, Regan JW, Caron MG, Lefkowitz RJ. 1988. Chimeric alpha 2-,beta 2-adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science 240:1310–16 [DOI] [PubMed] [Google Scholar]
- 49.Levy ED, Erba EB, Robinson CV, Teichmann SA. 2008. Assembly reflects evolution of protein complexes. Nature 453:1262–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li Y, Suino K, Daugherty J, Xu HE. 2005. Structural and biochemical mechanisms for the specificity of hormone binding and coactivator assembly by mineralocorticoid receptor. Mol. Cell 19:367–80 [DOI] [PubMed] [Google Scholar]
- 51.Liljas A, Laurberg M. 2000. A wheel invented three times: the molecular structures of the three carbonic anhydrases. EMBO Rep 1:16–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lunzer M, Golding GB, Dean AM. 2010. Pervasive cryptic epistasis in molecular evolution. PLOS Genet 6:e1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lynch M 2012. The evolution of multimeric protein assemblages. Mol. Biol. Evol 29:1353–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lynch M 2013. Evolutionary diversification of the multimeric states of proteins. PNAS 110:E2821–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marsh JA, Teichmann SA. 2014. Protein flexibility facilitates quaternary structure assembly and evolution. PLOS Biol 12:e1001870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marsh JA, Teichmann SA. 2015. Structure, dynamics, assembly, and evolution of protein complexes. Annu. Rev. Biochem 84:551–75 [DOI] [PubMed] [Google Scholar]
- 57.McKeown AN, Bridgham JT, Anderson DW, Murphy MN, Ortlund EA, Thornton JW. 2014. Evolution of DNA specificity in a transcription factor family produced a new gene regulatory module. Cell 159:58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, et al. 2003. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112:859–71 [DOI] [PubMed] [Google Scholar]
- 59.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. 2007. Crystal structure of an ancient protein: evolution by conformational epistasis. Science 317:1544–48First X-ray crystallographic structure of a resurrected protein revealed mechanisms for evolution of a new function.
- 60.Pauling L, Zuckerkandl E. 1963. Chemical paleogenetics: Molecular “restoration studies” of extinct forms of life. Acta. Chem. Scand 17:S9–16 [Google Scholar]
- 61.Perica T, Chothia C, Teichmann SA. 2012. Evolution of oligomeric state through geometric coupling of protein interfaces. PNAS 109:8127–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perica T, Kondo Y, Tiwari SP, McLaughlin SH, Kemplen KR, et al. 2014. Evolution of oligomeric state through allosteric pathways that mimic ligand binding. Science 346:1254346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perona JJ, Craik CS. 1997. Evolutionary divergence of substrate specificity within the chymotrypsin-like serine protease fold. J. Biol. Chem 272:29987–90 [DOI] [PubMed] [Google Scholar]
- 64.Perutz M 1983. Species adaptation in a protein molecule. Mol. Biol. Evol 1:1–28 [DOI] [PubMed] [Google Scholar]
- 65.Schwabe JWR, Chapman L, Finch JT, Rhodes D. 1993. The crystal structure of the estrogen receptor DNA-binding domain bound to DNA: how receptors discriminate between their response elements. Cell 75:567–78 [DOI] [PubMed] [Google Scholar]
- 66.Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. 2007. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure 15:299–311 [DOI] [PubMed] [Google Scholar]
- 67.Siddiq MA, Loehlin DW, Montooth KL, Thornton JW. 2017. Experimental test and refutation of a classic case of molecular adaptation in Drosophila melanogaster. Nature Ecol. Evol 1:0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stackhouse J, Presnell SR, McGeehan GM, Nambiar KP, Benner SA. 1990. The ribonuclease from an extinct bovid ruminant. FEBS Lett 262:104–6First study to resurrect and characterize an ancestral protein—in this case, a recent ribonuclease.
- 69.Starr TN, Thornton JW. 2016. Epistasis in protein evolution. Protein Sci. 25:1204–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thorne JL, Goldman N, Jones DT. 1996. Combining protein evolution and secondary structure. Mol. Biol. Evol 13:666–73 [DOI] [PubMed] [Google Scholar]
- 71.Thornton JW. 2004. Resurrecting ancient genes: experimental analysis of extinct molecules. Nat. Rev. Genet. 5:366–75 [DOI] [PubMed] [Google Scholar]
- 72.Thornton JW, Need E, Crews D. 2003. Resurrecting the ancestral steroid receptor: ancient origin of estrogen signaling. Science 301:1714–17 [DOI] [PubMed] [Google Scholar]
- 73.Tokuriki N, Tawfik DS. 2009. Protein dynamism and evolvability. Science 324:203–7 [DOI] [PubMed] [Google Scholar]
- 74.Ugalde JA, Chang BS, Matz MV. 2004. Evolution of coral pigments recreated. Science 305:1433. [DOI] [PubMed] [Google Scholar]
- 75.Venekei I, Szilagyi L, Graf L, Rutter WJ. 1996. Attempts to convert chymotrypsin to trypsin. FEBS Lett 379:143–47 [PubMed] [Google Scholar]
- 76.Voordeckers K, Brown CA, Vanneste K, van der Zande E, Voet A, et al. 2012. Reconstruction of ancestral metabolic enzymes reveals molecular mechanisms underlying evolutionary innovation through gene duplication. PLOS Biol 10:e1001446.Specific α-glucosidases evolved from a promiscuous ancestor through novel contacts that also exclude other substrates.
- 77.Wheeler LC, Lim SA, Marqusee S, Harms MJ. 2016. The thermostability and specificity of ancient proteins. Curr. Opin. Struct. Biol 38:37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wilson C, Agafonov RV, Hoemberger M, Kutter S, Zorba A, et al. 2015. Using ancient protein kinases to unravel a modern cancer drug’s mechanism. Science 347:882–86First study to use ancestral sequence reconstruction for understanding the genetic and structural basis of protein dynamics.
- 79.Wouters MA, Liu K, Riek P, Husain A. 2003. A despecialization step underlying evolution of a family of serine proteases. Mol. Cell 12:343–54 [DOI] [PubMed] [Google Scholar]
- 80.Yang Z 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol 39:306–14 [DOI] [PubMed] [Google Scholar]
- 81.Yang Z 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol 24:1586–91 [DOI] [PubMed] [Google Scholar]
- 82.Yang Z, Kumar S, Nei M. 1995. A new method of inference of ancestral nucleotide and amino acid sequences. Genetics 141:1641–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zuckerkandl E, Pauling L. 1965. Evolutionary divergence and convergence in proteins. Evol. Gen. Prot 97:97–166 [Google Scholar]