

Abstract
Background:
Boys with X-linked ectodermal dysplasia and immunodeficiency caused by mutations of nuclear factor-кB essential modulator have defects in innate and adaptive immunity, and some have colitis.
Objective:
We sought to determine whether curing the immune defect in such patients by means of allogeneic hematopoietic stem cell transplantation abolishes the susceptibility to colitis. Methods: A boy with X-linked hypohydrotic ectodermal dysplasia with immunodeficiency underwent allogeneic transplantation from a matched unaffected sibling identified by means of preimplantation genetic diagnosis. Toll-like receptor (TLR) function was assessed by measuring TLR agonist-induced cytokine production in whole blood tested in vitro. B-cell proliferation was measured by means of tritiated thymidine incorporation. Natural killer cell function was examined in PBMCs by means of K562 target cell lysis. Colitis severity was assessed clinically based on corticosteroid requirement and histology of large intestinal biopsy specimens.
Results:
Defects in cytokine production in response to TLR agonists, CD40-mediated proliferation, and natural killer cell cytotoxicity were all corrected after hematopoietic stem cell transplantation. Despite successful hematopoietic and immunereconstitution, the patient continued to have flares of colitis, often associated with bacterial infection.
Conclusions:
Our findings strongly suggest that nuclear factor-кB essential modulator deficiency intrinsic to the intestinal epithelium is sufficient to predispose to colitis, despite robust correction of immune defects. (J Allergy Clin Immunol 2008;122:1113–8.)
Keywords: Nuclear factor-кB essential modulator, Toll-like receptors, transplantation, colitis
X-linked hypohydrotic ectodermal dysplasia with immunodeficiency (X-ED-ID) caused by hypomorphic mutations in the gene enrolling nuclear factor-кB modulator (NEMO, IKBKG) and defective nuclear factor-кB (NF-кB) signaling is a rare but increasingly recognized disorder, with several dozen cases reported in the literature.1 Defects in the recognition of microbial components through Toll-like receptors (TLRs) and in natural killer (NK) cell cytotoxicity might be apparent before the onset of classical manifestations of ectodermal dysplasia. Boys with severe IKBKG mutations often die of overwhelming bacterial, viral, or mycobacterial infection.
Some patients with IKBKG mutations have colitis with histologic features similar to those of idiopathic inflammatory bowel disease. Colitis seen in patients with severe combined immunodeficiency or Wiskott-Aldrich syndrome, whose defects are confined to hematopoietically derived cells, is generally cured by allogeneic hematopoietic stem cell transplantation (HSCT). In addition to ectodermal defects, because NEMO is ubiquitously expressed, patients with IKBKG mutations likely have intestinal epithelial dysfunction. Thus their colitis might not be amenable to treatment with HSCT. A single previous report of immune reconstitution after HSCT of a boy with X-ED-ID who did not have colitis demonstrated correction of TNF-a production by PBMCs in response to TLR4 ligation by LPS and of IFN-g production in response to IL-18.2 We describe a patient with X-ED-ID and IKBKG mutation whose immune deficiency was comprehensively corrected by means of HSCT from a matched sibling but whose susceptibility to colitis persisted.
METHODS
Case report
A previously reported male patient with X-ED-ID and a T458G mutation in exon 4 of IKBKG3,4 was treated for cytomegalovirus (CMV) viremia and colitis with ganciclovir in infancy, which cleared the infection. A few months later, he had intractable CMV-negative colitis, frequent bacteremias, and failure to thrive, which persisted despite treatment with azathioprine and sulfasalazine. Multiple colonic biopsies throughout his course revealed neutrophilic infiltrate, cryptabscesses, glandulardestruction, andcryptregeneration (Fig 1, A) but with no evidence of CMV by means of histology, immunohistochemis- try, or spin-enhanced culture. At age 2 years and 7 months, the patient was treated with corticosteroids and had dramatic improvement. However, bloody diarrhea and biopsy-proved colitis recurred when the corticosteroid dose was reduced, and he was continued on prednisolone (0.7 mg/kg/d) to control his symptoms (Fig 1, B). Valganciclovir was given to suppress CMV reactivation during corticosteroid treatment.
FIG 1.
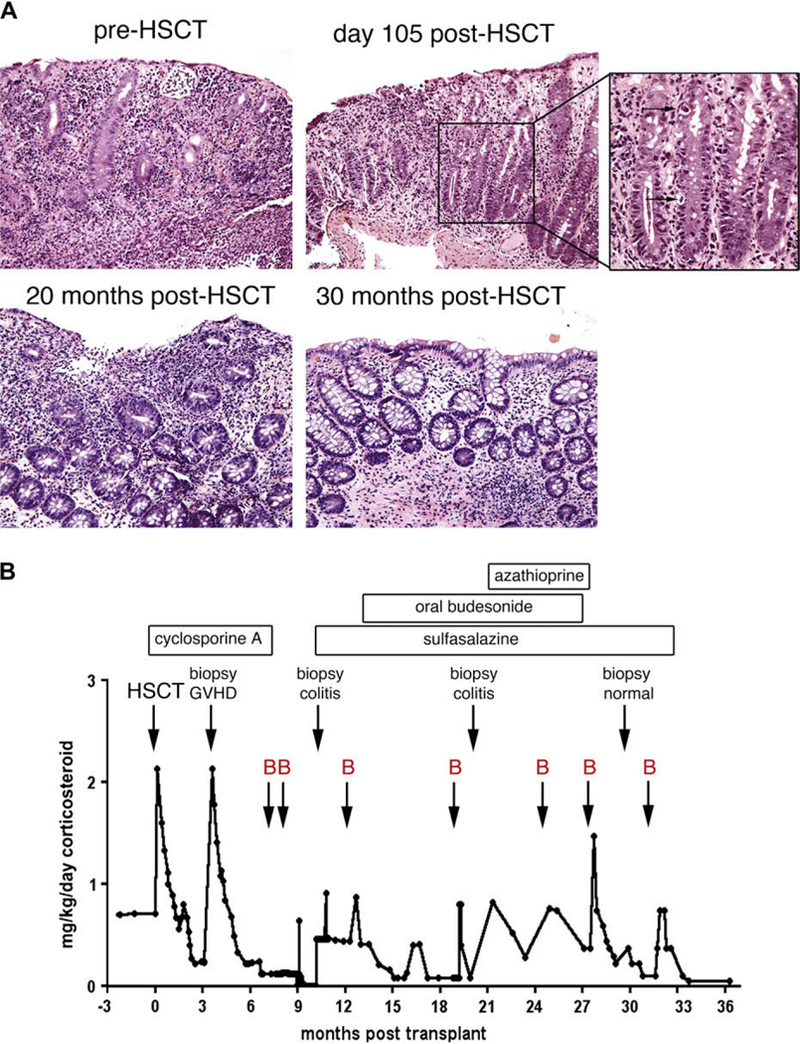
Clinical and histologic course of colitis after HSCT. A, Colonic biopsy specimens showing active colitis (before HSCT, 105 days after HSCT, and 20 months after HSCT), GVHD (105 days after HSCT; apoptosis is indicated by arrows in the inset), and resolution of both colitis and GVHD (30 months after HSCT). B, The dose of prednisolone equivalent is graphed as a function of time after HSCT with bacterial infections (arrows, B), relevant events, and treatments, as indicated.
In vitro fertilization with preimplantation genetic diagnosis (PGD) was performed to procure a fully matched sibling donor. Paternal sperm was enriched for X-chromosome carriers by using MicroSort technology,5 and single-cell PCR was used to identify 2 HLA-matched female embryos.6 A single female child, born when the patient was 4 years and 10 months old, was confirmed to be an HLA match and an IKBKG mutation carrier. The patient’s parents provided informed consent for the patient to undergo HSCT at age 5 years and 5 months.
Agents to treat colitis except corticosteroids were discontinued shortly before HSCT. Conditioning consisted of intravenous busulfan (every 6 hours ondays —9, —8, —7, and —6adjustedtoachievearea-under-the-curvedosing of 900 to1100 mmol-minutes) and cyclophosphamide (50 mg/kg body weight administered intravenously daily on days —4, —3, —2, and — 1). On day 0, the patient received sibling bone marrow containing 3.2 X 108 nucleated cells and 13.7 X 106 CD341 cells/kg of recipient body weight. He also received sibling cord blood previously cryopreserved by ViaCord (Cambridge, Mass) containing 4.3 X 107 nucleated cells and 1.4 X 105 CD341 cells/kg of recipient body weight. Intravenous cyclosporine A starting at day —2 and methylprednisolone (2 mg/kg/d) starting at day 15 were given for graft-versus-host disease (GVHD) prophylaxis, followed by slow taper (Fig 1, B). Because of asymptomatic CMV antigenemia detected by means of screening on admission, he was treated for 14 days before conditioning with intravenous ganciclovir (5 mg/kg) twice daily and intravenous foscarnet (60 mg/kg) three times daily until day 0. Foscarnet was continued twice daily during the pancytopenic period. He received cytomegalovirus immune globulin intravenous weekly.
Histology
Five-micrometer-thick sections of formalin-fixed, paraffin-embedded colonic mucosal biopsy specimens were stained with hematoxylin and eosin. Photomicrographs of representative images were obtained with an Olympus QColor5 digital camera and Olympus Qcapture software (Olympus, Center Valley, Pa).
Donor-recipient chimerism
For subset-specific analyses, anticoagulated blood was separated with Ficoll gradient. Granulocytes were positively selected from cell pellets by using magnetic beads directed against CD15 (Invitrogen, Carlsbad, Calif). T cells, B cells, and monocytes were positively selected from the buffy coat by using magnetic beads directed against CD3 (Stemcell Technologies, Vancouver, British Columbia, Canada), CD19 (Invitrogen), and CD14 (Miltenyi Biotec, Auburn, Calif), respectively. Immature dendritic cells were obtained by culturing CD14 monocytes in RPMI medium supplemented with 10% human serum, 300 U/mL GM-CSF, and 400 U/mL IL-4 for 3 days. DNA extracted from recipient cells was analyzed by means of short tandem repeat analysis, as previously described.7
Assessment of cellular function
All studies were performed with approval from the Committee for Clinical Investigation at Children’s Hospital Boston and with the informed consent of the patient’s parents. To measure responses to TLR agonists (InvivoGen, San Diego, Calif; except when indicated), whole citrated blood was treated with the diacylated macrophage-activating lipopeptide (TLR2/6; Alexis, San Diego, Calif), ultrapure LPS (TLR4; List Biologics, Carlsbad, Calif), Pam3Cys (TLR1/2), polyinosinic:polycytidylic acid (TLR3), imiquimod (TLR7), R-848 (TLR7/8), or oligodeoxynucleotide 2216 (ODN2216) (TLR9) with end-over-end rotation at 370C for 5 hours, and TNF-a levels in supernatants was measured by means of ELISA, as previously described.8 NK cell cytotoxicity and CD40-mediated proliferation of B cells was performed as previously described.3
RESULTS
Transplantation course
Neutrophil and platelet engraftment occurred on days +14 and +21, respectively. Donor chimerism of total white cells was 98.5% by means of fluorescent in situ hybridization for X and Y chromosomes (day 100) and 98% to 100% by means of short tandem repeat analysis of subsets, including CD3 T cells (8 months and 19 months), CD19 B cells (8 months and 19 months), CD15 granulocytes (19 months), CD14 monocytes (10 months), and in vitro differentiated immature dendritic cells (10 months).
Posttransplantation complications in general were mild. Symptomatic CMV reactivation occurred on day +57, and asymptomatic CMV antigenemia was detected on screening on day +79. Both were successfully treated with antiviral agents and cytomegalovirus immune globulin intravenous. While receiving low doses of steroids on day +105, the patient had bloody diarrhea. In contrast to previous biopsy specimens showing prominent neutrophilic infiltrate with microabscesses, colonic biopsy specimens at day +105 showed classic manifestations of GVHD,9 including crypt destruction, crypt regeneration, paucity of inflammation, and increased crypt apoptosis (Fig 1, A), with no evidence of CMV by means of histology, immunohistochemistry, or spin-enhanced culture and no CMV antigenemia. He was treated with corticosteroids (Fig 1, B), which were reduced to physiologic doses by day + 158. Cyclosporine was discontinued by day + 216. Intravenous immunoglobulin was discontinued 6 months after HSCT.
Correction of immunodeficiency after HSCT
Boys with X-ED-ID and IKBKG mutations have defects in responses of monocytes to LPS, NK cell cytotoxicity, and proliferation of B cells to CD40.3,10 We investigated the effect of allogeneic HSCT on these 3 functions of NEMO in our patient in comparison with adult and/or his sibling donor as control subjects. Because all TLRs in mice and in human subjects activate the NF-кB pathway, we tested the response of his blood cells to a comprehensive panel of TLR agonists. Before HSCT, stimulation of whole blood with agonists of TLR1/2 (Pam3Cys), TLR2/6 (macrophage-activating lipopeptide), TLR3 (polyinosinic:poly- cytidylic acid), TLR4 (LPS), TLR7 (imiquimod), TLR7 and TLR8 (R-848), and TLR9 (ODN2216) resulted in near absence of TNF-aproduction at multiple agonist concentrations. The responses were typically less than 1% of those of adult control cells assayed on the same day (Fig 2, A). At 4 to 8 months after HSCT, the response of the patient’s transplanted cells to TLR1/2, TLR2/ 6, and TLR9 was indistinguishable from that of normal adult control cells (Fig 2, B) or of the donor (Fig 2, C), whereas responses to TLR3, TLR4, TLR7, and TLR8 were greatly improved. Reconstitution of LPS-induced TNF-a production initially was the least complete (approximately 10% adult control) but was 10-fold to 100-fold improved relative to pre-HSCT evaluation. However, by 45 months after transplantation, the response to LPS had normalized (see Fig E1 in this article’s Online Repository at www.jacionline.org).
FIG 2.
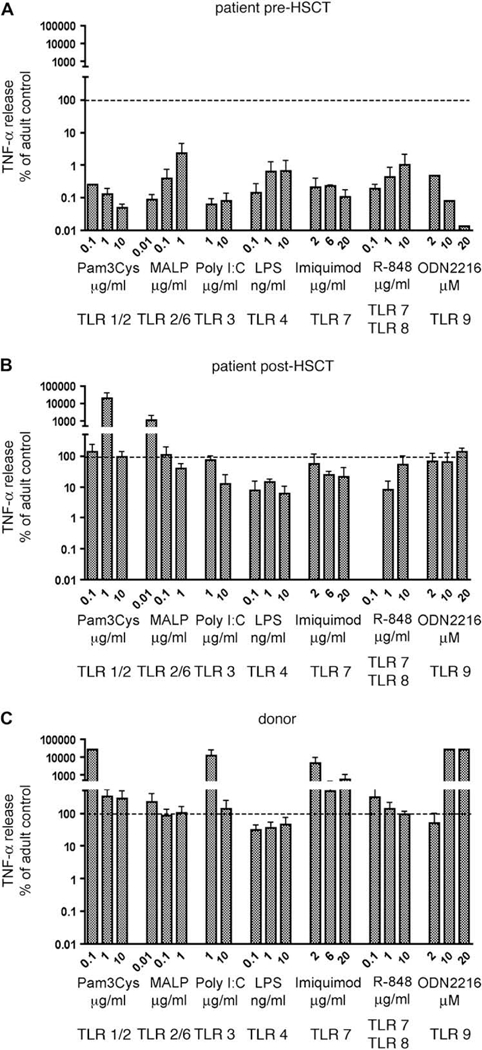
A-C, Cytokine production in responseto TLR ligands. TNF-a production is normalized relativeto adult control subjects assessed within each experiment. Data represent the mean plus SD of 2 to 3 independent assays performed at4to8 months after HSCT. MALP, Macrophage-activating lipopeptide; PolyI:C, polyinosinic:polycytidylic acid; ODN2216, oligodeoxynucleotide 2216.
FIG E1.
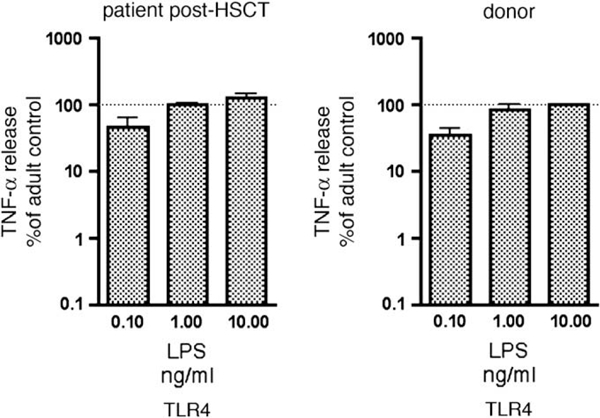
Cytokine production in response to TLR4 agonist. TNF production is normalized relative to adult control cells assessed within each experiment. Data represent mean plus SD of 2 independent assays performed at approximately 45 months after HSCT.
Before HSCT, the patient had a severe defect in NK cell cytotoxicity.3 In contrast, at 9 to 10 months after HSCT, his NK cell cytotoxicity was equivalent to that of the donor (Fig 3, A). Before HSCT, B cells failed to proliferate in response to CD40 stimulation,3 but at 9 to 10 months after HSCT, proliferation in response to CD40 and IL-4 stimulation was reconstituted to the level of both his sibling donor and adult control subject (Fig 3, B). Thus these 3 critically important defects in immune function of patients with mutations of IKBKG were fully reconstituted or vastly improved after allogeneic HSCT.
FIG 3.
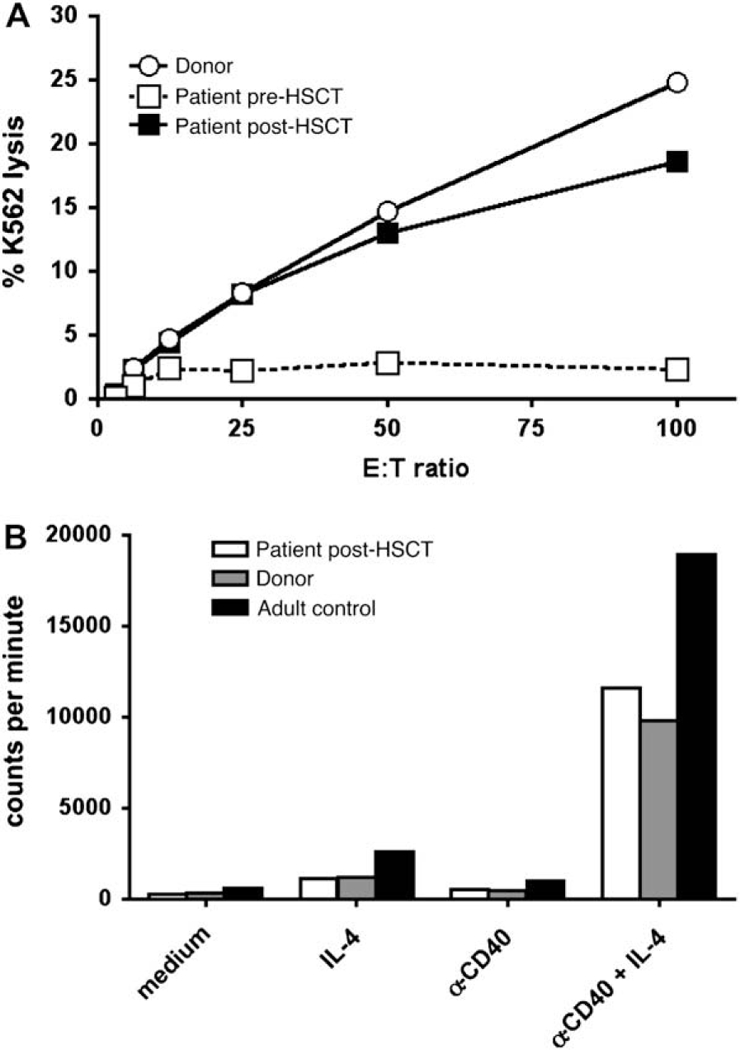
Restoration of B-cell and NK cell function after HSCT. A, NK cell cytotoxicity. B, Proliferation of PBMCs to anti-CD40 plus IL-4 measured by means of incorporation of tritiated thymidine. Data are representative of 2 experiments performed at 8 to 9 months after HSCT.
Reconstitution of lymphoid subsets followed the expected course after myeloablative transplantation (Table I). BeforeHSCT, T-cell proliferation to mitogens was normal, except to con- canavalin A,4 and were essentially normal when tested 16 months after HSCT (PHA: 86,607 vs 138,000 cpm for normal control cells; concanavalin A: 87,704 vs 99,500 cpm for normal control cells; and pokeweed mitogen: 30,547 vs 11,600 cpm for normal control cells). Because of intermittent corticosteroid therapy precluding an assessment of his antibody response to vaccines, immunizations after HSCT have not been completed.
TABLE I.
Lymphoid reconstitution after HSCT
| 3 mo | 15 mo | Normal range | |
|---|---|---|---|
| CD3 (cells/mm3) | 1198 | 3364 | 1000–2600 |
| CD4 (cells/mm3) | 508 | 1827 | 530–1500 |
| CD8 (cells/mm3) | 691 | 1530 | 330–1100 |
| CD19 (cells/mm3) | 28 | 1504 | 270–860 |
| CD16/CD56 (cells/mm3) | 141 | 98 | 70–480 |
| CD4/CD8 ratio | 0.73 | 1.19 | 0.9–3.70 |
Persistent susceptibility to colitis after HSCT
Before HSCT, the patient had symptoms of colitis when prednisolone was reduced to less than 0.7 mg/kg/d. In contrast, after HSCT, the patient was asymptomatic for weeks on as little as 0.15 mg/kg/d but had bloody diarrhea when he experienced bacterial infection (Fig 1, B). Infections included Clostridium difficile infection (7 months and 32 months), Sphingomonas pauci- mobilis-induced bacteremia requiring central line removal (8 months), Streptococcus pneumoniae-induced bacteremia (13 months), otitis media (19 months), cellulitis (25 months), suspected pneumonia with fever and hypoxia (27 months), and Escherichia coli-induced bacteremia (32 months). Biopsy specimens obtained at 11 and 20 months after transplantation showed chronic active colitis similar to that seen before HSCT and, importantly, did not show evidence of GVHD or CMV by means of histology, immunohistochemistry, or spin-enhanced culture (Fig 1, A, and data not shown). Colitis flares were treated with increased doses of corticosteroids, which were reduced once symptoms had abated (Fig 1, B). Flares continued despite treatment with alternate agents after HSCT that included sulfasalazine, oral budeso- nide, and azathioprine (Fig 1, B). At 30 months after HSCT, the patient was asymptomatic on physiologic replacement doses of corticosteroids and for the first time had no evidence of colitis on endoscopy (Fig 1, A). At 32 months after HSCT, he had recurrence of bloody diarrhea unresponsive to an increase in corticosteroids and was diagnosed with C difficile infection. Before initiation of treatment with nonabsorbable vancomycin, he had fever and E coli-induced bacteremia (Fig 1, B). Currently, 40 months after HSCT, the patient’s colitis is in remission, he is no longer receiving steroids, and he continues to receive nonabsorbable vancomycin. Thus bacterial infections of the respiratory tract, skin, and intestinal tract likely precipitated flares of clinical and histologic non-GVHD colitis in this patient after HSCT, requiring intermittent increases in corticosteroids, and recently resulted in bacteremia with an enteric organism. This strongly suggests that successful immune reconstitution could not compensate for NEMO deficiency in the intestinal epithelium, resulting in persistent susceptibility to infection with organisms likely entering through the gut
DISCUSSION
Boys with IKBKG mutation have immune defects that would be amenable to correction by means of HSCT. We show that defective responses of blood cells to a comprehensive panel of TLR agonists (Fig 2), virtually absent NK cell cytotoxicity (Fig 3, A), and low proliferation of B cells to CD40 stimulation (Fig 3, B) in a patient with a IKBKG mutation were all corrected by matched sibling allogeneic HSCT. Whether the relatively lower TLR4 responsiveness seen early after HSCT but that was found to be normal 3 years later is due to developmental differences between adults and young children,8 other modifier genes, or medication effect cannot be determined at this time.
A lack of NF-кB function could theoretically affect this patient’s ability to respond to genotoxic damage and tolerate cytotoxic conditioning. Agents such as topoisomerase inhibitors and doxorubicin are known to activate NF-кB, which in turn can prevent tumor death,11,12 raising the possibility that normal cells damaged by chemotherapy might rely on NF-кB for survival after exposure to cytotoxic agents. Recent evidence has shown that signaling pathways activated by means of DNA damage lead to activation of NEMO by means of phosphorylation and sumoylation, further strengthening the connection between genotoxicity and NEMO function.13,14 However, our patient did not have undue mucosal toxicity. Thus allogeneic HSCT corrected the innate immune defects in this patient with a IKBKG mutation without excess morbidity.
Despite replacement of the hematopoietic compartment and robust correction of innate and adaptive immune defects, our patient continued to have recurrent episodes of colitis requiring corticosteroid therapy. Although ultrasensitive testing for the presence of CMV by means of PCR analysis was not performed, the prompt response of the patient to steroids and the lack of progression of disease in the absence of antiviral therapy argue strongly against CMV-induced colitis as the cause. Furthermore, the histologic appearance and clinical course without other organ system involvement is inconsistent with GVHD.
The persistent susceptibility to colitis after successful immune reconstitution argues that factors extrinsic to hematopoietic cells were important in driving colitis in this patient. The normal innate immune system tolerates commensal bacteria, thereby protecting the intestinal epithelial barrier from cytokine-induced injury. Disruption of the epithelial barrier allows innate immune recognition of the indigenous microflora through TLR. The inflammation that ensues further compromises intestinal barrier function. This feed-forward cycle drives and sustains colitis.15,16 The critical importance of NEMO in intestinal epithelial barrier function has been recently demonstrated by using an intestinal epithelium-specific IKBKG-deficient mouse. These mice experience spontaneous severe colitis, despite a normal immune system, but colitis is abrogated when TLR signaling or TNF-a production is abolished.17 Our patient had debilitating steroid- dependent colitis complicated by bacteremia with gut-derived organisms before HSCT. After HSCT, he was intermittently asymptomatic on low doses of corticosteroids, but bacterial infection frequently triggered symptoms of colitis with bloody diarrhea (Fig 1, B). His development of E coli-induced bacteremia in the face of immune reconstitution as late as 32 months after transplantation implies ongoing susceptibility to breaches in the intestinal barrier, possibly because of an intrinsic inability of NEMO-deficient epithelial cells to maintain integrity. This suggests that defective NF-кB signaling in intestinal epithelium might be sufficient to drive colitis in the setting of human NEMO mutation, as in intestinal epithelium-specific IKBKG-deficient mice, and underscores the potential importance of NEMO in epithelial cells for maintenance of barrier function and protection against colitis in human subjects. Although we suspect that TNF-a elaboration plays a key role in barrier disruption, use of TNF-a blockade in this circumstance would be risky given the increased risk of serious infection.18 Reconstitution of gut-resident immune cells and their function could have lagged behind peripheral reconstitution and could have also contributed to continued colitis.
To our knowledge, this is the first report of the combined use of PGD and matched sibling HSCT to reconstitute innate and adaptive immunity in a patient with primary immunodeficiency. In this patient, for whom a fully matched, unrelated adult donor could not be identified, PGD allowed us to avoid additional toxicities associated with alternative donor-mismatched HSCT. Select patients who can be managed safely with supportive therapy during the time required for embryo generation and selection can clearly benefit from this approach.
Clinical implications:
We demonstrate that correction of primary immunodeficiency with a donor selected by means of PGD is safe and effective but might not abolish susceptibility to colitis in patients with X-ED-ID.
Acknowledgments
We thank the patient and his family and the doctors, nurses, and clinical staff who cared for the patient. We thank L. D. Notarangelo for critical review of the manuscript and K. Deering for technical assistance.
Supported by K08 AI50601 (S.-Y.P.), K08 AI050583 (O.L.), R01 AI 067353 (O.L.), USID-Net NIH N01AI22070 (J.S.O.), the Pennsylvania Department of Health (J.S.O.), R01 AI067946 (J.S.O.), R01 AI079731 (J.S.O.), and P01 AI035714 (R.S.G.).
Abbreviations used
- CMV
Cytomegalovirus
- GVHD
Graft-versus-host disease
- HSCT
Hematopoietic stem cell transplantation
- NEMO
Nuclear factor-кB essential modulator
- NF-кB
Nuclear factor-kB
- NK
Natural killer
- PGD
Preimplantation genetic diagnosis
- TLR
Toll-like receptor
- X-ED-ID
X-linked hypohydrotic ectodermal dysplasia with immunodeficiency
Footnotes
Disclosure of potential conflict of interest: S.-Y. Pai has received research support as a Principal Investigator from the Charles M. Hood Foundation and the National Institutes of Health. O. Levy has served as a consultant for XOMA, LLC, and Medimmune; has received research support from the National Institutes of Health, Dynavax, and the Dana Farber Cancer Institute; and has served as a legal consultant in a case regarding infectious diseases. J. N. Glickman has received research support from the National Institutes of Health (NIH). S. Nurko has received research grants from Sucampo Pharmaceuticals and TAP Pharmaceutical. R. S. Geha has received research support as a Principal Investigator from the NIH/National Institute of Allergy and Infectious Disease; the NIH/National Heart, Lung, and Blood Institute; and the March of Dimes. The rest of the authors have declared that they have no conflict of interest.
REFERENCES
- 1.Puel A, Picard C, Ku CL, Smahi A, Casanova JL. Inherited disorders of NF-kap-paB-mediated immunity in man. Curr Opin Immunol 2004;16:34–41. [DOI] [PubMed] [Google Scholar]
- 2.Tono C, Takahashi Y, Terui K, Sasaki S, Kamio T, Tandai S, et al. Correction of immunodeficiency associated with NEMO mutation by umbilical cord blood transplantation using a reduced-intensity conditioning regimen. Bone Marrow Transplant 2007;39:801–4. [DOI] [PubMed] [Google Scholar]
- 3.Orange JS, Brodeur SR, Jain A, Bonilla FA, Schneider LC, Kretschmer R, et al. Deficient natural killer cell cytotoxicity in patients with IKK-gamma/NEMO mutations. J Clin Invest 2002;109:1501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orange JS, Jain A, Ballas ZK, Schneider LC, Geha RS, Bonilla FA. The presentation and natural history of immunodeficiency caused by nuclear factor kappaB essential modulator mutation. J Allergy Clin Immunol 2004;113:725–33. [DOI] [PubMed] [Google Scholar]
- 5.Fugger EF, Black SH, Keyvanfar K, Schulman JD. Births of normal daughters after MicroSort sperm separation and intrauterine insemination, in-vitro fertilization, or intracytoplasmic sperm injection. Hum Reprod 1998;13:2367–70. [DOI] [PubMed] [Google Scholar]
- 6.Verlinsky Y, Rechitsky S, Sharapova T, Laziuk K, Barsky I, Verlinsky O, et al. Preimplantation diagnosis for immunodeficiencies. Reprod Biomed Online 2007;14: 214–23. [DOI] [PubMed] [Google Scholar]
- 7.Ballen KK, Spitzer TR, Yeap BY, McAfee S, Dey BR, Attar E, et al. Double unrelated reduced-intensity umbilical cord blood transplantation in adults. Biol Blood Marrow Transplant 2007;13:82–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy O, Zarember KA, Roy RM, Cywes C, Godowski PJ, Wessels MR. Selective impairment ofTLR-mediated innate immunity in human newborns: neonatal blood plasma reduces monocyte TNF-alpha induction by bacterial lipopeptides, lipopolysaccharide, and imiquimod, but preserves the response to R-848. J Immunol 2004;173:4627–34. [DOI] [PubMed] [Google Scholar]
- 9.Shulman HM, Kleiner D, Lee SJ, Morton T, Pavletic SZ, Farmer E, et al. Histopathologic diagnosis of chronic graft-versus-host disease: National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: II. Pathology Working Group Report. Biol Blood Marrow Transplant 2006;12:31–47. [DOI] [PubMed] [Google Scholar]
- 10.Doffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 2001;27:277–85. [DOI] [PubMed] [Google Scholar]
- 11.Wang CY, Cusack JC, Liu R, Baldwin AS. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kap-paB. Nat Med 1999;5:412–7. [DOI] [PubMed] [Google Scholar]
- 12.Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-kappaB response. Cell Death Differ 2006;13:773–84. [DOI] [PubMed] [Google Scholar]
- 13.Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell 2005;123:1079–92. [DOI] [PubMed] [Google Scholar]
- 14.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 2006;311:1141–6. [DOI] [PubMed] [Google Scholar]
- 15.Fritz JH, Le Bourhis L, Magalhaes JG, Philpott DJ. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends Immunol 2008;29:41–9. [DOI] [PubMed] [Google Scholar]
- 16.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007;448:427–34. [DOI] [PubMed] [Google Scholar]
- 17.Nenci A, Becker C, Wullaert A, Gareus R, van Loo G, Danese S, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007;446: 557–61. [DOI] [PubMed] [Google Scholar]
- 18.Bongartz T, Sutton AJ, Sweeting MJ, Buchan I, Matteson EL, Montori V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006;295:2275–85. [DOI] [PubMed] [Google Scholar]